
Abstract
Mitochondrial fatty acid oxidation disorders have been included in newborn screening programs worldwide since the implementation of tandem mass spectrometry-based screening. Disease-specific acylcarnitine profiles pinpoint at the respective enzyme defect; however, the diagnosis has invariably to be confirmed by enzyme assay and/or molecular analysis. Metabolic profiles can be normal in the anabolic state and, consequently, newborn screening may miss the diagnosis when performed outside the catabolic state on days 2 and 3 of life. Mitochondrial fatty acid oxidation disorders comprise four groups: (1) disorders of the entry of long-chain fatty acids into mitochondria, (2) intramitochondrial β-oxidation defects of long-chain fatty acids affecting membrane-bound enzymes, (3) β-oxidation defects of short- and medium-chain fatty acids affecting enzymes of the mitochondrial matrix, and (4) disorders of impaired electron transfer to the respiratory chain from mitochondrial β-oxidation. The main pathogenic mechanisms of fatty acid oxidation defects include toxic effects by accumulating acylcarnitine and acyl-CoA species and energy deficiency due to impaired fatty acid oxidation and ketone body formation. The respective disorders present with heterogeneous phenotypes. Before newborn screening (NBS) was introduced, the most common clinical presentations were hypoketotic hypoglycemia and sudden death, usually precipitated by an infection or fasting in the neonatal period or early childhood. In addition, severe cardiomyopathy and arrhythmias were leading clinical signs in the first months of life. Older children or adults may present with exercise- or illness-induced rhabdomyolysis. Patients can remain asymptomatic throughout life if they have mild defects and are not exposed to metabolic stress. Correlation of genotype and/or residual enzyme activity with disease phenotype has been reported for some defects, whereas in others additional disease modifiers are suspected. With newborn screening, disease incidence has significantly increased and the proportion of milder phenotypes has grown. Newborn screening greatly reduces the morbidity and mortality, though it does not eliminate early neonatal death in severe phenotypes. With respect to the heterogeneous clinical presentation, treatment needs to be tailored to the severity of the respective phenotype and the specific disorder.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Newborn Screening
- Carnitine Concentration
- Acylcarnitine Profile
- Fatty Acid Oxidation Disorder
- Electron Transfer Flavoprotein
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara SummaryMitochondrial fatty acid oxidation disorders have been included in newborn screening programs worldwide since the implementation of tandem mass spectrometry-based screening. Disease-specific acylcarnitine profiles pinpoint at the respective enzyme defect; however, the diagnosis has invariably to be confirmed by enzyme assay and/or molecular analysis. Metabolic profiles can be normal in the anabolic state and, consequently, newborn screening may miss the diagnosis when performed outside the catabolic state on days 2 and 3 of life. Mitochondrial fatty acid oxidation disorders comprise four groups: (1) disorders of the entry of long-chain fatty acids into mitochondria, (2) intramitochondrial β-oxidation defects of long-chain fatty acids affecting membrane-bound enzymes, (3) β-oxidation defects of short- and medium-chain fatty acids affecting enzymes of the mitochondrial matrix, and (4) disorders of impaired electron transfer to the respiratory chain from mitochondrial β-oxidation. The main pathogenic mechanisms of fatty acid oxidation defects include toxic effects by accumulating acylcarnitine and acyl-CoA species and energy deficiency due to impaired fatty acid oxidation and ketone body formation. The respective disorders present with heterogeneous phenotypes. Before newborn screening (NBS) was introduced, the most common clinical presentations were hypoketotic hypoglycemia and sudden death, usually precipitated by an infection or fasting in the neonatal period or early childhood. In addition, severe cardiomyopathy and arrhythmias were leading clinical signs in the first months of life. Older children or adults may present with exercise- or illness-induced rhabdomyolysis. Patients can remain asymptomatic throughout life if they have mild defects and are not exposed to metabolic stress. Correlation of genotype and/or residual enzyme activity with disease phenotype has been reported for some defects, whereas in others additional disease modifiers are suspected. With newborn screening, disease incidence has significantly increased and the proportion of milder phenotypes has grown. Newborn screening greatly reduces the morbidity and mortality, though it does not eliminate early neonatal death in severe phenotypes. With respect to the heterogeneous clinical presentation, treatment needs to be tailored to the severity of the respective phenotype and the specific disorder.
1 Introduction
Fat is an important source of energy and the body’s principal fuel store. In postnatal life fatty acids are used in preference to glucose by the heart. They are also the main fuel for skeletal muscle during sustained exercise.
Mitochondrial fatty acid oxidation involves four processes: (1) entry of fatty acids into mitochondria (the carnitine cycle), (2) mitochondrial β-oxidation via a spiral pathway utilizing membrane-bound enzymes, (3) mitochondrial β-oxidation of chain-shortened fatty acids using matrix enzymes, and (4) electron transfer to the respiratory chain.
The clinical presentation of these disorders is heterogeneous. Since implementation of newborn screening for fatty acid oxidation disorders (FAODs), milder phenotypes and asymptomatic patients with different genotypes are identified. Before newborn screening, FAODs have three characteristic clinical presentations:
-
1.
Acute hypoketotic hypoglycemia and encephalopathy accompanied by hepatomegaly and liver dysfunction precipitated by fasting or an infection. This is often described as a hepatic presentation or “Reye-like symptoms.” Presentation generally occurs in the first weeks and months of life.
-
2.
Cardiomyopathy (usually hypertrophic, but also dilated in later stages), arrhythmias, or conduction defects. The cardiac phenotype generally occurs in the first weeks and months of life.
-
3.
Myopathy, presenting either with weakness, with pain, or with acute rhabdomyolysis, precipitated by exercise or infection. The myopathic phenotype mainly occurs after infancy and in later childhood or adolescence.
A number of patients will never develop symptoms, either because they have a mild defect or because they are not exposed to the necessary environmental stress; it is not yet clear how many of the NBS-diagnosed patients will remain asymptomatic throughout life.
2 Metabolic Markers
Fasting hypoglycemia is primarily due to increased peripheral glucose consumption with a concomitant decreased production of ketones (Herrema et al. 2008); consequently the hypoglycemia is hypoketotic. Small amounts of ketone bodies can be synthesized, particularly in medium- or short-chain FAODs or if there is high residual enzyme activity, but the plasma concentrations are lower than expected for the degree of hypoglycemia. Hyperammonemia occurs in some severe defects, and lactic acidemia is particularly seen in LCHAD and MTP deficiencies and in MAD deficiency. Rhabdomyolysis is accompanied by highly elevated creatine kinase (CK) up to 100,000 U/l or higher. Hepatomegaly and steatosis resulting from endogenous lipolysis are reflected by increased liver transaminases (AST, ALT). CK and transaminases are very sensitive markers to indicate metabolic derangement in long-chain fatty acid oxidation defects. The toxic effects of accumulating long-chain acylcarnitines and acyl-CoA esters are likely responsible for arrhythmias in these disorders that are especially severe in disorders of the mitochondrial trifunctional protein (MTP and LCHAD deficiencies) and CACT deficiency (Corr et al. 1989). The development of retinopathy and peripheral neuropathy in LCHAD and MTP deficiencies is associated with the accumulation of long-chain hydroxyacylcarnitines.
The disease-specific acylcarnitine profile is used for newborn screening with tandem mass spectrometry on days 2 and 3 of life; postponing the blood sampling may yield false-negative results later on. Secondary carnitine deficiency may result in an otherwise normal acylcarnitine profile. Organic acid analysis in urine has previously been an important tool for the diagnosis of fatty acid oxidation defects and the assessment of dicarboxylic aciduria is highly diagnostic; however, nowadays acylcarnitine analysis is the primary diagnostic tool, invariably followed by enzyme (or molecular) analysis, since milder defects often present with a normal organic acid profile in urine.
Carnitine transporter Deficiency. The organic cation carnitine transporter OCTN2 is responsible for the carnitine uptake across the plasma membrane, particularly in heart, muscle, and kidney. Defects of the transporter lead to primary systemic carnitine deficiency with increased renal loss of carnitine and extremely low plasma carnitine concentrations. Parallel measurement of carnitine in blood and urine is the first diagnostic step. On newborn screening, low carnitine concentrations may also be found in newborns of vegan mothers or mothers with an undiagnosed OCTN2 deficiency. Asymptomatic adults with very low plasma carnitine concentrations have been identified. Therefore, carnitine measurement in the maternal plasma and urine has to be part of the diagnostic workup of a low carnitine on NBS.
Carnitine Palmitoyltransferase I (CPT I) Deficiency. Three different genetic isoforms of CPT I have been found in liver and kidney (CPT IA), muscle and heart (CPT IB), and brain (CPT IC). Only CPT IA deficiency has been identified in humans presenting in infancy with hypoketotic hypoglycemia and hepatopathy, induced by fasting or illness. Affected patients have an extremely high level of free carnitine in their blood spot with a concomitant decrease of the long-chain acylcarnitines.
Carnitine-Acylcarnitine Translocase (CACT) Deficiency. Most patients have presented in the neonatal period and died in infancy. This phenotype is characterized by severe ventricular arrhythmias that are most likely triggered by accumulating toxic fatty acid metabolites during catabolism. The acylcarnitine profile resembles that found in carnitine palmitoyltransferase II deficiency.
Carnitine Palmitoyltransferase II (CPT II) Deficiency. The most common form of this disorder presents with recurrent episodes of rhabdomyolysis and myoglobinuria precipitated by prolonged exercise, mainly in adolescents or young adults. The plasma CK often normalizes between episodes, but it may also remain moderately elevated, associated with chronic muscle weakness. Severe neonatal onset CPT II deficiency is lethal and is associated with congenital malformations, principally renal cysts and neuronal migration defects (North et al. 1995). The adult myopathic form of CPT II deficiency is biochemically difficult to diagnose since acylcarnitine profiles may be normal, even during metabolic derangement.
Very Long-Chain Acyl-CoA Dehydrogenase (VLCAD) Deficiency. VLCAD deficiency is the second most common fatty acid oxidation disorder in Europe and the USA, with a prevalence between 1:50,000 and 1:100,000. This is much higher than was assumed from the number of clinically manifesting patients. Neonatal screening has identified milder phenotypes with different genotypes and higher residual activities (Spiekerkoetter et al. 2010). A large number of patients are asymptomatic at diagnosis and during follow-up over up to 10 years.
Mitochondrial Trifunctional Protein (MTP), LKAT, and LCHAD Deficiencies. Isolated LKAT deficiency is rare and has only been reported in one patient. Patients with isolated LCHAD deficiency or general MTP deficiency irrespective of the localization of the mutation in the HADHA or HADHB gene present with similar heterogeneous phenotypes (Spiekerkoetter et al. 2004). Importantly, this is the only FAOD associated with retinopathy and peripheral neuropathy (Tyni et al. 1998). In neuromyopathic phenotypes, the acylcarnitine profile is often normal, even during an episode of rhabdomyolysis. Mothers who are heterozygous for LCHAD or MTP deficiency have an increased risk of HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets) and acute fatty liver of pregnancy (AFLP) during pregnancies when they are carrying an affected fetus (Wilcken et al. 1993).
Acyl-CoA Dehydrogenase 9 (ACAD9) Deficiency. ACAD9 is homologous to VLCAD and has dehydrogenase activity towards various long-chain acyl-CoA esters in vitro. Its physiological role is in the assembly of the mitochondrial respiratory chain complex I (Nouws et al. 2010).
Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency. MCAD deficiency presents with an incidence of approximately 1:10,000 in Europe, Australia, and the USA. Affected patients only present clinically if exposed to an appropriate environmental stress such as prolonged fasting or an infection with vomiting. Even before the era of newborn screening, many patients (affected siblings of symptomatic patients) remained asymptomatic (Wilcken et al. 2007). Blood C 8-carnitine is a highly specific marker, but may be only moderately elevated in mild phenotypes. The excretion of hexanoylglycine in urine is pathognomonic of MCADD; however, in milder phenotypes it is not detectable. Residual enzyme activity correlates with the genotype and the expected clinical phenotype. Compound heterozygous individuals with the c199T > C mutation on one allele present with residual activities in the range of heterozygotes, doubting a clinical relevance of this mutation.
Short-Chain Acyl-CoA Dehydrogenase (SCAD) Deficiency. There are two polymorphisms in the SCAD gene (c.625G > A and c.511C > T). In northern Europe, 6 % of the general population has one of these variants on both alleles. Biochemically the diagnosis relies on the finding of increased blood C4-carnitine and/or urine ethylmalonic acid. The clinical significance of SCAD deficiency is unclear. It may confer susceptibility to disease together with a second mitochondrial impairment (van Maldegem et al. 2006).
Short-Chain 3-Hydroxyacyl-CoA Dehydrogenase (SCHAD) Deficiency. SCHAD deficiency is associated with hypoglycemia due to hyperinsulinism, in contrast to other FAODs. SCHAD has a second role independent of β-oxidation, binding and inhibiting glutamate dehydrogenase (GDH): SCHAD mutations that prevent GDH binding lead to increased GDH activity and insulin secretion, particularly in response to leucine (Li et al. 2010). Increased plasma 3-hydroxy-C4-carnitine and urine 3-hydroxyglutaric acid are the accepted diagnostic parameters.
Electron Transfer Defects (Multiple Acyl-CoA Dehydrogenase Deficiency, MADD). This disorder results from defects of the electron transfer flavoprotein (ETF) or the ETF ubiquinone oxidoreductase (ETF:QO). Consequently, all acyl-CoA dehydrogenases will have a decreased activity in addition to glutaryl-CoA dehydrogenase and d-2-hydroxyglutarate dehydrogenase. The biochemical diagnosis is based on multiple elevated blood acylcarnitines and abnormalities of urine organic acids. Severely affected patients present in the first days of life and generally also exhibit congenital anomalies and facial dysmorphism. The malformations resemble those seen in CPT II deficiency and this phenotype is also mainly lethal in the neonatal period. The riboflavin-responsive phenotype has a good prognosis with riboflavin treatment. Mutations in the gene coding for ETF:QO have been identified in these patients (Olsen et al. 2007). Only recently, patients with a riboflavin transporter defect have been described also presenting as mild MAD deficiency and responding to riboflavin treatment.
3 Nomenclature
No. | Disorder | Alternative name | Abbreviation | Gene symbol | Chromosomal localization | Affected protein | OMIM no. | Subtype |
|---|---|---|---|---|---|---|---|---|
17.1.1 | Carnitine transporter deficiency | Carnitine uptake defect | OCTN2 | SLC22A5 | 5q31 | Organic cation carnitine transporter 2 | 212140 | All forms |
17.1.2 | Carnitine palmitoyltransferase I deficiency | Carnitine palmitoyl-CoA transferase I deficiency | CPT I | CPT Ia | 11q22.23 | Carnitine palmitoyltransferase | 255120 | All forms |
17.1.3 | Carnitine acylcarnitine translocase deficiency (30 patients) | CACT | SLC25A20 | 3p21.31 | Carnitine acylcarnitine translocase | 212138 | All forms | |
17.1.4 | Carnitine palmitoyltransferase II deficiency | Carnitine palmitoyl-CoA transferase II deficiency | CPT II | CPT II | 1p32 | Carnitine palmitoyltransferase II | 255110 | All forms |
17.2.1 | Very long-chain acyl CoA dehydrogenase deficiency | VLCAD | ACADVL | 17p13 | Very long-chain acyl-CoA dehydrogenase | 201475 | All forms | |
17.2.2 | Mitochondrial trifunctional protein deficiency | mTFP, MTP | HADHA, HADHB | 2p23 | Long-chain enoyl-CoA hydratase, long-chain 3-hydroxyacyl-CoA dehydrogenase, 3-ketoacyl-CoA thiolase | HADHA 600890; HADHB 143450 | All forms | |
17.2.3 | Isolated deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase | LCHAD | HADHA | 2p23 | Long-chain 3 hydroxyacyl-CoA dehydrogenase | 600890 | All forms | |
17.2.4 | Isolated deficiency of long-chain 3-ketoacyl CoA thiolase patient | LKAT | HADHB | 2p23 | Long-chain 3-ketoacyl-CoA thiolase | 143450 | All forms | |
17.2.5 | Acyl-CoA dehydrogenase 9 deficiency | Complex I assembly disorder | ACAD9 | ACAD9 | 3q26 | Acyl-CoA dehydrogenase 9 | 611126 | All forms |
17.3.1 | Medium-chain acyl CoA dehydrogenase deficiency | MCAD | ACADM | 1p31 | Medium-chain acyl CoA dehydrogenase | 201450 | All forms | |
17.3.2 | Short-chain acyl CoA dehydrogenase deficiency | Ethylmalonic aciduria | SCAD | ACADS | 12q22-qter | Short-chain acyl CoA dehydrogenase | 201470 | All forms |
17.3.3 | Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (13 patients) | SCHAD | HADH, SCHAD | 4q22–q26 | Short-chain 3-hydroxyacyl-CoA dehydrogenase | 231530 | All forms | |
17.4.1 | Multiple acyl-CoA dehydrogenase (ETF) deficiency | Glutaric aciduria type II, Electron transfer defect (ETF) | ETF, MAD | ETFA, ETFB | ETFA 15q23-q25, ETFB 19q13.3 | Electron transfer flavoprotein | ETFA 231680 ETFB 130410 | All forms |
17.4.2 | Multiple acyl-CoA dehydrogenase (ETF-DH) deficiency | Glutaric aciduria type II, ETF-ubiquinone oxidoreductase deficiency | ETF-DH, ETF-QO, MAD | ETFDH | 4q32-qter | ETF-ubiquinone oxidoreductase | 231675 | All forms |
17.4.3 | Riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency | Glutaric aciduria type II, Electron transfer defect (ETF) | ETF-DH, ETF-QO, MAD | ETFDH a | 4q32-qter | ETF-ubiquinone oxidoreductase | 231675 | All forms |
4 Metabolic Pathway
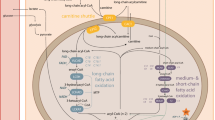
Fatty acids are activated to coenzyme A (CoA) esters in the cytoplasm, but long-chain fatty acids need to be esterified with carnitine in order to cross the inner mitochondrial membrane. Medium- and short-chain fatty acids can enter the mitochondria independent of carnitine. Carnitine mainly derives from dietary meat or is provided by hepatic endogenous synthesis. In vegans endogenous production is enhanced. By the high-affinity organic cation carnitine transporter 2 (OCTN2), carnitine is shuttled into the cytosol. At the outer mitochondrial membrane, the formation of acylcarnitine esters is catalyzed by carnitine palmitoyltransferase I (CPT I). The carnitine esters are then shuttled across the inner membrane by the carnitine acylcarnitine translocase (CACT). Carnitine palmitoyltransferase II (CPT II) is attached to the inner mitochondrial membrane and catalyzes the transfer back to CoA esters. The enzymes for the β-oxidation of long-chain substrates (C18-C14 fatty acids) are also membrane-bound and comprise the very long-chain acyl-CoA dehydrogenase (VLCAD) and three enzymes in the mitochondrial trifunctional protein complex (mTFP or MTP). This complex is composed of two subunits, harboring the long-chain enoyl-CoA hydratase (LCEH) and the long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) in the α-subunit and the long-chain 3 ketoacyl-CoA thiolase (LKAT) in the β-subunit. Each turn of the β-oxidation spiral involving four steps shortens the acyl-CoA by two carbons. These include two dehydrogenation reactions, linked respectively to flavin adenine dinucleotide (FAD) and nicotinamide adenine dinucleotide (NAD). The β-oxidation is catalyzed by enzymes of different chain length specificities. Medium- and short-chain enzymes such as medium-chain acyl-CoA dehydrogenase (MCAD), short-chain acyl-CoA dehydrogenase (SCAD), enoyl-CoA hydratase or crotonase, and short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) are located in the mitochondrial matrix. The liberated electrons are passed to the respiratory chain either directly (from NADH to complex I) or via two transfer proteins (from FADH2 to ubiquinone). Each action of an acyl-CoA dehydrogenase produces two electrons which are transferred to the electron transfer flavoprotein (ETF) and subsequently to ETF-dehydrogenase (ETF-DH, ETF-QO). Electron transfer plays a role in amino acid and choline metabolism in addition to mitochondrial β-oxidation. Acetyl-CoA residues released by β-oxidation can be either oxidized in the Krebs cycle or, utilized to synthesize ketone bodies in the liver, Flavin adenine dinucleotide (FAD), which is derived from riboflavin, acts as a cofactor in these reactions.

Transport of fatty acids into the mitochondrion, mitochondrial β-oxidation, and electron transfer (Modified according to Rinaldo et al. (2002))
5 Signs and Symptoms
6 Reference Values
Values of carnitine and acylcarnitines in dried blood spots and plasma differ (de Sain-van der Velden et al. 2013). The values also differ according to age, and in newborns there are reference values for the different days of life. Catabolism increases acylcarnitine concentrations also in healthy individuals, especially C2-carnitine, 3-hydroxybutyrylcarnitine, and the C12-C16 unsaturated carnitine esters (Costa et al. 1999). Reference values are given in Chap. 51 of this book dealing with the analysis of acylcarnitines in various body fluids.
Organic acid analysis in urine usually reveals a disease-specific profile that is not found in healthy individuals. Fasting significantly increases the excretion of disease-specific organic acids. The normal fasting urine may display a profile of organic acids as shown in the table below.
7 Pathological Values
An extremely wide variation in pathological values can be observed. In addition, patients can be missed at any time when the blood sample is taken at a moment when fatty acid oxidation is not requested such as postprandial blood samples. In general, adult and adolescent patients have a less prominent acylcarnitine profile than the neonatal/infantile presentations. Care should be taken in the interpretation of ketotic samples, since values can also be increased in healthy individuals.
Pathological values of acylcarnitines in newborn blood spot samples generally decrease in the first days of life and may normalize. Therefore, it will be inevitable that some isolated patients may be missed if screened too late. Importantly, also healthy newborns and children may present with abnormal acylcarnitine profiles suggesting a FAOD during severe catabolism. For VLCAD deficiency a false-positive rate of 1:2,600 is reported on newborn screening (Spiekerkoetter et al. 2010).
Reference pathological values are summarized by McHugh et al. and represent the 5th and 90th percentiles of pathological values taken from a large worldwide cohort of patients identified by newborn screening (McHugh et al. 2011). The table below summarizes pathological values when diagnosing clinically selected patients with FAO defects and exemplifies the variation of the levels of the clinically important acylcarnitines as well as that of free carnitine. Any patient with a primary defect of fatty acid beta oxidation may have free carnitine levels as low as those observed in the carnitine transporter defect OCTN2.
8 Diagnostic Tools and Flowchart
The investigation of a suspected FAOD starts by looking for abnormal acylcarnitines. Its diagnostic specificity can be increased by measuring the ratios of different acylcarnitines. Severe CPT II and CACT deficiencies have indistinguishable acylcarnitine profiles, as do LCHAD and MTP deficiencies. For CPT I deficiency, carnitine concentrations in dried blood spots are higher than in plasma, and diagnosis may be missed with measurement in plasma.
The clinical circumstances have a major effect on the acylcarnitine profile. Abnormalities are usually more marked during catabolism, but if the plasma-free carnitine concentration is very low, abnormal acylcarnitines may be hard to detect. Abnormalities may be masked by intravenous glucose or dietary treatment, such as the use of medium-chain triglycerides (MCT) in long-chain FAODs. An interpretation is especially difficult for samples obtained terminally or postmortem: these often show multiple raised acylcarnitine species, resembling MAD deficiency. The acylcarnitine profile can be completely normal in patients with high residual enzyme activity, such as mild VLCAD or MTP deficiencies. Abnormalities may, however, be detectable in samples collected after overnight fasting, exercise, or loading with carnitine.
If acylcarnitine analysis suggests a specific diagnosis, it has to be confirmed by enzyme assays and/or mutation analysis. If the metabolite results are nonspecific or if they are normal despite a strong clinical suspicion, it may be helpful to measure acylcarnitine production in vitro or flux through the pathway.
Quantitative acylcarnitine profiling may indicate the site of a defect if this is not clear from metabolite results. Acylcarnitines are analyzed by MS/MS after incubating fibroblasts or lymphocytes with fatty acids, labeled with stable isotopes (2H or 13C) (Ventura et al. 1999). This technique can identify most FAODs except carnitine transporter deficiency. CACT and severe CPT II deficiencies cannot be distinguished and respiratory chain defects sometimes mimic FAODs (Sim et al. 2002).
Fatty acid oxidation flux is measured by incubating cells with radiolabeled fatty acids and collecting the oxidation products (Olpin et al. 1997). This is useful in evaluating the severity of a disorder, but acylcarnitine profiling yields more diagnostic information. Severity of a disorder can also be best analyzed by direct enzyme activity measurement.
Urinary Organic Acids and Acylglycines
For many FAODs, urine organic acid analysis is normal when patients are well. During fasting or illness, however, medium-chain (and sometimes long-chain) dicarboxylic acids are elevated with little or no increase in ketone bodies. Dicarboxylic acids are formed whenever plasma-free fatty acid concentrations are increased, by β-oxidation in peroxisomes and ω-oxidation in microsomes, but normally they are accompanied by ketonuria. Dicarboxylic aciduria without ketonuria can also be seen in some respiratory chain defects. The analysis of acylcarnitines in the urine will disclose the presence of dicarboxylic acid carnitine esters that may have additional diagnostic significance. Defects of LCHAD and MTP deficiencies will show unusual hydroxydicarboxylic acids in addition, whereas MCADD is characterized by an abnormal excretion of several acylglycines such as hexanoylglycine, suberylglycine, and phenylpropionylglycine. The same acylglycines but also short branched-chain acylcarnitines will appear in MADD, in general accompanied by a variety of dicarboxylic acids such as ethylmalonic acid, glutaric acid, and d-2-hydroxyglutaric acid.
All mitochondrial FAODs show an autosomal recessive pattern of inheritance. There is molecular heterogeneity in all these disorders, but prevalent mutations have been identified. The relationship between genotype and phenotype varies in the different FAODs. In CPT II and VLCAD deficiencies, nonsense mutations on both alleles are generally associated with severe early-onset disease, whereas adult-onset rhabdomyolysis is associated with conservative missense mutations. In MCAD deficiency, the c.199T > C mutation is associated with significant residual activity and appears to be benign.
Enzyme assays are generally performed on cultured fibroblasts or lymphocytes (Wanders et al. 2010) and are available for all defects. Enzymology in lymphocytes allows a rapid confirmation of diagnosis. Moreover, for VLCAD and MCAD deficiencies, residual activity may allow some predictions with respect to the expected severity of the defect (Hoffmann et al. 2012).

Diagnostic flowchart
9 Specimen Collection
Test | Material | Handling | Transport | Pitfalls |
|---|---|---|---|---|
Acylcarnitines | Plasma, dried blood spot (DBS) | Room temperature(DBS) Frozen (plasma) | Normal mail | During anabolism eventually normal values, different profile with MCT diet, long-chain acylcarnitine accumulation also in healthy individuals during catabolism |
Carnitine | Plasma, dried blood spots (DBS) | Room temperature(DBS) Frozen (plasma) | Normal mail | Blood and tissue concentrations do not correlate; in CPT I deficiency carnitine is lower in plasma than in DBS; it may be missed on analysis in plasma |
Dicarboxylic acids | Spot urine | Frozen for longer storage | Normal mail | MCT diet and glucose infusion change the profile |
Free fatty acids | Plasma | Frozen | Frozen | Determine before the next food intake |
Enzyme assays in lymphocytes | EDTA plasma (2 ml) | Room temperature | Has to reach the laboratory within 48 h after withdrawal, room temperature | Poor quality of the lymphocytes (due to transport conditions, low temperature, etc.) may result in lower residual enzyme activities |
Enzyme assays in fibroblasts | Skin biopsy | Room temperature, in sterile 0.9 % sodium chloride, fibroblast culture | In culture medium | Fibroblast culture must grow 4–8 weeks until enzyme assays can be performed |
Molecular analysis | EDTA blood, dried blood spots | Room temperature | Normal mail | Delineation of only one mutation does not rule out deficiency of the enzyme |
10 Prenatal Diagnosis of Fatty Acid Oxidation Disorders
Prenatal diagnosis is available for all fatty oxidation disorders. Mutation analysis is the preferred technique, if the molecular defect is known in the index case. All enzymes of fatty acid oxidation are expressed in chorionic villus biopsies and amniocytes. Prenatal diagnosis is, therefore, also possible using enzyme assays. Chorionic villus biopsy can be performed at 11 + 0 gestational weeks, amniotic fluid test at 14 + 0 weeks.
Deficiency of | Prenatal diagnosis suggested | Remarks |
|---|---|---|
OCTN2 | − | Very favorable clinical outcome |
CPT I | − | Very favorable outcome |
CACT | + | Majority of patients die in the neonatal period due to severe cardiac arrhythmias |
CPT II | + | Suggested for severe neonatal phenotypes with congenital anomalies |
− | Myopathic phenotypes | |
VLCAD | − | Very favorable clinical outcome, many asymptomatic “patients,” skeletal myopathy needs to be discussed with the parents |
mTFP/MTP | + | Suggested for severe phenotypes, neonatal phenotypes generally lethal, irreversible neuropathy/retinopathy needs to be discussed with the parents in milder phenotypes |
(+) | ||
LCHAD | ± | Irreversible retinopathy/neuropathy needs to be discussed with the parents |
LKAT | + | Only 1 patient so far, died in the neonatal period |
ACAD 9 | + | Global mitochondrial dysfunction |
MCAD | − | Very favorable clinical outcome since screening |
SCAD | − | Only predisposition for disease |
SCHAD | ± | Phenotypes of different severity and response to treatment |
MAD | + | Suggested for severe neonatal/infantile phenotypes with/without congenital anomalies |
− | Myopathic phenotypes | |
Riboflavin-responsive MAD | − | Very favorable clinical outcome with riboflavin supplementation |
11 DNA Analysis and Prevalent Mutations
Molecular studies are used to confirm the diagnosis as an alternative to enzymology. This is satisfactory for well-defined mutations, such as the prevalent mutations seen in MCAD, LCHAD, and CPT II deficiencies. The pathogenicity of other sequence variants is sometimes hard to assess. Moreover, standard sequencing may miss some mutations, such as large deletions and those in introns that affect splicing.
Where enzymology is the confirmatory test, mutation analysis may also be undertaken to define the disorder more precisely, allow carrier testing, and simplify prenatal diagnosis.
Deficiency of | Prevalent mutations (Caucasian population) | Remarks | |
|---|---|---|---|
Mutation | Amino acid change | ||
OCTN2 | No prevalent mutation | ||
CPT I | c.1436C > T (Inuit population ) | p.P479L | 70 % of babies homozygous for c.1436C > T in the Inuit population (Canada and Greenland) |
CACT | No prevalent mutation | ||
CPT II | c.338C > T | p.S113L | Allele frequency (60 %) |
VLCAD | c.848T > C | p.V243A | Suggestive of mild VLCADD |
mTFP/MTP | No prevalent mutations | Many deletions and splice site mutations in the HADHA gene, most compound heterozygotes for c.1528G > C, and a second HADHA mutation have MTP deficiency | |
LCHAD | c.1528G > C | p.E474Q | Heterogeneous presentations despite homozygosity for c.1528G > C |
LKAT | No prevalent mutation | Only 1 patient reported | |
ACAD 9 | No prevalent mutation | ||
MCAD | c.985A > G | p.K329E | Classical MCADD, before screening: 80 % homozygosity |
c.199G > C | p.Y67H | Asymptomatic/(mild) variant, allele frequency: 6 % of mutant alleles in screened population, never found in a clinically diagnosed patient | |
SCAD | c.625G > A | p.G209S | Polymorphism with predisposition for disease, 625G > A: allele frequency: 22 % |
c.511C > T | p.171W | 511C > T: allele frequency, 3 % | |
SCHAD | No prevalent mutation | ||
MAD | No prevalent mutation | ||
Riboflavin-responsive MAD | No prevalent mutation | Mutations in the ETF-DH gene | |
Possibly also other genes affected | |||
12 Treatment
Prolonged fasting should be avoided in all FAODs in order to prevent acute metabolic decompensation. Frequent, regular feeds are recommended, especially during the first year of life, but subsequently overnight fasting will be tolerated in most disorders. Prolonged overnight fasting should be postponed until later childhood/adolescence especially in MTP and LCHAD deficiencies in order to reduce the risk of retinopathy and neuropathy as a consequence of accumulating toxic metabolites.
Dietary fat restriction is not indicated in MCAD deficiency and mild long-chain FAODs recently identified by newborn screening. Long-chain fat, however, needs to be restricted in severe long-chain FAODs and substituted by medium-chain triglycerides (MCT). The amount of dietary MCT depends on the severity of the phenotype. MTP and LCHAD deficiencies require dietary modification that is stricter (Spiekerkoetter et al. 2009). For symptomatic infants special MCT formulas exist. Anecdotal evidence suggests that a bolus of MCT before exercise can prevent rhabdomyolysis in patients with myopathic VLCAD deficiency. Studies in VLCAD-deficient mice also suggest that MCT should be given according to the energy demand, since MCT are otherwise elongated and stored as saturated long-chain fatty acids.
Carnitine treatment is undisputedly effective in patients with carnitine transporter deficiency. With a dose of 100 mg/kg/day, plasma concentrations may reach the lower normal range, but muscle carnitine concentrations remain less than 5 % of normal (Stanley et al. 1991). The value of carnitine supplementation in other FAODs is controversial. Plasma-free carnitine concentrations are often low, particularly after an acute illness, but tissue concentrations have seldom been measured. Carnitine treatment may even be harmful in long-chain FAODs, as it increases the concentrations of long-chain acylcarnitines, which are potentially arrhythmogenic (Primassin et al. 2008).
Patients with riboflavin-dependent MAD deficiency respond to treatment with riboflavin (100 mg/day). In moderately severe MAD deficiency, 3-hydroxybutyrate has been used with success in a few patients for the treatment of cardiomyopathy (Van Hove et al. 2003).
Bezafibrates (PPAR-α and PPAR-δ agonists) may be promising in the treatment of patients with myopathic CPT II or VLCAD deficiencies (Gobin-Limballe et al. 2007; Bonnefont et al. 2009).
Triheptanoin (C7 odd-chain fatty acid) has been substituted for MCT in a few patients with long-chain FAODs (Roe et al. 2002), but has not found a widespread application so far.
Emergency Treatment
Deficiency of | Emergency treatment | Pharmacological emergency treatment |
|---|---|---|
OCTN2 | Glucose i.v., oral glucose monomers (to avoid hypoglycemia) reach anabolism | l-carnitine i.v. (100–300 mg/kg/day) |
CPT I | Glucose i.v., oral glucose monomers (to avoid hypoglycemia) reach anabolism: <3 years: 10–12 mg/kg/min 3–10 years: 8–10 mg/kg/min >10 years: 5–8 mg/kg/min Oral MCT, i.v. MCT generally not available (special preparations) In case of severe decompensation reach anabolism with use of insulin ACAD 9: cave lactate | No carnitine, d,l-3-hydroxybutyrate in case of severe cardiomyopathy Pharmacological treatment of arrhythmias |
CACT | ||
CPT II | ||
VLCAD | ||
mTFP/MTP | ||
LCHAD | ||
LKAT | ||
ACAD 9 | ||
MCAD | Glucose i.v., oral glucose monomers (to avoid hypoglycemia) reach anabolism No MCT | |
SCAD | Glucose i.v. in case of hypoglycemia | |
SCHAD | Glucose i.v. | Glucagon, somatostatin diazoxide (treatment of hyperinsulinism) |
MAD | Glucose i.v., oral glucose monomers (to avoid hypoglycemia) reach anabolism | d,l-3-hydroxybutyrate in case of severe cardiomyopathy |
Riboflavin-responsive MAD | Glucose i.v., oral glucose monomers (to avoid hypoglycemia) | Riboflavin (100–300 mg/day) |
Standard Treatment
Deficiency of | Dietary treatment | Pharmacological treatment |
|---|---|---|
OCTN2 | Normal diet, regular meals, avoid catabolism | l-carnitine p.o. (100–300 mg/kg/day) |
CPT I | Normal diet, rarely MCT supplementation in severe phenotypes | No general carnitine supplementation |
CACT | MCT modified diet, strictly avoid catabolism (cave arrhythmias!) | |
CPT II | Severe phenotypes: Carbohydrate-enriched diet (65–75 % of total calories), fat restriction (25 % of total calories, 10–15 % MCT, 4 % essential fatty acids, up to 10 % LCT) Regular meals After 1st year of life: glucose polymers at night Myopathic phenotypes: Fat-reduced or normal diet MCT prior to exercise Regular meals, normal overnight fasting tolerance Asymptomatic patients: no dietary interventions | |
VLCAD | ||
mTFP/MTP | Strict long-chain fat reduction MCT supplementation (see CPT II, VLCAD) Docosahexaenoic acid (200–400 mg/kg/day) supplementation to prevent retinopathy is controversial | |
LCHAD | ||
LKAT | ||
ACAD 9 | No recommendations available | |
MCAD | No dietary intervention, regular meals | |
SCAD | No dietary intervention, regular meals | |
SCHAD | No dietary intervention, regular meals | Glucagon, somatostatin, diazoxide (treatment of hyperinsulinism) |
MAD | Fat-reduced and less protein-reduced diet or normal diet, regular meals | |
Riboflavin-responsive MAD | Normal diet, regular meals | Riboflavin (100–300 mg/day) |
Experimental Treatment
Treatment | Mechanism of treatment | Current status | Disorders |
|---|---|---|---|
Bezafibrate | PPAR-α and PPAR-δ agonists, stimulate residual enzyme activity in milder phenotypes | Successful in isolated patients, clinical trials going on | Myopathic CPT II and VLCAD deficiencies |
Triheptanoin | Odd-chain C7 fatty acid, anaplerotic action by provision of propionyl-CoA | Successful in isolated patients, clinical trials going on | Myopathic CPT II and VLCAD deficiencies |
References
Bonnefont JP, Bastin J, Behin A et al (2009) Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med 360:838–840
Corr PB, Creer MH, Yamada KA et al (1989) Prophylaxis of early ventricular fibrillation by inhibition of acylcarnitine accumulation. J Clin Invest 83:927–936
Costa CC, de Almeida IT, Jakobs C, Poll-The BT, Duran M (1999) Dynamic changes of plasma acylcarnitine levels induced by fasting and sunflower oil challenge test in children. Pediatr Res 46:440–444
de Sain-van der Velden MGM, Diekman EF, Jans JJ et al (2013) Differences between acylcarnitine profiles in plasma and bloodspots. Mol Genet Metab 110:116–121
Gobin-Limballe S, Djouadi F, Aubey F et al (2007) Genetic basis for correction of very-long-chain acyl-coenzyme A dehydrogenase deficiency by bezafibrate in patient fibroblasts: toward a genotype-based therapy. Am J Hum Genet 81:1133–1143
Herrema H, Derks TG, van Dijk TH et al (2008) Disturbed hepatic carbohydrate management during high metabolic demand in medium-chain acyl-CoA dehydrogenase (MCAD)-deficient mice. Hepatology 47:1894–1904
Hoffmann L, Haussmann U, Mueller M et al (2012) VLCAD enzyme activity determinations in newborns identified by screening: a valuable tool for risk assessment. J Inherit Metab Dis 35:269–277
Li C, Chen P, Palladino A et al (2010) Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem 285:31806–31818
McHugh DMS, Cameron C, Abdenur JE et al (2011) Clinical validation of cut-off target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med 13:230–254
North KN, Hoppel CL, De Girolami U et al (1995) Lethal neonatal deficiency of carnitine palmitoyltransferase II associated with dysgenesis of the brain and kidneys. J Pediatr 127:414–420
Nouws J, Nijtmans L, Houten S et al (2010) Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab 12:283–294
Olpin SE, Manning NJ, Pollitt RJ et al (1997) Improved detection of long-chain fatty acid oxidation defects in intact cells using [9,10-3H]oleic acid. J Inherit Metab Dis 20:415–419
Olsen RK, Olpin SE, Andresen BS et al (2007) ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 130:2045–2054
Primassin S, ter Veld F, Mayatepek T, Spiekerkoetter U (2008) Carnitine supplementation induces acylcarnitine production in tissues of very long-chain acyl-CoA dehydrogenase-deficient mice, without replenishing low free carnitine. Pediatr Res 63:632–637
Rinaldo P, Matern D, Bennett MJ (2002) Fatty acid oxidation disorders. Annu Rev Physiol 64:477–502
Roe CR, Sweetman L, Roe DS et al (2002) Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest 110:259–269
Sim KG, Carpenter K, Hammond J et al (2002) Acylcarnitine profiles in fibroblasts from patients with respiratory chain defects can resemble those from patients with mitochondrial fatty acid beta-oxidation disorders. Metabolism 51:366–371
Spiekerkoetter U, Khuchua Z, Yue Z et al (2004) General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatr Res 55:190–196
Spiekerkoetter U, Lindner M, Santer R et al (2009) Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis 32:498–505
Spiekerkoetter U, Haussmann U, Mueller M et al (2010) Tandem mass spectrometry screening for very long-chain acyl-CoA dehydrogenase deficiency: the value of second-tier enzyme testing. J Pediatr 157:668–673
Stanley CA, DeLeeuw S, Coates PM et al (1991) Chronic cardiomyopathy and weakness or acute coma in children with a defect in carnitine uptake. Ann Neurol 30:709–716
Tyni T, Kivela T, Lappi M et al (1998) Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: a new type of hereditary metabolic chorioretinopathy. Ophthalmology 105:810–824
Van Hove JL, Grunewald S, Jaeken J et al (2003) D, L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD). Lancet 361:1433–1435
van Maldegem BT, Duran M, Wanders RJ et al (2006) Clinical, biochemical, and genetic heterogeneity in short-chain acyl-coenzyme A dehydrogenase deficiency. JAMA 296:943–952
Ventura FV, Costa CG, Struys EA et al (1999) Quantitative acylcarnitine profiling in fibroblasts using [U-13C] palmitic acid: an improved tool for the diagnosis of fatty acid oxidation defects. Clin Chim Acta 281:1–17
Wanders RJ, Ruiter JP, IJlst L et al (2010) The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J Inherit Metab Dis 33:479–494
Wilcken B, Leung KC, Hammond J et al (1993) Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency. Lancet 341:407–408
Wilcken B, Haas M, Joy P et al (2007) Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet 369:37–42
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Spiekerkoetter, U., Duran, M. (2014). Mitochondrial Fatty Acid Oxidation Disorders. In: Blau, N., Duran, M., Gibson, K., Dionisi Vici, C. (eds) Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-40337-8_17
Download citation
DOI: https://doi.org/10.1007/978-3-642-40337-8_17
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-40336-1
Online ISBN: 978-3-642-40337-8
eBook Packages: MedicineMedicine (R0)