
Abstract
Cellulose cannot melt and is not soluble in common organic solvents. The first part of this chapter is a review of the main aspects of cellulose dissolution. Research results obtained in several EPNOE laboratories are then described. This includes the mechanisms of the dissolution of native cellulose fibres, solution properties in sodium hydroxide water and ionic liquids, stabilisation of cellulose in N-methylmorpholine-N-oxide–water and mixtures of cellulose with other polysaccharide or lignin in solution.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 Introduction
Cellulose is a linear, semi-flexible polymer (see Chap. 2) which arranges itself when condensed into crystalline and noncrystalline phases. This is the case when cellulose is biosynthesised in plants or other organisms or when it is regenerated from a solution. As such, it is following the general rules that are applicable to long-chain molecules. Among these rules are the facts that noncrystalline phase (so-called amorphous phase) has different degrees of order and organisation, that long chains are more difficult to be dissolved than short ones for thermodynamic reasons and that they can entangle at high enough molar mass and concentration.
Since cellulose cannot melt, dissolution is a major issue. Many reviews have been devoted to cellulose dissolution (e.g. Warwicker et al. 1966; Liebert 2010). Cellulose solutions are used for processing directly cellulose in the form of fibres, films, membranes or other not too bulky objects like sponges or for performing chemical derivatisation (see Chap. 9). Since cellulose chains have no specific features, dissolving cellulose should then happen as it is occurring for any other flexible or semi-flexible polymer, and solutions should behave as normal polymer solutions. This is indeed almost the case with one major difference from most synthetic polymers: Cellulose is “synthesised” by nature in a complex environment where many other compounds (lignin, hemicellulose, fats, proteins, pectins, etc.) are present and interacting more or less strongly with cellulose chains. In addition and due to the biosynthesis mechanisms, the organisation of chains is usually complex, like in the secondary plant cell walls where cellulose chains are forming a sort of self-composite with many differently oriented layers. Due to the importance of the field of polymer dissolution in materials engineering where dissolution is used in many industrial areas (drug delivery, pulp and paper, membranes, recycling, etc.), there is a good knowledge of the mechanisms at stake when a solid polymer is placed in contact with a solvent (Miller-Chou and Koenig 2003). As a general basis, polymer chains will go into solution through the interface between the solid polymer and the solvent and will pass several phases as shown on Fig. 5.1.

Basic mechanisms for a solid polymer part situated at the interface between polymer and solvent
When the solid phase is placed in contact with the solvent (Fig. 5.1a), the solvent is swelling the solid phase at the interface which goes above Tg (Fig. 5.1b), and this swelling is increasing up to the point of disentanglement (Fig. 5.1c). Chains can then move out of the swollen phase to the solvent phase (Fig. 5.1d) and the solubilisation front can advance inside the solid material (Fig. 5.1e). Such a scheme is indeed what is occurring when a regenerated cellulose fibre is placed in a solvent (Chaudemanche and Navard 2011). In this case, the swelling and dissolution mechanisms of dry, never-dried and rewetted lyocell fibres prepared from solutions in N-methylmorpholine-N-oxide were investigated by redissolving these cellulose fibres in the same solvent varying the contents of water (from monohydrate to 24 % w/w). As for any synthetic polymer fibre, a radial dissolution starting from the outer layers was observed.
It is usually said that cellulose is difficult to be dissolved and that a rather limited number of solvents are available. However, this is the case for many polymers. Dissolution is favoured since the entropy will increase in a solution state through the contributions of several entropic factors like the entropy of mixing, the entropy of conformation mobility and, if applicable, the entropy gain due to counter ions. The major difficulty for dissolving a polymer is well known and is due to the very large decrease of entropy that is happening when dissolving of the polymer is compared to its parent monomer, because of its long-chain character. If considering, for example, very common polymers like polyethylene or polypropylene, the number of the possible solvents is not larger than the one for cellulose. Since these polymers are melting, it is also true that efforts placed in developing their solvents were much smaller than the efforts for finding solvents for cellulose. There are solvents for cellulose, but they are not the simple, common organic solvents we are used of for the vast majority of polymers. A recent series of papers (Glasser et al. 2012a, b) discussed these issues with questions around the reason why cellulose is not soluble in water, Lindman et al. 2010 arguing that the main reason is not the existence of hydrogen bonds but due to hydrophobic interactions
5.1.1 Derivatising Pathways
Dissolving cellulose has always been a search since cellulose was isolated in the nineteenth century. Many compounds have been tested and several commercial pathways have been implemented to produce regenerated cellulose. Most of them are in fact going through cellulose derivatives or complexes (Liebert 2010). Many compounds and methods are able to produce derivatives that are then chemically reverted to cellulose after processing. After 7 years of research, one of the first to be industrially implemented in 1890 was the use of nitrocellulose by Count Hilaire de Chardonnet. Another example is the Fortisan process, not any more in production, which used cellulose acetate–acetone solutions for producing cellulose acetate that were saponified in caustic soda to obtain cellulose fibres (Segal and Eggerton 1961). Cellulose carbamate, obtained by the reaction of cellulose with urea, is another example. The first to report this reaction were Hill and Jacobsen (1938). But it was only at the beginning of the 1960s that Sprague and Noether (1961) found that this reaction leads to a cellulose derivative they named cellulose carbamate. The potential for using this derivative for producing fibres was explored by the Finnish company Neste Oy. Cellulose is treated with urea to produce cellulose carbamate that is dissolved in NaOH–water. After processing this solution, cellulose is recovered after an acidic treatment. Despite its interest and further developments (Kunze and Finck 2005), there is no industrial use at the present time.
The only large scale production of cellulose products through a cellulose derivative is the viscose process made from cellulose xanthate, dating from first patent on the use of cellulose xanthate preparations and subsequent regeneration by Cross et al. (1892). Since that time, after some first economical and technical difficulties, the viscose process proved to be a very efficient and powerful method for producing fibres, films and sponges, still in use today despite its difficulty for controlling air and water pollution. The viscose process is based on the treatment of cellulose fibres from wood or cotton with sodium hydroxide and carbon disulphite, forming a cellulose derivative called cellulose xanthate which has the interesting property to be soluble in sodium hydroxide–water mixtures. The solution can then be shaped, followed by an acid or a thermal treatment that reverts the cellulose derivative back to cellulose, with a noticeable change of cellulose crystal structure (cellulose I to cellulose II transformation). Among the many key chemical reactions and physical processes that are characterising this process, the dissolution step of the cellulose xanthate fibres into the sodium hydroxide–water mixture is of great importance since it is controlling the quality of the subsequent processing. As can be imagined, there has been numerous scientific works studying, for example, the influence of cellulose origin, purity, molecular weight, xanthate group distribution (Russler et al. 2005, 2006) or dissolution conditions, temperature (Musatova et al. 1972), soda concentration, etc., on the efficiency of the process. The way cellulose xanthate dissolves in NaOH–water (LeMoigne and Navard 2010a) is a good illustration of the difficulty to go through a gel phase before entering into the solution phase. Cellulose xanthate with 61 % CS2 placed in NaOH 8 % water is producing a very viscous, gelly phase that will disperse only during shear through the extraction of cellulose xanthate viscous solution phase filaments as can be seen on Fig. 5.2. The dispersion and complete dissolution of this viscous phase can only be achieved by a strong flow.

Dissolution of cellulose xanthate in NaOH 8 %–water under flow (shear rate about 20 s−1, flow from right to left) observed with the contra-rotating rheometer at two successive dissolution times: (a) 150 s and (b) 190 s. Cellulose xanthate fibres are producing a highly viscous phase that has difficulties to be dispersed into the solvent. Dispersion and final dissolution happen through the production of a thin filament of viscous phase that is extracted by the solvent friction
5.1.1.1 Complexing Agents
Many aqueous-based compounds are complexing cellulose. They are not leading to a real solution, i.e. a dispersion of cellulose chains in a solvent. The first agent was discovered by Schweitzer in the middle of 1800s. He found that it is possible to prepare clear solutions when cellulose is mixed in a solution of copper salts and ammonia. This process called now cuprammonium is leading to good regenerated cellulose objects (fibres and membranes) but the cost of solvent and of depollution is very high. Today, the production is limited to a few tens of thousands tons per year. However, this class of compounds is very interesting in the sense that the cellulose complex can be easily studied. Two compounds of this class, cuoxen (Cu(NH2(CH2)2NH2)2[HO]2) and cadoxen (Cd(NH2(CH2)2NH2)2[HO]2), are still widely used to give an indication of the cellulose molar mass through the measurement of the intrinsic viscosity (these methods being normalised like in ASTM D1795 and ASTM D4243, for example). These metal complexes dissolve cellulose by deprotonating and coordinatively binding C2 and C3 hydroxyl groups (Liebert 2010). Many other inorganic salt hydrates combined or not with water have also been found to dissolve cellulose whilst complexing it. A list of aqueous salts is given in Liebert (2010). As an example (Hattori et al. 2004), ethylenediamine/thiocyanate salts have been shown to solubilise cellulose DP210 up to 16 %, leading to the formation of a mesophase for the highest concentrations.
5.1.2 Non-derivatising Compounds
There are not so many classes of compounds that are leading to more or less well-dispersed but non-derivatised cellulose solutions. Aside the fact that the increase of entropy for long chains going into solution is low, two other reasons are also at stake for cellulose. The first reason is chain rigidity. Degrees of freedom for undergoing chain conformational changes are very limited for a cellulose chain that can only turn around its 1,4 links. A lot of work was performed from 1950s till 1970s for estimating the mean conformation parameters. Benoît 1948 calculated the mean-square end-to-end distance of cellulose chains in the unperturbed state, showing that it is much larger than chains with cis or trans free rotations. A large number of papers were then devoted to the theoretical approaches needed to measure various conformation parameters of cellulose and cellulose derivatives by light and neutron scattering (Gupta et al. 1976), flow birefringence (Noordermeer et al. 1975) and viscometry (Holt et al. 1976). Aside debates on chain statistics theories, all data show that the cellulose backbone is semi-rigid. This can be appreciated in Table 5.1 where the persistence length of cellulose and cellulose derivatives is markedly larger than the one of polyolefins. Polymers having a rigid backbone have a further difficulty in going into solution since there is a further loss of conformational entropy increase compared to flexible polymer, when dissolved. A second reason is the large amount of intra- and inter-hydrogen bond connections present in cellulose, needed solvents able to break them and leading to a high tendency to self-aggregation in solution.
The last point of interest before detailing the properties of cellulose solutions is whether these mixtures of cellulose and a solvent give a true solution, i.e. if the state is a homogeneous dispersion of individual cellulose chains in the solvent. This is a critical issue with all cellulose solutions where nanoscale cellulose agglomerates (often called prehump, or micro- or nanogels) are often present in the solution. The method for ascertaining the presence of cellulose agglomerates in solution is light scattering (Seger et al. 1996). Many papers are showing that indeed, aggregates are present in what is usually considered as good, non-derivatising solutions. Drechsler et al. (2000) showed that cellulose is molecularly dispersed only in special mixtures of N-methylmorpholine-N-oxide (NMMO)/water with diethylenetriamine. The M w dependence of the radii of gyration follows a power law behaviour with an exponent equal to the theoretical renormalisation group value for flexible, linear chains in a good solvent (i.e. 0.6). But solutions in pure N-methylmorpholine-N-oxide/water are not showing this. Röder and Morgenstern (1999) plotted Guiner–Zimm plots that showed that aggregates of the order of several million g/mol were observed. Aggregates comprising several 100 chains were present together with small aggregates, giving a bimodal structure. Activation of cellulose, i.e. pretreatments like NaOH or ammonia, was effective for decreasing the size of aggregates, but did not change the fact that aggregates were present. The same applies to solutions in N,N-dimethylacetamide/lithium chloride where aggregates with sizes above 100 nm are present (Röder et al. 2000), even if these results must be taken with care regarding the very important effect of water on chain aggregation (Potthast et al. 2002). Cellulose aggregates with a radius of gyration of 230 nm were found in solutions of NaOH–urea (Chen et al. 2007). All these results show that attaining molecularly dispersed cellulose solutions is very difficult, probably owing to the ability of cellulose to build intermolecular hydrogen bonds in water-based solutions.
5.1.2.1 Phosphoric Acid-Based Solvents
The possibility to dissolve cellulose in phosphoric acid has been known from a long time, starting probably with a patent filled by British Celanese (1925). It is only in the 1980s that interest for this solvent emerged again (Turbak et al. 1980). More recently, Boerstel et al. (2001) and Northolt et al. (2001) showed that concentrated solutions are anisotropic at rest and that their spinning is giving high modulus (44 GPa) and high strength (1.7 GPa) cellulose fibres. Despite that this solvent is not used, it may have an industrial potential to give specialty products.
5.1.2.2 LiCl-Based Solvents
The possibility to dissolve cellulose in a non-aqueous mixture of N,N-dimethylacetamide (DMAc) and LiCl was first published in 1979 by Charles McCormick (1979). It is a widely used tool to analyse cellulose chains. For example, McCormick et al. (1985) measured cellulose chain dimensions and conformation parameters, showing that this solvent is not degrading cellulose chains and is able to dissolve up to 15 % of cellulose. These authors proposed that the dissolution mechanism involves hydrogen bonding of the hydroxyl protons of cellulose with the chloride ions. They showed that concentrated solutions have a lyotropic character, but this must be taken with care since this liquid crystalline order is obtained by shearing, and not at equilibrium. In another paper, Matsumoto et al. (2001) compared the solution behaviour of plant and bacterial cellulose solution rheology in this solvent. Strangely, they found that plant cellulose behaves as a flexible polymer, while bacterial cellulose is like a rod-like chain. In a similar work, Tamai et al. (2003) reported that X-ray scattering data showing that the solutions have the characteristic of a two-phase system could explain the rod-like character of bacterial cellulose if it is not well dissolved and stays in bundles. Ramos et al. (2005a) found that not all cellulose can be easily dissolved down to molecular scale in this class of solvent and that activation is sometimes needed, as for cotton linters that can only dissolve if previously mercerised. This solvent is also used for preparing cellulose derivatives, sometimes in various ammonium variants like dimethylsulfoxide/tetrabutylammonium fluoride (Ramos et al. 2005b). None of the solvents of this class are commercially exploited to produce cellulose products.
5.1.2.3 N-Methylmorpholine-N-Oxide/Water
In 1939, Graenacher and Salman applied for a patent (Graenacher and Sallman 1939) where they described the possibility to dissolve cellulose in amine oxides, with aliphatic and cycloaliphatic amine oxides giving 7–10 % solutions of cellulose at 50–90 °C. This discovery was not exploited until the end of the 1960s when a series of patents (Johnson 1969; Franks and Varga 1979; McCormick and Lichatowich 1979) disclosed that one member of this series of compounds, N-methylmorpholine-N-oxide (NMMO) mixed with water, is able to dissolve cellulose (see Chap. 5 for more details). The most recent phase diagram of the mixture of NMMO and water is given in Fig. 5.3.

NMMO–water phase diagram (adapted from Biganska O, Navard P (2003) Phase diagram of a cellulose solvent: N-methylmorpholine-N-oxide – water mixtures, Polymer 44: 1035-1039)
It is only within a rather limited range of composition and temperature that NMMO–water can dissolve cellulose. At too low water concentration, below 10 %, the required temperature for melting the solvent is very high and a very strong degradation of cellulose and solvent occurs, leading to very dangerous exothermic transitions. The instability of the solutions with cellulose has been studied in depth by the group of Rosenau (Rosenau et al. 2001). Above about 25 %, the mixture is not a solvent anymore (Cuissinat and Navard 2006a). Spinning cellulose in NMMO monohydrate is now a commercial process called lyocell by the Lenzing AG company in Austria or Alceru by Smart Fiber Company in Germany. Fibres are mainly used in the textile industry. Their main drawback is their strong tendency for fibrillation in the wet state (Fig. 5.4), thought to be due to their very high chain orientation (Ducos et al. 2006). More details can be found in Chap. 5 “Cellulose Products from Solutions: Film, Fibres and Aerogels”.

Fibrillated cellulose fibres prepared from NMMO–water solution [adapted from Ducos et al. (2006)]
Two other classes of solvent have been widely studied, ionic liquids and NaOH–water. They are described in details in next paragraphs.
5.1.2.4 Rheology of Solutions
Rheology of cellulose solutions has been investigated from a very long time due to its use for measuring molar masses and to assess the behaviour of the cellulose chain in solution. To this end, the measurement of only the viscosity of dilute solutions has been performed extensively. Generally, in the dilute regime, noncharged polymers present a Newtonian flow behaviour and absence of viscoelasticity. The viscosity of polymer solutions depends on the concentration and size of the dissolved macromolecules, as well as on the solvent quality and temperature, supplying information about chain dimensions in solution, molecular shape, polymer–polymer interactions, excluded volume effects governed by polymer–solvent interactions and chain stiffness.
Generally, the relationship between the intrinsic viscosity [η] and dilute solution viscosity η takes the form of a power series in concentration c:
where \( {k_1} \), \( {k_2} \), \( {k_3} \), etc., are dimensionless constants. In order to evaluate the intrinsic viscosity, the theoretical analysis of the hydrodynamics of macromolecules with different flexibilities led to different relationships (Lovell 1989). The most frequently used one is the Huggins equation:
\( {k_{\mathrm{H}}} \) is referred as the Huggins dimensionless constant and is correlated to the size and shape of polymer segments, as well as to hydrodynamic interactions between different segments of the same polymer chain. \( {k_{\mathrm{H}}} \) equals to 0.5 means that solvent is theta solvent (no excluded volume expansion, polymer coils like ideal chains and excess chemical potential of mixing between a polymer and a theta solvent being zero); below 0.5 the solvent is thermodynamically good and above the solvent is bad.
Equation (5.2) is an approximation of (5.1), and it is applicable only for \( [\eta ] \cdot c < 1 \). At higher concentrations, the experimental data show an upward curvature when plotted according to (5.2).
The Kraemer equation is an approximation of the Huggins equation, from which it may be derived assuming \( {\eta_{\mathrm{sp}}} < < 1 \):
Usually, the experimental data are plotted according to both Huggins and Kraemer equations, and the correct evaluation of \( [\eta ] \) is made by double extrapolation to infinite dilution. Theory predicts that \( {k_{\mathrm{H}}} + {k_{\mathrm{K}}} \cong 0.5 \) when the approximation is satisfactory valid. From a practical point of view, the determination of molar masses is performed in cadoxen, cuoxam or cupriethylenediamine according to norms (Kasaai 2002). The analysis of the viscosity is also giving indications on the rigidity and hydrodynamic behaviour of cellulose (Danilov et al. 1970; Kasaai 2002; Zhou et al. 2004).
Rheology of more concentrated solutions requires good solvents like N-methylmorpholine-N-oxide or ionic liquids (Blachot et al. 1998; Gericke et al. 2009b). It shows that cellulose is behaving as a normal semi-flexible polymer with a Newtonian region at low shear rate followed by a shear-thinning, non-linear region at high shear rates.
5.2 Macroscopic Mechanisms of the Dissolution of Native Cellulose Fibres
When placed in a swelling agent or a solvent, natural cellulose fibres show a nonhomogeneous swelling. The most spectacular effect of this nonhomogeneous swelling is the ballooning phenomenon where swelling takes place in some selected zones along the fibres (Fig. 5.5). This heterogeneous swelling has been observed and discussed long ago by Nägeli (1864), Pennetier (1883), Flemming and Thaysen (1919), Marsh (1941), Hock (1950) or Tripp and Rollins (1952). One explanation for this phenomenon is that the swelling of the cellulose present in the secondary wall is causing the primary wall to extend and burst. According to this view, the expanding swollen cellulose pushes its way through the tears in the primary wall, the latter rolls up in such a way as to form collars, rings or spirals which restrict the uniform expansion of the fibre and forms balloons as described by Ott et al. (1954). This explanation assumes that cellulose is in a swollen state in each of the balloons.

Gossypium hirsutum cotton fibre swollen by ballooning in N-methylmorpholine-N-oxide with 20 % of water w/w
Further studies of Chanzy et al. (1983), Cuissinat and Navard (2006a, b, 2008a) and Cuissinat et al. (2008b, c) have shown that the dissolution mechanism is strongly dependent on the solvent quality. Cuissinat and Navard performed observations by optical microscopy of free-floating fibres between two glass plates for a wide range of solvent quality (as an example, N-methylmorpholine-N-oxide (NMMO) with various amounts of water, w/w). They identified four main dissolution modes for wood and cotton fibres as a function of the quality of the solvent (the quality of the solvent decreases from mode 1 to mode 4):
Mode 1: Fast dissolution by fragmentation, occurring in good solvent (e.g. in NMMO with <17 % of water, 90 °C)
Mode 2: Swelling by ballooning and full dissolution, occurring in moderately good solvent (e.g. in NMMO with 19–24 % water, 90 °C)
Mode 3: Swelling by ballooning and no complete dissolution, occurring in bad solvent (e.g. in NMMO with 25–35 % water, 90 °C)
Mode 4: Low homogeneous swelling and no dissolution, occurring in very bad solvent (e.g. in NMMO with more than 35 % water, 90 °C)
These mechanisms also have been observed with NaOH–water with or without additives (Cuissinat and Navard 2006a), ionic liquids (Cuissinat and Navard 2006b) and for a wide range of plant fibres (Cuissinat and Navard 2008a) and some cellulose derivatives that had been prepared without dissolution (Cuissinat and Navard 2008, 2008b). From all these studies, it is shown that the key parameter in the dissolution mechanism is the morphology of the fibre. Indeed, as long as the original wall structure of the native fibre is preserved, the dissolution mechanisms are similar for wood, cotton, other plant fibres and some cellulose derivatives. Ballooning, often observed when cellulose native fibres are dissolving, originated from the specific cut followed by the rolling of the primary wall (LeMoigne et al. 2010a).
5.2.1 Gradient of Solubility in the Various Locations of the Cell Wall
In order to investigate in more depth the influence of the primary wall on the dissolution mechanism, cotton fibres with different maturity were studied (LeMoigne 2008). The cotton fruit is a capsule (commonly called cotton boll) composed of 4 or 5 carpels, each of them bearing about 10 seeds. Each cotton fibre is produced by the outgrowth of a single epidermal cell of the seed coat. A cotton fibre is a single cell, mainly made of cellulose microfibrils arranged in concentric walls. Fibre development can be divided in five main growth stages: initiation, elongation, transition, development and maturation (Figs. 5.6 and 5.7):


-
The initiation stage corresponds to the differentiation of epidermal cells into fibre cells and takes place around 2 or 3 days preanthesis.
-
The elongation stage corresponds to the synthesis of the primary wall and takes place between 1 and 15 days postanthesis (DPA)
-
The transition stage corresponds to the end of the primary wall synthesis and to the beginning of the secondary wall deposition (S1 wall) and takes place between 15 and 25 DPA. At this stage, secondary wall deposition is initiated while the cell is still elongating.
-
The development stage corresponds to the massive deposition (without elongation) of cellulose forming the main body of the secondary wall (S2 wall) and takes place between 25 and 50 DPA.
-
After about 50 days, the cotton bolls are mature, and it opens. After opening, the cotton fibres dry out. This stage is called maturation.
The dissolution of fibres taken at the elongation stage turned out to be impossible in both good and moderately good solvents. In NMMO with 16 and 20 % water, the reaction is very slow and leads to a uniform gel-like material with no measurable swelling. The elongation stage fibres dissolve only in the very good solvent like NMMO monohydrate (13.3 % of water). The reaction leads to a uniform gel-like material. A subsequent regeneration in water shows that this material was partially dissolved. These results show that the primary wall of cotton fibres is very resistant to dissolution in solvents that, as will be seen below, are dissolving the secondary wall. Another important observation is that the ballooning phenomenon is not present with these fibres. This is fully in agreement with the common explanation which attributes the balloons to the swelling of the secondary wall causing the extension and the bursting of the primary wall. Without secondary wall, there are no balloons (note that mature fibres without primary walls are also not showing balloons). At the transition stage, ballooning starts due to the presence of the secondary cellulose-based walls and a small number of balloons can be observed. These balloons are much smaller (swelling ratio around 200–300 %) than those observed for mature cotton fibres (swelling ratio around 450–600 %). At the mature stages, swelling and dissolution proceeds like described above.
These dissolution experiments on cotton fibres at different growth stages show that there is a gradient of dissolution capacity from the inside to the outside of the fibre. For fibres with enough cellulose inside the secondary wall, the inside of the fibre is the easiest part to dissolve. The dissolution mechanism is fragmentation where weak parts of the wall are quickly dissolved, leaving rod-like fragments floating, which completely dissolve later. Ballooning appears in fibres having a secondary wall, at least being at the transition stage. Balloons are formed by the expansion of the secondary wall due to the dissolution. When the secondary wall is swelling by ballooning, the primary wall breaks in localised places and rolls up to form helices and surrounds fibre sections that cannot be swollen. The primary wall does not dissolve easily and even sometimes does not dissolve at all as it is occurring in bad solvents like NaOH–water. The study of well-characterised cotton fibres in terms of growth stage has shown that most recent deposited cell wall layers (S2 wall) are most easily dissolved. The absence of non-cellulosic polysaccharide networks in younger wall layers may explain their higher dissolution capacity.
5.2.2 Overall Dissolution Mechanism
As was shown (LeMoigne 2008), the secondary S2 wall is the easiest to dissolve as compared to the external walls which contain larger amount of non-cellulosic components. The solvent goes inside the fibre through the primary wall which is permeable to the solvent but not easy to dissolve and not extensible. Optical observations show that the primary wall breaks (Fig. 5.8) in one or more places under the pressure of the S1 wall swelling and then rolls up. Finally, balloons are formed due to the large swelling of the S1 wall. The primary wall that is cut and rolled up forms threads (seen as thin lines along the balloon surface) and collars (regroupment of the rolled primary wall around the fibre diameter, preventing fibres to swell and form collars).

Left side: initiation of the breakage of the primary wall leading to its rolling. Right side: rolling of the two sides of the primary wall leading to a double helical thread surrounding the balloon
The selection between threads and collars is depending on the way the primary wall is broken, i.e. depends on the shape of the initial cut. When cut occurs on the whole circumference of the fibre, the primary wall rolls up along the fibre direction in the two opposite directions and forms only collars. If cut is more local and directed along the fibre axis, the primary wall rolls up perpendicularly to the fibre axis and forms one or more threads attached to two collars. The collars block the swelling of the fibre, forming what has been called the unswollen sections (actually a region between two balloons) by Cuissinat and Navard (2006a). When the S2 wall is fully dissolved, the swelling of the balloons reaches its maximum size. The balloon is thus formed of the dissolved S2 wall cellulose inside an undissolved membrane composed of the swollen S1 wall, surrounded by one or more threads of the primary wall and delimited by two collars.
5.3 Cellulose–Sodium Hydroxide–Water Solutions
To prepare solutions of cellulose in a NaOH–water mixture is very attractive. It is simple, with reagents that are easy to recycle and cheap. It is thus not surprising that it attracted attention. Already in the 1930s, it was found that cellulose is soluble in NaOH–water mixtures in a certain range of rather low NaOH concentrations and low temperatures, but that this dissolution was difficult, with only partial dissolution of most untreated cellulose samples. Due to these difficulties, effects of other chemicals in NaOH–water were tested. It was found at that time that addition of compounds like ZnO or urea was helping dissolution (Davidson 1937).
The history of the relations between cellulose and sodium hydroxide dates back in the nineteenth century. It is the discoveries of the viscose process where a cellulose derivative is dissolved in NaOH–water and of mercerisation that were the real starts of the use of NaOH in the cellulose industry. In three instances, groups of researchers tried later to use NaOH–water for processing cellulose fibres. The first group in Japan worked in the 1980s and found that steam-exploded cellulose was more readily soluble. More recently, two processes based on enzymatically treated pulps for one and additives for the second were developed, but with no industrial development up to now. Difficulties of mixing (use of low temperatures), low stability of the mixtures, low maximum concentration of cellulose and moderate mechanical properties of the regenerated fibres are the main factors that explain why this dissolution method is not used. Other methods where cellulose is molecularly dispersed like N-methylmorpholine-N-oxide or ionic liquids are favoured over NaOH–water at the present time.
5.3.1 Mercerisation
Mercerisation is a process named after its inventor, John Mercer (The Editors 1903). Patented between 1844 and 1850, mercerisation is the treatment of native cellulose by concentrated (18–20 wt%) caustic alkaline solution. After immersion in the NaOH solution and then washing, the initial cotton fabric has improved properties like better lustre and smoothness, improved dye intake, improved mechanical properties and improved dimension stability. Mercerisation has been an industrial process from the beginning of the twentieth century up to now. Mercerisation is strongly acting on the cell wall cotton morphology, changing, for example, the crystalline structure from the native cellulose I to cellulose II (Okano and Sarko 1985a, b). Mercerisation is not a dissolution but a complex change of morphology and crystalline structure that occurs in a derivatised (production of various alkali–cellulose complexes) and highly swollen state. The accessibility of –OH groups depends on the crystallinity of cellulose (Tasker et al. 1994), the highly crystalline cellulose being more difficult to mercerise (Chanzy and Roche 1976).
5.3.2 Dissolution in NaOH–Water
In the 1930s, Davidson (1934, 1936) studied cellulose dissolution, and he is to our knowledge the first to report dissolution, not swelling as in the mercerisation process. He looked for optimal conditions to dissolve modified cotton, termed hydrocellulose, which was cellulose hydrolysed in strong acid conditions, which is decreasing its molar mass. Such a product would be called microcrystalline cellulose now. Davidson showed that a decrease of temperature improves cellulose dissolution, as illustrated in Fig. 5.9.

Hydrocellulose solubility versus NaOH concentration and solution temperature. Adapted from Davidson (1934)
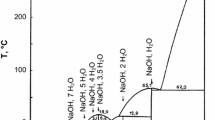
Davidson found a maximum solubility at a very large value, 80 %, probably due to the low molar mass of his hydrolysed cellulose, that solubility occurred in a narrow range of NaOH–water concentration (he reported 10 % of NaOH) and that dissolution was possible at the low temperature of –5 °C. He noticed that solubility increases when the chain length decreases, leading Davidson to deduce that it will not be possible to dissolve unmodified (i.e. high molar mass) cellulose. With these results, we must consider that Davidson is the real inventor of cellulose dissolution in NaOH–water mixtures. However, it is often Sobue (Sobue et al. 1939) who is cited when cellulose dissolution in NaOH–water is concerned. The reason is that Sobue explored the whole ramie cellulose–NaOH–water ternary phase diagram, based on the work of his group and previously published data on cellulose–alkali mixtures by Saito (1939). He noticed that in a narrow range of NaOH, water and temperature, cellulose can be dissolved. The dissolution range was NaOH concentrations of 7–10 % and in low temperature range (−5 to +1 °C). They refer to this region and cellulose state by the term “Q-state”. This phase diagram, reported in all reviews, is plotted on Fig. 5.10.

NaOH–water–cellulose phase diagram, adapted from Sobue et al. (1939). The circle locates the dissolution range
Apparently, the discovery of cellulose dissolution was not a major event and not many scientific papers reported its study. A revival of an interest in cellulose dissolution in NaOH–water came from Japan in the middle of 1980s. A team of Japanese researchers from Asahi Chemical Industry Co. made a breakthrough in the dissolution of cellulose in dilute aqueous solutions of sodium hydroxide. In a series of papers, Kamide et al. (1984, 1987, 1990, 1992), Yamashiki et al. (1988, 1990a, b, c, 1992), Yamada et al. (1992), Matsui et al. (1995), Yamane et al. (1996a, b, c, d) extensively studied cellulose dissolution mechanisms using different approaches, focusing on methods able to lead to produce materials, mainly fibres and films. The first paper by Kamide et al. (1984) reported that regenerated cellulose from a cuprammonium solution and ball-milled amorphous cellulose where intramolecular hydrogen bonds are completely broken or weakened dissolves in aqueous alkali and that the solution is stable over a long period of time. They identified the hydrogen bond intramolecular (O3–H…O5′) as the one needing to be weakened. The authors found that if crystallinity has a role, the more crystalline cellulose being the more difficult to dissolve, as found already by Davidson in 1934, it is not by far the only factor governing dissolution. The authors conclude that “in other words, the solubility behaviour cannot be explained by only the concepts of ‘crystal-amorphous’ or ‘accessible-inaccessible’”. The lack of a strong correlation between the amount of amorphous phase and solubility was confirmed later (Kamide et al. 1992). The authors deduced that these solutions were molecularly dispersed. The solution was found to be a theta solvent at 40 °C. Cellulose has the behaviour of a semi-rigid chain (flexibility in NaOH–water lying between those in cadoxen and iron–sodium tartrate) having a partially free draining behaviour. The unperturbed dimensions decreased with temperature increase. A detailed study of cellobiose–NaOH interactions was conducted by Yamashiki et al. (1988). Based on 1H and 23Na NMR results, they proposed a model for explaining NaOH–cellulose interactions. The number of water molecules solvated to a NaOH molecule is maximum at 4 °C in the range 0–15 % of NaOH, decreasing at this temperature from 11 water molecules at very low concentration to 8 water molecules at 15 % of NaOH. The authors concluded that provided that the proper intermolecular hydrogen bonds have been broken, the factor that controls dissolution is the structure of the alkali. If, as hypothesised, weakening intermolecular hydrogen bonds is a prerequisite for good solubility in NaOH–water, the same group turned towards steam explosion in order to avoid having to use regenerated cellulose. The solubility of steam-exploded pulp was found to be very high, always with a maximum solubility in the temperature–NaOH concentration window found by the first investigators (Davidson and Sobue), as shown in Fig. 5.11.

Dependence of solubility Sa of alkali-soluble cellulose as a function of alkali concentration, at four temperatures (−5, 4, 15 and 30 °C). Plot adapted from Yamashiki et al (1990d)
X-ray studies showed that dissolution occurs in this 8–9 % of NaOH in water at low temperature without any conversion of cellulose into Na-cell I, and that dissolution may occur first in the amorphous parts, resulting in a transparent, molecularly dispersed solution (Kamide et al. 1990). The processing and properties of fibres and films produced by this method have been studied and reported in several papers (Yamane et al. 1996a, b, c, d). Starting from previous knowledge, authors developed an industrial method for dissolving cellulose starting from steam-exploded cellulose pulp, wet pulverisation to increase the surface of cellulose particles, pretreatment in NaOH–water with 2–6 % NaOH at −2 °C and high-speed mixing followed by a dissolution at 6–9 % NaOH–water at −2 °C at 12,000 rpm during 1 mn (Yamane et al. 1996a). Tensile strength of maximum 2 g/den, elongation about 20 % (Yamane et al. 1994) and Young’s modulus of about 110 g/den in the dry state were reported in Yamane et al. (1996c, d). These values are comparable to viscose fibres and inferior to lyocell or rayon fibres. Isogai and Atalla (1998) found difficult to dissolve microcrystalline cellulose using the procedure developed by the Asahi group. They found that dissolution of a large variety of cellulose origins (microcrystalline cellulose, cotton linter, softwood bleached and unbleached kraft pulp, groundwood pulp) and treatments (mercerised, regenerated) can be better performed if a cellulose solution in 8.5 % NaOH is frozen at −20 °C before thawing at room temperature while adding water to reach 5 % NaOH in the solvent. With this procedure, solutions with microcrystalline cellulose were stable at room temperature. Other cellulose samples were partially soluble. The authors found that the presence of hemicellulose did not seem to be a factor influencing dissolution since most hemicellulose fractions were soluble in NaOH– water mixtures. Molar mass was supposed to be the key point for explaining solubility, the higher masses being more difficult to dissolve, a fact explained by the authors though the concept of cellulose chain coherent domains.
However, despite huge efforts for understanding cellulose dissolution in NaOH–water and the processing of these solutions, this process did not reach an industrial stage. The reasons are multiple, but the stability of the spinning dope was a difficulty that prevented the use of this route for preparing regenerated cellulose objects. Increasing stability (i.e. preventing gelation) and improving the quality of the solution were then looked at by the addition of various compounds.
Additives like urea, thiourea or zinc oxide have been known to influence mercerisation from a long time. Davidson (1937) found that the addition of small amounts of zinc oxide to a solution of sodium hydroxide in water increases its swelling and dissolution capability towards cellulose. Additives like urea and thiourea were also studied and used from a long time for improving the viscose process or understanding Na-cell formation (e.g. Harrison 1928).
It was rather straightforward to investigate if the addition of compounds like urea, thiourea or ZnO would improve the state of dissolution of cellulose in NaOH. The first to report such attempts with thiourea is Laszkiewicz (Laszkiewicz and Cuculo 1993) following a previous work on mercerisation with additives like thiourea, ZnO, urea and aluminium hydroxide (Laszkiewicz and Wcislo 1990). He found that addition of thiourea improves the solubility of a cellulose III sample in NaOH–water. Later, the same author reported that the addition of 1 % urea in a solution of bacterial cellulose in NaOH–water allows dissolution of this cellulose if DP is lower than 560 (Laszkiewicz 1998). The Institute of Biopolymers and Chemical Fibres in Poland together with other research groups mainly in Finland developed methods to activate cellulose either by hydrothermal or enzymatic treatments (Ciechańska et al. 1996, 2007; Mikolajczyk et al. 2002; Struszczyk et al. 2000; Wawro et al. 2009). They developed a process called Biocelsol (see a detailed description in Chap. 5) where cellulose is treated by enzymatic activations and dissolved in NaOH–water with the addition of ZnO (Vehviläinen et al. 2008). Typical conditions are cellulose concentration of 6 %, 7.8 % NaOH and 0.84 % of ZnO. The best fibres have tenacity of 1.8 cNdtex-1 with 15 % elongation at break.
Starting in 2000, the group of L. Zhang revisited the dissolution of cellulose in NaOH–water using derivatives (Zhou and Zhang 2000; Zhang et al. 2002, 2004; Zhou et al. 2002a, b, 2004; Weng et al. 2004; Cai and Zang 2005). The preparation of solutions was inspired by Isogai and Atalla (1998) adding first urea and later thiourea, inspired by the work of Laszkiewicz (Laszkiewicz and Wcislo 1990; Laszkiewicz and Cuculo 1993; Laszkiewicz 1998). Solutions with 4–8 % were prepared and were stable enough to allow spinning fibres and preparing membranes. Cellulose cannot be dissolved at temperatures above 10 °C in NaOH–urea–water, and the lower the molar mass was, the higher was the dissolution yield (Qi et al. 2008a). They found that treating mechanically the cellulose fibres (Valley beating machine) was only slightly decreasing molar mass but decreasing more substantially crystallinity, which resulted in an increase of dissolution yield. The authors deduced that crystallinity is an important factor for increasing solubility, not in agreement with previous results (Kamide et al. 1984, 1992). One explanation for this discrepancy is that mechanical beating is also changing the structure of the fibres, mainly removing its primary wall and thus increasing accessibility to the solvent (LeMoigne 2008; LeMoigne and Navard 2010b).
The addition of urea (Zhou and Zhang 2000) has two advantages. It increases the dissolution yield, i.e. a larger fraction of a given pulp is dissolved and the solution is more stable (gelation delayed in time). Whether there is or not complexation between urea, NaOH and cellulose was a matter of debate. Kunze and Fink (2005) found that there exists a specific urea–NaOH–cellulose complex different from the usual Na–cellulose complex by NMR, in the range of 0–8 % NaOH and 15–40 %, both at −25 °C and room temperature. Such complex is also formed when treating cellulose with only urea. As found by the Asahi group in the 1980s for cellulose dissolved in only NaOH, intramolecular hydrogen bonds are destroyed in the NaOH–urea–water mixture (Zhang et al. 2001). Further NMR work (Zhou et al. 2004) suggests that an interaction between urea and NaOH exists in the solution and plays a role in the solvation of cellulose, improving dissolution. Interactions between urea and cellulose block the self-association of cellulose, preventing gelation. But cellulose chains form aggregates of radius of gyration 200–300 nm (Chen et al. 2007), comparable to what is classically found for cellulose solutions (Röder and Morgenstern 1999; Röder et al. 2000). Chen et al. suggests that the polar amine and carbonyl groups of urea can be considered as hydrogen-bonding donors and acceptors, respectively, for the cellulose molecules. The presence of NaOH allows hydrogen bonds to be formed between urea and cellulose. Helped by DSC measurements, Cai and Shang (2005) suggest that when cellulose is placed in NaOH–urea/water at low temperatures, alkali hydrates, urea hydrates and free water surround the cellulose molecules as shown in Fig. 5.12 destroying intra- and intermolecular hydrogen bonds, solvating the cellulose molecules and protecting it with a sort of “overcoat”, preventing reaggregation.

Schematic dissolution process of the cellulose in LiOH/urea and NaOH/urea aqueous solutions pre-cooled to −10 °C: (a) cellulose bundle in the solvent, (b) swollen cellulose in the solution and (c) transparent cellulose solution. With kind permission from Springer Science + Business Media: Cai and Zang (2005), Fig. 5.10
Further studies by Zhang et al. (Cai et al. 2008; Lu et al. 2011; Lue et al. 2011) show that in NaOH (as well as LiOH)–urea–water solutions, cellulose is present as isolated molecules and as aggregates. The fraction of aggregates increases with increasing temperature above −12 °C. Based on NMR, FT-IR, SANS, TEM and WAXS, Cai et al. (2008) found that low temperatures promote the formation of hydrogen-bonded networks of NaOH, urea and water, with the formation of these complexes being favoured by low temperatures. Using SANS, they suggest that NaOH is weakening the association of urea with water, a result found at the same time by Egal et al. (2008) using calorimetry. The chemical shifts of carbon atoms are similar in NaOH–urea–water and NaOH–water environments, showing that urea does not interact with cellulose, a result in agreement with Egal et al. findings (Egal et al. 2008). Cai et al. suggest a model where NaOH hydrates are hydrogen-bonded to cellulose molecules and that urea hydrates are bonded to NaOH hydrates at the surface of cellulose–NaOH complex. This is making a sort of envelope protecting cellulose chains to aggregate. These authors claim that this arrangement called inclusion complex is a non-stable arrangement that is slowly displaced with time and when increasing temperature, leading to the formation of large aggregates of radius of gyration larger than 200 nm (Lu et al. 2011; Lue et al. 2011).
Some authors claimed that using both urea and thiourea is improving further dissolution (Jin et al. 2007). 13C NMR shows that this mixture is a direct solvent and that NaOH, urea and thiourea are all bound to cellulose which “brings cellulose molecules into aqueous solution to a certain extent” and prevents gelation. Solutions of up to 6 % of DP 510 can be prepared and are stable below 0 °C. NMR suggests similar interactions among NaOH, urea, thiourea and water in the solvent and the cellulose solution. The structure of the solvent does not change with the introduction of cellulose. The authors suggest that NaOH hydrates bound to cellulose to form a protective layer preventing cellulose chain aggregation. This solvent was studied also by Zhang et al. (2011) and Kihlman et al. (2011)
The properties of fibres prepared from solutions in NaOH–water–urea (Cai et al. 2004) and NaOH–water–thiourea (Ruan et al. 2004) give mechanical properties of about tensile strength of 1 cN/dtex and elongation at break of about 15 % for thiourea. A comparison between fibres prepared in the 1990s in Japan, in China by the group of Zhang and in Poland by the Biocelsol process is given in Table 5.2. It shows that processing in NaOH–water system brings properties lower than the ones of lyocell and viscose. One of the reasons is that solubility is low, with maximum cellulose concentration being in the order of 7–8 % (see below the results of Centre de Mise en Forme des Matériaux for an explanation of this concentration). Only low molar masses can be dissolved at these maximum concentrations. In addition, the stability of the solution is posing problems. These solutions are gelling and gelation is faster with temperature increase and when concentration and/or molar mass is large. Weng et al. (2004) and Ruan et al. (2008) studied gelation kinetics and found gelation to be an irreversible physical phenomenon. All these features are not helping in preparing a good spinning dope with high enough cellulose molar mass and concentration able to produce fibres with good mechanical properties. The fact that a cooling much below room temperature is needed is hampering the industrial use of this process.
We present below part of the work that has been conducted in Centre de Mise en Forme des Matériaux, Mines Paristech/CNRS/Armines, on cellulose–NaOH-solution properties, with and without additives. The use of NaOH–water to prepare cellulose materials is presented in Chap. 5.
5.3.2.1 Structure of Cellulose–NaOH–Water Solutions (Roy et al. 2001; Egal et al. 2007)
In order to understand better what is controlling the solubility of cellulose in NaOH–water, binary NaOH/water and the ternary cellulose/NaOH/water phase diagrams at NaOH concentrations in the region of cellulose dissolution (7–10 % NaOH below 0 °C) were studied by DSC. To avoid the complexity usually associated with natural fibres directly extracted from the cell wall (Cuissinat and Navard 2006b), microcrystalline cellulose Avicel®PH-101 (FMC Corporation, mean degree of polymerisation of 170) was used. NaOH was dissolved in water at concentrations around 12 % and cooled down to −6 °C. At the same time but in another vessel, water was added to cellulose pulp for cellulose swelling and the cellulose/water system was left at +5 °C for about 2 h. Cold NaOH/water solution was added to this swollen-in-water cellulose in such a proportion that in 100 g of final solution there were 0.5–7.6 g of cellulose and 7.6 or 8 g of NaOH. The weight proportions between the components in 100 g solution will be noted as Xcellulose–YNaOH–water which means X g of cellulose, Y g of NaOH and (100 − X − Y) grams of water. Cellulose–NaOH–water mixtures were placed into a thermobath at −6 °C and stirred at ~1,000 RPM for 2 h. Then the solutions were removed from the bath and stored at +5 °C.
DSC melting thermograms of NaOH + H2O solutions at T < 0 °C and C NaOH = 0–20 wt% are showing two peaks, characteristic of a eutectic phase diagram, as been shown by Cohen-Addad et al. (1960) and Rollet and Cohen-Addad (1964). The melting peak at low temperature, around −33/−34 °C, is independent on the NaOH concentration and is the trace of the melting of the crystalline eutectic mixture, composed of one metastable sodium pentahydrate and four water molecules (NaOH·5H2O; 4H2O). Its melting enthalpy is ΔH eut, pure = 187 J/g, measured at C NaOH = 20 %. The high temperature peak corresponds to the melting of ice. The higher the sodium hydroxide concentration, the lower the ice melting temperature because of the decrease of ice fraction in solution. Applying the level rule on NaOH/water phase diagram, it is possible to calculate the fractions of the eutectic mixture f eut, calc and of ice f ice, calc at any NaOH concentration. It was straightforward to determine the corresponding fractions from the experimental data, f eut, exp and f ice, exp, i.e. from experimentally measured melting enthalpies ΔH eut and ΔH ice at a given NaOH concentration, and to know the melting enthalpies of pure compounds (ice and eutectic), ΔH eut, pure and ΔH ice, pure, respectively. Fractions calculated and determined from experimental data coincide within experimental errors. This match is good for the enthalpy of the eutectic compound which has a well-defined melting peak. The match is less good for the melting of ice which occurs over a large range of temperatures with a peak having a “tail” at low temperatures difficult to extract from the baseline.
Structure of Cellulose–NaOH–Water Solutions at T < 0 °C: An example of DSC melting thermograms for solutions of different cellulose concentrations is given in Fig. 5.13.

DSC melting thermograms of XAvicel/7.6NaOH/water solutions with X = 0, 0.5, 4, 5 and 7.6 g in 100-g solution. Dashed line corresponds to X = 0 (solution without cellulose). Curves are shifted vertically for clarity. Reprinted with permission from Egal et al. (2007). Copyright 2007 American Chemical Society
Whatever the concentrations of cellulose and sodium hydroxide are, the melting temperature of the peak at lower temperatures is constant. It coincides with the melting temperature of the eutectic mixture in pure aqueous sodium hydroxide solution of 7.6 % but with a systematic shift of about 1 °C towards lower temperatures. This means two things. First, the same eutectic mixture (NaOH·5H2O; 4H2O) is present in cellulose–sodium hydroxide aqueous solutions in this region of temperatures and NaOH concentrations, owing to the fact that the temperature shift is very small. The presence of cellulose is not changing its composition. Second, because there is a change in molecular environment due to the presence of cellulose, there is a slight shift in the melting temperature. However, the presence of cellulose is drastically decreasing the amount of the water–NaOH eutectic mixture, as can be seen in Fig. 5.13.
The higher is the cellulose concentration, the smaller is the amount of eutectic compound that can crystallise and then melt at −34 °C. Since NaOH is present only in the eutectic compound, the decrease of its melting enthalpy allows the calculation of the number of NaOH molecules linked to cellulose and thus not able to participate to the NaOH·5H2O crystal fraction of the eutectic mixture. The fact that ΔH eut is reaching zero at a certain cellulose concentration means that all sodium hydroxide molecules have been trapped by the cellulose chains. This corresponds to the dissolution limit since there is no more NaOH molecules able to solvate any additional cellulose chain that could be brought in the mixture. At this dissolution limit, we can calculate the proportion between cellulose anhydroglucose unit (AGU) and NaOH molecules. In our case, the eutectic peak disappears when weight ratio M cell/M NaOH = 1. In moles, this proportion is four NaOH per AGU (m AGU = 162 g/mol and m NaOH = 40 g/mol). It is also possible to calculate the proportion between linked AGU and NaOH at any weight ratio of the components. The amount of NaOH molecules linked to one anhydroglucose unit as a function of the weight ratio M cell/M NaOH for the systems Avicel–7.6NaOH–water (dark squares) and Avicel–8NaOH–water is shown in Fig. 5.14 (open squares). At M cell/M NaOH > 0.4–0.5, the proportion between AGU and NaOH molecules is constant and equal to the limit of cellulose dissolution which is four NaOH per AGU. At low cellulose concentrations, M cell/M NaOH < 0.25, up to 20 NaOH molecules seem to be linked to one anhydroglucose unit.

Number of NaOH molecules linked to one anhydroglucose unit, calculated from melting enthalpy data, as a function of cellulose/NaOH weight ratio in solution. Dark squares: cellulose/7.6 NaOH/water, open squares: cellulose/8 NaOH/water, crosses: data from Kuo and Hong (2005) for NaOH concentrations of 4.5, 6.2 and 7.9 %. Reprinted with permission from Egal et al. (2007). Copyright 2007 American Chemical Society
DSC experiments and careful analysis of experimental data allowed understanding the thermodynamic behaviour and structure of microcrystalline cellulose–sodium hydroxide aqueous solutions at temperatures below 0 °C, in the region of cellulose dissolution. It was possible to determine the limit of cellulose dissolution in NaOH–water as being at least four NaOH molecules per one anhydroglucose unit or the weight ratio of cellulose/NaOH being one. If the concentration of cellulose is higher, it will not be dissolved. Because cellulose can be dissolved only in a narrow range of sodium hydroxide concentrations, from 7 to 8–10 %, this means that maximal amount of cellulose that can be dissolved in NaOH–water solutions is 8–10 %. This result is compatible with what is found experimentally when preparing cellulose/NaOH/water solutions for processing.
When the amount of water is too large (below 6–7 % of NaOH), we can speculate that the size of the hydrate is too large to penetrate the cellulose fibres. When the amount of water is low (NaOH concentrations higher than 18–20 %), NaOH is forming Na–cellulose crystals (mercerisation process). When the amount of water is in between these two cases (i.e. NaOH is 7–10 %), NaOH hydrates penetrate into the cellulose fibres and bind to each chain, but without forming Na–cellulose crystal. This pushes the detachment of individual chains out of the cellulose fibre and makes a solution. At these concentrations, there is an unstable equilibrium between NaOH hydrates bound to each other and to cellulose, which makes all the entities to be in solution.
5.3.2.2 Viscosity of Solutions of Cellulose in NaOH–Water (Roy et al. 2003)
The shear rheology of a microcrystalline cellulose dissolved in a 9 % NaOH aqueous solution was studied in the steady and oscillatory modes. Dilute solutions, below 0.8–1.2 % (the value of the critical overlap concentration depending on temperature, as it will be shown in the following), flow like Newtonian fluids. Semi-dilute solutions below 5 % show a Newtonian plateau and a shear-thinning regime. Above 5 %, in the studied shear rate range, the Newtonian plateau disappears. The flow index n of viscosity–shear rate curves (\( \eta \sim {\dot{\gamma}^n} \)) in the shear-thinning region monotonously increases with temperature from 0.09 to 0.12 for the 3 % cellulose solution and from 0.13 to 0.17 for the 5 % cellulose solution. These values are 4–5 times smaller than the ones of either typical “normal” polymer solutions (experimental data or theoretical predictions both for flexible and rigid chain polymers) or, in particular, polysaccharides like guar gum, λ-carrageenan, hyaluronate (see, e.g. Morris et al. 1981) or entangled physical networks (Clark and Ross-Murphy 1987). The viscosity–shear rate dependencies of cellulose–9 % NaOH solutions do not also obey the empirical law found by Morris (1990) and Haque and Morris (1993) for several polysaccharides. The flow of cellulose–9 % NaOH solutions is close to the one of suspensions with a flow index lower than 0.5. Indeed, as shown by several authors (Röder and Morgenstern 1999, 2000; Drechsler et al. 2000; Schulz et al. 2000), the dynamic and static light scattering studies of cellulose–NMMO, viscose and several cellulose derivative solutions demonstrated that cellulose chains can aggregate in solutions. Thus, the very low flow index value of cellulose–9 % NaOH solutions is an indirect proof that cellulose in 9 % NaOH aqueous solution is also aggregated, as was proved later by light scattering (Chen et al. 2007).
It was also shown that cellulose intrinsic viscosity [η] decreases with temperature increase. As a consequence, the overlap concentration, C*, which is a critical parameter for gelation, is also temperature dependent: C* = 0.83 % for cellulose–9 % NaOH below 20 °C and C* = 1.25 % for cellulose–9 % NaOH at 40 °C.
Gelation of Cellulose–NaOH Solutions Seen by Dynamic Rheology (Roy et al. 2003; Egal 2006, Gavillon and Budtova 2008; Budtova et al. 2010): Gelation is the transition between a fluid liquid state and an elastic solid state. According to the gelation theory of Winter and Chambon (1986), in the fluid-state viscous modulus G′ ~ ω a and elastic modulus G″ ~ ω b (with ω being angular frequency), ideally the gel point is reached when a = b = 0.5. The development of the slopes of G′ = f(ω) and G″ = f(ω) curves in time is thus a privileged method for looking at gelation kinetics. This works well for chemically cross-linked networks but may be difficult to apply for more complicated systems like physical gels. In the latter case a simplified approach is used: G′ and G″ evolution as a function of time t is monitored at a fixed frequency and gel point is considered to be reached when G′(t) = G″(t).
The evolution of G′(t) and G″(t) at various cellulose concentrations and temperatures T was recorded and the simplified approach, as described above, was used to deduce gelation time as a function of solution temperature and cellulose concentration. At t = 0, G″ is larger than elastic modulus G′: The system is in solution state and behaves like a viscous liquid. With time, G′ increases more rapidly than G″: It crosses G″ at a certain gelation time t gel and becomes larger than G″. The system gradually transforms from a viscous liquid to an elastic network. It should be noted that while the exponent a decreases with time as expected (it should ideally reach a = 0 or G′ = const indicating the formation of a stable network), G′ of cellulose–NaOH–water solutions never reached the ideal constant value. Together with G″, G′ was slowly increasing in time after the gel point. The formed gel is opaque; the reason could be a microphase separation into polymer-rich and polymer-poor phases, as it occurs for some gelling polysaccharide solutions such as methylcellulose. In a few hours after gel point, the experimental points become scattered because of syneresis: Solvent is released from the gel and the sample slides leading to bad data reproducibility.
Whatever is the temperature, the gelation of semi-dilute cellulose–NaOH solutions takes place, being faster at higher temperatures. Gelation time t gel exponentially decreases with a temperature increase t gel ∼ exp(−aT), varying from 0.35 to 0.4 for 8–9 % NaOH–water solvent.
Gel strength at the gel point G gel = G′ = G″ was studied as a function of cellulose concentration (Gavillon and Budtova 2008). It was found that G gel is power law dependent on cellulose concentration with exponent being from 3 to 4. This is due to a progressive increase of the number of contacts between cellulose chains leading to a stronger network structure. Such a strong power law concentration dependence is not typical for gelling polysaccharides, which is reported to be square dependent on polymer concentration.
The thermally induced gelation of cellulose–NaOH solutions can be interpreted as follows. As shown for dilute solutions, solvent thermodynamic quality decreases with temperature increase. This leads to the preferential cellulose–cellulose and not cellulose–solvent interactions. In dilute solutions below polymer overlap concentration, the coils contract. Above the overlap concentration, gelation occurs via intra-chain interactions. Both time and temperature are acting on cellulose–NaOH–water solutions in the same “destabilising” way.
5.3.2.3 Structure of Cellulose/NaOH–Urea–Water Solutions (Egal 2006; Egal et al. 2008)
Cellulose is better dissolving in NaOH–water when a certain amount of urea is added. In order to understand the mechanisms of this dissolution and the interactions between the components, the binary phase diagram of urea/water, the ternary urea–NaOH–water phase diagram and the influence of the addition of microcrystalline cellulose in urea–NaOH–water solutions were studied by DSC.
Binary Urea/Water Phase Diagram: The full urea–water phase diagram was plotted using DSC experiments and X-ray scattering. Apart for 30 % urea where only one peak is present, all DSC thermograms reveal two melting peaks more or less well separated. The first one, at low temperature, has the same position whatever the urea concentration from 6 to 30 % is. At the onset of the peak, the temperature is about −12.5 °C. This peak corresponds to the melting of urea–water eutectic mixture. The second one, at higher temperatures, is shifted towards lower temperatures when the urea content increases. This peak corresponds to the gradual melting of free ice. The peak of free ice disappears at about 30 % of urea. This means that the urea–water eutectic mixture corresponds to 30 % of urea. As the molar masses of urea—CO(NH2)2—and water are 60 g/mol and 18 g/mol, respectively, 30 % of urea in weight corresponds to about a molar ratio \( {n_{{{{\mathrm{H}}_{2}}{\mathrm{O}}}}}/{n_{\mathrm{urea}}} = 7.8 \). Thus, the total urea–water eutectic mixture consists of 1 urea and 8H2O molecules. This is confirmed by X-ray scattering that shows that at higher urea concentrations, above 30 %, only two types of crystals are present, ice and crystalline urea.
Ternary NaOH–Water–Urea Phase Diagram: The composition of cellulose solvents studied was in the range from 7.6 NaOH–6 urea–water to 7.6 NaOH–25 urea–water. DSC melting thermograms were plotted for several urea concentrations. Four peaks can be seen on Fig. 5.15.

Peak no. 1 corresponds to the melting of the metastable eutectic mixture NaOH·5H2O + 4H2O. Peak no. 2 appears at about −30 °C, but it is not always present. It corresponds to the melting of the stable eutectic mixture NaOH·7H2O +2H2O that was not present in NaOH/water solution when heated/cooled with a low temperature rate (Pickering 1893; Cohen-Adad et al. 1960). It seems that the presence of urea in a 7.6 NaOH/water is favouring the crystallisation of this stable eutectic, while this is not occurring in the same conditions of cooling and heating rates without urea. Since we have no access to conditions where only the stable eutectic compound is present without urea, we do not know its pure melting enthalpy. In the following, we will use the same specific enthalpy for the stable eutectic compound as the known one of the metastable eutectic compound (187 J/g). Peak no. 3 appears as soon as urea is added. This peak should correspond to the melting of a urea compound. As will be seen below, peak no. 3 represents the melting of urea eutectic mixture and peak no. 4, the melting of free ice. The temperature of the melting peak no. 4 decreases when urea concentration increases from 0 to 14 g of urea in 100 g of solution. This peak corresponds to the gradual melting of free ice.
We have shown that urea does not react with NaOH molecules and does not modify the NaOH eutectic. Thus we assume in the presence of NaOH, urea and water behave as if they were alone: They should form the eutectic mixture containing one urea and eight water molecules, as determined for the binary urea/water solution. The fact that peak no. 3 corresponds to the melting of the urea eutectic mixture can be checked by plotting the melting temperature of both peaks as a function of urea concentration. It shows that peaks no. 3 and no. 4 are joining at a composition corresponding at 7.6 NaOH–18 urea–water. If the assumption of noninteraction between NaOH and urea is correct, this means that at this composition there should be two independent, coexisting eutectic compounds, NaOH + water and urea +water, with no more free water in solution. In a 100-g solution containing 7.6-g NaOH + 18-g urea + 74.4-g water, there are 4.13 moles of water, 0.19 moles of NaOH which corresponds to 0.19 moles of NaOH eutectic mixture (there is one NaOH mole per mole of the NaOH eutectic compound) and 0.3 moles of urea. The NaOH eutectic mixture is not modified by the addition of urea, and it contains 1 NaOH and 9 H2O molecules. 1.71 moles of water is thus trapped in the NaOH eutectic mixture, and it remains 4.13 − 1.71 = 2.42 moles of water. The urea eutectic mixture is composed of 1 urea and 8 H2O molecules, and thus, 0.3 × 8 = 2.4 moles of water is trapped in the urea eutectic. There is no more water in the system (the remaining 0.02 moles of free water is within the experimental error of determination of the crossing composition). As a conclusion, 100 g of 7.6 NaOH/18 urea/water solution contains 18 g of urea and 43.56 g of water, i.e. the local concentration of urea in water is \( \tfrac{{18}}{{18 + 43.56}} \times 100\% = 29.2\% \) which corresponds to the composition of the urea eutectic mixture, as shown above for urea/water binary solution. At this composition, the solution is composed of only the two eutectic mixtures: NaOH + water and urea + water. These results show that both NaOH and urea eutectic mixtures are formed independently and thus are competing for water. NaOH, urea and water behave as in their binary systems.
Solutions of Cellulose in NaOH–Urea–Water: The addition of cellulose does not modify or change the position of the peaks as compared to the case without cellulose. The DSC melting curves have three peaks. The peak at the lowest temperature, at about −35 °C, corresponds to the melting of the NaOH + water eutectic mixture. The next one, at about −25 °C, corresponds to the melting of the urea + water eutectic mixture. The peak at the highest temperature, at about 7 °C, corresponds to the melting of free ice. Whatever the concentration of cellulose is, the melting temperature of the NaOH + water eutectic mixture is equal to the one obtained for a pure 7.6 NaOH–6 urea–water (without cellulose). During the dissolution process, cellulose is trapping some NaOH molecules that cannot participate anymore to the formation of the eutectic mixture. It is remarkable to see that the addition of cellulose is trapping the same amount of NaOH with or without urea. The addition of urea does not change the interactions between cellulose and NaOH. Whatever the concentration of cellulose is, the peak of the urea + water eutectic mixture is seen. The enthalpy of the urea eutectic does not depend on cellulose concentration. The addition of cellulose does not change the urea–water interactions. Since the melting enthalpy of free ice is approximately constant for all cellulose concentrations (the same was observed for cellulose–7.6 NaOH–water without urea, see Egal 2006; Egal et al. 2007), we conclude that the amount of free ice remains the same with and without cellulose.
Three main facts are obtained from these series of experiments and their interpretations:
-
1.
NaOH and urea do not interact when mixed together with water. Their eutectic mixtures are formed independently.
-
2.
Urea and cellulose do not interact when mixed with NaOH and water since the urea + water eutectic is not changed.
-
3.
The interactions between NaOH hydrates and cellulose are not changed by the presence of urea (the decrease of the eutectic peak enthalpy of NaOH + water with cellulose content is the same with or without urea).
This means that urea is interacting neither with NaOH nor with cellulose and is not changing the NaOH–cellulose interactions. A possible origin of the role of urea as a dissolution promoter can be that it strongly interacts with water. Cellulose chains have the tendency to aggregate as soon as the proportion 4 NaOH/AGU does not hold any more for any reason (temperature increase, cellulose concentration increase). The addition of urea is decreasing the amount of free water, thus helping cellulose chains to stay in solution.
5.3.2.4 Influence of ZnO on Cellulose–NaOH–Water Solution Properties (Liu et al. 2011)
Zinc oxide is an additive improving cellulose dissolution known since the pioneering work of Davidson (1937). Recently, it was revisited for processing fibres (Vehviläinen et al. 2008), and Yang et al. reported that the maximal cellulose dissolution is reached at 0.5 % ZnO in the mixture of 7 % NaOH–12 % urea. The reason of the improved solubility was supposed to be the stronger hydrogen bonds between cellulose and Zn(OH) 2−4 as compared with cellulose–NaOH and cellulose–urea. The goal of our work was to make a comprehensive investigation of the influence of ZnO on used Avicel® PH-101 microcrystalline cellulose–NaOH–water solution properties and especially on its gelation delaying effect using viscometry and rheology.
Cellulose solutions were prepared following the procedure described in Egal et al. (2007, 2008). pH strongly influences ZnO solubility (Liu and Piron 1998). It was shown that ZnO is practically insoluble in water (solubility below 10−6 g L−1) and becomes more soluble in strong acidic or basic media (Liu and Piron 1998). In our work the preparation of solutions is started by making 18–20 % NaOH–water (pH ≈ 14.7) and adding a certain amount of ZnO. At this pH, ZnO can be dissolved up to 27 g L−1 (Liu and Piron 1998). The pH of the final cellulose solutions is 14.3 in which ZnO solubility decreases to about 4 g L−1. Thus, the saturation of ZnO is reached in the final solutions containing cellulose and part of ZnO is in suspended state. Seen by optical microscopy, dispersions of undissolved ZnO were homogeneous, with two-size populations, i.e. ZnO particles of about 1 μm and a few aggregates of about 10–20 μm. Calculations of sedimentation times imposed to limit ZnO concentrations to 1.5 %.
Different concentrations of components in cellulose solutions will be noted as X % cellulose–Y % NaOH–Z % ZnO–water, in which X % is the weight concentration of cellulose, calculated as X % = 100 × M cell/(M cell + M water + M NaOH + M ZnO) where M i is the weight of each component. Y and Z % are the weight concentrations of NaOH and ZnO, respectively, in solvent only, calculated as Y % = 100 × M NaOH/(M water + M NaOH + M ZnO) and Z % = 100 × M ZnO/(M water + M NaOH + M ZnO). The concentration of NaOH in the final solution was fixed to 8 %, and the concentration of ZnO was varied from 0 to 1.5.
A steady decrease of the intrinsic viscosity [η] with increasing temperature was observed for solutions with and without ZnO (here 0.7 wt%) and is consistent with the results reported above for cellulose of another molecular weight dissolved in 8 % NaOH–water without any additive (Egal 2006) and in 9 % NaOH–water (Roy et al. 2003). The drop of [η] signifies that the thermodynamic quality of solvent decreases with temperature. The presence of ZnO did not bring any noticeable change to the intrinsic viscosity of cellulose. When 0.7 % ZnO is mixed with 8 % NaOH–water, only part of ZnO is dissolved. The unaffected value of [η] indicates that neither dissolved nor suspended ZnO influences the conformation and behaviour of cellulose chains at molecular level in dilute region. As shown above, gelation of cellulose–NaOH–water solution is thermally induced and irreversible. The results on gelation time of cellulose–8 % NaOH–water solutions at temperatures from −5° to 50 °C at various cellulose and ZnO concentrations are summarised in Table 5.3.
Gelation is significantly delayed in the presence of ZnO: For example, a 6 % cellulose solution at 20 °C is gelling in 15 min in 8 % NaOH–water and in 36 h in the presence of 1.5 % ZnO. The results presented in Table 5.1 also show that gelation time depends on solution temperature and cellulose and ZnO concentrations
Influence of Temperature on Cellulose– NaOH–Water Gelation in the Presence of ZnO: Gelation time of cellulose solutions was first studied at low temperatures, around −5 to +5 °C to check if the sudden decrease of gelation time with temperature decrease below −0 °C, reported for cellulose–7 % NaOH–12 % urea (Cai and Zhang 2006) system, was seen in our solutions. We did not observe any decrease of gelation time with temperature decrease in this temperature range (Table 5.1). Gelation cannot be studied below −5 °C as far as water in 8 % NaOH–water starts crystallising at this temperature (the end of water melting peak was recorded at −6 to −4 °C when solutions were heated from −60 °C to room temperature, Egal et al. 2007).
As shown above (Roy et al. 2003; Gavillon and Budtova 2008), gelation time is exponentially temperature dependent at given cellulose concentration t gel = D exp(−aT) where D and a are adjustable constants. In the range of cellulose and ZnO concentrations studied, the exponent a varies from 0.28 to 0.46. Similar results were obtained for the same microcrystalline cellulose dissolved in (7.6–9) % NaOH–water (a = 0.35–0.4) (Gavillon and Budtova 2008; Roy et al. 2003). In the presence of ZnO, solutions are gelling with temperature with the same physical mechanisms (with the same exponent) as without ZnO.
Influence of Cellulose Concentration on Gelation: Increasing polymer concentration facilitates gelation, since chains are closer to each other, allowing an easier formation of a network. The influence of cellulose concentration on gelation time in the presence of ZnO is given in Fig. 5.4. The inverse of t gel as a function of cellulose concentration C cell presented in a double logarithmic plot shows that there is a power law relation between these two parameters. A power law dependence of gelation time on carrageenan concentration was suggested by Ross-Murphy (1991):
where C 0 is the critical concentration above which gelation could happen, n′ is the number of polymer chains involved in a junction zone (assumed to be 2), K is a rate constant and p is a percolation exponent estimated to be around 2. For cellulose–NaOH–ZnO–water solutions, the overlap concentration C*, which could be considered as critical concentration for gelation, is around 1 %, depending on temperature (Liu et al. 2011). C* of the same microcrystalline Avicel cellulose in 9 % NaOH–water was reported to be 0.83 % below 20 °C and 1.25 % at 40 °C (Roy et al. 2003). Considering these values, (5.3) becomes t gel ≈ k C cell −n , where k is a rate constant and n is a kinetic exponent. The exponent n is related to the gelation kinetics as well as to the organisation of the junction zones in gel. The values of n obtained here show that they are more or less constant (within the experimental errors) and equal to 9 ± 2, with or without ZnO, strongly suggesting that junction characteristics are not strongly influenced by the addition of ZnO.
Influence of ZnO Concentration on Gelation: Table 5.1 shows that the increase of ZnO concentration delays gelation. We measured the gelation time of 6 % cellulose–8 % NaOH–water at various ZnO contents, 0, 0.7 and 1.5 % from 10 to 45 °C. A master plot can be obtained by shifting two data sets, for 0.7 and 1.5 % ZnO, towards the one without ZnO (Fig. 5.16). The same shape of the initial curves indicates that despite ZnO efficiency in delaying gelation, the increase in ZnO concentration does not change gelation kinetics.

Gelation time versus temperature for 6 % cellulose–8 % NaOH–water solutions with 0, 0.7 and 1.5 % ZnO shifted towards 0 % ZnO by 6.5 °C for 0.7 % ZnO and by 14 °C for 1.5 % ZnO. Filled triangle: without ZnO; open square with 0.7 % ZnO; open circle with 1.5 % ZnO
We demonstrated that ZnO is quite efficient in delaying gelation of cellulose–NaOH–water solutions and it does not have much influence on either gelation kinetic order or the junction zones in cellulose gels. In addition, the addition of ZnO does not change the properties of cellulose at the molecular level and does not improve the thermodynamic property of solvent towards cellulose. To dissolve cellulose, at least four NaOH molecules per one anhydroglucose unit are needed (Egal et al. 2007), with NaOH hydrates breaking cellulose intra-chain hydrogen bonds and linking to cellulose chains. The same is valid for cellulose–NaOH–water solutions containing urea (Egal et al. 2008). As shown many times, cellulose–NaOH–water solutions are not stable and cellulose chains tend to aggregate, leading to gelation. We are suggesting that when ZnO is suspended in NaOH–water solutions, a “network” of tiny particles is formed. The surface of particles is hydrolysed and a layer of hydroxide is formed (≡Zn–OH) attracting water molecules. When dissolved, Zn(OH) −3 and Zn(OH) 2−4 ions are formed (Reichle et al. 1975; Degen and Kosec 2000) that also trap water. Thus, ZnO may play the role of a water “binder”, strongly decreasing the amount of free water around the cellulose chains that may drive chain aggregation. ZnO stabilises the solution by keeping water far from the cellulose chains, as urea does (Egal et al. 2008). This could explain why ZnO only delays the gelation but does not change cellulose dissolution, solvent quality and gelation kinetics in NaOH–water solutions.
5.3.2.5 Cellulose Coagulation Kinetics from Cellulose–NaOH–Water Solutions (Gavillon and Budtova 2008; Sescousse and Budtova 2009; Sescousse et al. 2011a)
Shaping cellulose from NaOH–water-based solvents requires coagulation of cellulose, a process driven by diffusion coefficient of NaOH from cellulose solution or gel into the regenerating bath. The time of reaching complete coagulation is controlled by kinetics of cellulose solvent release into the bath which in turn depends on coagulation conditions and sample geometry. Obviously, coagulation time and rate are important parameters for cellulose processing. The understanding of the mechanisms governing coagulation kinetics opens the ways in controlling final cellulose morphology.
Coagulation kinetics of cellulose from cellulose–8 % NaOH–water gelled solutions wad studied by Gavillon and Budtova (Gavillon and Budtova 2008; Sescousse et al. 2011a). Coagulation of cellulose was performed in a bath of cellulose nonsolvent, water or alcohol, with a controlled volume and temperature. The diffusion coefficient of cellulose solvent (NaOH) from a sample towards the coagulation bath was studied as follows. A sample of Avicel–NaOH–water gel or Solucell–NMMO solution of a given weight and volume was placed in the regenerating bath at a fixed temperature. The proportion between sample/bath weights was kept constant and equal to ten. The amount of NaOH released into the bath was measured as a function of time by potentiometry and titration
The experimental data, i.e. the increase of NaOH amount in the coagulation bath as a function of time and in different conditions (various cellulose concentration, regenerating bath liquid, bath temperature), was analysed using Fick approach. It is widely applied in drug release field and formation of membranes due to phase separation and was already used to describe the kinetics of cellulose coagulation from cellul ose–NMMO–water solutions (Biganska and Navard 2005). The applicability of Fick approach was checked by plotting the cumulative amount of substance M(t) (NaOH) released in time t as a function of \( \sqrt {t} \). The amount of a substance released in time from a semi-infinite plane can be described as follows:
where M is the amount of substance released at t = ∞ (here M coincides with the amount of substance in the initial sample), D is diffusion coefficient and l is half sample thickness because diffusion takes place from its both sides.
If the diffusion coefficient is constant, several simplifications are used to determine D from the slope of \( \displaystyle\frac{{M(t)}}{M} = f\left( {\sqrt {{\frac{t}{{{l^2}}}}} } \right) \) curves:
-
(a)
Early time approximation (\( 0 \le \frac{{M(t)}}{M} \le 0.4 \)):
$$ \frac{{M(t)}}{M} = 4{\left( {\frac{{Dt}}{{\pi {l^2}}}} \right)^{{1/2}}} $$ -
(b)
Late time approximation (\( 0.4 \le \frac{{M(t)}}{M} \le 1 \)):
$$ \frac{{M(t)}}{M} = 1 - \frac{8}{{{\pi^2}}}\exp \left( {\frac{{ - {\pi^2}Dt}}{{{l^2}}}} \right) $$ -
(c)
Half time (\( \displaystyle\frac{{M(t)}}{M} = 1/2 \)) approximation: Here the diffusion coefficient is calculated at the point where \( \displaystyle\frac{{M(t)}}{M} = 1/2 \); it is equal to \( D = \displaystyle\frac{{0.049}}{{{{(t/{l^2})}_{{1/2}}}}} \) where \( {(t/{l^2})_{{1/2}}} \) is the abscissa when\( \displaystyle\frac{{M(t)}}{M} = 1/2 \). The experimental data can be then fitted with formula 1 with n = 0.
The initial state of the Avicel–NaOH–water samples is a gel and the final state is coagulated swollen cellulose. During coagulation, the state of cellulose changes: A phase separation takes place. Different types of models explaining the transport behaviour of solutes in hydrogels or in porous media were tested (Gavillon and Budtova 2008). These models mainly include free-volume theory, hydrodynamic and obstruction approaches and their combinations as well.
The analysis of the influence of cellulose concentration on NaOH diffusion coefficients shows that “porous membrane” (i.e. free-volume or hydrodynamic approach) and not “hydrogel-obstruction” approach must be used for the understanding and interpretation of cellulose coagulation from cellulose–NaOH–water gels. The applicability of a free-volume or hydrodynamic approach is caused by the phase separation process occurring during coagulation of cellulose from cellulose–NaOH gels placed in a nonsolvent liquid. The interpretation of NMMO diffusion and thus of cellulose coagulation should also be with the “porous membrane” approach, but it is complicated by the high concentration of NMMO in the sample, by the presence of NMMO not linked to cellulose at low cellulose concentrations and the change of NMMO phase state during coagulation. An increase of the size of the diffusing entity due to NMMO dragging water molecules during diffusion should also be considered. NaOH activation energy obtained from diffusion experiments coincides with the activation energy calculated from the rheological experiments for cellulose– NaOH–water and pure NaOH–water solutions. This result confirms the suggestion made in Roy et al. (2003) that cellulose–NaOH–water solutions are in fact suspensions of NaOH hydrates with or without cellulose.
The influence of cellulose coagulation conditions on NaOH diffusion coefficient was studied by Sescousse and Budtova (2009). Gel discs were placed in 100 mL acetic acid regenerating bath of 0.1 mol L−1, and pH evolution in time was recorded. The measured diffusion coefficients do not depend on the gelation mode (increasing temperature or time) within the experimental errors. It was noted that gels were different in terms of hardness after “gentle” gelation (50 °C—2 h; 80 °C—15 min) as compared to “hard” gelation conditions (50 °C—20 h; 80 °C—90 min). Long heating times coupled with elevated temperatures lead to at least two phenomena: sample slight weight loss (syneresis due to microphase separation and probably cellulose degradation) and increase of cellulose–cellulose bondings. Both phenomena induce gel “densification” and are due the decrease of solvent thermodynamic quality with heating (Roy et al. 2003; Egal 2006). The obtained independence of the diffusion coefficient on gelation conditions shows that as well as for cellulose coagulation in water from cellulose–NaOH–water gel (Gavillon and Budtova 2007), the coagulation kinetics in an acid bath should be described with “membrane” approaches, like hydrodynamic or free-volume theories, that are not sensitive to gel structure on the molecular level. The diffusion coefficient of NaOH from non-gelled solution is three times lower than that from cellulose–NaOH–water gels, due to lower local cellulose concentration in gel “pores” and thus lower medium viscosity, as compared with the homogeneous cellulose concentration in solution. This result means that cellulose coagulation from cellulose–NaOH–water solution will take longer time than from the gel of the same concentration and geometry.
Diffusion coefficients were measured as function of acetic acid concentration in coagulation bath. They do not depend on acetic acid concentration within experimental errors. This means that coagulation of cellulose in water or in an acid bath from cellulose–NaOH gels of the same size and in the same temperature conditions will take the same time.
NaOH diffusion coefficient from 5 % cel lulose–8 % NaOH–water gel to 100 mL of 0.1 mol L−1 acetic acid regenerating bath was determined for five bath temperatures: 22, 28, 37, 45 and 65 ° C. Solution gelation was performed at 80 °C during 15 min. As expected, the diffusion coefficient decreases with temperature increase, varying from 3.2 × 10−4 mm2 s−1 at 22 °C to 7 × 10−4 mm2 s−1 at 65 °C. The diffusion coefficient values obtained were plotted versus inverse temperature and an activation energy of 16 ± 3 kJ mol−1was calculated using Arrhenius law.
5.3.2.6 Influence of Cellulose Pulp Enzymatic Activation on Solubility in NaOH–Water (LeMoigne et al. 2010b)
The enzymatic treatment is a way to modify the composition of cellulose fibre and/or cellulose molecular weight in order to increase the accessibility of cellulose chains to chemical reagents. Enzymatic treatments were performed on three dissolving sulphite wood pulp samples named VHV-S (intrinsic viscosity 1,430 ml/g), SA (intrinsic viscosity 880 ml/g) and sample called LV-U (bleached sulphite pulp, intrinsic viscosity 350 ml/g) provided by Borregaard (Norway). A mixture of two commercial enzymes was used, Celluclast 1.5 L (cellulase derived from Trichoderma reesei, purchased by Novozymes, Denmark, used classically for the hydrolysis of lignocellulosic biomass feedstocks. This enzyme contains a broad spectrum of cellulolytic enzyme activities, like cellobiohydrolases and endo-1,4-glucanases) and Econase HC400 (highly concentrated liquid formulation of endo-1.4-beta-xylanase produced by Trichoderma reesei and standardised to minimum enzyme activity of 400,000 BXU/g as well as side activities like beta-glucanase, purchased by AB enzymes, Finland).
The non-treated and treated pulps were dissolved in an 8 % NaOH–water solution to test their alkaline solubility. The solutions were prepared as follows: 132 g of 12 % NaOH 12 %–water was stored at −6 °C. 2 g of pulp was added to 66 g of distilled water and stored 1 h at 4 °C. Then, 12 % NaOH–water and cellulose–water solutions were mixed together during 2 h at −6 °C and 1,000 rpm giving 200 g of a solution of 1 % cellulose in 8 % NaOH–water. These 1 % cellulose solutions were directly centrifuged to isolate the insoluble fraction.
The enzyme mixture used has two effects at short peeling times: (1) a digestion of the primary wall which is seen by the near absence of ballooning and (2) a destructuration action in the inside of the fibre which is seen by the large decrease of DP. At long peeling time, the external walls are totally digested and the fibre structure is totally destructured, as seen by the absence of birefringence.
The observation by optical microscopy of the insoluble material clearly shows that the enzyme mixture used has two effects at short peeling times: (1) a digestion of the primary wall which is seen by the near absence of ballooning and (2) a destructuration action in the inside of the fibre which is seen by the large decrease of DP. At long peeling times, the external walls are totally digested and the fibre structure is totally destructured, as seen by the absence of birefringence. Non-treated samples mostly stay in a low swelling or ballooned state even after 2 h of mixing.
The SA sample contains fibres with high swelling ratio which can be related to the more severe digestion of the fibres during the original sulphite pretreatment. Several fibres are cut in sections due to the shearing involved by the mixing, and few fragments can also be observed. After 2 min of peeling, the shape of the insoluble parts is changing drastically. Only highly swollen sections and flat rings are observed. The removal of the primary wall by the enzymatic peeling gives a large swelling of the fibres and a subsequent cutting in flat rings. This cutting in flat rings was observed when putting and submitting fibres to shearing in 8 % NaOH– water, independently of the origin and the preparation mode of the wood pulp. With fibres without primary wall, the large swelling was shown to be sufficient to dismantle the fibres in highly swollen sections and flat rings. At longer peeling times, the insoluble material is made of small fragments implying that the fibres were totally destructured by the enzyme treatment.
The amount of insoluble material follows a linear relationship as a function of intrinsic viscosity and crystallinity. A simple way to explain the evolution of the alkaline solubility after the enzymatic treatment is thus to associate the increase of solubility to the decrease of the DP and of the crystallinity. However, the morphological study of the insoluble parts gives a more comprehensive interpretation on the influence of the structural changes due to the enzymatic treatment on the alkaline solubility. Figure 5.17 gives the amount of insoluble material of the non-treated (A, B) and 2 min enzymatically treated (C, D) SA and VHV-S pulps and of the LV-U pulp (E). By following the dotted line number 1 of Fig. 5.10, an interesting result is found. Despite the large difference of intrinsic viscosity of the two original SA and VHV-S pulps (880 ml/g and 1,430 ml/g, respectively), the amount of insoluble material is nearly the same. The DP is thus not the only parameter that governs the alkaline solubility after enzymatic treatment. This result was already found in the early work of the Asahi group (Kamide et al. 1984, 1992). The dotted line number 2 of Fig. 5.17 shows that the amount of insoluble material of the treated VHV-S sample (D, black diamond) is nearly two times lower than the original SA sample (A, grey square) whilst keeping the same intrinsic viscosity. This observation has to be related with the removal of the external walls that occur during the first minutes of treatment. The external walls are still present in the original SA sample, while they have been removed in the 2-min treated VHV-S sample. The removal of the external walls allows a higher swelling and thus eases the dismantlement and the fragmentation of the fibres during the solution preparation. The dotted line 3 shows that the LV-U sample (E) which was not enzymatically treated presents similar amount of insoluble material as compared to the treated SA and VHV-S samples (C, D), while its intrinsic viscosity is more than two times lower. The enzyme destructuration allows having a better solubility by favouring the fragmentation of the fibres during the dissolution.

Final amount of insoluble material versus intrinsic viscosity for the non-treated SA and VHV-S pulps (A, B), the 2 min treated SA and VHV-S pulps (C, D) and a non-treated LV-U pulp (E). From LeMoigne (2008)
The removal of the external walls and the macrostructural destructuration of the fibres must thus be considered as key factors in the dissolution of wood fibres in 8 % NaOH–water.
The enzymatic treatment is thus leading to a fast and large decrease of degree of polymerisation and of crystallinity, showing that the enzymes do not act simply on the fibre surface. The alkaline solubility of the different treated samples was investigated in a NaOH 8 %–water solution. As expected from thermodynamic considerations, there is a direct correlation between the solubility and the degree of polymerisation. However, aside thermodynamics, the removal of the external walls and the macrostructural destructuration of the fibres are key factors in the improvement of the dissolution of wood cellulose fibres. At constant intrinsic viscosities of the cellulose materials, the alkaline solubility is almost two times higher when the external walls are removed. The macrostructural destructuration of the fibre by the enzymes allows preserving a high degree of polymerisation while keeping a good alkaline solubility.
5.4 Cellulose–Ionic Liquid Solutions
As described above, several solvents have been successfully used for cellulose dissolution for making films and fibres and as homogeneous reaction media for synthesising cellulose derivatives. However, most of them are either toxic, or volatile, or with limited thermal stability, or needing rather high temperatures for the dissolution, or not allowing cellulose dissolution with reasonably high concentrations. There is thus a continuous search for new “green” cellulose solvents both for making objects from coagulated cellulose and also for the chemical derivatisation under homogeneous conditions. In this context, ionic liquids (IL) have been suggested as promising cellulose solvents. The first mention of cellulose dissolution in IL, quaternary ammonium salts, was published in US patent by Graenacher (1934). At the beginning of the twenty-first century, it was reported that cellulose can be dissolved at rather high concentrations (up to 15–20 %) without any pre-activation, in imidazolium-based ionic liquids, such as 1-butyl-3-methylimidazolium chloride (BMIMCl), 1-ethyl-3-methylimidazolium acetate (EMIMAc) and 1-allyl-3-methylimidazolium chloride (AMIMCl) (Swatloski et al. 2002; Zhang et al. 2005). Due to their ionic structure, IL possesses several advantages over common solvents: They are non-volatile, with high thermal stability, and have good dissolution properties for a large variety of chemical compounds. An interesting advantage is that the properties of IL may be easily tuned by varying anions or cations.
The dissolution of cellulose in ionic liquids is simple and occurs without a derivatisation step, which makes ionic liquids very attractive. Wet-spinning process can be used to obtain cellulose fibres; their properties were reported to be very similar to the ones known for lyocell fibres (Kosan et al. 2008a, b). Various novel cellulose materials can be prepared from cellulose–ionic liquid solutions, such as all-cellulose composites (Zhao et al. 2009), porous aerogel-like cellulose materials (Tsioptsias et al. 2008; Sescousse et al. 2011b), bioactive cellulose films (Turner et al. 2004) and functionalised cellulose microparticles (Lin et al. 2009). It is also possible to perform chemical reactions in homogeneous conditions (El Seoud et al. 2007): Cellulose sulphates, acetates and laurates and furoates were obtained (Gericke et al. 2009a; Barthel and Heinze 2006; Kohler and Heinze 2007). Characterisation of cellulose/IL solutions and understanding their properties and various practical applications have been summarised in recent reviews (Pinkert et al. 2009; Zhu et al. 2006).
Besides using IL as a reaction medium for synthesising cellulose derivatives, the research in cellulose–IL area can be divided into the following directions: understanding of (micro)structure of cellulose–IL solutions including molecular modelling, studying of solution properties including use of co-solvents and making materials for various applications. For example, using 1H and 13C NMR-technique cellobiose solvation in EMIMAc was recently reported (Zhang et al. 2010). Based on the analysis of the concentration dependences of chemical shifts for cellobiose and EMIMAc, authors concluded on hydrogen bonding between the anhydroglucose hydroxyl groups and both the [EMIM]+ and [Ac]− ions (Zhang et al. 2010). A screening of more than 2000 ionic liquids with COSMO-RS approach (conductor-like screening model for realistic solvation) points to the anion as mainly being responsible for the respective dissolving power (Kahlem et al. 2010). Molecular dynamic simulations predict that 1-n-butyl-3-methylimidazolium acetate (BMIMAc) should have the strongest capability to break cellulose intra- and intermolecular hydrogen bonds (Gupta et al. 2011).
The understanding of cellulose–IL solutions rheological properties and cellulose hydrodynamic properties in these solvents is a need for a successful processing and for chemical derivatisation as well. Solution viscoelastic shear and extensional flow properties as a function of cellulose concentration and solution temperature were reported (Kosan et al. 2008a, b; Sammons et al. 2008; Collier et al. 2009; Haward et al. 2012). An extended study of the shear rheological properties of cellulose–AMIMCl (Kuang et al. 2008), cellulose–EMIMAc (Gericke et al. 2009b) and cellulose–BMIMCl solutions (Sescousse et al. 2010a, b) in dilute and semi-dilute states was performed. Cellulose intrinsic viscosity in EMIMAc was obtained for cellulose of various molecular weights in a large temperature range, from 0° to 100 °C, and the first attempt to determine Mark–Kuhn–Houwink constants was made (Gericke et al. 2009b).
It is known that the viscosity of imidazolium-based ionic liquids used to dissolve cellulose is much higher than the one of conventional organic solvents. This is a disadvantage for their use for cellulose processing and also as a reaction medium. For the latter case, dimethylformamide (DMF) and dimethylsulfoxide (DMSO) were used as co-solvents to decrease the overall viscosity (Gericke et al. 2009a, 2011). No decrease of solvent dissolution power was observed. These co-solvents and dimethylacetamide were mixed with EMIMAc, and cellulose solution’s spinnability via electro-spinning was studied (Hairdelin et al. 2012). Shear rheological properties of cellulose solutions in AMIMCl and BMIMCl mixed with DMSO were investigated (Le et al. 2012). Cellulose concentration regimes and viscosities in these binary solvents were compared with the same parameters in pure EMIMAc. It was shown that IL-DMSO seems to be cellulose θ-solvent as suggested for EMIMAc in Gericke et al. 2009b, and the conformation of cellulose is not changed with the addition of DMSO not only in the dilute regime but also in the entanglement regime. The use of co-solvents opens new ways of tuning cellulose–IL solution viscosity. Still a lot of research has to be done to characterise and understand cellulose–IL-co-solvent solution properties.
Below we present results on the properties of cellulose–ionic liquid solutions performed in CEMEF/Mines ParisTech in collaboration with EPNOE partners and with School of Physics and Astronomy, University of Leeds, UK. Most of the results described below have been published in Gericke et al. (2009b), Sescousse et al. (2010a, b), Lovell et al. (2010) and Le et al. (2012).
5.4.1 Materials and Methods
Four celluloses of different molecular weight were used: microcrystalline cellulose with DP 180 and 300 (cell 170 and cell 300 in the following), spruce sulphite pulp with DP 1000 (cell 1000) and bacterial cellulose with DP 4420 (cell 4420). Ionic liquids were EMIMAc and BMIMCl. All products were purchased from Sigma-Aldrich except bacterial cellulose which was produced in Research Centre for Medical Technology and Biotechnology GmbH, Germany. Cellulose was dried at 50 °C in vacuum prior to use. Solvent and cellulose were mixed under nitrogen and stirred in a sealed vessel at 80 °C for at least 24 h to ensure complete dissolution. Clear solutions were obtained; they were stored at room temperature and protected against moisture absorption.
Rheological measurements on cellulose– EMIMAc solutions were performed on a Bohlin Gemini rheometer equipped with plate–plate geometry and a Peltier temperature control system. Steady-state viscosity of cellulose– BMIMCl solutions was measured using ARES rheometer from TA Instruments, with plate–plate geometry. In order to prevent moisture uptake and evaporation at elevated temperatures for both systems, a small quantity of low viscosity silicon oil was placed around the borders of the measuring cell. Because BMIMCl and cellulose–BMIMCl solutions are solid at room temperature (melting temperature of BMIMCl is around 70 °C as given by the manufacturer), the solutions were heated prior to measurements up to 140 °C and kept for 10 min, and viscosity–shear rate dependences were recorded at fixed temperatures, from 130° down to 70 °C.
Pulsed-Field Gradient 1H NMR Spectroscopy. These experiments were performed in the group of Dr. M. E. Ries, School of Physics and Astronomy, University of Leeds, UK. Self-diffusion coefficients of both the [EMIM]+ and [Ac]− ions were determined by a pulsed-field gradient 1H NMR technique using a widebore Avance II NMR spectrometer (Bruker BioSpin) operating at a proton resonant frequency of 400 MHz. A specialised Diff60 diffusion probe (Bruker BioSpin) capable of producing a maximum field gradient of 24 T m−1 was employed in the experiments. More details are given in Lovell et al. (2010).
5.4.2 Steady-State and Intrinsic Viscosity of Cellulose–EMIMAc and Cellulose–BMIMCl Solutions
Steady-state viscosity of cellulose–EMIMAc and cellulose–BMIMCl solutions was measured at various concentrations and temperatures. For all systems a Newtonian plateau was recorded for at least two decades of shear rates (Gericke et al. 2009b; Sescousse et al. 2010a, b). Viscosity mean values were calculated for each concentration and temperature and were used in the following analysis. An example of viscosity η versus cellulose concentration for cell 300 and cell 1,000 dissolved in EMIMAc at 20° and 100 °C is presented in Fig. 5.18. Solution viscosity decreases with increasing temperature, which is typical for classical polymer solutions and for cellulose dissolved in other solvents. Two regions on each viscosity–concentration dependence can be seen: a linear one in dilute regime and power law η ∼ C n above the overlap concentration C*. The exponent n was calculated for all cellulose–EMIMAc solutions; the values ranged from about 4 at low temperatures (0–40 °C) to 2.5–3 at high temperatures (60–100 °C). Similar values were reported for cellulose–LiCl/DMAc (n = 3 for bacterial cellulose and n = 4 for cotton linters and dissolving pulp) (Matsumoto et al. 2001).

Viscosity as a function of cellulose concentration for solutions of cell 300 and cell 1,000 in EMIMAc at 20° and 100 °C; the slopes for the concentrated region and indicated at the corresponding data. Adapted with permission from Gericke et al. (2009b). Copyright 2009 American Chemical Society
Cellulose intrinsic viscosity [η] was determined for solutions studied at different temperatures (Fig. 5.19). The intrinsic viscosity strongly decreases with temperature increase, for all samples investigated. This is a direct indication of a decrease in the thermodynamic quality of EMIMAc. A similar temperature influence on the size of cellulose macromolecules was observed for cellulose dissolved in 9 % NaOH–water (Roy et al. 2003). Cellulose–NaOH–water solutions are known to gel with temperature increase when polymer concentration exceeds the overlap concentration; no gelation or phase separation was observed for celluloses dissolved in imidazolium-based ionic liquids, unless they contain water.

Intrinsic viscosity of three cellulose samples dissolved in EMIMAc as a function of temperature. Lines are to guide the eye. Adapted with permission from Gericke et al. (2009b). Copyright 2009 American Chemical Society
Cellulose intrinsic viscosities obtained in EMIMAc were compared with the ones in other solvents, such as in cupriethylenediamine hydroxide (Cuen), in LiCl–DMAc, in 9 % NaOH–water and in cadoxene (Gericke et al. 2009b). In Cuen cellulose intrinsic viscosity was higher than in EMIMAc for the same cellulose sample, and in NaOH–water it was very similar. The overall conclusion is that EMIMAc is thermodynamically not a better solvent for cellulose as compared with the other ones. The exponent in Mark–Houwink equation relating intrinsic viscosity and molecular weight varied between 0.4 and 0.6 indicating that EMIMAc seems to be cellulose theta solvent.
The viscosity of cellulose–BMIMCl solutions was compared with the one of cellulose–EMIMAc (Sescousse et al. (2010a, b)). The properties of these imidazolium-based solvents are very different: EMIMAc is a room-temperature liquid with no glass transition or melting/crystallisation temperatures reported, while BMIMCl is solid at room temperature and melts around 60–70 °C. Because of the difference in solvent viscosity, the viscosity of cellulose–BMIMCl solution is 5–6 times higher than the one of cellulose–EMIMAc solution in the same conditions. If plotting the relative viscosities for both types of cellulose solutions in dilute regime, this difference disappears (Fig. 5.20).

Relative viscosities as a function of temperature for cell 180-EMIMAc (crosses) and cell 180-BMIMCl (dark points) solutions with 0.5 % (1) and 0.25 % (2) cellulose. The error bars of 10 % are shown only for 0.5 % cellulose–BMIMCl solutions in order not to overload the graph. Adapted with permission from Sescousse et al. (2010a). Copyright 2010 American Chemical Society
The comparison between cellulose intrinsic viscosity in EMIMAc and in BMIMCl for the same cell 180 is demonstrated in Fig. 5.21. BMIMCl solvent behaves in an extremely similar way to EMIMAc. In overall, dilute regime of the studied solutions is at polymer concentrations below 1 % for room temperatures (only for cellulose–EMIMAc solution, cellulose–BMIMCl is solid) and slightly lower than 2 % for the temperatures above 80 °C for both solutions.

Cell 180 intrinsic viscosity as a function of temperature for EMIMAc (crosses) and BMIMCl (triangles) solvents. Adapted with permission from Sescousse et al. (2010a). Copyright 2010 American Chemical Society
5.4.3 Influence of Water on Cellulose–EMIMAc Viscosity
It is known that imidazolium-based ionic liquids are extremely hygroscopic: They absorb humidity from the air not only modifying solvent viscosity but completely changing solution thermodynamics as far as water is cellulose nonsolvent. Cellulose processing on the pilot scale, as it is described in Kosan et al. (2008a, b), involves water evaporation from cellulose–IL–water slurry in a close-loop cycle of cellulose dissolution/coagulation. Water is removed from the slurry up to the moment when cellulose is dissolved in the ionic liquid. It is thus extremely important, both for cellulose shaping and chemical derivatisation as well, to understand how water influences cellulose–IL solution properties on the molecular level.
First, EMIMAc–water was prepared as a “new solvent” by mixing the components in different proportions. The viscosity of EMIMAc–water mixtures was analysed. Viscosity–shear rate dependence for EMIMAc–water at various water concentrations and temperatures was measured. In all cases studied, a Newtonian flow was recorded within at least two decades of the shear rates. Mean Newtonian viscosity values were determined, plotted as a function of water content and compared with viscosity calculated according to the logarithmic additive mixing rule. A noticeable difference between calculated and measured viscosity was obtained (Le et al. 2012). It should also be noted that heat is produced during mixing of EMIMAc and water (Le et al. 2012); it is undoubtfully an exothermal reaction suggesting, together with viscosity, special interactions between water and EMIMAc. This is an important result to keep in mind for the analysis of cellulose behaviour in ionic liquids in the presence of water.
An example of steady-state viscosity of a dilute cellulose–EMIMAc solution for various water contents is shown in Fig. 5.22. At water concentrations below 15 wt%, a Newtonian plateau was always found for 1 % cellulose solutions and lower concentrations. However, with 15 wt% water content and above the flow of dilute cellulose solution is shear thinning, clearly demonstrating that the properties of solutions changed despite the fact that they are transparent and no non-dissolved particles were seen with the optical microscope. We hypothesise that above 15 % water cellulose is not completely dissolved in EMIMAc; it exists as a sort of swollen agglomerates or “micro-gels” that are flowing as a “suspension”. These soft species consisting of highly swollen cellulose chains are deforming and structuring under shear leading to viscosity decrease with the increase of shear rate. If increasing cellulose concentration in EMIMAc–15 wt% water mixture to 3 % cellulose, non-dissolved crystals can be seen with polarised optical microscopy (Le et al. 2012). 15 % of water in EMIMAc thus seems to be the maximal limit of water allowing the dissolution of 1 % cellulose in these conditions.

Cellulose intrinsic viscosity as a function of water content in the solvent EMIMAc–water is presented in Fig. 5.23 for some selected temperatures. The intrinsic viscosity first increases with water concentration, goes through a maximum at about 8–10 wt% water and then decreases. The decrease of cellulose intrinsic viscosity at high water content, above 10 wt%, was expectable: Polymer coil should contract when a large amount of nonsolvent is added. The increase of polymer intrinsic viscosity with the addition of nonsolvent is more complicated to interpret. When a polymer is dissolved in a mixed solvent of different thermodynamic quality towards the polymer, various cases may occur. The presence of a bad or a nonsolvent does not necessarily decrease binary solvent thermodynamic quality. For example, a mixture composed of a poor and nonsolvent may become a good solvent: The second viral coefficient changes the sign from negative to positive and goes through a maximum as a function of solvent composition (Masegosa et al. 1984). A convex shape of the intrinsic viscosity versus solvent composition, with solvent being a mixture of good + poor, poor + θ-solvent or good + nonsolvent, had already been reported for synthetic polymers (Pouchly and Patterson 1976; Pingping et al. 2006; Budtov et al. 2010). Such a deviation from the classical Flory lattice theory was interpreted by the preferential solvation introduced as “local” solvent composition being different from the “bulk” composition (Dondos and Benoit 1973; Hong and Huang 2000).

Considering the results obtained on the flow of dilute cellulose–EMIMAc–water solutions (Fig. 5.22), the presence of non-dissolved cellulose in 3 wt% cellulose–EMIMAc–15 wt% water and the fact that water and EMIMAc are interacting (exothermal mixing, deviation from the additive viscosity), the following interpretation of the increase of cellulose intrinsic viscosity with the increase of water content (left-hand part of Fig. 5.23) was suggested (Le et al. 2012). Cellulose and water are competing for EMIMAc. Because of strong EMIMAc–water interactions, cellulose is self-associating, probably via inter- and intramolecular hydrogen bonding, and aggregates are formed. It is the hydrodynamic size of these aggregates and not of one molecule that is determined with the intrinsic viscosity, as suggested in the schematic presentation in Fig. 5.24. The higher the water concentration, the larger the size of cellulose aggregates up to such water content at which cellulose becomes hardly soluble leading to a further strong decrease of the intrinsic viscosity.

5.4.4 Influence of Cellulose on the Diffusion of [EMIM]+ and [Ac]− Ions in Cellulose–EMIMAc Solutions
In order to better understand cellulose–ionic liquid solution microscopic properties (transport phenomena, polymer–solvent interactions), diffusion-ordered two-dimensional NMR spectroscopy (DOSY) was used to measure ions’ self-diffusion coefficients and correlate their mobility with cellulose concentration and solution temperature (Lovell et al. 2010). First, the change in the chemical shifts of each EMIMAc spectral band on cellulose concentration was recorded and analysed. The obtained cellulose concentration dependence of chemical shifts in cellulose–EMIMAc solutions was similar to the ones reported in Zhang et al. (2010) for cellobiose dissolved in EMIMAc. It was thus confirmed that cellulose dissolution in EMIMAc proceeds via hydrogen bonding and coordination of [EMIM]+ and [Ac]− ions with cellulose hydroxyl groups.
The diffusion coefficients of [EMIM]+ and [Ac]− ions at different cellulose concentrations and solution temperatures were calculated. The smaller anion was observed to diffuse slower than the larger cation (Lovell et al. 2010), a result already reported for other imidazolium-based ionic liquids (Noda et al. 2001; Tokuda et al. 2006) and also confirmed by molecular dynamic simulations (Tsuzuki et al. 2009, Urahata and Ribeiro 2005). The faster diffusion of a larger cation was explained by anisotropic diffusion of imidazolium ring in the directions of the ring plane as compared with the direction orthogonal to it (Urahata and Ribeiro 2005). For the case of cellulose–EMIMAc solution, the ratio between the diffusion coefficients of anion to cation is smaller than one, and it increases with temperature (Fig. 5.25) (Lovell et al. 2010). It is interesting to note that the presence of cellulose increases the difference between the diffusivity of anion and cation, and this ratio becomes less sensitive to temperature (Fig. 5.25).

The ratio of [Ac]− to [EMIM]+ diffusion coefficients at various cell 180 concentrations as a function of temperature. Lines correspond to linear approximations. Error bars are shown only for EMIMAc data in order not to overload the graph. Reprinted with permission from Lovell et al. (2010). Copyright 2010 American Chemical Society
The influence of cellulose concentration on the diffusion of ions was analysed with different theoretical approaches developed for polymer solutions. Obstruction, hydrodynamic and free-volume models were tested for fitting experimental data (Lovell et al. 2010). For example, by applying the obstruction model we realised that the notion of bound solvent and hydration shell around cellulose macromolecule should be considered to match theoretical predictions with experimental data. The proportion [EMIM]+:[Ac]−:hydroxyl = 1:1:1 was taken as a background approach. It gives the ratio of 5 between the volume fraction of cellulose and volume fraction of cellulose with bound solvent. With this ratio, different variations of the obstruction model approach fit, more or less, the experimental data (Lovell et al. 2010). The result obtained predicts that when cellulose concentration in EMIMAc reaches ≈ 27 wt%, all solvent molecules are bound in the primary solvation shell. This cellulose concentration seems to be the maximal possible to be dissolved in EMIMAc (Lovell et al. 2010). In the next section it will be shown that this prediction is confirmed by cellulose–EMIMAc–DMSO phase diagram.
The free-volume model was found not to give a satisfactory agreement with the experimental data for both ions’ diffusion dependence on cellulose concentration. On the contrary, the hydrodynamic model as developed by Phillies (1986) showed a good agreement with the obtained experimental data (Lovell et al. 2010).
5.4.5 Phase Diagram and Solubility Limit of Cellulose in EMIMAc in the Presence of DMSO
DMSO (as well as DMF) is used to perform chemical reactions on cellulose in the homogeneous conditions. The main reason is that these co-solvents strongly decrease ionic liquid viscosity. They can also be interesting candidates for cellulose processing, allowing matching solution viscosity to the desired values. However, as far as DMSO is not cellulose solvent, the key question for processing is how the maximal cellulose concentration soluble in EMIMAc–DMSO depends on DMSO content. The goal of this work was to answer this question and build a phase diagram of cellulose in EMIMAc–DMSO.
Cellulose–EMIMAc–DMSO solutions were prepared at various EMIMAc–DMSO proportions and different cellulose concentrations and carefully analysed with optical microscopy using polarised and nonpolarised light in the view of the presence of non-dissolved cellulose. The magnification used was such that 5–10 μm objects were easily detectable. With this method, it is not possible to judge if the dissolution occurred on the molecular level; however, a phase diagram can be built in the first rough approximation. The results are presented in Fig. 5.26 as cellulose concentration in the system versus weight fraction of DMSO in the mixture EMIMAc–DMSO. Open points correspond to completely transparent (in nonpolarised light) or completely dark (in polarised light) solutions; crosses correspond to the cases when even a few small non-dissolved particles were detected.

Phase diagram for cell 180 in EMIMAc–DMSO. Solid line corresponds to the maximal cellulose concentration soluble in EMIMAc–DMSO calculated supposing 1 anhydroglucose unit binding 2.4 mol of EMIMAc; dashed lines correspond to AGU binding 3 mol (lower) or 2 mol (higher) of EMIMAc
The proportion between the maximal cellulose concentration dissolved at a given co-solvent fraction and the corresponding EMIMAc concentration in the solution was estimated. We supposed that neither cellulose nor EMIMAc interact with DMSO. The amount of EMIMAc moles per anhydroglucose unit (AGU) turned out to be a constant value around 2.4, not depending on DMSO concentration in solution, within the experimental errors. It seems that indeed EMIMAc and DMSO as well as cellulose and DMSO are not interacting. The theoretical maximal concentration of cellulose soluble in EMIMAc–DMSO was calculated for various cases: C cell max = 2, 2.4 and 3 mol of EMIMAc. The results are shown in Fig. 5.26: solid line for C cell max = 2.4 mol EMIMAc and upper and lower dashed lines for C cell max = 2 and 3 mol EMIMAc, respectively. C cell max = 3 mol EMIMAc overestimates the amount of EMIMAc needed to dissolve cellulose as far as the corresponding dashed line is passing through the well-dissolved region. 2 mol of EMIMAc per one AGU underestimates the amount if EMIMAc needed to dissolve cellulose; the corresponding line intersects the region that contains non-dissolved cellulose. We can thus conclude that one AGU needs about 2.3–2.5 mol of EMIMAc to be dissolved. This means that maximal cellulose concentration possible to dissolve in EMIMAc is around 28–30 wt%, the values predicted by NMR (Lovell et al. 2010).
5.4.6 Conclusions
Ionic liquids are a class of powerful cellulose solvents; they open new opportunities in cellulose processing and performing homogeneous chemical reactions. In our research we are trying to understand the fundamental properties of cellulose–ionic liquid solutions (polymer–solvent interaction, solvent thermodynamic quality, cellulose dissolution limit), and we are using ionic liquids for making highly porous cellulose materials, aerocellulose, as described in Chap. 5.
5.5 Stabilisation of Cellulose: NMMO Solutions (Lyocell Process)
Quality of the cellulose pulp and dissolution conditions determine the spinning behaviour and the performance of the finished material. Unfortunately, it is not possible to evaluate the suitability of pulps for the cellulose–NMMO process called lyocell only by analysis data such as intrinsic viscosity, molecular weight and their distribution or the content of heavy metals, ash or carbonyl and carboxyl groups. Although cellulose is more or less chemically identical in all pulps, there are significant differences in their physical states, mainly influenced by origin and extraction process of the pulps. Therefore, standard conditions are required to maintain dissolution and spinning quality, especially with the aim of upscaling (Michels and Kosan 2000). Solution quality, solution state and thermostability are important factors for characterising cellulose solutions for using them in the lyocell process.
5.5.1 Solution Quality and Solution State
Solution quality refers to the “spinning stability”, which guarantees continuous fibre shaping of large spinning masses under technical conditions. Possible inhomogeneities like undissolved pulp residues can lead to clumps in the jet holes followed by permanent breaks of the filament. Consequently, the maximum diameter of particles is in direct relation to the spinnable fineness. To ensure a fineness of 1.3 dtex (B-type fibre), the spinning dope should not contain any undissolved particles >15 μm. A visual observation of the solution quality gives a with a detection threshold down to ~20 μm. Therefore, particle analysis by laser deflection must be used to provide information about the content and the distribution of particles in the range from 0.5 to 175 μm. Kosan and Michels (1999) have shown that the particle content depends on pulp origin and pretreatment and that the undissolved particle distribution is determined by the activation and solution conditions such as shear flow present in the mixing reactor, dissolution temperature and time.
Whereas solution quality describes the microscopic and submicroscopic range of undissolved particles, the solution state characterises its molecular range. Using the relatively easy accessible rheological parameters zero shear viscosity η 0, storage G′ and loss modulus G″ and other parameters derived from the ones mentioned above, width of the molecular weight distribution of linear polymers in concentrated solutions can be determined (Michels 1998). Zero shear viscosity is a function of the molecular weight, concentration and temperature. By monitoring G′ and G″ as a function of angular speed ω at a reference temperature (usually 85 °C), master curves of a deformation function can be calculated (Fig. 5.27). The quotient of zero shear viscosity and viscosity at the “crossover” point where η # = G′/ω # = G″/ω # characterise the viscoelastic behaviour of the dope and correspond to the molecular inhomogeneity parameter U η = η 0/η # − 1. Measurements of different pulp origins give inhomogeneity parameter U η of 2.5–4.2 for cotton, 4.9–8.0 for wood and 6.8–9.5 for cellulose mixtures.

Master curve of a deformation function for a cotton pulp as a function of angular speed (frequency) (DP 445, 13.2 % in NMMO) at 85 °C
The relaxation time, calculated from the deformation function, is proportional to the molecular weight. Figure 5.28 displays the weighted relaxation time spectra of different wood pulp/NMMO solutions with varying DP and inhomogeneities. Lower molar mass fraction of the pulp causes increased inhomogeneities connected with low relaxation times and, consequently, leads to worse fibre performances. Short-chain fractions accelerate the relaxation and do not contribute to the entangled cellulose network. Both inhomogeneity and relaxation time have a direct effect on the range of suitable spinning conditions and on the mechanical parameters of the spun fibres or filaments because they determine the kinetics of fibre shaping. Temperature dependence of the relaxation time explains the significant influence of the spinning temperature on spinning stability and fibre properties.

Relaxation time spectra weighted of different wood pulp/NMMO solutions with varying DP and inhomogeneities
5.5.2 Thermostability of Cellulose Solutions
Cellulose dissolution and shaping of the solutions are always connected with an alteration of the morphological structure accompanied by chain length reduction. Origin of the pulp, the nature of the solvent and the process conditions determine the decrease of the degree of polymerisation (DP). Whereas the DP reduction in systems like DMAc/LiCl is quite low, ionic solvents such as NMMO or ionic liquids (ILs) are involved in chemical reactions provoking degradation of both polymer and solvent. Therefore, a technical upscaling of a fibre-shaping technology is only possible if the process is stabilised, as for the lyocell process, which breakthrough arose from the introduction of NaOH/propyl gallate system, which suppresses the radical formation of NMMO and chain scission of cellulose (Buijtenhuijs et al. 1986).
Although the dissolution of cellulose in NMMO, being a non-derivatising solvent, and its fibre spinning are entirely physical processes, chemical alterations may appear under industrial conditions. High temperatures during cellulose dissolution and long residence times of the solutions during the spinning process result in discoloration and degradation of NMMO and cellulose. These reactions lower the recovery rate of the solvent and decrease the product performance (Taeger et al. 1985; Rosenau et al. 2001). A mass-related accumulation of the reaction mixture in the course of production increases the risk of exothermic reactions, so-called thermal runaway reactions, with an erratic temperature and pressure rise. Both chemical stabilisation and special technological features may prevent exothermic reaction and deflagrations (Firgo et al. 1994).
NMMO/cellulose segregation occurs as soon as the hydrogen bond system, which is considered as being responsible for cellulose solubility, is deteriorated (Novoselov et al. 1997; Cibik 2003). Local enrichment of NMMO and generated boundary surfaces of crystallised cellulose represent potential sites for subsequent reactions. NMMO is well known as an oxidising reagent in organic chemistry. Further, high amounts of many additives are able to disrupt hydrogen bonds by changing the polarity of the cellulose/NMMO system. In Fig. 5.29 the factors influencing the thermal stability are summarised.

Detailed ESR spectroscopy analysis of degradation processes in cellulose/NMMO solutions and, hence, formation of radical types have been carried out by Konkin et al. (2006) and Wendler et al. (2008). It shows that nitroxyl-type radicals R–NO∙R′ and oxymethylene species ∙O–CH2~ and ∙CH2–O~ (R11) at the first step were detected under irradiation as a result of ring degradation by a first-order reaction pathway (Scheme 5.1). Proposed NMMO ring degradation mechanism includes three variants (Ia, Ib, Ic) of di-radical types. The second step as a second-order reaction is invisible by ESR because the products are not radicals. However, they have to be potential precursors of further radical formations by step III under the continued laser illumination and were studied by high-performance liquid chromatography (HPLC). Further on, methyl ∙CH3 and formyl ∙CHO radicals are formed at the third step.

Suggested reaction pathway leading to degradation of NMMO–cellulose mixtures
Analytical investigations of lyocell dopes with regard to the thermostability are commonly provided by rheological, calorimetric, chromatographic and spectroscopic methods (Taeger et al. 1985, Rosenau et al. 2001). Especially, measurements of the pressure gradient and the generation of chromophores give information about the time dependence behaviour of the sample when kept at a constant temperature (Wendler et al. 2005a). Beside the type of cellulose, its concentration plays an important role. Higher cellulose concentrations in the solution mean more reactive end groups and, as a consequence, higher concentrations of degradation products measured. Chromatographic methods revealed morpholine, N-methylmorpholine, N-formylmorpholine and formaldehyde as the main products resulting from reactions of the N–O bond. The activation of the most labile structure in the molecule occurs by protonation, complex formation with metals or O-alkylation. The cleavage of the NMMO ring structure as a consequence leads to resonance-stabilised nitroxyl radicals and stable ring degradation products. Acetaldehyde, 2-methylaminoethanol, 2-ethylaminoethanol and N,N-diethylhydroxylamine were detected by HPLC and attributed to cleavage products. Figure 5.30 displays the individual fingerprints of ring degradation products for solutions with different cellulose concentrations and the corresponding onset temperature.

Ring degradation products and the corresponding onset temperature (T on) of cellulose/NMMO solutions with varying cellulose concentrations
The first thermal activity of a solution indicated as the onset temperature (T on) is preferably measured by calorimetric methods and determined by plotting the deviation of pressure with respect to time (dp/dt) versus temperature. Cellulose/NMMO solutions give onset temperatures in the range 130–160 °C. Together with DSC mini-autoclave measurements with a sample weight of 2 g and analysis of the slope of pressure, it gives information about gas evolution even at lower temperatures and shows larger differentiations resulting in a more accurate characterisation of the influences of stabilisers and additives.
In order to prevent segregation and degradation of cellulose and NMMO, a stabiliser has to adjust the pH value, to scavenge by-products and to complex heavy metals. Propyl gallate commonly used as phenolic antioxidant in plastics, elastomers, fuels, etc. (Klemchuk 1985) was first introduced as a standard stabiliser preferably applied for trapping of radicals by forming relatively stable phenoxy radicals (Brandner and Zengel 1980). Further patents proposed reductants, e.g. amines, sulphites, aldehydes, sugars (Lukanoff and Schleicher 1981), bases (Franz et al. 1983), phosphonates (Laity 1983), sterically hindered phenols (Michels and Mertel 1984) and radical scavengers like 3,3′-thiodipropanol or carbon tetrabromide (Guthrie and Manning 1990). The combination of alkaline and antioxidant stabilisation (NaOH, propyl gallate) is generally accepted as the most efficient mode (Kalt et al. 1993). However, propyl gallate may interact with cellulose end groups and form coloured reaction products.
Up to now, no stabilisation system can be assessed as an ideal practical solution. The mentioned stabilisation procedures exclusively concern cellulose/NMMO solutions without any additive. Substances with functional groups or surface-active materials additionally cause degradation processes. TITK investigations of solutions modified with an IER and stabilised with NaOH and propyl gallate show decreased thermal stability leading to exothermic reactions during extrusion. Similar results were obtained in case of the presence of activated charcoal (Wendler et al. 2005b). Finally, it turned out that low thermal stability and large reduced viscosity will result in unstable spinning performance.
Table 5.4 summarises different bases, amines, complexing agents and phenolic oxidants employed as stabilisers in TITK under real time conditions. The measurements of the onset temperature (T on) were taken as indicator for thermal stability. Commonly used stabilisers like propyl gallate; Irganox, a phenolic hydrazine stabiliser in plastics processing or the morpholine/HCHO system show high T on for unmodified solutions but fail to work for solutions modified with reactive charcoal or ion exchange resins. Therefore new stabilisers able to weaken surface reactions must be found. Additives such as activated charcoal or carbon black may have a serious effect as heterogeneous catalysts increasing degradation.
TITK investigations using a novel stabilising system with the combination of iminodiacetic acid sodium salt (ISDB) each covalently bound to a styrene/divinylbenzene copolymer exhibit favourable stabilising properties of cellulose/NMMO solutions modified with carboxyl group containing surface-active additives (Büttner et al. 2003). To estimate the effects of the polymeric stabilising system, the chelating ionogenic groups of ISDB in combination with the amine reaction potential of BSDB have to be taken into account. The iminodiacetic acid group forms coordination bonds between its donor atom nitrogen and metals. On the other hand, the partly sodium-neutralised carboxylic groups act as a buffer keeping constant pH values and thus maintaining the hydrogen bond system between cellulose and NMMO. Benzylamine of BSDB as a primary amine is able to bind carbon dioxide, acids and aldehydes. Additionally, BSDB may deactivate highly reactive intermediates, such as the aminiumyl radical, by scavenging these compounds within the polymeric skeleton.
Calorimetric, spectroscopic and rheological measurements were used to describe the stabilisation effects of ISDB/BSDB on modified solutions compared to solutions without stabilisers and the “state-of-the-art” stabiliser NaOH/PG by Wendler et al. (Wendler et al. 2006, 2008). Table 5.5 summarises an extract of the investigated additives. Since it is not possible to present here detailed examinations, only T on is used to evaluate the thermal stability when the new stabiliser system is applied. Generally, a shift of T on to higher values is registered on an average increase of 4.5 °C in a range from 0 to 8 °C.
5.6 Cellulose–Other Polysaccharide Blends
Multicomponent systems (polymer blends, alloys, composites, interpenetrating networks) have been receiving more and more attention owing to their scientific interest and practical applications. Blending is an efficient technique for the development of novel materials with improved properties (Guo 1999; Cazacu and Popa 2004). Especially cellulose materials offer several advantages when combined with synthetic or natural polymers due to their low density, high modulus, strength and high stiffness, little damage during processing, small requirements of processing equipment, biodegradability and relatively low price (Zadorecki and Michell 1989; Joly et al. 1996). Hydrogen bonding between –OH groups are recognised as an important factor in providing driving forces for the attainment of thermodynamic miscibility in many polymer blend systems where polysaccharides are present (Nisho 1994). Desired coupling of different polysaccharides’ functionalities by joint or separate dissolving allows the shaping of the arising solution and solidification towards the polysaccharide blend (Taeger et al. 1997).
Most reports in literature concern the combination of a polysaccharide (including derivatives) and a synthetic polymer. There exist only few reports about polysaccharide/polysaccharide blends from different R&D groups, e.g. combination of pure cellulose with chitosan (Hasegawa et al. 1992), starch, xanthan, locust bean gum (LBG), guar gum, tragacanth gum, chitosan (Taeger et al. 1997), chitin (Pang et al. 2003; Kondo et al. 2004), sodium alginate (Kim et al. 2007) or carrageenan (Prasad et al. 2009). Some interesting studies dealt with hemicelluloses as structure regulators of native cellulose (Atalla et al. 1993; Zhang and Tong 2007). Recently, the incorporation of 15 different polysaccharides into a cellulose matrix using three solvents was studied to evaluate the interactions of polysaccharides or mixtures of those in solutions and the solid state after shaping (Wendler et al. 2010). In the following main results of that comprehensive study are inserted.
Various polysaccharides were chosen to find new structured biopolymer blends bearing adjustable properties, as shown in (Fig. 5.31). Three solvent routes for dissolving were followed: (1) sodium hydroxide–water (NaOH), (2) N-methylmorpholine-N-oxide monohydrate (NMMO) and (3) ionic liquids (ILs). By studying structure–property relations with a variety of analytical methods, new possibilities for the design of interesting cellulose-based fibre materials can be opened.

Expected effects of selected blend polysaccharides on cellulose properties
Dissolution screenings yielded promising polysaccharides which were used for the preparation of cellulose/polysaccharide solutions and, consequently, for shaping of blends with cellulose. Chitin and chitosan were not soluble in any of the three solvents. Dissolution was possible in the case of carboxymethylcellulose, cellulose carbamate, xylan, xanthan and the mannans, guar gum, locust bean gum and tragacanth gum. In opposite to NaOH and NMMO, the EMIMac ionic liquid is not able to dissolve polysaccharides with functional groups like carrageenan or methylhydroxyethylcellulose above only about 1 %.
Solubility and miscibility with cellulose were evaluated by microscopy, DSC, particle analysis and rheology. To evaluate the states of solutions, microscopic images between cross polarisers were taken at room temperature. Most of the pure polysaccharide solutions were transparent and clear. Interestingly, only one polysaccharide solution—cellulose carbamate (15 %) in EMIMAc—showed an anisotropic behaviour which is of special importance for shaping of carbamate in ionic liquids (Rademacher et al. 1986). In the case of combination with cellulose and EMIMAc, xanthan, tragacanth gum and LBG, anisotropic structures could be observed as shown in Fig. 5.32 and interpreted as liquid crystalline (Kosan et al. 2008a, b; Chanzy et al. 1982). Usually, cellulose solutions in NMMO with polymer concentration higher than 20 % and molar ratios water/NMMO lower than one were found to be anisotropic.

Microscopic images of cellulose/blend polysaccharide solutions in EMIMac. Left: cellulose/LBG (97.5/2.5); middle: cellulose/LBG (95/5); right: cellulose/tragacanth gum (95/5)
Different types of the polysaccharide blends can also be shown for NMMO solutions. Figure 5.33 shows cellulose/xylan solutions and reveals the presence of particles of xylan in the range of 2–4 μm suggesting immiscibility with cellulose. However, it has to be noticed that these xylan particles are still present at the cellulose/xylan ratio 0:100, meaning that the presence of these particles is not due to the immiscibility between xylan and cellulose chains but to the interaction of xylan with the NMMO monohydrate. A possible explanation for the observed formation of xylan particles is the hydrophobic character of xylan chains related to the amount of side chains such as arabinose (Sternemalm et al. 2008) that can involve aggregation of the chains in aqueous solvent such as NMMO monohydrate. In comparison, cellulose/xanthan solutions reveal a continuous phase for all concentration ratios. It was not possible to distinguish the xanthan phase from the cellulose phase in the solutions.

Microscopic images of cellulose/xylan solutions at 90 °C with a concentration ratio of 75/25 (a) 50/50 (b), 25/75 (c) and 0/100 (d) after image analysing (xylan particles are surrounded)
Rheological investigations of cellulose/xylan solutions reveal no entanglement or interactions between cellulose and xylan chains. This is confirmed by a decay of zero shear viscosity accompanied by a decreasing relaxation time with increasing xylan amount. The rheological study of cellulose/xanthan solutions shows the inverse phenomenon compared to xylan, exhibiting a large increase of zero shear viscosity and relaxation time with xanthan concentration, mainly due to the high molar mass of xanthan chains (up to two millions).
Fibres in form of a multifilament yarn were manufactured from cellulose–other polysaccharide solutions in NaOH, NMMO and EMIMac using a wet-spinning method. As expected, textile physical properties are lower than unmodified fibres with slightly better properties for fibres spun in NMMO and EMIMac (Table 5.6). Residue analysis was carried out to determine the uptake rates of various polysaccharides which were added to the cellulose spinning dope during fibre production. The contents of arabinose, galactose, mannose, xylose and glucose in pure polysaccharide and in the blended fibre were measured. The content of the polysaccharide blended with cellulose in the fibres increases in the order NaOH–NMMO–EMIMac. This content depends rather on solvent than on type of polysaccharide and technology. Even though NMMO and EMIMac fibres were spun at the same equipment, the latter case favours higher amounts of the blended polysaccharide. It can be concluded that the coagulation of polymers from NMMO is different, meaning that the hydrogen bond system between cellulose chains and the blended polysaccharide is weaker than inside the cellulose skeleton. The release of the blended polysaccharide out off the skeleton might be easier.
Spinning of fibres from all the solvents was possible even when using the higher molar polysaccharides. It has to be pointed out that the insertion of fine-milled polysaccharides into the cellulose is the crucial factor as previously described for the modified lyocell process where organic or inorganic compounds are inserted (see Chap. 5). Nevertheless, the morphology of blended solutions is of great interest to prevent clogging of filters and to control the final morphology and properties of the regenerated fibres.
X-ray diffraction measurements of the coagulated cellulose/polysaccharide blend fibres were performed to evaluate crystal orientation. Tenacity of cellulose fibres depends on chain orientation in the amorphous regions, whereas modulus depends on crystallinity and crystallite orientation. All examined fibre types give a cellulose II crystal structure. Main lattice planes are (1–10) and (110/020), which appear at the equator as discrete spots with small arcs of varying amplitude indicating different crystallite orientation. Figure 5.34 shows, for example, flat film diagrams of a cellulose/gum tragacanth fibre. Among the polysaccharide blends of the same fibre type, there are less significant differences between solvents, with the exception of the EMIMac fibres, whilst between polysaccharide blends, differences are larger. NMMO fibres have the highest orientation compared to EMIMac and NaOH fibre types.

X-ray flat film diagrams of cellulose/gum tragacanth fibre: left EMIMac, middle NaOH, right NMMO
From AFM analysis, it can be concluded that immiscibility state is not changed by the spinning, whereas the fibres exhibit distinctive differences in surface topography with regard to the solvent. Intertwining areas of different components can be differentiated. In general, on 1 × 1 μm sample size, surface roughness of fibres is increasing in the following sequence: EMIMac ≤ NMMO < NaOH (Fig. 5.35). Most fibres show irregular surfaces with fibrillar bundles or grainy structures of two phases as in the case of xanthan or tragacanth.

AFM of surface topography on 1 × 1 μm sample size for pure cellulose (above) and cellulose/xanthan (below) fibres from EMIMac (left, roughness 2.0 nm), NMMO (middle, 4.8 nm) and NaOH (right, 20.6)
Contact angle measurements revealed only minor changes compared to the unmodified cellulose fibres. It has to be pointed out that the standard deviation of all samples is relatively high; the reason could be the sample non-hom ogeneity. The water contact angles of cellulose/polysaccharide fibres from EMIMac and NaOH are almost the same, i.e. 83–88°, while blend fibres from NMMO are slightly more hydrophilic, i.e. values are ranging from 73 to 76°. However, water contact angles are comparatively higher than viscose (68°) or modal fibre types (77°) (Persin et al. 2002).
Electrokinetic investigations (zeta potential measurements, ZP) were carried out to study the surface charges of the blended fibres. Apart from some exceptions, NMMO and EMIMac fibres show plateau values ranged from −7 to –10 mV, whereas the ZP values of NaOH fibres are in regions between –7 and –2 mV. The latter values can be seen in connection with the distinctive higher water retention, lower crystallite orientation and even much higher roughness as AFM measurements revealed. Indeed, water retention values of NMMO and EMIMac fibres are ranging between 40 and 54 % in comparison to main higher values obtained for the NaOH fibres (77–93 %).
Even though X-ray measurements revealed only little changes in the crystalline orientation of the blended fibres, further experiments were done to find out how the disturbed morphology of cellulose in blended fibres affects the accessibility, reactivity and adsorption properties. Mainly, the volume and inner surface areas of the voids play the decisive role in all cellulose heterogeneous chemical reactions (Fink et al. 1998). Consequently, sorption behaviour studied after special treatment of the fibres with distilled water buffered to varying pH value is enhanced for fibres from NaOH compared to EMIMac and NMMO fibres, which can be related to the higher surface roughness of the surface and lower crystallinity. Furthermore, dyeability tests show the same trends regard to the fibre types. Compared to the reference pure cellulose fibres, dye affinity of the blend fibres is clearly enhanced. As expected, fibres with carboxymethylcellulose and xanthan exhibit the highest distribution coefficient and monolayer sorption capacity for the cationic dye used. It appears that the solvent and/or the spinning process exerted an influence on the accessibility of carboxyl groups.
As was shown, a variety of parameters have an impact on the performance of the blend fibres, mainly the type of spinning technology used, the nature of the solvent and the type of polysaccharide mixed with cellulose. It can be concluded that what matters is the type of polymer added to cellulose more than the miscibility or not character of the mixture.
5.7 Cellulose–Lignin Mixtures in Solution (Sescousse et al. 2010b)
Cellulose and lignin are the main components of plant fibres. The synergy of their properties, together with hemicellulose, results in all varieties and special properties of natural fibres. For most of applications, cellulose and lignin have to be separated. During the separation, cellulose and lignin are in contact being in solution or suspension state (e.g. in papermaking); it is thus very important to know if they interact or not. Some applications suggest using lignin as a component in blends with polysaccharides. If cellulose or its derivatives are mixed with lignin, will the final material demonstrate the synergetic properties as it occurs in a natural fibre? The answer depends on the type of lignin, common solvent and mixing conditions used.
Literature reports some interactions between cellulose and lignin when they coexist in a natural fibre. It was shown that lignin is oriented preferentially parallel to the surface of the cell wall (Terashima and Seguchi 1988; Atalla and Agarwal 1985). Carbohydrates seem to form complexes with lignin as a unit of secondary cell wall structure (Chesson 1993). Molecular dynamics studied the growth and deposition of lignin on a cellulose surface in a cell wall. These studies supported the above-mentioned experimental finding and demonstrated not only a certain spatial organisation of lignin around cellulose microfibrils but also gave some evidences of links between two polymers (Houtman and Atalla 1995; Perez et al. 2004; Besombes and Mazeau 2005). However, the nature of these links was not specified.
In papermaking, lignin should be separated from cellulose. Possible redeposition of lignin onto cellulose fibres during cooking, washing and bleaching is of great importance, and thus, the compatibility and interactions between cellulose and lignin must be known. Maximova et al. (2001, 2004) studied the absorption of lignin by cellulose fibres and showed that it is absorbed due to capillary forces; lignin can be then easily removed by washing with water. They concluded that there was no true molecular adsorption of lignin onto fibres under various pH and ionic strengths investigated.
Blending of lignin with cellulose and cellulose derivatives has been extensively studied in the nineties (see, e.g. Rials and Glasser 1989, 1990; Dave et al. 1992; Dave and Glasser 1997). Fibres and films were usually prepared by mixing polymers in a common solvent followed either by solvent evaporative drying (film casting) or washing (fibres wet spinning). In the case of organosolv lignin–hydroxypropyl cellulose (HPC) films, miscible amorphous blends at low organosolv lignin concentrations were prepared and phase separation above 50 % of lignin was observed (Rials and Glasser 1990). For the mixtures of organosolv lignin with ethyl cellulose or with cellulose acetate butyrate (CAB), similar results were reported, but lignin content at which phase separation occurred was lower, 5–10 %. Fibres were spun from organosolv lignin–CAB mixtures and regenerated in water (Glasser et al. 1998). SEM and TEM results showed pore-size increase with the increase of organosolv lignin content. Analysing the above-mentioned results obtained on organosolv lignin–cellulose derivatives mixtures, Glasser et al. (1998) claim that there is an evidence of strong intermolecular interactions between the two components. Cellulose and lignin were mixed in dimethylacetamide/LiCL solvent and mixture viscosity was studied by Glasser et al. (1998): An increase of mixture dynamic elastic modulus with the addition of lignin was recorded, and it was explained by strong “secondary interactions” between the components.
Work performed in Centre de Mise en Forme des Matériaux, France, had the objective to study how the addition of lignin influences the properties of cellulose–8 % NaOH–water solutions. Certain simplifications, as compared with the naturally occurring systems, were made: We used microcrystalline cellulose and organosolv lignin. Mixture viscosity was compared to the result obtained with various cases known for polymer pairs forming interpolymer complexes or making an immiscible blend. Gelation of cellulose–8 % NaOH solution in the presence of lignin was investigated. Finally, dry lignocellulose materials were obtained according to the procedure used to prepare aerocellulose (Gavillon and Budtova 2008; Sescousse and Budtova 2009). Samples were analysed in terms of porosity and amount of lignin.
5.7.1 Cellulose–Lignin Mixtures in Aqueous 8 % NaOH
Microcrystalline cellulose Avicel PH101 was of DP 180. Organosolv lignin (“lignin” in the following, from vTI-Institute for Wood Chemistry, Hamburg, Germany) was from Norway spruce pulp. The molecular weight is M w = 7,300 g mol−1 with polydispersity of 4.1, as determined with GPC using polyethylene glycol calibration. This lignin is soluble in aqueous solutions of neutral and high pH. Cellulose–NaOH solutions were prepared as described in Gavillon and Budtova (2008) and Sescousse and Budtova (2009). Briefly, an aqueous solution of 12 % wt NaOH was cooled down to −6 °C. Cellulose was swollen in distilled water in a certain proportion and kept at 5 °C. 12 % NaOH–water and swollen-in-water cellulose were mixed at −6 °C with a stirring rate of 1,000 rpm for 2 h. The ready solutions of various cellulose concentrations in 8 % NaOH–water were stored at 5 °C to avoid aging. Lignin was dissolved in aqueous 8 % NaOH by stirring for a few hours at room temperature. Cellulose–lignin mixtures in 8 % NaOH–water were prepared by simply mixing ready solutions in different proportions. All mixtures prepared were visually homogeneous and did not show any phase separation within 1 week storage time in refrigerator. Two types of mixtures were prepared: (a) series of mixtures with the total polymer concentration being constant and composition varied and (b) mixtures with cellulose concentration being constant and lignin concentration (and thus total polymer concentration) varied.
To prepare dry samples, cellulose–lignin–8 % NaOH–water mixtures were poured into cylindrical moulds with dimensions of about 3–5 cm height and 2 cm diameter and kept at 65 °C for 2 h. These conditions were chosen to ensure cellulose gelation (Roy et al. 2003; Gavillon and Budtova 2008). The gels were then placed in coagulation bath of either distilled water or 0.1 and 1 mol L−1 acetic acid. Bath liquid was regularly changed until pH did not vary anymore indicating that all NaOH is washed out. The samples were then rinsed with water to remove acid and then washed in acetone to remove water which is not compatible with CO2. Swollen-in-acetone samples were dried in supercritical CO2 conditions as described earlier (1 L autoclave, 80 bar, 35 °C) by (Gavillon and Budtova 2008; Sescousse and Budtova 2009). After depressurisation (4 bar per hour at 37 °C), light brown samples of aero-lignocellulose were extracted and analysed.
The flow of cellulose–(7–9 %)NaOH–water solutions and viscosity–concentration–temper ature dependences have been studied extensively (Roy et al. 2003; Egal 2006; Gavillon and Budtova 2008). It was shown that above the overlap concentration solutions are gelling with time and temperature increase. Gelation time is exponentially temperature dependent, as shown by Roy et al. (2003) and Gavillon and Budtova (2008). Viscosity of cellulose solutions in conditions far from gel point is Newtonian below the overlap concentration and slightly shear thinning above it. The flow of lignin–8 % NaOH–water was investigated for 6–20 % lignin concentrations in the temperature interval from 0° to 35 °C. In all cases studied lignin solutions behave like Newtonian fluids; an example for 20 % lignin–8 % NaOH–water is shown in Fig. 5.36. This is what should be expected due to lignin low molecular weight. The activation energies Ea were calculated from viscosity–temperature dependences: Ea15% lign = 16 ± 2 kJ mol−1 and Ea20% lign = 18 ± 2 kJ mol−1.

One way to estimate the interactions between the components in a common solvent is to study how mixture viscosity varies with its composition. The experimental data η exp obtained should be then compared with a theoretical dependence of mixture additive viscosity η add calculated at different compositions which assumes the absence of special interactions between the components (i.e. polymer1–polymer2 interactions are the same as the interactions between the macromolecules of the same type). For the mixtures where the concentration of at least one of the components is above the overlap concentration, the additive viscosity η add must be calculated according to the logarithmic rule below:
where φ 1 and φ 2 are the weight fractions at which the components are present in the mixture (φ 1 + φ 2 = 1); η 1 and η 2 are the components’ viscosities at φ 1 =1 and φ 2 = 1, respectively.
In order to plot the experimental dependence of cellulose–lignin mixture viscosity as a function of mixture composition, the flow of cellulose– lignin–8 % NaOH–water of various compositions with total polymer concentration of 6 % was studied at 15 °C. These conditions were chosen as a compromise between not too low viscosity of lignin solution and not too fast gelation of cellulose solutions (e.g. 7–8 % cellulose solutions are gelling irreversibly within a few minutes as soon as they are extracted from the thermobath where they were prepared at −6 °C). Gelation of cellulose and cellulose–lignin mixtures was studied by measuring G′ and G″ at 15 °C at frequency 1 Hz and stress 1 Pa. As it will be shown in the following, gelation of all 6 % mixtures studied at 15 °C is slow enough to be neglected for this type of measurements: The duration of one flow experiment was less than 30–40 min, while gelation takes more than 10 h. Because the mixtures showed a slightly shear-thinning flow, like observed previously for cellulose–(7–9)%NaOH–water solutions (Roy et al. 2003; Egal 2006; Gavillon and Budtova 2008), the viscosity values to be plotted as a mixture composition were taken at the shear rate of 10 s−1.
The viscosity of 6 % mixtures at various compositions is shown in Fig. 5.37 together with the calculated additive dependence. The experimental points coincide with the additive dependence demonstrating the evidence of the absence of any special interaction between cellulose and lignin in 8 % NaOH–water that could lead to the formation of new bonds. This important finding means that not only there are no bonds built between cellulose and lignin molecules but that these polymers coexist in the common solvent and may phase separate as soon as their total concentration becomes high enough. Electrostatic repulsive interactions between cellulose and lignin could be one of the reasons of the absence bonds built between the components as far as both polymers should be negatively charged in 8 % NaOH–water.

Viscosity versus mixture composition for mixtures with 6 % total polymer concentration. Points are experimental data; line corresponds to the additive dependence calculated according to (5.1) (see details in the text). With kind permission from Springer Science + Business Media: Sescousse et al. (2010b), Fig. 5.3
It was shown (Roy et al. 2003; Egal 2006; Gavillon and Budtova 2008) that cellulose–(7–9)%NaOH–water solutions are irreversibly gelling with the increase of temperature, time and concentration. It was interesting to check if and how the presence of lignin influences gelation of cellulose–8 % NaOH–water solutions. Gelation time, t gel, was chosen as one of the important parameters characterising gelation. It was determined from the evolution of elastic G′ and viscous G″ moduli at a fixed temperature, 15 °C, for various mixture compositions and taken at the moment when G′ = G″.
Gelation time of cellulose–lignin mixtures is shown in Fig. 5.38 for three mixtures containing 5, 5.5 and 6 % of cellulose. The amount of lignin was varied. Despite that low molecular weight lignin solutions in 8 % NaOH–water are not gelling, lignin presence in the mixture, even in low amounts, accelerates cellulose–8 % NaOH gelation (see a drastic decrease in gelation time for 5 % cellulose–1 % lignin as compared to 5 % cellulose–0 % lignin or with the increase of lignin content in the mixture of 6 % cellulose with 0.5, 1, 1.5 and 2 % lignin, Fig. 5.38).

Gelation time as a function of lignin concentration in the mixtures containing 5 % (1), 5.5 % (open point) and 6 % (2) cellulose. Lines are given to guide the eye. The circle shows the mixtures with the same total polymer concentration, 6 %. With kind permission from Springer Science + Business Media: Sescousse et al. (2010b), Fig. 5.5
Another way of looking at the same data gives a key in the understanding of what is happening in cellulose–lignin mixtures. Let us consider mixtures with the same total polymer concentration, 6 %: 6 % cellulose, 5.5 % cellulose–0.5 % lignin and 5 % cellulose–1 % lignin (shown with a circle in Fig. 5.38). On one hand, a decrease of cellulose concentration from 6 to 5 % in solutions without lignin leads to a strong increase in gelation time, as expected. On the other hand, gelation of all 6 % mixtures, pure 6 % cellulose, 5.5 % cellulose–0.5 % lignin and 5 % cellulose–1 % lignin takes practically the same time which is surprising because 0.5 and 1 % lignin solution has a very low viscosity and cannot be considered as a gelling component. A most probable explanation is that due to possible electrostatic repulsion between the polymers, lignin “helps” the formation of cellulose-rich and cellulose-poor domains, thus leading to cellulose–lignin microphase separation. The increase of local cellulose concentration facilitates gelation; lignin can thus be considered as gelation promoter for cellulose–8 % NaOH solutions. The result obtained on gelation also suggests lignin and cellulose incompatibility in 8 % NaOH–water. Cellulose–lignin gels should thus be much more heterogeneous as compared with pure cellulose–8 % NaOH–water gels.
5.7.2 Coagulation of Cellulose from Cellulose–Lignin–8 % NaOH–Water Gels and Aero-Lignocellulose Morphology
Cellulose from cellulose–NaOH–water gels can be coagulated if placing a gel into a liquid which is a nonsolvent for cellulose (water, aqueous acid solutions, alcohols). A 3D object made of cellulose “network” with nonsolvent liquid filling the pores is formed. Because lignin is not gelling and is not bound to cellulose, the question is what will happen with lignin during cellulose coagulation. The answer depends on the type of liquid used in coagulation bath. Qualitative observations are easy as far as lignin gives a dark brown colour either to the samples or to the coagulation bath. When water bath was used, it became brown when cellulose–lignin gel was placed in it. This is because lignin was washed out from cellulose as far as lignin used is water-soluble. The higher the bath acidity, the lower the amount of lignin washed out from the sample during coagulation because lignin is not soluble in acidic media and it coagulates inside the cellulose network. In the latter case, samples remain dark: The higher the bath acidity, the darker the samples.
If cellulose–lignin–8 % NaOH–water gels are very heterogeneous, coagulated samples should also be of very heterogeneous porosity. In order to study the porosity of swollen-in-nonsolvent coagulated cellulose, two ways of drying that preserve the pores against collapse can be used: either freeze drying or drying under supercritical conditions. We used the same way of drying in supercritical CO2 as reported for the preparation of ultralight and highly porous pure cellulose, aerocellulose (Gavillon and Budtova 2008; Sescousse and Budtova 2009). It was shown that aerocellulose can be obtained from cellulose–8 % NaOH–water solutions and gels after cellulose coagulation in water, acid or alcohol bath, washing in acetone and drying in supercritical CO2.
New aero-lignocellulose with various lignin contents and of various porosities were obtained using the same approach as described above for aerocellulose preparation (see Chap. 5). SEM images of dry ligno-aerocellulose coagulated in the baths of two different acid concentrations are shown in Fig. 5.39. All the other preparation parameters were identical. The lower is acid bath concentration (Fig. 5.39a), the more lignin was washed out and thus larger pores are obtained.

SEM images of aero-lignocellulose from cellulose–lignin mixtures: regenerated in 0.1 mol L−1 (a) and in 1 mol L−1 acetic acid bath. The scale bar is 2 μm. Concentration of cellulose and lignin in the initial mixture was 4 and 4.3 %, respectively. Gelation was at 65 °C for 2 h. With kind permission from Springer Science + Business Media: Sescousse et al. (2010b), Fig. 5.7
The proportion between cellulose and lignin in two dry samples was determined in vTI-Institute for Wood Chemistry, Hamburg, Germany, for the aero-lignocelluloses made from the same initial mixture with high initial lignin content but coagulated in different baths: one of 0.1 mol L−1 acetic acid and the other of 1 mol L−1. The initial mixture was 4 % cellulose–8.6 % lignin, giving the initial proportion lignin/cellulose = 2.15. Sample preparation was identical except acetic acid concentration in coagulation bath. The amount of lignin in the aero-lignocelluloses was determined as a Klason and as acid-soluble lignin using UV spectrometer. The amount of acid-soluble fraction was very low, below 5–3 % of Klason lignin. The proportion between lignin and cellulose obtained was 0.14 for the dilute acid bath and 0.33 for the concentrated one. In other words, keeping in mind that the amount of cellulose in wet and dry samples does no change, 82 % of lignin that was mixed with cellulose was then washed out during coagulation in 0.1 mol L−1 acetic acid bath and 65 % was washed out in 1 mol L−1 bath. It is a direct proof that cellulose and lignin macromolecules are not bound together. The reason could be, as mentioned above, the electrostatic repulsive interactions.
The influence of lignin concentration on aero-lignocellulose is reflected by sample density: 0.135 and 0.1 g cm−3 for sample with the initial cellulose/lignin proportion in the mixture 4 % cellulose–3.3 % lignin and 4 % cellulose–8.6 % lignin, respectively. Coagulation bath was 0.1 mol L−1 acetic acid. Different densities are due to large voids that are formed because of washed-out lignin during coagulation: The higher the lignin concentration in the mixture, the more are there large pores. However, the specific surface obtained with BET analysis for these two and other samples is practically the same within the experimental errors, around 200 ± 20 m2 g−1. The reason is that large pores do not make any significant contribution to pores’ surface. Similar values were obtained for aerocelluloses prepared from pure cellulose solutions in 8 % NaOH–water (Gavillon and Budtova 2008) and in N-methylmorpholine-N-oxide (Innerlohinger et al. 2006).
Conclusions
Mixtures of cellulose and organosolv lignin in a common solvent, 8 % NaOH–water, were prepared. Mixture viscosity coincides with the one calculated according to the mixing rule, thus suggesting the absence of any bonds made between cellulose and lignin. The presence of lignin accelerates cellulose gelation, possibly due to the formation of cellulose-rich domains. Highly heterogeneous porous samples were obtained via cellulose coagulation and drying under CO2 supercritical conditions. A significant part of lignin was washed out during coagulation confirming the repulsion between cellulose and lignin in NaOH– water solvent. The higher the lignin concentration in the mixture, the larger pores were obtained in the dry material.
References
Adusumali RB, Reifferscheid M, Weber H, Roeder T, Sixta H, Gindl W (2006) Mechanical properties of regenerated cellulose fibres for composites. Macromol Symp 244:119–125
Atalla RH, Agarwal UP (1985) Raman microprobe evidence for lignin orientation in the cell wall of native tissue. Science 227:636–638
Atalla RH, Hackney JM, Uhlin I, Thompson NS (1993) Hemicelluloses as structure regulators in the aggregation of native cellulose. Int J Biol Macromol 15:109–112
Barthel S, Heinze T (2006) Acylation and carbanilation of cellulose in ionic liquids. Green Chem 8:301–306
Benoît H (1948) Calcul de l’écart quadratique moyen entre les extrémités de diverses chaînes moléculaires de type usuel. J Polym Sci 3:376–388
Besombes S, Mazeau K (2005) The cellulose/lignin assembly assessed by molecular modeling. Part 2: seeking for evidence of organization of lignin molecules at the interface with cellulose. Plant Physiol Biochem 43:277–286
Biganska O, Navard P (2005) Kinetics of precipitation of cellulose from cellulose-NMMO-water solutions. Biomacromolecules 6:1949–1953
Blachot JF, Brunet N, Cavaille JY, Navard P (1998) Rheological behaviour of cellulose/(N-methylmorpholine N-oxyde-water) solutions. Rheol Acta 37:107–114
Boerstel H, Maatman H, Westerink JB, Koenders BM (2001) Liquid crystalline solutions of cellulose in phosphoric acid. Polymer 42:7371–7379
Brandner A, Zengel HG (1980) German Patent 303468
British Celanese (1925) GB 263810
Budtov VP, Bel’nikevich NG, Litvinova LS (2010) Thermodynamics and viscosities of dilute polymer solutions in binary solvents. Polym Sci Ser A 52:362–367
Buijtenhuijs FA, Abbas M, Witteveen AJ (1986) The degradation and stabilization of cellulose dissolved in N-methylmorpholine-N-oxide (NMMO). Papier 40:615–619
Büttner T, Graneß G, Wendler F, Meister F, Dohrn W (2003) German Patent 2003 (TITK) DE 10331342
Cai J, Zang L (2005) Rapid dissolution of cellulose in LiOH/urea and NaOH:urea aqueous solutions. Macromol Biosci 5:539–548
Cai J, Zhang LN (2006) Unique gelation behavior of cellulose in NaOH/Urea aqueous solution. Biomacromolecules 7:183–189
Cai J, Zhang L, Zhou J, Li H, Chen H, Jin H (2004) Novel fibres prepared from cellulose in NaOH:urea aqueous solutions. Macromol Rapid Commun 25:1558–1562
Cai J, Zhang L, Zhou J, Qi H, Chen H, Kondo T, Chen X, Chu B (2007) Multifilament fibers based on dissolution of cellulose in NaOH/urea aqueous solution: structure and properties. Adv Mater 19:821–825
Cai J, Zhang L, Liu S, Liu Y, Xu X, Chen X, Chu B, Guo X, Xu J, Cheng H, Han C, Kuga S (2008) Dynamic self-assembly induced rapid dissolution of cellulose at low temperatures. Macromolecules 41:9345–9351
Cazacu G, Popa VI (2004) Blends and composites based on cellulose materials. In: Dumitriu S (ed) Polysaccharide: structural diversity and functional versatility, vol 2. Dekker, New York, pp 1141–1177
Chanzy H, Roche E (1976) Fibrous transformation of Valonia cellulose I into cellulose II. J Appl Polym Symp 28:701
Chanzy H, Nawrot S, Peguy A, Smith P, Chevalier J (1982) Phase behavior of the quasiternary system N-methylmorpholine-N-oxide, water, and cellulose. J Polym Sci 20:1909–1924
Chanzy H, Noe P, Paillet M, Smith P (1983) Swelling and dissolution of cellulose in amine oxide/water systems. J Appl Polym Sci 37:239–259
Chaudemanche C, Navard P (2011) Influence of fibre morphology on the swelling and dissolution mechanisms of Lyocell regenerated cellulose fibres. Cellulose 18:1–15
Chen X, Burger C, Wan F, Zhang J, Rong L, Hsiao B, Chu B, Cai J, Zhang L (2007) Structure study of cellulose fibers wet-spun from environmentally friendly NaOH-urea aqueous solutions. Biomacromolecules 8:1918–1926
Chesson A (1993) Mechanistic model of forage cell wall degradation. In: Jung HG, Buxton DR, Hatfield RD, Ralph J (eds) Forage cell wall structure and digestibility. UAS Wisconsin, Madison, p 358
Cibik T (2003) Untersuchungen am System NMMO/H2O/Cellulose. PhD Thesis, Technical University of Berlin
Ciechańska D, Wawro D, Stęplewski W, Wesolowska E, Vehvilonen M, Nousiainen P, Kamppuri T, Hroch Z, Sandak, Janicki J, Włochowicz A, Rom M, Kovalainen A (2007) Ecological method of manufacture of the cellulose fibres for advanced technical products. In Edana conference, Nonwovens Research Academy, 29–30 Mar 2007, University of Leeds, UK
Clark AH, Ross-Murphy SB (1987) Structural and mechanical properties of biopolymer gels. Adv Polym Sci 83:57–192
Cohen-Adad R, Tranquard A, Peronne R, Negri P, Rollet AP (1960) Le système eau-hydroxyde de sodium. Comptes Rendus de l’Académie des Sciences, Paris, France, 251 (part 3), pp 2035–2037
Cross CF, Bevan EJ (1892) Improvements in Dissolving Cellulose and Allied Compounds. British patent no. 8,700
Collier JR, Watson JL, Collier BJ, Petrovan S (2009) Rheology of 1-butyl-methylimidazolium chloride cellulose solutions. II. Solution character and preparation. J Appl Polym Sci 111:1019–1027
Cuissinat C, Navard P (2006a) Swelling and dissolution of cellulose, Part I: free floating cotton and wood fibres in N-methylmorpholine-N-oxide – water mixtures. Macromol Symp 244:1–18
Cuissinat C, Navard P (2006b) Swelling and dissolution of cellulose, Part II: free floating cotton and wood fibres in NaOH water-additives systems. Macromol Symp 244:19–30
Cuissinat C, Navard P (2008) Swelling and dissolution of cellulose, Part III: Plant fibres in aqueous systems. Cellulose 15:67–74
Cuissinat C, Navard P, Heinze T (2008a) Swelling and dissolution of cellulose, Part IV: Free floating cotton and wood fibres in ionic liquids. Carbohydr Polym 72:590–596
Cuissinat C, Navard P, Heinze T (2008b) Swelling and dissolution of cellulose, Part V: Cellulose derivatives fibres in aqueous systems and ionic liquids. Cellulose 15:75–80
Danilov SN, Samsonova TI, Bolotnikova LS (1970) Investigation of solutions of cellulose. Russ Chem Rev 39:156–168
Dave V, Glasser WG (1997) Cellulose-based fibres from liquid crystalline solutions: 5. Processing and morphology of CAB blends with lignin. Polymer 38:2121–2126
Dave V, Glasser WG, Wilkies GL (1992) Evidence of cholesteric morphology in films of cellulose acetate butyrate by transmission electron microscopy. Polym Bull 29:565–570
Davidson GF (1934) The dissolution of chemically modified cotton cellulose in alkaline solutions. Part I: In solutions of NaOH, particularly at T°C below the normal. J Text Inst 25:T174–196
Davidson GF (1936) The dissolution of chemically modified cotton cellulose inalkaline solutions. Part II: A comparison of the solvent action of solutions of Lithium, Sodium, Potassium and tetramethylammonium hydroxides. J Text Inst 27:T112–T130
Davidson GF (1937) The solution of chemically modified cotton cellulose in alkaline solutions. III. In solutions of sodium and potassium hydroxide containing dissolved zinc, beryllium and aluminum oxides. J Text Inst 28:2
Degen A, Kosec M (2000) Effect of pH and impurities on the surface charge of zinc oxide in aqueous solution. J Eur Ceram Soc 20:667–673
Dondos A, Benoit H (1973) The relationship between the unperturbed dimensions of polymers in mixed solvents and the thermodynamic properties of the solvent mixture. Macromolecules 6:242–245
Drechsler U, Radosta S, Vorwerg W (2000) Characterization of cellulose in solvent mixtures with N-methylmorpholine N-oxide by static light scattering. Macromol Chem Phys 201:2023–2030
Ducos F, Biganska O, Schuster KS, Navard P (2006) Influence of the Lyocell fibre structure on their fibrillation. Cell Chem Technol 40(5):299–311
Egal M (2006) Structure and properties of cellulose/NaOH aqueous solutions, gels and regenerated objects. PhD thesis, Ecole des Mines de Paris/Cemef, Sophia-Antipolis, France
Egal M, Budtova T, Navard P (2007) Structure of aqueous solutions of microcrystalline cellulose/sodium hydroxide below 0 °C and the limit of cellulose dissolution. Biomacromolecules 8:2282–2287
Egal M, Budtova T, Navard P (2008) The dissolution of microcrystalline cellulose in sodium hydroxide-urea aqueous solutions. Cellulose 15:361–370
El Seoud OA, Koschella A, Fidale LC, Dorn S, Heinze T (2007) Applications of ionic liquids in carbohydrate chemistry: a window of opportunities. Biomacromolecules 8:2629–2647
Fink H-P, Weigel P, Purz H-J (1998) Formation of lyocell-type fibres with skin-core structure. Lenz Ber 78:41–44
Fink HP, Gensrich J, Rihm R (2001) Structure and properties of CarbaCell-type cellulosic fibres. In: Proceedings of the 6th Asian textile conference, Hong-Kong, 22–24 Aug 2001
Fink H-P, Weigel P, Purz HJ, Ganster J (2001b) Structure formation of regenerated cellulose materials from NMMO-solutions. Prog Polym Sci 26(9):1473–1524
Firgo H, Eibl K, Kalt W, Meister G (1994) Kritishe fragen zur zukunft der NMMO-technologie. Lenz Ber 9:81–90
Flemming N, Thaysen AC (1919) On the deterioration of cotton on wet storage. Biochem J 14:25–29
Franks NA, Varga JK (1979) Process for making precipitated cellulose. US Patent 4,145,532
Franz H, Reusche P, Schoen W, Wiesener E, Taeger E, Schleicher H, Lukanoff B (1983) (AdW Teltow, TITK) German Patent 218104, 17 Oct 1983
Gavillon R, Budtova T (2007) Kinetics of cellulose regeneration from cellulose-NaOH-water gels and comparison with cellulose-N-methylmorpholine-N-oxide-water solutions. Biomacromolecules 8:424–432
Gavillon R, Budtova T (2008) Aerocellulose: new highly porous cellulose prepared from cellulose-NaOH aqueous solutions. Biomacromolecules 9:269–277
Gericke M, Liebert T, El Seoud O, Heinze T (2011) Tailored media for homogeneous cellulose chemistry: ionic liquid/co-solvent mixtures. Macromol Mater Eng 296:83–493
Gericke M, Liebert T, Heinze T (2009a) Interaction of ionic liquids with polysaccharides, 8 – synthesis of cellulose sulfates for polyelectrolyte complex formation. Macromol Biosci 9:343–353
Gericke M, Schlufter K, Liebert T, Heinze T, Budtova T (2009b) Rheological properties of cellulose/ionic liquid solutions: from dilute to concentrated states. Biomacromolecules 10:1188–1194
Glasser WG, Rials TG, Kelley SS, Dave V (1998) Studies of the molecular interaction between cellulose and lignin as a model for the hierarchical structure of wood. In: Heinze TJ, Glasser WG, Rojas O (eds) Cellulose derivatives. Modification, characterization and nanostructures. ACS symposium series 688 Chapter 19, pp 265–282
Glasser WG, Atalla RH, Blackwell J, Brown R Jr, Burchard W, French AD, Klemm DO, Nishiyama Y (2012a) About the structure of cellulose: debating the Lindman hypothesis. Cellulose. doi:10.1007/s10570-012-9691-7
Glasser WG, Atalla RH, Blackwell J, Brown R Jr, Burchard W, French AD, Klemm DO, Navard P, Nishiyama Y (2012b) Erratum to: about the structure of cellulose: debating the Lindman hypothesis. Cellulose. doi:10.1007/s10570-012-9702-8
Graenacher C (1934) Cellulose solution, US patent 1943176, 9 Jan 1934
Graenacher C, Sallman R (1939) Cellulose solutions. US Patent 2179181
Guo Q (1999) Thermosetting Polymer Blends: Miscibility, Crystallization, and Related Properties. In: Shonaike GO, Simon G (eds) Polymer blends and alloys, Marcel Dekker: New York, Chap. 6, pp 155–187
Gupta AK, Cotton JP, Marchal E, Burchard W, Benoit H (1976) Persistence length of cellulose tricarbanilate by small angle neutron scattering. Polymer 17:363–366
Gupta KM, Hu Z, Jiang J (2011) Mechanistic understanding of interactions between cellulose and ionic liquids: A molecular simulation study. Polymer 52:5904–5911
Guthrie JT, Manning CS (1990) The cellulose/N-methylmorpholine-N-oxide/H2O system: degradation aspects. In: Kennedy JF, Phillips GO, Williams PA (eds) Degradation Aspects, Cellulose Sources and Explotation. Ellis Horwood, New York, pp 49–57
Hairdelin L, Thunberg J, Perzon E, Westman G, Walkenstrom P, Gatenholm P (2012) Electrospinning of cellulose nanofibers from ionic liquids: the effect of different cosolvents. J Appl Polym Sci 125:1901–1909
Haque A, Morris E (1993) Thermogelation of methylcellulose. Part I: Molecular structures and processes. Carbohydr Polym 22:161–173
Harrison W (1928) Manufacture of carbohydrate derivatives. US Patent 1,684, 732
Hasegawa M, Isogai A, Onabe T, Usada M, Atalla RH (1992) Characterization of cellulose–chitosan blend films. J Appl Polym Sci 45:1873–1879
Hattori K, Abe E, Yoshide T, Cuculo JA (2004) New solvents for cellulose. II Ethylenediamine/thiocyanate salt system. Polym J 36:123–130
Haward SJ, Sharma V, Butts CP, McKinley GH, Rahatekar SS (2012) Shear and extensional rheology of cellulose/ionic liquid solutions. Biomacromolecules 13:1688–1699
Hill JW, Jacobsen RA (1938) US patent 2,134,825
Hock CW (1950) Degradation of cellulose as revealed microscopically. Text Res J 20:141–151
Holt C, Mackie W, Sezllen DB (1976) Configuration of cellulose trinitrate in solution. Polymer 17:1027–1034
Hong PD, Huang HT (2000) Effect of co-solvent complex on preferential absorption phenomenon in polyvinyl alcohol ternary solutions. Polymer 41:6195–6204
Houtman CJ, Atalla RH (1995) Cellulose-lignin interactions. A computational study. Plant Physiol 107:997–984
Innerlohinger J, Weber HK, Kraft G (2006) Aerocellulose: aerogels and aerogel-like materials made from cellulose. Macromol Symp 244:126–138
Isogai A, Atalla RH (1998) Dissolution of cellulose in aqueous NaOH solutions. Cellulose 5:309–319
Jin H, Zha C, Gu L (2007) Direct dissolution of cellulose in NaOH:thiourea/urea aqueous solutions. Carbohydr Polym 342:851–858
Johnson DL (1969) Compounds dissolved in cyclic amine oxides. US Patent 3,447,939
Joly C, Kofman M, Gauthier RJ (1996) Polypropylene/cellulose fiber composites chemical treatment of the cellulose assuming compatibilization between the two materials. J Macromol Sci Pure Appl Chem A33(12):1981–1996
Kahlem J, Masuch K, Leonhard K (2010) Modelling cellulose solubilities in ionic liquids using CPSMO-RS. Green Chem 12:2172–2187
Kalt W, Männer J, Firgo H (1993) (Lenzing) PCT Int. Appl. 9,508,010, 14 Sep1993
Kamide K, Saito M (1983) Persistence length of cellulose and cellulose derivatives in solution. Makromol Chem Rapid Commun 4:33–39
Kamide K, Saito M (1987) Cellulose and cellulose derivatives: recent advances in physical chemistry. Adv Polym Sci 83:1–56
Kamide K, Okajima K, Matsui T, Kowsaka K (1984) Study on the solubility of cellulose in aqueous alkali solution by deuteration IR and 13C NMR. Polym J 16–12:857–866
Kamide K, Saito M, Kowsaka K (1987) Temperature dependence of limiting viscosity number and radius of gyration for cellulose dissolved in aqueous 8 % sodium hydroxide solution. Polym J 19:1173–1181
Kamide K, Yasuda K, Matsui T, Okajima K, Yamashiki T (1990) Structural change in alkali-soluble cellulose solid during its dissolution into alkaline solutions. Cellulose Chem Technol 24:23–31
Kamide K, Okajima K, Kowsaka K (1992) Dissolution of natural cellulose into aqueous alkali solution: role of super-molecular structure of cellulose. Polym J 24–1:71–96
Kasaai M (2002) Comparison of various solvents for determination of intrinsic viscosity and viscometric constants for cellulose. J Appl Polym Sci 86:2189–2193
Kihlman M, Wallberg O, Stigsson L, Germgard U (2011) Dissolution of dissolving pulp in alkaline solvents after stream explosion pretreatments. Holzforschung 65:613–617
Kim IS, Kim JP, Kwak SY, Ko YS, Kwon YK (2006) Novel regenerated cellulosic material prepared by an environmentally-friendly process. Polymer 47:1333–1339
Kim J, Wang N, Chen Y, Lee S-K, Yun G-Y (2007) Electroactive-paper actuator made with cellulose/NaOH/urea and sodium alginate. Cellulose 14:217–223
Klemchuk PP (1985) Antioxydants. In: Gerhartz W, Yamamoto YS (eds) Ullmann’s encyclopedia of industrial chemistry, vol A3. Weinheim, VCH, pp 91–111
Kohler S, Heinze T (2007) Efficient synthesis of cellulose fuorates in 1-N-butyl-3-methylimidazolium chloride. Cellulose 14:489–495
Kondo T, Kasai W, Brown RM (2004) Formation of nematic ordered cellulose and chitin. Cellulose 11:463–474
Konkin A, Wendler F, Roth H-K, Schroedner M, Bauer R-U, Meister F, Heinze T, Aganov A, Garipov R (2006) Electron spin resonance study of radicals generated in cellulose/N-methylmorpholine solutions after flash photolysis at 77 K. Magn Reson Chem 44:594–605
Kosan B, Michels C (1999) Chem Fibers Int 49:50–54
Kosan B, Michels C, Meister F (2008a) Dissolution and forming of cellulose with ionic liquids. Cellulose 15:59–66
Kosan B, Schwikal K, Hesse-Ertelt S, Meister F (2008) In: Proceedings 8th international symposium alternative cellulose – manufacturing, forming, properties, Rudolstadt, Germany, 03–04 Sept 2008
Kuang QL, Zhao JC, Niu YH, Zhang J, Wang ZG (2008) Celluloses in an ionic liquid: the rheological properties of the solutions spanning the dilute and semidilute regimes. J Phys Chem B 112:10234–10240
Kunze J, Fink HP (2005) Structural changes and activation of cellulose by caustic soda solution with urea. Macromol Symp 223:175–187
Kuo Y-N, Hong J (2005) Investigation of solubility of microcrystalline cellulose in aqueous NaOH. Polym Adv Technol 16:425–428
Laity PR (1983) (Courtaulds) PCT International Application 8,304,415, 7 June 1983
Laszkiewicz B (1998) Solubility of bacterial cellulose and its structural properties. J Appl Polym Sci 67:1871–1876
Laszkiewicz B, Cuculo JA (1993) Solubility of cellulose III in sodium hydroxide solution. J Appl Polym Sci 50:27–34
Laszkiewicz B, Wcislo P (1990) Sodium cellulose formation by activation process. J Appl Polym Sci 39:415–425
Le KA, Sescousse R, Budtova T (2012) Influence of water on cellulose-EMIMAc solution properties: a viscometric study. Cellulose 19:45–54
LeMoigne N (2008) Mécanismes de gonflement et de dissolution des fibres de cellulose. Thèse de doctorat. Ecole Nationale Supérieure des Mines de Paris. Sophia Antipolis, France
LeMoigne N, Montes E, Pannetier C, Höfte H, Navard P (2008) Gradient in dissolution capacity of successively deposited cell wall layers in cotton fibers. Macromol Symp 262:65–71
LeMoigne N, Bikard J, Navard P (2010a) Contraction and rotation and contraction of native and regenerated cellulose fibres upon swelling and dissolution: the role of stress unbalance. Cellulose 17(3):507–519
LeMoigne N, Jardeby K, Navard P (2010b) Structural changes and alkaline solubility of wood cellulose fibers after enzymatic peeling treatment. Carbohydr Polym 79:325–332
Liebert TF (2010) Cellulose solvents-remarkable history, bright future. In: Liebert TF, Heinze TJ, Edgazr KJ (eds) Cellulose solvents: for analysis, shaping and chemical modification, ACS Symposium Series 1033, Oxford Press University, pp 3–54
Lin C-X, Zhan H-Y, Liu M-H, Fu S-Y, Lucia LA (2009) Novel preparation and characterisation of cellulose microparticles functionalised in ionic liquids. Langmuir 25:10116–10120
Lindman B, Karlström G, Stigsson L (2010) On the mechanism of dissolution of cellulose. J Mol Liq 156(1):76–81
Liu Y, Piron DL (1998) Study of tin cementation in alkaline solution. J Electrochem Soc 145:186–190
Liu W, Budtova T, Navard P (2011) Influence of ZnO on the properties of dilute and semi-dilute cellulose-NaOH-water solutions. Cellulose 18:911–920
Lovell PA (1989) Dilute solution viscometry. In: Colin B, Colin C (eds) Comprehensive polymer science, the synthesis, characterization, reactions and applications of polymers, vol I, Polymer characterization. Pergamon Press, Oxford
Lovell CS, Walker A, Damion RA, Radhi A, Tanner SF, Budtova T, Ries ME (2010) Influence of cellulose on ion diffusivity in 1-ethyl-3-methyl-imidazolium acetate cellulose solutions. Biomacromolecules 11:2927–2935
Lu A, Liu Y, Zhang L, Potthast A (2011) J Phys Chem B 115:12801–12808
Lue A, Liu Y, Zhang L, Potthast A (2011) Light scattering study on the dynamic behaviour of cellulose inclusion complex in LiOH/urea aqueous solution. Polymer 52:3857–3864
Lukanoff B, Schleicher H (1981) (AdW Teltow) German Patent 158656, 27 Apr 1981
Marsh JT (1941) The growth and structure of cotton, Mercerising. Chapman & Hall, London
Masegosa RM, Prolongo MG, Hernandez-Fuentes I (1984) Preferential and total sorption of poly(methyl methacrylate) in the cosolvent formed by acetonitrile with pentyl acetate and with alcohols (1-butanol, 1-propanol, and methanol). Macromolecules 17:1181–1187
Matsui T, Sano T, Yamane C, Kamide K, Okajima K (1995) Structure and morphology of cellulose films coagulated from novel cellulose/aqueous sodium hydroxide solutions by using aqueous sulphuric acid with various concentrations. Polym J 27–8:797–812
Matsumoto T, Tatsumi D, Tamai N, Takaki T (2001) Solution properties of celluloses from different biological origins in LiCl-DMAc. Cellulose 8:275–282
Maximova N, Osterberg M, Koljonen K, Stenius P (2001) Lignin adsorption on cellulose fibre surfaces: effect on surface chemistry, surface morphology and paper strength. Cellulose 8:113–125
Maximova N, Stenius P, Salmi J (2004) Lignin uptake by cellulose fibres from aqueous solutions. Nord Pulp Pap Res J 19:135–145
McCormick CL, Lichatowich DK (1979) Homogeneous solution reactions of cellulose, chitin, and other polysaccharides to produce controlled-activity pesticide systems. J Polym Sci Polym Lett Ed 17(8):479–484
McCormick CL, Callais PA, Hutchinson BH (1985) Solution studies of cellulose in lithium chloride and N,N-dimethylacetamide. Macromolecules 18:2394–2401
Michels C (1998) Beitrag zur Bestimmung von Molmasseverteilungen in Cellulosen aus rheologischen Daten. Determination of the mole-mass distributions of cellulose, using rheological data. Das Papier 52(1):3–8
Michels C, Kosan B (2000) Chem Fibers Int 50:556–561
Michels C, Mertel H (1984) (TITK) German Patent 229708, 13 Dec 1984
Mikolajczyk W, Struszczyk H, Urbanowski A, Wawro D, Starostka P (2002) Process for producing fibres, film, casings and other products from modified soluble cellulose. Poland, Patent no. WO 02/22924 (21 mars 2002)
Miller-Chou B, Koenig JL (2003) A review of polymer dissolution. Prog Polym Sci 28:1223–1270
Morris ER (1990) Shear-thinning of “random coil” polysaccharides: characterisation by two parameters from a simple linear plot. Carbohydr Polym 13:85–96
Morris ER, Cutler AN, Ross-Murphy S, Rees DA, Price J (1981) Concentration and shear rate dependence of viscosity in random coil polysaccharide solutions. Carbohydr Polym 1:5–21
Musatova GN, Mogilevskii EM, Ginzberg MA, Arkhangelskii DN (1972) The dissolution temperature of cellulose xanthate. Fibre Chem 2:451–453
Nägeli C (1864) Ueber den inneren Bau der vegetabilischen Zellenmem- branen Sitzber. Bay. Akad. Wiss. Munchen 1:282–323
Nisho Y (1994) Hyperfine composites of cellulose with synthetic polymers. In: Gilbert RD (ed) Cellulosic polymers, blends and composites. Hanser Publishers, New York, pp 95–113
Noda A, Hayamizu K, Watanabe M (2001) Pulsed-gradient spin-echo H-1 and F-19 NMR ionic diffusion coefficient, viscosity, and ionic conductivity of non-chloroaluminate room-temperature ionic liquids. J Phys Chem B 105:4603–4610
Noordermeer JWM, Daryanani R, Janeschitz-Kriegl H (1975) Flow birefringence studies of polymer conformation: cellulose tricarbanilate in two characteristics solvents. Polymer 16:359–369
Northolt MG, Boerstel H, Maatman H, Huisman R, Veurink J, Elzerman H (2001) The structure and properties of cellulose fibres spun from an anisotropic phosphoric acid solution. Polymer 42:8249–8264
Novoselov NP, Tret’yak VM, Sinel’nikov EV, Saschina ES (1997) Russ J Gen Chem 67(3):430–434
Okajima K, Yamane C (1997) Cellulose filament spun from cellulose aqueous NaOH solution system. Cell Commun 4:7–12
Ott E, Spurlin HM, Grafflin MW (1954) In Cellulose and cellulose derivatives (Part 1). Interscience Publisher, New York, p 353
Pang F-J, He C-J, Wang Q-R (2003) Preparation and properties of cellulose/chitin blend fiber. J Appl Polym Sci 90:3430–3436
Pennetier G (1883) Note micrographique sur les altérations du cotton. Bull Soc Ind Rouen 11:235–237
Persin Z, Stana-Kleinschek K, Kreze T (2002) Hydrophilic/hydrophobic characteristics of different cellulose fibres monitored by tensiometry. Croatica chemica acta 75(1):271–280
Perez DDS, Ruggiero R, Morais LC, Machado AEH, Mazeau K (2004) Theoretical and experimental studies on the adsorption of aromatic compounds onto cellulose. Langmuir 20:3151–3158
Phillies GDJ (1986) Universal scaling equation for self-diffusion by macromolecules in solution. Macromolecules 19:2367–2376
Pickering SU (1893) The hydrates of sodium, potassium and lithium hydroxides. J Chem Soc 63:890–909
Pingping Z, Yuanli L, Haiyang Y, Xiaoming C (2006) Effect of non-ideal mixed solvents on dimensions of poly(N-vinylpyrrolidone) and poly(methyl methacrylate) coils. J Macromol Sci Part B Phys 45:1125–1134
Pinkert A, Marsh KN, Pang S, Staiger MP (2009) Ionic liquids and their interactions with cellulose. Chem Rev 109:6712–6728
Potthast A, Rosenau T, Buchner R, Röder T, Ebner G, Bruglachner H, Sixta H, Kosma P (2002) The cellulose solvent system/N,N-dimethylacetamide/lithium chloride revisited: the effect of water on physicochemical properties and chemical stability. Cellulose 9:41–53
Pouchly J, Patterson D (1976) Polymers in mixed solvents. Macromolecules 9:574–579
Prasad K, Kaneko Y, Kadokawa J (2009) Novel Gelling Systems of κ-, ι- and λ-Carrageenans and their composite gels with cellulose using ionic liquid. Macromol Biosci 9:376–382
Qi H, Chang C, Zhang L (2008a) Effects of temperature and molecular weight on dissolution of cellulose in NaOH/urea aqueous solutions. Cellulose 15:779–787
Qi H, Cai J, Zhang L, Nishiyama Y, Rattaz A (2008b) Influence of finishing oil on structure and properties of multifilament fibers from cellulose dope in NaOH/urea aqueous solution. Cellulose 15:81–89
Rademacher P, Bauch J, Puls J (1986) Investigations of the wood from pollution-affected spruce. Holzforschung 40:331–338
Ramos LA, Assaf JM, El Seoud OA, Frollini E (2005a) Influence of the supramolecular structure and physicochemical properties of cellulose on its dissolution in a lithium chloride/N,N-dimethylacetamide solvent system. Biomacromolecules 6:2638–2647
Ramos LA, Frollinni E, Heinze T (2005b) Carboxymethylation of cellulose in the new solvent dimethylsulfoxide/tetrabutylammonium fluoride. Carbohydr Polym 60:259–267
Reichle RA, McCurdy KG, Hepler LG (1975) Zinc hydroxide – solubility product and hydroxyl–complex stability-constants from 12.5-75 °C. Can J Chem 53:3841–3845
Rials TG, Glasser WG (1989) Multiphase materials with lignin. VI. Effect of cellulose derivative structure on blend morphology with lignin. Wood Fiber Sci 21:80–90
Rials TG, Glasser W (1990) Multiphase materials with lignin: 5. Effect of lignin structure on hydroxypropyl cellulose blend morphology. Polymer 31:1333–1338
Röder T, Morgenstern B (1999) The influence of activation on the solution state opf cellulose dissolved in N-methylmorpholine N-oxyde-monohydrate. Polymer 40:4143–4147
Röder T, Morgenstern B, Glatter O (2000) Light scattering studies on solutions of cellulose in N,N-dimethylacetamide/lithium chloride. Lenz Ber 79:97–101
Rollet AP, Cohen-Adad R (1964) Les systèmes “eau-hydroxyde alcalin”. Revue de Chimie Minérale 1:451
Rosenau T, Potthast A, Sixta H, Kosma P (2001) The chemistry of side reactions and by-product formation in the system NMMO/cellulose (Lyocell process). Progr Polym Sci 26:1763–1837
Ross-Murphy SB (1991) Concentration dependence of gelation time. In: Dickinson E (ed) Food polymers, gels and colloids. Royal Society of Chemistry, Cambridge, pp 357–368
Roy C, Budtova T, Navard P, Bedue O (2001) Structure of cellulose-soda solutions at low temperatures. Biomacromolecules 2:687–693
Roy C, Budtova T, Navard P (2003) Rheological properties and gelation of aqueous cellulose-NaOH solutions. Biomacromolecules 4:259–264
Ruan D, Zhang L, Zhou J, Jin H, Chen H (2004) Structure and properties of novel fibers spun from cellulose in NaOH/thiourea aqueous solution. Macromol Biosci 4(12):1105–1112
Ruan D, Lue A, Zhang L (2008) Gelation behaviours of cellulose solution dissolved in aqueous NaOH-thiourea at low temperature. Polymer 49:1027–1036
Russler A, Lange A, Potthast A, Rosenau T, Berger-Nicoletti E, Sixta H, Kosma P (2005) A novel method for analysis of xanthate group distribution in viscoses. Macromol Symp 223:189–200
Russler A, Potthast A, Rosenau T, Lange T, Saake B, Sixta H, Kosma P (2006) Determination of substituent distribution of viscoses by GPC. Holzforschung 60:467–473
Saito G (1939) Das verhalten der zellulose in alkalilösungen. I. Mitteilung. Kolloid-Beihefte 29:365–454
Sammons RJ, Collier JR, Rials TG, Petrovan S (2008) Rheology of 1-butyl-methylimidazolium chloride cellulose solutions. I. Shear rheology. J Appl Polym Sci 110:1175–1181
Schulz L, Seger B, Burchard W (2000) Structures of cellulose in solution. Macromol Chem Phys 201:2008–2022
Segal L, Eggerton F (1961) Some aspects of the reaction between urea and cellulose. Text Res J 31:460–471
Seger B, Aberle T, Burchard W (1996) Solution behaviour of cellulose and amylose in iron-sodium tartrate (FeTNa). Carbohydr Polym 31(1–2):105–112
Sescousse R, Budtova T (2009) Influence of processing parameters on regeneration kinetics and morphology of porous cellulose from cellulose-NaOH-water solutions. Cellulose 16:417–426
Sescousse R, Le KA, Ries ME, Budtova T (2010a) Viscosity of cellulose-imidazolium-based ionic liquid solutions. J Phys Chem B 114:7222–7228
Sescousse R, Smacchia A, Budtova T (2010b) Influence of lignin on cellulose-NaOH-water mixtures properties and on Aerocellulose morphology. Cellulose 17:1137
Sescousse R, Gavillon R, Budtova T (2011a) Wet and dry highly porous cellulose beads from cellulose-NaOH-water solutions: influence of the preparation conditions on beads shape and encapsulation of inorganic particles. J Mater Sci 46:759–765
Sescousse R, Gavillon R, Budtova T (2011b) Aerocellulose from cellulose-ionic liquid solutions: preparation, properties and comparison with cellulose-NaOH and cellulose-NMMO routes. Carbohydr Polym 83:1766–1774
Sobue H, Kiessig H, Hess K (1939) The cellulose-sodium hydroxide-water system as a function of the temperature. Z Physik Chem B 43:309–328
Sprague BS, Noether HD (1961) The relationship of fine structure to mechanical properties of stretched saponified acetate fibers. Text Res J 31:858–865
Sternemalm E, Höije A, Gatenholm P (2008) Effects of arabinose substitution on the material. Properties of arabinoxylan films. Carbohydr Res 343:753–757
Struszczyk H, Wawro D, Starostka P, Mikolajscyk W, Urbanowski A (2000) EP 1317573 B1 “Process for producing fibres, film, casings and other products from modified soluble cellulose”, 13/09/2000
Swatloski RP, Spear SK, Holbrey JD, Rogers RD (2002) Dissolution of cellulose with ionic liquids. J Am Chem Soc 124:4974–4975
Taeger E, Franz H, Mertel H, Schleicher H, Lang H, Lukanoff B (1985) Formeln Fasern Fertigware 4:14–22
Taeger E, Berghof K, Maron R, Meister F (1997) Eignshaftsänderungen im Alceru-faden durch zweitpolymere. Lenz Ber 76:126–131
Tamai N, Oano H, Tatsumi D, Matsumoto T (2003) Differences in rheological properties of plant and bacrterial cellulose in LiCl//N,N-dimethylacetamide. J Soc Rheol Jap 31(3):119–130
Tasker S, Baadyal JPS, Backson SCE, Richards RW (1994) Hydroxyl accessibility in celluloses. Polymer 35(22):4717–4721
Terashima N, Seguchi Y (1988) Heterogeneity in formation of lignin. IX. Factors affecting the formation of condensed structures in lignin. Cell Chem Technol 22:147
The Editors of “Dyer and Calico Printer” (1903) Mercerisation: a practical and historical manual, vol I. Heywood and Company Ltd., London
Tokuda H, Ishii K, Susan M, Tsuzuki S, Hayamizu K, Watanabe M (2006) Physicochemical properties and structures of room-temperature ionic liquids. 3. Variation of cationic structures. J Phys Chem B 110:2833–2839
Tripp VW, Rollins ML (1952) Morphology and chemical composition of certain components of cotton fiber cell wall. Anal Chem 24:1721–1728
Tsioptsias C, Stefopoulos A, Kokkinomalis I, Papadopoulou L, Panayiotou C (2008) Development of micro- and nano-porous composite materials by processing of cellulose with ionic liquids and supercritical CO2. Green Chem 10:965–971
Tsuzuki S, Shinoda W, Saito H, Mikami M, Tokuda H, Watanabe M (2009) Molecular dynamics simulations of ionic liquids: cation and anion dependence of self-diffusion coefficients of ions. J Phys Chem B 113:10641–10649
Turbak AF, Hammer RB, Davies RE, Hergert HL (1980) Cellulose solvents. Chem Tech 10:51–57
Turbak AF, El-Kafrawy A, Snyder FW, Auerbach AB (1981) Solvent system for cellulose, US Patent 4,302,252
Turner MB, Spear SK, Holbrey JD, Rogers RD (2004) Production of bioactive cellulose films reconstituted from ionic liquids. Biomacromolecules 5:1379–1384
Urahata SM, Ribeiro MCC (2005) Single particle dynamics in ionic liquids of 1-alkyl-3-methylimidazolium cations. J Chem Phys 122:024511–024520
Vehviläinen M, Kamppuri T, Rom M, Janicki J, Ciechanska D, Grönqvist S, Sioika-Aho M, Christoffersson K, Nousiainen P (2008) Effect of wet spinning parameters on the properties of novel cellulosic fibres. Cellulose 15:671–680
Warwicker JO, Jeffries R, Colbran RL, Robinson RN (1966) A review of the literature on the effect of caustic soda and other swelling agents on the fine structure of cotton. St Ann’s Press, Manchester, 93
Wendler F, Graneß G, Heinze T (2005a) Characterization of autocatalytic reactions in modified cellulose/NMMO solutions by thermal analysis and UV/VIS spectroscopy. Cellulose 12(4):411–422
Wendler F, Kolbe A, Meister F, Heinze T (2005b) Thermostability of lyocell dopes modified with surface – active additives. Macromol Mater Eng 290:826–832
Wendler F, Graneß G, Büttner R, Meister F, Heinze T (2006) A novel polymeric stabilizing system for modified lyocell solutions. J Polym Sci Part B Polym Phys 44:1702
Wendler F, Konkin A, Heinze T (2008) Studies on the stabilization of modified lyocell solutions. Macromol Symp 262:72–84
Wendler F, Meister F, Wawro D, Wesolowska E, Ciechanska D, Saake B, Puls J, Le Moigne N, Navard P (2010) Polysaccharide Blend Fibres Formed from NaOH, N-Methylmorpholine-N-oxide and 1-Ethyl-3-methylimidazolium acetate. Fibers Text Eastern Eur 18(79):21–31
Weng L, Zhang L, Ruan D, Shi L, Xu J (2004) Thermal gelation of cellulose in a NaOH/thiourea aqueous solution. Langmuir 20(6):2086–2093
Winter HH, Chambon F (1986) Analysis of linear viscoelasticity of a crosslinking polymer at the gel point. J Rheol 30(2):367–382
Yamada H, Kowsaka K, Matsui T, Okajima K, Kamide K (1992) Nuclear magnetic study on the dissolution of natural and regenerated celluloses onto aqueous alkali solutions. Cell Chem Technol 26:141–150
Yamane C, Saito M, Kowsaka K, Kataoka N, Sagara K, Kamide K (1994) New cellulosic filament yarn spun from cellulose/aq NaOH solution. In: Proceedings of ’94 cellulose R&D, 1st annual meeting of the Cellulose Society of Japan (Cellulose Society of Japan, ed.) Tokyo, pp 183–188
Yamane C, Saito M, Okajima K (1996a) Industrial preparation method of cellulose-alkali dope with high solubility. Sen’I Gakkaaishi 52–6:310–317
Yamane C, Saito M, Okajima K (1996b) Specification of alkali soluble pulp suitable for new cellulosic filament production. Sen’I Gakkaaishi 52–6:318–324
Yamane C, Saito M, Okajima K (1996c) Spinning of alkali soluble cellulose-caustic soda solution system using sulphuric acid as coagulant. Sen’I Gakkaaishi 52–6:369–377
Yamane C, Saito M, Okajima K (1996d) New spinning process of cellulose filament production from alkali soluble cellulose dope-net process. Sen’I Gakkaaishi 52–6:378–384
Yamashiki T, Kamide K, Okajima K, Kowsaka K, Matsui T, Fukase H (1988) Some characteristic features of dilute aqueous alkali solutions of specific alkali concentration (2.5 mol l-1) which possess maximum solubility power against cellulose. Polymer J 20(6):447–457
Yamashiki T, Kamide K, Okajima K (1990a) New cellulose fibres from aq. alkali cellulose solution. In: Kennedy JF, Phillips GO, Williams PA (eds) Cellulose sources and exploitation. Ellis Horwood Ltd., New York, pp 197–202
Yamashiki T, Matsui T, Saitoh M, Okajima K, Kamide K (1990b) Characterisation of cellulose treated by the steam explosion method. Part 1: Influence of cellulose resources on changes in morphology, degree of polymerisation, solubility and solid structure. Br Polym J 22:73–83
Yamashiki T, Matsui T, Saitoh M, Okajima K, Kamide K (1990c) Characterisation of cellulose treated by the steam explosion method. Part 2: Effect of treatment conditions on changes in morphology, degree of polymerisation, solubility in aqueous sodium hydroxide and supermolecular structure of soft wood pulp during steam explosion. Br Polym J 22:121–128
Yamashiki T, Saitoh M, Yasuda K, Okajima K, Kamide K (1990d) Cellulose fibre spun from gelatinized cellulose/aqueous sodium hydroxide system by the wet-spinning method. Cell Chem Technol 24:237–249
Yamashiki T, Matsui T, Kowsaka K, Saitoh M, Okajima K, Kamide K (1992) New class of cellulose fiber spun from the novel solution of cellulose by wet spinning method. J Appl Polym Sci 44:691–698
Zadorecki P, Michell AJ (1989) Future prospects for wood cellulose as reinforcement in organic polymer composites. Polym Compos 10:69–77
Zhang H, Tong M (2007) Influence of hemicelluloses on the structure and properties of Lyocell fibers. Polym Eng Sci 47:702–706
Zhang L, Ruan D, Zhou J (2001) Structure and properties of regenerated cellulose films prepared from cotton linters in NaOH/urea aqueous solution. Ind Eng Chem Res 40:5923–5928
Zhang L, Ruan D, Gao S (2002) Dissolution and regeneration of cellulose in NaOH/thiourea aqueous solution. J Polym Sci Part B 40:1521–1529
Zhang H, Wu J, Zhang J, He J (2005) 1-allyl-3-methylimidazolium chloride room temperature ionic liquid: a new and powerful nonderivatizing solvent for cellulose. Macromolecules 38:8272–8277
Zhang S, Li FX, Yu JY (2009) Preparation of cellulose/chitin blend bio-fibres via direct dissolution. Cell Chem Technol 43:393–398
Zhang JM, Zhang H, Wu J, Zhang J, He JS, Xiang JF (2010) NMR spectroscopic studies of cellobiose solvation in EmimAc aimed to understand the dissolution mechanism of cellulose in ionic liquids. Phys Chem Chem Phys 12:1941–1947
Zhang S, Li FX, Yu JY (2011) Kinetics of cellulose regeneration from cellulose-NaOH/thiourea/urea/H2O system. Cell Chem Technol 45:5
Zhao Q, Yam RCM, Zhang B, Yang Y, Cheng X, Li RKY (2009) Novel all-cellulose ecocomposites prepared in ionic liquids. Cellulose 16:217–226
Zhou J, Zhang L (2000) Solubility of cellulose in NaOH/Urea aqueous solution. Polym J 32(10):866–870
Zhou J, Zhang L, Cai J, Shu H (2002a) Cellulose microporous membranes prepared from NaOH/urea aqueous solution. J Memb Sci 210:77–90
Zhou J, Zhang L, Shu H, Chen F (2002b) Regenerated cellulose films from NaOH/urea aqueous solution by coagulating with sulphuric acid. J Macromol Sci Phys B41(1):1–15
Zhou J, Zhang L, Cai J (2004) Behaviour of cellulose in NaOH/urea aqueous solution characterized by light scattering and viscosimetry. J Polym Sci Part B Polym Phys 42:347–353
Zhu SD, Wu YX, Chen QM, Yu ZN, Wang CW, Jin SW, Ding YG, Wu G (2006) Dissolution of cellulose with ionic liquids and its application: a mini-review. Green Chem 8:325–327
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag/WIen
About this chapter
Cite this chapter
Navard, P., Wendler, F., Meister, F., Bercea, M., Budtova, T. (2012). Preparation and Properties of Cellulose Solutions. In: Navard, P. (eds) The European Polysaccharide Network of Excellence (EPNOE). Springer, Vienna. https://doi.org/10.1007/978-3-7091-0421-7_5
Download citation
DOI: https://doi.org/10.1007/978-3-7091-0421-7_5
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-0420-0
Online ISBN: 978-3-7091-0421-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)