
Abstract
Despite major advances in treatment, chronic heart failure (CHF) remains one of the leading causes of mortality and morbidity in the western world. Although the stimuli for CHF are wide ranging, the underlying pathophysiological response, known as cardiac remodeling, is broadly similar. The process of cardiac remodeling is characterized by a series of initially adaptive structural and functional alterations which become progressively maladaptive and ultimately lead to CHF. Oxidative stress and reactive oxygen species (ROS) play a well-established role in the development and progression of cardiac remodeling. ROS include both free radicals (e.g., superoxide, hydroxyl) and oxidants (e.g., peroxynitrite, hydrogen peroxide) and exert important physiological actions, in addition to their well-documented harmful effects. Increased ROS production has been widely reported in experimental and clinical CHF and has been linked to several key remodeling processes such as cardiomyocyte hypertrophy, apoptosis, contractile dysfunction, and extracellular matrix remodeling. It is now apparent that myocardial ROS function as key regulators of intracellular signaling that are capable of altering the activity of numerous molecules and pathways to exert subtle modulatory effects on cardiac remodeling. Indeed, several key signaling pathways involved in CHF pathogenesis, including MAPKs, Akt, NF-kB, and AP-1 are known to be redox sensitive, with varying thresholds for activation. The downstream effects of ROS appear to be critically dependent upon a number of factors including the source of ROS, cellular localization, temporal regulation, and stimulus for production, which dictate the highly specific and targeted phenotypic changes associated with cardiac remodeling. Although the precise mechanisms underlying ROS-dependent modulation of myocardial signaling remain unknown, it is plainly evident that a more complete delineation of these pathways in cardiac remodeling and CHF holds clear potential for the identification of improved therapeutic targets.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Cardiomyocyte hypertrophy
- Chronic heart failure
- Extracellular matrix remodeling
- Mitochondria
- NADPH oxidases
- Reactive oxygen species
- Serine/threonine protein kinases
- Transcription factors
Introduction
Chronic heart failure (CHF) affects up to 2 % of the adult population in the western world and is the leading cause of morbidity and mortality (Jessup and Brozena 2003). The incidence of CHF is rapidly increasing, and despite improved treatment with agents such as ACE inhibitors and β-blockers, it carries a poor prognosis, with reported 1-year mortality rates of up to 45 % (Jessup and Brozena 2003; Dolezal 2006). The causes of CHF are often multifactorial and include myocardial ischemia, hypertension, cardiotoxicity, valvular heart disease, and cardiomyopathy, although the underlying pathophysiological response, collectively known as cardiac remodeling, is broadly similar (Swynghedauw 1999; Jessup and Brozena 2003). The process of cardiac remodeling is characterized by substantial alterations in myocardial structure, which are initially adaptive in maintaining or improving contractile function. However, progressive remodeling leads to increased wall stress, cardiac chamber dilatation, contractile dysfunction, and ultimately CHF (Brilla et al. 1990; Swynghedauw 1999). Initially, cardiac remodeling was considered to occur consequent of compensatory mechanisms producing direct toxic actions on the heart; however, accumulating evidence indicates that the process itself significantly contributes to disease progression in heart failure (Kurrelmeyer et al. 1998; Mann 1999). Despite improved clinical management of CHF with agents such as ACE inhibitors and β-adrenoceptor blockers, which have been shown to attenuate cardiac remodeling and confer morbidity/mortality benefits, there remains a substantial incidence of CHF even in optimally treated patients (Francis 2001). An improved and detailed understanding of the mechanisms underlying the pathogenesis of CHF is therefore essential in the search for novel therapeutic CHF targets.
Oxidative Stress and Cardiac Remodeling
It is now well established that oxidative stress and reactive oxygen species (ROS) play a key role in the development and progression of cardiac remodeling, with increased ROS production being widely reported in both experimental and clinical CHF (McMurray et al. 1993b; Hill and Singal 1996; Dieterich et al. 2000; Date et al. 2002; Heymes et al. 2003). At the cellular level, ROS have been linked to several key processes including cardiomyocyte hypertrophy, apoptosis, contractile dysfunction, extracellular matrix remodeling (ECM), inflammation, and vascular remodeling, each of which will be discussed in detail in this section.
Cardiomyocyte Hypertrophy
One of the most characteristic features of the remodeled heart is cardiomyocyte hypertrophy, which occurs in response to increased cardiac load. At the molecular level, stretching of the cardiomyocyte results in local neurohumoral activation, e.g., norepinephrine, angiotensin II (Ang II), endothelin-1 (ET-1), and significant alterations in gene expression which are largely responsible for the hypertrophic response (Swynghedauw 1999; Cohn et al. 2000). While it is thought to be initially adaptive by normalizing wall stress, progressive cardiomyocyte hypertrophy becomes maladaptive resulting in increased wall stress, ventricular dilatation, contractile dysfunction, and eventually development of CHF (Brilla et al. 1990; Swynghedauw 1999). Indeed, left ventricular hypertrophy (LVH) itself is a major independent risk factor for ischemic CHF and sudden death and is one of the most potent predictors of adverse cardiovascular outcomes in hypertensive patients (Gradman and Alfayoumi 2006). Over recent years, oxidative stress has become inexorably linked with the development and progression of cardiomyocyte hypertrophy. For example, increased ROS production has been widely reported in both experimental and clinical LVH (Treasure et al. 1993; Dhalla et al. 1996; Date et al. 2002; Yasunari et al. 2004). Increased ROS production is also implicated in the hypertrophic response of isolated cardiomyocytes induced by various stimuli including norepinephrine, Ang II, ET-1, phenylephrine (PE), tumor necrosis factor-α (TNF-α), and cyclic stretch (Nakamura et al. 1998; Pimentel et al. 2001; Hirotani et al. 2002). Furthermore, inhibition of the endogenous antioxidants, Cu-Zn superoxide dismutase (SOD) and thioredoxin, also induces oxidative stress and cardiomyocyte hypertrophy in vitro and in vivo, respectively (Siwik et al. 1999; Yamamoto et al. 2003). Similarly, the development and progression of experimental pressure-overload LVH was inhibited by either treatment with the antioxidants, vitamin E, N-acetylcysteine, and hemeoxygenase-1, or overexpression of endogenous catalase or thioredoxin-2, supporting a role for ROS production in cardiac hypertrophy in vivo (Dhalla et al. 1996; Byrne et al. 2003; Hu et al. 2004; Widder et al. 2009; Qin et al. 2010).
Extracellular Matrix Remodeling
One of the defining characteristics of cardiac remodeling is a marked increase in interstitial fibrosis, which is thought to be largely responsible for increased myocardial stiffness and the development of diastolic dysfunction in CHF (Litwin and Grossman 1993; Weber et al. 1994; Swynghedauw 1999). The process of ECM remodeling involves a complex reorganization and delicate balance between collagen deposition and degradation (Swynghedauw 1999). The main driving force for matrix turnover is a family of zinc-containing endoproteinases, known as matrix metalloproteinases (MMPs), whose activity and expression is markedly altered in CHF (Spinale et al. 2000; Creemers et al. 2001). The activity of MMPs is controlled by TIMPs (tissue inhibitors of MMPs), which are also dysregulated in CHF and work in tandem to control collagen deposition and accumulation (Li et al. 1998; Sun and Weber 2000). MMP activation not only facilitates the initial development of fibrosis but is also implicated in subsequent left ventricular (LV) dilatation which occurs during the transition to CHF (Peterson et al. 2001; Spinale 2002). The key role of MMPs in cardiac remodeling is underlined by studies in which pharmacological inhibition or genetic manipulation was found to attenuate LV dilatation and contractile dysfunction in experimental CHF (Matsusaka et al. 2006; Morita et al. 2006). The process of ECM remodeling is known to be particularly redox sensitive, with key underlying processes, such as fibroblast proliferation, collagen synthesis, and MMP activation/expression, being modulated by ROS (Kunsch and Medford 1999; Siwik et al. 2001; Zhang et al. 2002). Indeed, several of the main pathophysiological stimuli that drive interstitial fibrosis and matrix turnover, such as Ang II, cytokines, and cyclic load, also stimulate intracellular ROS production (Kunsch and Medford 1999; Sano et al. 2001). Furthermore, the development of cardiac interstitial fibrosis driven specifically by Ang II or aldosterone and associated MMP activation have been demonstrated to be mediated by ROS (Bendall et al. 2002; Sun et al. 2002; Johar et al. 2006). Chronic treatment with the antioxidants dimethylthiourea, probucol, and apocynin is also reported to attenuate the progression of cardiac fibrosis in vivo, confirming an important role for ROS in this setting (Kinugawa et al. 2000; Sia et al. 2002; Park et al. 2004).
Contractile Dysfunction
Structural remodeling of the cardiomyocyte and extracellular matrix is known to have a significant impact on cardiac contractile function. However, the development and progression of CHF is also characterized by marked effects on cardiomyocyte biology, such as alterations in membrane protein expression, excitation-contraction coupling, myofilament calcium sensitivity, and organization of the cytoskeleton, together with significant changes in the extracellular matrix (as discussed above), all of which contribute to the onset of contractile dysfunction and subsequent transition to CHF (Swynghedauw 1999). It is well established that contractile dysfunction in CHF is associated with oxidative stress and increased ROS production (McMurray et al. 1993a; Mallat et al. 1998; MacCarthy et al. 2001; Byrne et al. 2003). Indeed, in a murine model of pressure overload, treatment with the antioxidants, N-acetylcysteine and tempol, reduced ROS production and cardiac contractile dysfunction, whereas global deficiency of extracellular SOD exacerbated myocardial oxidative stress and contractile dysfunction (Grieve et al. 2006; Chess et al. 2008; Lu et al. 2008). In isolated cardiomyocytes, elevated ROS levels have been shown to suppress contractile function which is restored by antioxidants (Ye et al. 2004; Dong et al. 2006; Li et al. 2008; Rueckschloss et al. 2010). At the cellular level, ROS have been reported to impair calcium transients, modulate opening of ryanodine receptors, decrease L-type calcium channel current, and inhibit activity of the sarcoplasmic reticulum Ca2+-ATPase (SERCA), all of which are important regulators of cardiomyocyte contractility (Gill et al. 1995; Xu et al. 1997; Kawakami and Okabe 1998; Zima et al. 2004; Rueckschloss et al. 2010). The precise mechanisms by which ROS modulate Ca2+ handling and contractile function in cardiomyocytes are unknown. However, recent studies suggest an important role for oxidative protein modifications of thiols on SERCA, the sodium-Ca2+ exchanger, and ryanodine receptor 2 (Belevych et al. 2007; Lancel et al. 2010; Kuster et al. 2010).
Cardiomyocyte Apoptosis
The major cause of cardiomyocyte death during cardiac remodeling is apoptosis, which not only results in significant loss of contractile tissue, but also stimulates compensatory cardiomyocyte hypertrophy and reparative fibrosis, all of which contribute to associated contractile dysfunction (Swynghedauw 1999). Apoptosis is a highly regulated form of programmed cell death occurring by two main pathways, both of which converge on caspase-3 activation resulting in nuclear degradation and characteristic DNA fragmentation (van Empel et al. 2005; Anilkumar et al. 2009). The intrinsic mitochondrial pathway involves opening of the mitochondrial permeability transition pore (MPTP), typically in response to stimuli such as ischemia and hypoxia, resulting in the release of several proteins, including cytochrome c, which complexes with caspase-9, triggering activation of caspase-3. The extrinsic pathway is stimulated by binding of specific ligands, such as Fas or TNF-α, to death receptors resulting in formation of the death-inducing signaling complex (DISC) and subsequent activation of caspase-8 and caspase-3 (van Empel et al. 2005). Although cardiomyocyte apoptosis is widely reported to occur at very low levels in both experimental and clinical CHF, it is thought to make a key contribution to the development and progression of cardiac remodeling (Kang and Izumo 2000). This is exquisitely demonstrated by a recent study in which conditional cardiac-specific activation of caspase-8 was found to result in severe dilated cardiomyopathy, despite only 0.23 % of the cardiomyocytes undergoing apoptosis (Wencker et al. 2003). It is well established that ROS can stimulate cardiomyocyte apoptosis (von Harsdorf et al. 1999). For example, oxidative stress has been continually associated with cardiomyocyte apoptosis occurring in response to several in vivo stimuli including ischemia, pressure overload, and cardiotoxicity (Looi et al. 2008; El Azzouzi et al. 2010; Zhao et al. 2010). Furthermore, hydrogen peroxide (H2O2) causes cardiomyocyte apoptosis both in vitro and in vivo, and the process has been shown to be sensitive to treatment with antioxidants (Aoki et al. 2002; Kwon et al. 2003; Qin et al. 2006; Qin et al. 2007, 2010). In addition to directly initiating apoptosis, ROS have been reported to exert several other important actions including mediation of apoptotic signaling, modulation of antiapoptotic systems, transcriptional regulation of apoptosis-relevant genes, and causation of direct mitochondrial damage (Anilkumar et al. 2009).
Inflammation
Over recent years, inflammation has emerged as an increasingly important player in the cardiac remodeling process (Heymans et al. 2009; Oikonomou et al. 2011). The first suggestion of the importance of inflammatory activation came from initial reports of raised plasma cytokine levels in CHF patients (Rauchhaus et al. 2000; Deswal et al. 2001). Although subsequent experimental studies have indicated that anti-inflammatory therapies may be beneficial in CHF (Zhou et al. 2006; Westermann et al. 2007), large-scale clinical trials of TNF-α blockade in this setting have provided disappointing results and may even be detrimental (Chung et al. 2003; Mann et al. 2004). Nonetheless, it is clear that increased inflammatory cell infiltration is a hallmark of CHF, which appears to play a key role in regulating underlying processes such as ECM remodeling and cardiomyocyte apoptosis (Zhou et al. 2006; Westermann et al. 2011). Although the underlying mechanisms remain unclear, it is evident that oxidative stress and increased ROS production are important in both mediating inflammatory cell activation and the ensuing myocardial response in experimental and clinical CHF (Machida et al. 2003; Chiang et al. 2006; Mercuro et al. 2007; Gao et al. 2008; Zhao et al. 2010). Furthermore, cardiac inflammation and remodeling are inhibited by antioxidants, underlining a key role for ROS in this setting (Muller et al. 2000; Xu et al. 2011).
Vascular Remodeling
Coronary angiogenesis is tightly linked to cardiac growth during development and is considered to be an important component of the adaptive response to myocardial stress (Giordano et al. 2001; Hilfiker-Kleiner et al. 2004). Indeed, it is thought that an imbalance between vascular and myocardial remodeling may be a key factor in determining progression to CHF (Shiojima et al. 2005). Myocardial vascular endothelial growth factor (VEGF) production appears to be one of the key regulators of angiogenesis in CHF, which is itself controlled by transcription factors such as hypoxia-inducible factor-1α (HIF-1α) and GATA4 (Shiojima et al. 2005; Heineke et al. 2007; Sano et al. 2007). Interestingly, it now appears that expression and activity of these pro-angiogenic factors may be redox sensitive. For example, VEGF is known to stimulate ROS production which plays a key role in downstream pro-angiogenic signaling, which is also attenuated by antioxidant treatment (Colavitti et al. 2002; Ushio-Fukai et al. 2002). Furthermore, ROS can activate upstream HIF-1α both directly and indirectly via phosphatase inhibition, thereby controlling activity of pro-angiogenic factors such as VEGF and erythropoietin (Kietzmann and Gorlach 2005; Bonello et al. 2007). Of particular interest is a recent study demonstrating that cardiomyocyte-derived H2O2 specifically promotes increases in myocardial capillary density, thereby conferring protection against adverse cardiac remodeling and dysfunction in response to pressure overload (Zhang et al. 2010).
ROS as Signaling Molecules
It is well established that ROS production and oxidative stress play a key role in the pathophysiology of cardiac remodeling, with increased ROS levels widely reported in both experimental and clinical CHF (Beswick et al. 2001; MacCarthy et al. 2001; Landmesser et al. 2002; Heymes et al. 2003). ROS in the cardiovascular system include the free radicals, superoxide (O2 •–) and hydroxyl (•OH), in addition to the powerful oxidants, peroxynitrite (ONOO−) and hydrogen peroxide (H2O2), and these are summarized in Fig. 40.1. Although the harmful cardiovascular effects of ROS are well documented, it is becoming increasingly apparent that ROS play a critical role in normal physiological processes, in particular as specific regulators of intracellular signaling pathways (Finkel 2011). For example, fertilization of a sea urchin egg results in a large increase in H2O2, which, rather than damaging the organism, serves to facilitate development of a protective embryonic matrix (Wong et al. 2004). In addition, inhibition of epidermal growth factor (EGF)-induced H2O2 production in A431 human epidermoid carcinoma cells resulted in marked attenuation of associated tyrosine phosphorylation of a range of key cellular proteins (Bae et al. 1997). Furthermore, downregulation of mitochondrial complex III in human fibroblasts prevented hypoxia-induced stabilization of HIF-1α, suggesting a crucial role for mitochondrial ROS in the setting of oxygen sensing (Brunelle et al. 2005). In the heart, ROS signaling has been shown to play a key physiological function in both cardiac development via regulation of proliferation and differentiation (Sauer et al. 2000) and regulation of excitation-contraction coupling via redox modulation of the ryanodine receptor (Zima and Blatter 2006; Hidalgo and Donoso 2008; Santos et al. 2011). It is now widely recognized that ROS are capable of altering the activity of numerous intracellular molecules and signaling pathways to exert subtle modulatory effects that can induce highly specific phenotypic changes. In health, the production and activity of ROS are tightly regulated by several endogenous antioxidant systems, which serve to maintain physiological ROS levels sufficient for basal signaling. These antioxidant systems are both enzymatic and nonenzymatic in nature. Enzymatic antioxidants include superoxide dismutase (SOD), catalase, and glutathione peroxidase (Sies 1997). Superoxide dismutases, of which there are three isoforms (cytosolic Cu/Zn-SOD, mitochondrial Mn-SOD, and extracellular SOD), represent the major mechanism by which excess O2 •– is degraded to H2O2 (Giordano 2005). Thioredoxin and thioredoxin reductase together form an additional enzymatic antioxidant and redox regulatory system that has been implicated in a wide variety of ROS-related processes (Nordberg and Arner 2001). Thioredoxin and thioredoxin reductase can catalyze the regeneration of many antioxidant molecules, including ubiquinone (Q10), lipoic acid, and ascorbic acid, and as such constitute an important antioxidant defense mechanism. Examples of nonenzymatic antioxidants include vitamins C and E and β-carotene as well as glutathione, all of which work in combination to regulate ROS levels (Chen et al. 2012). In the normal physiological situation, ROS and endogenous antioxidants therefore act in concert to maintain normal signaling processes. However, in pathological conditions oxidative stress may occur due to an imbalance between ROS generation and antioxidant activity. Increased ROS levels then exert deleterious oxidative damage to DNA, proteins, lipids, membranes, and macromolecules, which underlie many of the changes associated with adverse cardiac remodeling (Cave et al. 2005). It is becoming increasingly clear that the source and location of cellular ROS production is critical in determining their downstream effects. Once thought of as nonspecific signaling molecules, it is now apparent that ROS signaling may be regulated in both a spatial and temporal manner to exert highly targeted effects which dictate distinct phenotypic changes. In this regard, important potential enzymatic sources of ROS in the cardiovascular system will now be discussed.

Schematic representation of the major ROS-producing pathways in the cardiovascular system. Oxidases convert oxygen to superoxide (O2 •–), which may then be either dismutated to hydrogen peroxide (H2O2) by superoxide dismutase (SOD) or rapidly react with nitric oxide (NO) to form highly toxic peroxynitrite (ONOO−). H2O2 can then be further converted to (1) hydroxyl radical (OH•−) after reaction with Fe2+ or (2) H2O and O2 by catalase or glutathione peroxidase (GSH-Px) via the oxidation of glutathione (GSH) to glutathione disulphide (GSSG)
Cardiovascular Sources of ROS
Xanthine Oxidase
The xanthine oxidase enzyme is a form of xanthine oxidoreductase, which is interconvertible with xanthine dehydrogenase, and produces O2 •– as a by-product of the terminal steps of purine metabolism. Specifically, xanthine oxidase catalyzes the single electron reduction of molecular oxygen to O2 •– by promoting the conversion of hypoxanthine to xanthine and then uric acid. Basal levels of xanthine oxidase are low, although increases in expression and xanthine oxidase-mediated ROS production have been demonstrated in both experimental (Ekelund et al. 1999; Ukai et al. 2001) and clinical end-stage CHF (Leyva et al. 1998; Cappola et al. 2001). In addition, temporal alterations in xanthine oxidoreductase expression have been reported in rat models of left and right ventricular failure, suggesting that this enzyme system may be important in the transition from compensation to CHF (De Jong et al. 2000). Furthermore, several studies have documented beneficial effects of the xanthine oxidase inhibitor, allopurinol, in experimental models of pacing-induced CHF, dilated cardiomyopathy, and chronic myocardial infarction (MI), with reported improvements in LV contractile function, calcium cycling, myocardial efficiency, and adverse cardiac remodeling (Engberding et al. 2004; Stull et al. 2004; Duncan et al. 2005; Minhas et al. 2006; Saliaris et al. 2007). These effects appear to translate to the clinical setting as allopurinol treatment has been shown to result in acute improvement in myocardial efficiency and reduced mortality in CHF patients (Cappola et al. 2001; Wei et al. 2009) although larger scale clinical studies have reported limited benefits (Gavin and Struthers 2005; Pacher et al. 2006).
NADPH Oxidases
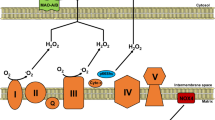
NADPH oxidases are a family of ROS-generating enzymes which are established as a major source of cardiac O2 •– production (Li et al. 2002; Byrne et al. 2003). The original NADPH oxidase was first described in neutrophils, where it produces high levels of ROS during phagocytosis (the “respiratory burst”) by catalyzing the single electron reduction of molecular oxygen, using NADPH as the electron donor (Griendling et al. 2000). Interestingly, unlike other sources, the primary function of NADPH oxidases appears to be ROS generation (Lambeth 2004). Seven NADPH oxidase family members have now been identified (Nox1-5 and Duox1-2), which are each based on a distinct catalytic subunit with tissue-specific expression (Lambeth. 2004; Brown and Griendling 2009). The major cardiac isoforms appear to be Nox2 and Nox4 (see Fig. 40.2), which are expressed in cardiomyocytes, fibroblasts, and endothelial cells (Anilkumar et al. 2008). The prototypic Nox2 NADPH oxidase is comprised of a membrane-bound catalytic core consisting of one Nox2 and one p22phox subunit and several regulatory cytosolic subunits (p47phox, p67phox, p40phox, and Rac1), which associate with the heterodimer upon activation by stimuli such as Ang II, ET-1, VEGF, and TNF-α (Griendling et al. 2000; Lambeth. 2004). In contrast, Nox4 NADPH oxidase is comprised of a single Nox4/p22phox heterodimer localized to the perinuclear endoplasmic reticulum and mitochondria and is constitutively active, being mainly regulated by protein level (Block et al. 2009; Brown and Griendling 2009). Interestingly, emerging data indicates that Nox2 NADPH oxidase may produce primarily O2 •–, whereas Nox4 generates mainly H2O2 (Martyn et al. 2006; Bedard and Krause 2007; Dikalov et al. 2008; Takac et al. 2011). Importantly, H2O2 is much more stable than O2 •– and can diffuse widely, which may account for distinct effects of different ROS forms on cellular redox regulation and underlie the apparent complexity of Nox signaling. Under physiological conditions, Nox NADPH oxidases generate low levels of ROS which are thought to be involved in basal intracellular signaling (Brown and Griendling 2009). However, it is well established that under pathological conditions, myocardial NADPH oxidase activity and expression is markedly increased and contributes significantly to CHF development and progression (Byrne et al. 2003; Heymes et al. 2003; Looi et al. 2008; Zhao et al. 2010). Specifically, NADPH oxidase-derived ROS have been shown to be critically involved in several of the cellular processes involved in cardiac remodeling, including interstitial fibrosis, MMP activation, cardiomyocyte hypertrophy, contractile dysfunction, and apoptosis (MacCarthy et al. 2001; Sun et al. 2002; Park et al. 2004; Rude et al. 2005; Qin et al. 2006). Interestingly, recent studies suggest that Nox2 and Nox4 NADPH oxidases exert differential effects on the heart which are dependent on the stimulus for remodeling. Studies in Nox2-deficient mice found that Nox2 NADPH oxidase was critical for cardiomyocyte remodeling and interstitial fibrosis induced by MI, Ang II, and doxorubicin (Bendall et al. 2002; Looi et al. 2008; Zhao et al. 2010). In contrast, cardiac hypertrophy in response to chronic pressure overload was unaltered in Nox2-deficient mice which showed elevated Nox4 expression, although Nox2 was essential for the development of contractile dysfunction and fibrosis (Byrne et al. 2003; Grieve et al. 2006). Interestingly, further studies found that mice with cardiomyocyte-targeted Nox4 overexpression, which exhibit elevated levels of H2O2, showed reduced LV hypertrophy, dilatation, and fibrosis with preserved contractile function after both pressure overload and MI, indicating that Nox4 NADPH oxidase may be cardioprotective, at least in this setting (Zhang et al. 2009; Zhang et al. 2010). However, a recent study reported that Nox4 NADPH oxidase-derived ROS promote cardiac remodeling associated with type 1 diabetes (Maalouf et al. 2012). The role of NADPH oxidases in cardiac remodeling is therefore complex, with differences in regulation, activation, subcellular localization, and ROS generation appearing to dictate distinct isoform-specific actions (Dikalov et al. 2008; Brown and Griendling 2009).

Structure of cardiac NADPH oxidases. Nox2 and Nox4 NADPH oxidases are expressed throughout the heart. They both contain a membrane-bound core (Nox2 or Nox4 together with p22phox) but have differing activities and functions. Nox2 NADPH oxidase activity requires the association of several regulatory cytosolic subunits (p47phox, p67phox, p40phox, Rac) and is thought to generate mainly superoxide using NADPH as the electron donor. In contrast, Nox4 is constitutively active and is thought to primarily produce H2O2
Mitochondria
Mitochondria represent not only an important enzymatic source of ROS but also a major target for ROS-mediated damage in the myocardium. Under normal physiological conditions, low levels of O2 •– are produced as a by-product of mitochondrial respiration via single electron transport from FADH and NADH to molecular oxygen, catalyzed by carriers located on the inner mitochondrial membrane (as shown in Fig. 40.3). These small amounts of ROS are then easily scavenged by endogenous antioxidant systems, primarily Mn-SOD (Sawyer and Colucci 2000). Due to their increased energy requirement cardiomyocytes have a particularly high density of mitochondria, and uncoupling of the mitochondrial respiratory chain in pathological conditions can consequently result in marked elevation in ROS production (Sorescu and Griendling 2002). Indeed, increased production of mitochondrial-derived ROS has been reported in several experimental models of cardiac remodeling and CHF. For example, in canine hearts subjected to chronic rapid ventricular pacing, ROS generation by mitochondrial complex I was shown to be markedly increased, resulting primarily in the formation of •OH (Ide et al. 1999; 2000). In a murine model of chronic MI, increases in ROS production were associated with mitochondrial DNA damage and defective electron transport, together with parallel increases in mitochondrial levels of 8-oxo-dGTPase, which is protective against ROS-mediated DNA damage and may be a marker of mitochondrial oxidative stress (Ide et al. 2001; Tsutsui et al. 2001). Furthermore, gene-modified mice with mitochondrial-targeted overexpression of catalase or cardiac-specific overexpression of the mitochondrial antioxidant, thioredoxin-1, demonstrated reduced cardiac remodeling in response to chronic pressure overload, while mitochondrial peroxiredoxin-3 overexpression protected against post-MI ventricular remodeling (Yamamoto et al. 2003; Matsushima et al. 2006; Dai et al. 2012). Data relating to mitochondrial ROS production in clinical CHF are scarce, although one study reported markedly increased expression of Mn-SOD in failing hearts of patients with nonischemic dilated cardiomyopathy, in parallel with elevated production of O2 •– (Sam et al. 2005).

Generation of O 2 •– by mitochondria. The mitochondrial electron transport chain is a sequence of carrier complexes, I, II, III, and IV, which transport electrons gradually to O2. Coenzyme Q (CoQ) shuttles the electrons between complex I or II and III and further between III and IV. CoQ is a major source of O2 •– in pathologic states because some of the electrons, which are transported from NADH or other compounds to O2, “escape” when CoQH• reacts with O2 to form O2 •–. This O2 •– can then either exert direct detrimental effects on the mitochondria itself or mediate more widespread actions via activation of redox-sensitive signaling pathways. FMN flavin mononucleotide, FAD flavin adenine dinucleotide [Figure adapted from Sorescu and Griendling (2002)]
Dysfunctional NOS
In addition to its well-established role in regulation of vascular function, nitric oxide (NO) is also known to limit the development of experimental and clinical CHF by exerting specific effects on cardiac remodeling (GISSI-3. 1994; Ritchie et al. 1998; Kim et al. 1999). Indeed, ventricular remodeling in response to both chronic pressure overload and MI was exacerbated in gene-modified mice lacking global expression of endothelial NO synthase (NOS3) (Scherrer-Crosbie et al. 2001; Ruetten et al. 2005). Conversely, cardiomyocyte-specific NOS3 overexpression resulted in attenuation of pressure-overload-induced cardiac remodeling (Buys et al. 2007). However, under pathological conditions NOS3 may become uncoupled due to oxidation of its essential cofactor tetrahydrobiopterin (BH4) by ROS (Landmesser et al. 2003). Uncoupling of NOS in CHF leads not only to a significant reduction in NO bioavailability but also to further increases in ROS production. Under physiological conditions, NOS3 catalyzes the conversion of L-arginine and oxygen to NO and citrulline. However, increased ROS production or deficiency of BH4 or L-arginine may cause NOS3 to become uncoupled and produce O2 •– rather than NO (Stuehr et al. 2001; Landmesser et al. 2003). Furthermore, uncoupled NOS3 has also been reported to potentiate mitochondrial ROS production by increasing mitochondrial nitration (Sud et al. 2008). In addition, NO itself, levels of which may be increased in CHF, can react with ROS leading to production of the powerful oxidant, peroxynitrite, and nitrosative stress (Stuehr et al. 2001; Landmesser et al. 2003; Nava et al. 1995). Indeed, decreased BH4 levels in experimental hypertension and pressure overload have been shown to lead to NOS3 uncoupling and consequent increases in myocardial ROS production, cardiac remodeling, and contractile dysfunction (Takimoto et al. 2005; Silberman et al. 2010). The development of these changes was attenuated by co-treatment with BH4, which was also able to reverse preexisting cardiac hypertrophy and fibrosis (Takimoto et al. 2005; Moens et al. 2008; Silberman et al. 2010). However, recent data suggests that the clinical use of BH4 in this setting may be limited by in vivo oxidation resulting in dose-dependent increases in competing levels of BH2 (Moens et al. 2008; Crabtree et al. 2009).
Monoamine Oxidases
Monoamine oxidases (MAOs) are emerging as a significant source of ROS within the cardiovascular system. There are two isoforms, MAO-A and MAO-B, which are expressed on the outer mitochondrial membrane and catalyze the oxidative deamination of monoamines resulting in H2O2 production (Kaludercic et al. 2011). In the context of CHF, MAO-A is of particular interest as it primarily catabolizes epinephrine, serotonin, and norepinephrine, all of which are potent stimuli of cardiac remodeling. Indeed, recent studies have documented an important role for cardiac MAO-A-derived ROS both in vitro and in vivo. In rat neonatal and H9c2 cardiomyocytes, stimulated MAO-A-dependent ROS production was associated with cell apoptosis and hypertrophy, which were inhibited by the specific MAO-A inhibitor, clorgyline (Pchejetski et al. 2007; Kaludercic et al. 2010). In addition, myocardial MAO-A expression is increased in a Dahl salt-sensitive rat model of experimental CHF (Kong et al. 2005). Furthermore, chronic MAO-A inhibition has been found to protect against the development of pressure-overload-induced LV contractile dysfunction and dilatation, while acute ischemia-reperfusion injury was reduced in MAO-A-deficient mice (Pchejetski et al. 2007; Kaludercic et al. 2010). Although the majority of studies to date have focused on the MAO-A isoform, myocardial expression of MAO-B is also reported to be increased in dogs with chronic rapid ventricular pacing-induced CHF (Ojaimi et al. 2007), suggesting that ROS produced by this isoform may also play an important role in cardiac remodeling.
Myeloperoxidase
Myeloperoxidase is a major enzymatic source of ROS produced by activated leukocytes, including neutrophils and monocytes, which play a key role in myocardial inflammation and subsequent modulation of cardiac remodeling (Hampton et al. 1998; Heymans et al. 1999). Myeloperoxidase generates oxidants such as hypochlorous acid from H2O2, chloride ions, and monochloramines that are cytotoxic and are produced under physiological conditions as part of the respiratory burst of host defense which functions to destroy invading bacteria and pathogens (Klebanoff 2005). Similar to NADPH oxidases, this enzyme plays a key basal function, but levels are increased in the ischemic heart, where it has been shown to play an important role in ventricular dilatation and rupture post-MI via oxidation of plasminogen activator inhibitor-1 (PAI-1), an established mediator of ischemic cardiac remodeling (Heymans et al. 1999; Askari et al. 2003; Nahrendorf et al. 2008). Indeed, increased circulating myeloperoxidase levels have been suggested as both a predicative and diagnostic marker of acute heart failure (Sinning et al. 2008; Tang et al. 2011). Myeloperoxidase is also known to combine with amino acids to produce reactive aldehydes, such as acrolein, which may be important in the pathophysiology of cardiac remodeling (Srivastava et al. 2002; Ismahil et al. 2011). Interestingly, a recent study reported that myeloperoxidase-deficient mice were specifically protected against chronic post-MI remodeling but not acute infarct remodeling, suggesting that myeloperoxidase-derived oxidants do not mediate tissue necrosis after MI but may be an important mediator of heart failure progression (Vasilyev et al. 2005).
Cross Talk Between ROS Sources
Interestingly, it is becoming increasingly apparent that cross talk between different cardiovascular ROS sources and the emerging concept of ROS-induced ROS release are likely to play a significant role in overall regulation of cardiac remodeling (Kimura et al. 2005; Dikalov 2011). Given the highly specific nature of ROS signaling, it is thought that such interactions may exert spatial and temporal control over regional ROS production and facilitate localized amplification of ROS-dependent cellular processes (Zinkevich and Gutterman 2011). For example, mitochondrial ROS have been shown to stimulate phosphatidylinositol 3-kinase (PI3K)-mediated Rac1 translocation resulting in activation of Nox1, whereas converse activation of mitochondrial O2 •– production occurs in response to NADPH oxidase-derived ROS in a Ca2+-dependent manner (Lee et al. 2006; Hawkins et al. 2007). Indeed, cross talk between mitochondria and NADPH oxidase-derived ROS is thought to play a key role in Ang II-mediated vascular redox responses (Wosniak et al. 2009). Furthermore, NADPH oxidases are known to modulate the activity of xanthine oxidase by regulating its conversion from xanthine dehydrogenase, and to oxidize BH4 which results in uncoupling of NOS3 and conversion of this NO-producing enzyme to one that generates primarily O2 •– (Landmesser et al. 2003; McNally et al. 2003). Such cross talk between different ROS sources adds further complexity to ROS signaling and is likely to underlie many of their highly specific actions in the setting of cardiac remodeling and CHF.
Redox-Sensitive Pathways in Remodeling and Failure
It is clear that a number of ROS sources, produced in response to multiple stimuli, are important in regulating important aspects of cardiac remodeling and CHF (as summarized in Fig. 40.4). Indeed, several key signaling pathways involved in disease pathogenesis have been identified as being redox sensitive. Although the precise underlying mechanisms remain to be established, it is clear that ROS are capable of exerting highly focused actions on the remodeling phenotype via specific modulation of defined signaling pathways, and these will now be discussed in detail.

Summary of the actions of major cardiac ROS sources in CHF. Several major cardiac ROS sources may be produced in response to multiple stimuli, which then exert important downstream actions on specific components of the cardiac remodeling phenotype, ultimately leading to CHF development. Ang II angiotensin II, Dox doxorubicin, ECM extracellular matrix, ET-1 endothelin-1, MI myocardial infarction, NE norepinephrine, PE phenylephrine; TNFα tumor necrosis factor-α
Serine/Threonine Protein Kinases
Protein kinases are major regulators of signal transduction that catalyze phosphorylation of other proteins, thereby controlling their activity. Primary targets of protein kinases are transcription factors which modulate intracellular signaling via specific alteration of downstream gene expression/activity (Force et al. 1999). The most important protein kinases in the heart are the serine/threonine subfamily, which act by promoting phosphorylation of the OH group of serine or threonine residues on target proteins (Cheng and Force 2010). Of these, mitogen-activated protein kinases (MAPKs) and protein kinase B (Akt) play a well-established role in cardiac remodeling and CHF and are known to be particularly sensitive to redox modulation (Feuerstein and Young. 2000; Sugden and Clerk 2006).
Mitogen-Activated Protein Kinases (MAPKs)
Cardiac MAPK Signaling
Mitogen-activated protein kinases (MAPKs) represent a large family of proteins that play important roles in signal transduction within the heart, where they are involved in regulation of a number of biological processes such as cell motility, survival, apoptosis, proliferation, and differentiation. In cardiomyocytes, MAPK signaling is promoted by a number of stimuli, including G-protein-coupled receptor (GPCR) activation, receptor tyrosine kinases, oxidative stress, and growth factors, and contains a number of sequentially acting kinases which ultimately result in phosphorylation and activation of terminal effector kinases which transduce specific cellular actions (Robinson and Cobb 1997; Bueno et al. 2000). Several MAPK family subgroups have been identified, of which the major mammalian types appear to be extracellular signal-regulated kinases (ERK1/2), c-Jun NH2-terminal kinases (JNK1, 2 and 3), and p38MAPK (α, β, δ and γ), all of which play key roles during both cardiac development and in disease (Robinson and Cobb 1997; Zhang et al. 2007). Indeed, activity of ERK1/2, JNK, and p38MAPK has been reported to be increased in both experimental and clinical CHF (Cook et al. 1999). However, the involvement of MAPKs in this setting appears to be particularly complicated as different components within each pathway may exert positive or negative effects during disease progression and may be damaging in one situation while beneficial in another (Rose et al. 2010). Nonetheless, it is clear that MAPK activation plays a key role in the pathogenesis of CHF and is critically involved in regulation of specific aspects of cardiac remodeling (Sano et al. 2001).
ROS Modulation of MAPK Signaling in Cardiac Remodeling
MAPKs are the most investigated family of signal transducer proteins in relation to ROS-dependent modulation of cardiac remodeling, with the three major mammalian MAPK subgroups being implicated in controlling important aspects of the underlying pathophysiological processes. However, the precise role that ROS modulation of MAPK signaling plays in overall regulation of cardiac remodeling remains controversial, with the current evidence highlighting species-, stimulus-, and model-specific effects of ROS-dependent activation of the major MAPK pathways. It is well established that several MAPKs are activated in a ROS-dependent manner in the heart. For example, cardiac p38MAPK signaling has been reported to be activated by both xanthine oxidase-derived ROS and by mitochondrial-derived ROS generated under hypoxic conditions (Kulisz et al. 2002; Gaitanaki et al. 2006). Increased NADPH oxidase expression/activity and O2 •– production has also been shown to be associated with activation of ERK1/2 and p38MAPK and JNK1/2 in an experimental guinea-pig model of chronic pressure overload (Li et al. 2002). To date, the majority of experimental studies in relation to ROS-dependent modulation of MAPK signaling in cardiac remodeling and CHF have been performed in vitro. In isolated rat cardiomyocytes, both ET-1- and PE-induced hypertrophy were associated with increased ERK activity, which were both attenuated by acute antioxidant treatment, suggesting a key role for ROS-dependent modulation of ERK signaling in this setting (Amin et al. 2001; Tanaka et al. 2001). Further studies in these cells have suggested that NADPH oxidase-derived ROS may specifically activate the ERK1/2 signaling pathway (Xiao et al. 2002). Similarly, hypertrophy of neonatal rat cardiomyocytes induced by Rac1, which is known to be critical for Nox2 NADPH oxidase activity, was found to involve activation of both ERK1/2- and JNK-dependent signaling pathways (Clerk et al. 2001). Additional studies employing the same cell model have demonstrated that both H2O2- and aldosterone-induced apoptosis and GPCR agonist-induced hypertrophy are associated with ROS-dependent activation of Apoptosis signal-regulating kinase-1 (ASK-1), an upstream regulator of MAPK signaling (Hirotani et al. 2002; Hayashi et al. 2008; Zhang et al. 2011a). Interestingly, it appears that H2O2 may exert differential concentration-dependent actions on cardiomyocyte remodeling. Low concentrations of H2O2 (10 μM) were found to induce hypertrophy of primary rat ventricular cardiomyocytes, with associated activation of ERK1/2 but not JNK, p38MAPK, or Akt, whereas increased concentrations (100–200 μM) produced an apoptotic response which was related to additional activation of JNK, p38MAPK, and Akt with a further activation of ERK1/2 (Kwon et al. 2003). Complementary studies performed in H9c2 cardiomyocytes indicated that ROS-induced apoptosis was associated with activation of JNK, but not ERK or p38MAPK pathways, and that specific inhibition of JNK signaling resulted in significant attenuation of ROS-mediated cell death (Turner et al. 1998). In the same cell model, Ang II-induced cardiomyocyte apoptosis was shown to involve NADPH oxidase-dependent activation of p38MAPK (Qin et al. 2006). Studies such as these serve to highlight the co of ROS signaling and suggest that differential MAPK activation may play an important role in regulation of cardiomyocyte remodeling. Interestingly, it appears that ROS may also activate MAPK signaling in a paracrine manner, as Ang II-dependent ROS production in isolated rat cardiac fibroblasts was found to stimulate cardiomyocyte hypertrophy via IL-6-induced activation of p38 MAPK, ERK1/2, and JNK-1 (Sano et al. 2001). MMP-2, which plays a key role in regulating extracellular matrix turnover, was also shown to be activated in H9c2 cardiomyocytes exposed to doxorubicin via NADPH oxidase-mediated JNK, p38MAPK, and ERK1/2 signaling (Spallarossa et al. 2006). Furthermore, chronic isoproterenol infusion in rats induced myocardial ROS production and collagen accumulation, which was associated with activation of ASK-1, ERK1/2, JNK, and p38MAPK (Zhang et al. 2005), supporting the suggestion that ROS-dependent modulation of MAPK signaling may be important in regulation of extracellular matrix remodeling. In addition to exerting specific actions on cardiac remodeling, other studies indicate that ROS-dependent modulation of MAPK signaling may also be important in vascular remodeling, which is known to be a central factor in CHF progression. For example, NADPH oxidase-dependent ROS generation in response to ischemia/reperfusion has been shown to promote in vitro angiogenesis and angiogenic growth factor expression in porcine coronary artery endothelial cells via activation of ERK1/2 and Akt pathways (Chen et al. 2007). Interestingly, mitochondrial ROS generation in porcine aortic endothelial cells under hypoxic conditions was found to occur upstream of ERK2 activation and further NADPH oxidase-mediated ROS production (Schäfer et al. 2003). Furthermore, ROS-dependent activation of p38MAPK is thought to represent a key step in Ang II-induced hypertrophy of rat vascular smooth muscle cells (VSMC) (Ushio-Fukai et al. 1998) and may also be important in the development of atherosclerosis, with additional involvement of NADPH oxidase-dependent JNK activation (Viedt et al. 2000).
Akt
Cardiac Akt Signaling
Akt (also known as protein kinase B, PKB) is a protein kinase belonging to the serine/threonine family, which is stimulated by various receptor tyrosine kinases in a PI3K-dependent manner. In the myocardium, Akt activation has been shown to be regulated by a variety of physiological factors (e.g., exercise training, insulin/nutritional status) and under pathological conditions, such as chronic pressure overload, where it plays an important role in regulation of cardiomyocyte growth and hypertrophy (Walsh 2006). However, the precise function that Akt signaling plays in regulation of cardiac remodeling remains controversial, with several lines of evidence suggesting a protective role, while others indicate that Akt activation may exert maladaptive actions in this setting. For example, Ang II-induced H9c2 cardiomyocyte hypertrophy has been shown to be associated with Akt activation which was abolished by pretreatment with a recombinant adenoviral vector targeting Akt (Hingtgen et al. 2006). Furthermore, in a mouse model of chronic pressure overload, cardiac hypertrophy and associated Akt activation was attenuated by intramyocardial injections of an Akt-targeting viral vector (Hingtgen et al. 2010). However, it has also been reported that chronic blockade of Akt signaling in rats exacerbates pathological hypertrophy in response to both MI and pressure overload but inhibits physiological hypertrophy induced by exercise training (Buss et al. 2012). In addition, intracoronary gene delivery of Akt1 appears to be protective against the development of experimental doxorubicin-induced cardiac dysfunction (Taniyama and Walsh 2002), suggesting that Akt activation may play an adaptive role.
ROS-Dependent Akt Activation in Cardiac Remodeling
Although the precise actions of Akt signaling in cardiac remodeling and CHF remain incompletely understood, it is clear that Akt activation in this setting is redox sensitive and subject to differential regulation by several ROS types and sources. Interestingly, ROS have been shown to both promote and inhibit Akt activation, which are likely to contribute to their highly specific actions on distinct components of the cardiac remodeling phenotype, further illustrating the complexity of ROS signaling in this situation. Several studies have shown ROS to mediate activation of Akt, thereby exerting maladaptive actions on key remodeling processes. For example, H2O2-induced H9c2 cardiomyocyte apoptosis was found to occur via activation of the PI3K-Akt pathway, by promotion of disulfide bond formation between the Cys-297 and Cys-311 residues of Akt and consequent dephosphorylation, effects which were inhibited by both antioxidant treatment and endogenous overexpression of glutaredoxin (Murata et al. 2003; Zhu et al. 2011). Similarly, agonist-induced hypertrophy of both rat neonatal cardiomyocytes and H9c2 cells is associated with Akt activation and has been shown to be both dependent upon increased Nox2 NADPH oxidase activity and sensitive to overexpression of endogenous SOD (Hingtgen et al. 2006; Häuselmann et al. 2011). Ang II-induced endothelial dysfunction also appears to be dependent on activation of both Nox2 NADPH oxidase and Akt signaling (Thakur et al. 2010). Further evidence for specific activation of Akt signaling by ROS comes from a recent study reporting that development of pressure-overload cardiac hypertrophy in rats was dependent on both increased ROS generation and phosphorylation of glycogen synthase kinase-3β (GSK-3β), a known substrate of Akt, with established pro-hypertrophic actions (Planavila et al. 2008).
ROS-Dependent Inactivation of Akt in Cardiac Remodeling
Although the above studies clearly indicate that ROS may exert their established maladaptive remodeling actions via activation of Akt signaling, other contrasting reports suggest that these effects may occur secondary to negative regulation of Akt by ROS. Interestingly, doxorubicin-induced cardiotoxicity was exacerbated in transgenic mice overexpressing Nrdp1 via enhanced ROS-mediated inhibition of Akt signaling (Zhang et al. 2011b), while cardiac hypertrophy in a rat model of experimental hyperthyroidism was associated with increased H2O2 activity generation and downregulation of the Akt pathway (Fernandes et al. 2011). Similarly, elevated ROS production in rats subjected to chronic MI was accompanied by decreased phosphorylation of both Akt and GSK-3β in association with significant cardiac hypertrophy and contractile dysfunction (Schenkel et al. 2010). Furthermore, norepinephrine-induced apoptosis of rat neonatal cardiomyocytes was shown to involve ROS-mediated inhibition of Akt signaling (Fu et al. 2006), suggesting that Akt may confer cardioprotective actions. Indeed, in the setting of acute ischemia/reperfusion injury, increased ROS generation is regarded as being an important trigger for several cardioprotective mechanisms, which in this case involves activation, rather than inhibition of Akt (Wang et al. 2011).
Transcription Factors
Transcription factors are proteins which bind to specific DNA sequences on target genes, thereby mediating important downstream effects of many intracellular signaling pathways (Latchman. 1997). Several transcription factors are known to be activated during cardiac remodeling and CHF, including nuclear factor-κB (NF-κB), activator protein-1 (AP-1), nuclear factor of activated T cells (NFAT), myocyte enhancer factor-2 (MEF-2), cAMP response element-binding protein (CREB), GATA-4/6, and CCAAT-enhancer-binding protein β (C/EBPβ), and to play important roles in this setting (Watson et al. 2010; Boström et al. 2010; Liu et al. 2012). Of these, NF-κB and AP-1 appear to be particularly redox sensitive and to play a significant part in signal transduction during the development and progression of cardiac remodeling.
Nuclear Factor-κB (NF-κB)
Cardiac NF-κB Signaling
The NF-κB mammalian superfamily consists of five members separated into two groups based on divergence of the C terminus, known as the “Rel” subfamily (RelA, RelB, c-Rel) and the “NF-κB” subfamily (p50, p52). Rel subfamily members possess C-terminal transcriptional activating domains (TADs) which recruit basal transcriptional machinery to target promoter/enhancer regions, whereas the NF-κB subfamily members are deficient in TAD and thus require dimerization of Rel subfamily members for transcriptional activation (Gordon et al. 2011). Activation of NF-κB triggers its release from the inhibitory protein, IκB, which facilitates translocation to the nucleus. This can occur through various signaling mechanisms that have been subdivided into canonical and noncanonical pathways, with the former usually stimulated by TNF-α activating the most common NF-κB dimer between p65 and p50, while the latter activates a RelB/p52 dimer. Within the heart, the canonical pathway represents the most investigated aspect of NF-κB signaling, with little information available concerning the role of the noncanonical pathways. However, there is some evidence for the existence of cross talk within NF-κB signaling as ligands for the noncanonical pathway have been shown to activate the canonical pathway. Activation of NF-κB signaling has become inexorably linked with the development and progression of cardiac remodeling and CHF in a number of settings and is considered to be the major driver of myocardial inflammatory signaling via activation of downstream mediators such as TNF-α, iNOS, COX-2, IL-1β, and IL-6 (Gordon et al. 2011; Hamid et al. 2011). In the clinical setting, increased NF-κB activation has been demonstrated in peripheral blood leukocytes from CHF patients, while a genetic polymorphism which promotes NF-kB activity and is associated with a more severe CHF phenotype has also been identified (Frantz et al. 2004; Jankowska et al. 2005; Santos et al. 2010). Activation of NF-κB signaling has also been reported in commonly employed murine experimental models of CHF, such as coronary artery ligation, pressure overload, and streptozotocin diabetes (Hamid et al. 2009; Zelarayan et al. 2009; Rajesh et al. 2010). Gene-modified mice deficient in the c-Rel NF-κB subunit were protected against Ang II-induced cardiac hypertrophy and fibrosis, while mice with cardiac-specific myotrophin overexpression exhibited myocardial hypertrophy which was markedly attenuated by in vivo silencing of NF-κB (Gupta et al. 2008; Gaspar-Pereira et al. 2012). Furthermore, targeted disruption of the p50 subunit of NF-κB in mice subjected to experimental MI was found to reduce reactive cardiac hypertrophy, fibrosis, chamber dilatation, and contractile dysfunction (Frantz et al. 2006; Kawano et al. 2006). Similarly, in the same model, chronic treatment with IMD-0354, a phosphorylation inhibitor of IκB, attenuated post-MI geometric remodeling and associated inflammation (Onai et al. 2007). Indeed, NF-κB is known to mediate pro-inflammatory signaling in cardiac remodeling associated with both diabetes and MI, where it is activated by TNF-α and IL-1 (Rajesh et al. 2010; Van der Heiden et al. 2010). Although the above studies and numerous others suggest that activation of NF-κB promotes pathological cardiac remodeling, a recent report indicates that NF-κB signaling may also be protective. Interestingly, mice with cardiac-specific deletion of IKKβ, a subunit of the IKK complex which is essential for NF-κB activation, demonstrated significantly worse contractile dysfunction, hypertrophy, fibrosis, and apoptosis in response to chronic pressure overload compared to controls (Hikoso et al. 2009), suggesting that IKKβ/NF-κB signaling may perform a protective function, at least in this setting.
ROS Modulation of NF-κB Signaling in Cardiac Remodeling
Over 20 years ago, H2O2 was shown to activate NF-κB in human T cells at micromolar concentrations (Schreck et al. 1991). Since this initial report of redox modulation of NF-κB signaling, it has become apparent that NF-κB may be activated by various ROS types from different sources and that this plays a key role during the development and progression of cardiac remodeling. For example, in neonatal rat cardiomyocytes, glycated proteins, which are associated with diabetic cardiomyopathy, were found to increase NF-κB activity via activation of Nox2 NADPH oxidase-derived O2 •– (Zhang et al. 2006). In isolated perfused rat hearts, xanthine oxidase-derived ROS induced contractile dysfunction which was associated with NF-κB activation (Turan et al. 2005). Furthermore, in obese db/db diabetic mice, increased mitochondrial free radical production and parallel LV dilatation and contractile dysfunction were attenuated by the NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) (Rajesh et al. 2010). NF-κB also appears to exert important ROS-dependent actions on specific components of the cardiac remodeling phenotype. Hypertrophy of rat neonatal cardiomyocytes in response to both GPCR agonists and TNF-α is reported to be mediated via NF-κB activation, while sustained NF-κB activity was shown to cause apoptosis of H9c2 cardiomyocytes (Higuchi et al. 2002; Hirotani et al. 2002; Hamid et al. 2009). Furthermore, Ang II-induced cardiac fibrosis in mice is associated with Nox2 NADPH oxidase-dependent NF-κB activation (Johar et al. 2006). Similarly, aldosterone-induced cardiac fibrosis and inflammation were attenuated by chronic treatment with PDTC, a specific inhibitor of NF-κB (Sun et al. 2002). Interestingly, exogenous and endogenous antioxidants appear to be effective at targeting NF-κB signaling and reducing experimental cardiac remodeling. For example, GPCR-induced cardiomyocyte hypertrophy in rat neonatal cells was significantly reduced by a number of antioxidants, including N-acetylcysteine and vitamin E, which was associated with parallel decreases in NF-κB activity (Hirotani et al. 2002). Intramyocardial gene transfer of Cu/Zn SOD in a murine model of pressure overload has also been demonstrated to ameliorate NF-κB activation and cardiac hypertrophy (Hingtgen et al. 2010). Similarly, augmented pressure-overload-induced contractile dysfunction and remodeling observed in mice with cardiac-specific deletion of IKKβ was thought to occur secondary to reduced expression/activity of manganese SOD (Hikoso et al. 2009). Furthermore, PDTC, which is not only a pharmacological inhibitor of NF-κB but also a potent antioxidant, attenuated the development of cardiac hypertrophy in rats subjected to chronic pressure overload and those with spontaneous hypertension (Li et al. 2004; Gupta et al. 2005).
Activator Protein-1 (AP-1)
Cardiac AP-1 Signaling
AP-1 is a heterodimer with the major components, c-Jun and c-Fos, which is activated by multiple stimuli (e.g., cytokines, growth factors, stress), and is involved in a number of physiological processes including cell proliferation, differentiation, and apoptosis. The wide-ranging actions of AP-1 may be explained by its complex structure, consisting of dimeric basic region-leucine zipper (bZIP) proteins belonging to Jun, Fos, ATF, and JDP subfamilies which recognize TPA response elements of several target genes (Shaulian and Karin 2002; Hess et al. 2004). Activation of AP-1 has been demonstrated in experimental models of MI, pressure overload, and isoproterenol-induced cardiac hypertrophy and in human CHF patients with ischemic or dilated cardiomyopathy (Takemoto et al. 1999; Frantz et al. 2003; Chen et al. 2009; Garlie et al. 2011). In addition, Ephx2 was recently identified as a susceptibility gene for experimental CHF in spontaneously hypertensive rats, which is thought to act by increasing AP-1 binding and activation (Monti et al. 2008). Furthermore, mutation of the consensus binding site for AP-1 on the AT1a receptor, which is a major mediator of cardiac Ang II signaling, was shown to abolish pressure-overload-induced hypertrophy in rats (Herzig et al. 1997). Importantly, AP-1 activation is associated with several aspects of the cardiac remodeling process. In rat neonatal cardiomyocytes, hypertrophy induced by the GPCR agonists, ET-1 and PE, was linked to c-Jun-dependent activation of AP-1 (Irukayama-Tomobe et al. 2004; Hilfiker-Kleiner et al. 2006). Indeed, the ET-1 gene itself has binding sites for AP-1 within its promoter region, suggesting that AP-1 may also contribute to ET-1 induction (Sakai et al. 2002). Isolated rat hearts subjected to acute ischemia/reperfusion also demonstrated a progressive increase in AP-1 activity which was associated with apoptotic cell death (Maulik et al. 2000). Furthermore, cardiac fibrosis both induced by streptozotocin treatment and in transgenic rats overexpressing the human renin and angiotensinogen genes was associated with AP-1 activation (Fiebeler et al. 2001; Aragno et al. 2008). Pro-inflammatory IL-18, which mediates MMP induction and extracellular matrix degradation, also activates AP-1 in adult rat cardiomyocytes (Reddy et al. 2010).
ROS Modulation of AP-1 Signaling in Cardiac Remodeling
It is well established that AP-1 activation is redox sensitive, with ROS, in particular O2 •– and H2O2, reported to induce expression of the AP-1 components, c-Fos and c-Jun (Peng et al. 1995; Cheng et al. 1999; Yang et al. 2005). AP-1 may also be activated secondary to upstream redox-sensitive modulation of the major MAPKs, JNK, ERK, and p38MAPK (Karin 1995; Taimor et al. 2001; Shaulian and Karin 2002), which themselves play an important role in ROS signaling in cardiac remodeling and CHF (see above). It is clear that ROS specifically derived from several important cardiac sources are capable of activating AP-1. In rat cardiomyocytes, the pro-hypertrophic GPCR agonist Ang II was found to increase AP-1 activity via activation of Nox2 NADPH oxidase, which occurred upstream of TGFβ1 induction and was dependent on p38MAPK (Wenzel et al. 2001; Wu et al. 2005). Mitochondrial-derived O2 •– also regulates TNF-α-induced activation of both NF-κB and AP-1 in human myelogenous leukemia cells (Higuchi et al. 2002), while xanthine oxidase-induced rat cardiac fibroblast proliferation and associated induction of MMP gene expression is mediated by increased AP-1 activity (Xie et al. 2009). ROS-dependent activation of AP-1 also appears to play a key role in modulating specific aspects of cardiac remodeling. For example, Ang II-induced hypertrophic growth of rat neonatal cardiomyocytes was found to be dependent on ROS-mediated induction of AP-1 activity and expression of c-Fos and c-Jun (Li et al. 2005). Interestingly, norepinephrine used at pro-hypertrophic and proapoptotic concentrations in H9c2 cardiomyocytes caused differential induction of the AP-1 components, FosB and Fra-1, which was ROS dependent (Gupta et al. 2006), indicating that distinct redox regulation of AP-1 signaling may play an important role in cardiac remodeling, at least in this setting. Ang II-induced proliferation of neonatal rat cardiac fibroblasts is also dependent on ROS, which mediate AP-1 binding to the promoter region of ET-1 (Cheng et al. 2003). An important function for ROS-induced AP-1 activation in cardiac fibrosis is supported by data from a mouse model of cardiac ischemia-reperfusion injury indicating that MMP-2 induction in this situation occurs through the AP-1 components FosB and JunB and is dependent on hydroxyl radical generation (Alfonso-Jaume et al. 2006). Furthermore, cardiac fibrosis and ROS production in STZ-treated rats was associated with parallel AP-1 activation which was attenuated by dehydroepiandrosterone (DHEA), which is known to prevent hyperglycemia-induced oxidative damage (Aragno et al. 2008). Acute ischemia-reperfusion in dogs also leads to ROS-dependent endothelial induction of pro-inflammatory monocyte chemotactic protein-1 (MCP-1) via activation of AP-1 and NF-κB (Lakshminarayanan et al. 2001). The apparent importance of redox modulation of AP-1 activity in cardiac remodeling is confirmed by several studies demonstrating that associated activation of AP-1 may be attenuated by antioxidants. In experimental models of cardiac ischemia-reperfusion, both recombinant adenoviruses expressing Cu/ZnSOD or Mn-SOD and the oxygen radical scavengers N-(2-mercaptopropionyl)-glycine (MPG) and DMTU inhibited acute AP-1 activation with parallel reductions in infarct size and cardiomyocyte apoptosis (Maulik et al. 1999; Fan et al. 2002; Yang et al. 2003). In addition, norepinephrine-induced expression of the AP-1 components FosB and Fra-1 in H9c2 cardiomyocytes was suppressed by both the H2O2 scavenger, catalase, and the SOD mimetic, MnTMPyP (Jindal and Goswami 2011). Interestingly, however, basal expression of FosB and Fra-1 was increased by MnTMPyP but inhibited by catalase, indicating that the precise nature of AP-1 regulation in this setting may be dependent on the type of ROS produced.
Novel Redox-Sensitive Signaling Pathways
In addition to the established redox-sensitive serine/threonine protein kinase and transcription factor-mediated pathways discussed above, it is becoming apparent that several other ROS-dependent signaling pathways may also be involved in cardiac remodeling. For example, CD40 ligand, which is structurally related to TNF-α and expressed in T cells and platelets, is known to bind CD40 on B cells, macrophages, endothelial cells, vascular smooth muscle cells, and cardiomyocytes and so may mediate processes such as inflammatory activation and extracellular matrix remodeling (Grewal and Flavell 1997; Seko et al. 1998). Indeed, CD40 ligand is increased in patients with both acute and chronic heart failure (Ueland et al. 2005). Intriguingly, CD40 ligand activation is known to induce ROS production (possibly via NADPH oxidases) and induction of several downstream pathways including PI3 kinase, p38, Akt, ERK1/2, and NF-κB and so may function as a master regulator of redox signaling in the heart (Chakrabarti et al. 2005; Chen et al. 2008; Xia et al. 2010). Furthermore, ROS have been shown to mediate cardiac hypertrophy induced by ouabain and related glycosides via modulation of Na+/K+ ATPase activity and induction of Src-, Ras-, and p42/44 MAPK-dependent signaling (Xie et al. 1999; Tian et al. 2003; Xie and Xie 2005). Indeed, administration of a monoclonal antibody against the cardiotonic steroid, marinobufagenin, which may act via similar mechanisms, reverses cardiac fibrosis in rats with chronic renal failure via a ROS-dependent mechanism (Haller et al. 2012).
Summary and Clinical Perspective
Taken together, it is clear that ROS-mediated modulation of downstream signaling plays a key role in regulating important components of the cardiac remodeling phenotype (as summarized in Fig. 40.5). Although the precise nature of these ROS-dependent actions remains unclear, it is becoming increasingly apparent that the underlying signaling mechanisms are extremely complex and that a number of factors, such as stimulus, type of ROS produced, concentration, cellular localization, and local antioxidant balance, need to be considered. Redox modulation of intracellular signaling appears to be particularly complicated, as evidenced by reports of concentration-dependent induction of MAPKs and AP-1 components by ROS, and differential activation or inactivation of Akt signaling, which may exert either adaptive or maladaptive actions on cardiac remodeling. Paradoxically, however, the complex nature of ROS signaling in this setting may offer hope for the development of novel specifically targeted strategies for the more effective treatment of CHF. Numerous investigations have reported beneficial effects of antioxidant treatment on experimental cardiac remodeling and CHF in animal models (Dhalla et al. 1996; Byrne et al. 2003; Hamblin et al. 2007). Some clinical studies have shown antioxidants such as α-tocopherol and ascorbic acid to reduce oxidative stress (Ghatak et al. 1996; Rössig et al. 2001), while others have reported cardiovascular benefits including reduced rate of nonfatal MI in patients with coronary artery disease (Stephens et al. 1996) and decreased cardiovascular events and mortality in women (Lee et al. 2005; Cook et al. 2007). However, the findings of clinical antioxidant trials in terms of long-term effects on patient symptoms and clinical outcome have been generally disappointing (Yusuf et al. 2000; Heart Protection Study Collaborative Group 2002), with some studies even reporting an increased risk of CHF (Lonn et al. 2002; Marchioli et al. 2006). Given the apparent complexity of myocardial ROS signaling, it is perhaps not surprising that such nontargeted antioxidant approaches have had limited success. Not only do ROS play a critical physiological function as regulators of basal intracellular signaling, but they also alter the activity of numerous pathways to exert subtle modulatory effects on cardiac remodeling, which may be either adaptive or maladaptive. In this regard, it would clearly be attractive to selectively target detrimental aspects of the remodeling phenotype, such as fibrosis or cardiomyocyte apoptosis, while maintaining beneficial functions of ROS. Therefore, it is abundantly evident that a detailed understanding of the precise mechanisms by which ROS interact with key genes in order to specifically modulate downstream signaling in the heart is essential to facilitate the identification of novel therapeutic targets for more effective treatment of adverse cardiac remodeling and CHF.

Summary of the major redox-sensitive signaling pathways in cardiac remodeling. ROS modulation of downstream signaling plays a key role in mediating both adaptive and maladaptive cardiac remodeling. ROS-dependent signaling in this setting is extremely complex and is influenced by a number of factors. However, it appears that in general, mitogen-activated protein kinases (MAPK) and activator protein-1 (AP-1) are positively regulated by ROS and exert detrimental actions on cardiac remodeling, whereas Akt and nuclear factor-κB (NF-κB) may be both positively and negatively influenced by ROS and govern both adaptive and maladaptive aspects of cardiac remodeling. ERK extracellular signal-regulated kinase, JNK c-Jun NH2-terminal kinase
References
Alfonso-Jaume MA, Bergman MR, Mahimkar R, Cheng S, Jin ZQ, Karliner JS, Lovett DH (2006) Cardiac ischemia-reperfusion injury induces matrix metalloproteinase-2 expression through the AP-1 components FosB and JunB. Am J Physiol Heart Circ Physiol 291:H1838–H1846
Amin JK, Xiao L, Pimental DR, Pagano PJ, Singh K, Sawyer DB, Colucci WS (2001) Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol 33:131–139
Anilkumar N, Weber R, Zhang M, Brewer A, Shah AM (2008) Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler, Thromb Vascular Biol 28:1347–1354
Anilkumar N, Sirker A, Shah AM (2009) Redox sensitive signaling pathways in cardiac remodeling, hypertrophy and failure. Front Biosci 14:3168–3187
Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, Izumo S (2002) Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J Biol Chem 277:10244–10250
Aragno M, Mastrocola R, Alloatti G, Vercellinatto I, Bardini P, Geuna S, Catalano MG, Danni O, Boccuzzi G (2008) Oxidative stress triggers cardiac fibrosis in the heart of diabetic rats. Endocrinology 149:380–388
Askari AT, Brennan ML, Zhou X, Drinko J, Morehead A, Thomas JD, Topol EJ, Hazen SL, Penn MS (2003) Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction. J Exp Med 197:615–624
Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG (1997) Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. J Biol Chem 272:217–221
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Gyorke S (2007) Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J 93:4083–4092
Bendall JK, Cave AC, Heymes C, Gall N, Shah AM (2002) Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105:293–296
Beswick RA, Dorrance AM, Leite R, Webb RC (2001) NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension 38:1107–1111
Block K, Gorin Y, Abboud HE (2009) Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci USA 106:14385–14390
Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Gorlach A (2007) Reactive oxygen species activate the HIF-1α promoter via a functional NFκB site. Arterioscler, Thromb Vascular Biol 27:755–761
Boström P, Mann N, Wu J, Quintero PA, Plovie ER, Panáková D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A (2010) C/EBPβ controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell 143:1072–1083
Brilla C, Pick R, Tan L, Janicki J, Weber K (1990) Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 67:1355–1364
Brown DI, Griendling KK (2009) Nox proteins in signal transduction. Free Radic Biol Med 47:1239–1253
Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS (2005) Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 1:409–414
Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF (2000) The MEK1–ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J 19:6341–6350
Buss SJ, Riffel JH, Malekar P, Hagenmueller M, Asel C, Zhang M, Weiss C, Katus HA, Hardt SE (2012) Chronic Akt blockade aggravates pathological hypertrophy and inhibits physiological hypertrophy. Am J Physiol Heart Circ Physiol 302:H420–H430
Buys ES, Raher MJ, Blake SL, Neilan TG, Graveline AR, Passeri JJ, Llano M, Perez-Sanz TM, Ichinose F, Janssens S (2007) Cardiomyocyte-restricted restoration of nitric oxide synthase 3 attenuates left ventricular remodeling after chronic pressure overload. Am J Physiol Heart Circ Physiol 293:H620–H627
Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM (2003) Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res 93:802–805
Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, Marban E, Hare JM (2001) Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation 104:2407–2411
Cave A, Grieve D, Johar S, Zhang M, Shah AM (2005) NADPH oxidase-derived reactive oxygen species in cardiac pathophysiology. Philos Trans R Soc Lond B Biol Sci 360:2327–2334
Chakrabarti S, Varghese S, Vitseva O, Tanriverdi K, Freedman JE (2005) CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler Thromb Vasc Biol 25:2428–2434
Chen AF, Chen DD, Daiber A, Faraci FM, Li H, Rembold CM, Laher I (2012) Free radical biology of the cardiovascular system. Clin Sci (Lond) 123:73–91
Chen C, Chai H, Wang X, Jiang J, Jamaluddin MS, Liao D, Zhang Y, Wang H, Bharadwaj U, Zhang S, Li M, Lin P, Yao Q (2008) Soluble CD40 ligand induces endothelial dysfunction in human and porcine coronary artery endothelial cells. Blood 112:3205–3216
Chen JX, Zeng H, Tuo QH, Yu H, Meyrick B, Aschner JL (2007) NADPH oxidase modulates myocardial Akt, ERK1/2 activation, and angiogenesis after hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol 292:H1664–H1674
Chen DD, Dong YG, Liu D, He JG (2009) Epigallocatechin-3-gallate attenuates cardiac hypertrophy partly via MAPKs signals in hypertensive rats. Clin Exp Pharmacol Physiol 36:925–932
Cheng H, Force T (2010) Why do kinase inhibitors cause cardiotoxicity and what can be done about it? Prog Cardiovasc Dis 53:114–120
Cheng TH, Cheng PY, Shih NL, Chen IB, Wang DL, Chen JJ (2003) Involvement of reactive oxygen species in angiotensin II-induced endothelin-1 gene expression in rat cardiac fibroblasts. J Am Coll Cardiol 42:1845–1854
Cheng T, Shih N, Chen S, Wang D, Chen J (1999) Reactive oxygen species modulate endothelin-I-induced c-fos gene expression in cardiomyocytes. Cardiovasc Res 41:654–662
Chess DJ, Xu W, Khairallah R, O'Shea KM, Kop WJ, Azimzadeh AM, Stanley WC (2008) The antioxidant tempol attenuates pressure overload-induced cardiac hypertrophy and contractile dysfunction in mice fed a high-fructose diet. Am J Physiol Heart Circ Physiol 295:H2223–H2230
Chiang WC, Chien CT, Lin WW, Lin SL, Chen YM, Lai CF, Wu KD, Chao J, Tsai TJ (2006) Early activation of bradykinin B2 receptor aggravates reactive oxygen species generation and renal damage in ischemia/reperfusion injury. Free Radic Biol Med 41:1304–1314
Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT (2003) Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure. Circulation 107:3133–3140
Clerk A, Pham FH, Fuller SJ, Sahai E, Aktories K, Marais R, Marshall C, Sugden PH (2001) Regulation of mitogen-activated protein kinases in cardiac myocytes through the small G protein Rac1. Mol Cell Biol 21:1173–1184
Cohn JN, Ferrari R, Sharpe N (2000) Cardiac remodeling - concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol 35:569–582
Colavitti R, Pani G, Bedogni B, Anzevino R, Borrello S, Waltenberger J, Galeotti T (2002) Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J Biol Chem 277:3101–3108
Cook NR, Albert CM, Gaziano JM, Zaharris E, MacFadyen J, Danielson E, Buring JE, Manson JE (2007) A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: results from the Women's Antioxidant Cardiovascular Study. Arch Intern Med 167:1610–1618
Cook SA, Sugden PH, Clerk A (1999) Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease. J Mol Cell Cardiol 31:1429–1434
Crabtree MJ, Tatham AL, Al-Wakeel Y, Warrick N, Hale AB, Cai S, Channon KM, Alp NJ (2009) Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status. J Biol Chem 284:1136–1144
Creemers EE, Cleutjens JP, Smits JF, Daemen MJ (2001) Matrix metalloproteinase inhibition after myocardial infarction a new approach to prevent heart failure? Circ Res 89:201–210
Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS (2012) Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res 93:79–88
Date M, Morita T, Yamashita N, Nishida K, Yamaguchi O, Higuchi Y, Hirotani S, Matsumura Y, Hori M, Tada M (2002) The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricular hypertrophy in in vivo murine pressure-overload model. J Am Coll Cardiol 39:907–912
De Jong J, Schoemaker R, De Jonge R, Bernocchi P, Keijzer E, Harrison R, Sharma H, Ceconi C (2000) Enhanced expression and activity of xanthine oxidoreductase in the failing heart. J Mol Cell Cardiol 32:2083–2089
Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL (2001) Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103:2055–2059
Dhalla AK, Hill MF, Singal PK (1996) Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol 28:506–514
Dieterich S, Bieligk U, Beulich K, Hasenfuss G, Prestle J (2000) Gene expression of antioxidative enzymes in the human heart: increased expression of catalase in the end-stage failing heart. Circulation 101:33–39
Dikalov S (2011) Crosstalk between mitochondria and NADPH oxidases. Free Radic Biol Med 51:1289–1301
Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK (2008) Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med 45:1340–1351
Dolezal T (2006) Review: Imidapril in heart failure. J Renin Angiotensin Aldosterone Syst 7:146–154
Dong F, Zhang X, Ren J (2006) Leptin regulates cardiomyocyte contractile function through endothelin-1 receptor–NADPH oxidase pathway. Hypertension 47:222–229
Duncan JG, Ravi R, Stull LB, Murphy AM (2005) Chronic xanthine oxidase inhibition prevents myofibrillar protein oxidation and preserves cardiac function in a transgenic mouse model of cardiomyopathy. Am J Physiol Heart Circ Physiol 289:H1512–H1518
Ekelund UE, Harrison RW, Shokek O, Thakkar RN, Tunin RS, Senzaki H, Kass DA, Marban E, Hare JM (1999) Intravenous allopurinol decreases myocardial oxygen consumption and increases mechanical efficiency in dogs with pacing-induced heart failure. Circ Res 85:437–445
El Azzouzi H, Van Oort RJ, Van Der Nagel R, Sluiter W, Bergmann MW, De Windt LJ (2010) MEF2 transcriptional activity maintains mitochondrial adaptation in cardiac pressure overload. Eur J Heart Fail 12:4–12
Engberding N, Spiekermann S, Schaefer A, Heineke A, Wiencke A, Muller M, Fuchs M, Hilfiker-Kleiner D, Hornig B, Drexler H (2004) Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation 110:2175–2179
Fan H, Sun B, Gu Q, Lafond-Walker A, Cao S, Becker LC (2002) Oxygen radicals trigger activation of NF-κB and AP-1 and upregulation of ICAM-1 in reperfused canine heart. Am J Physiol Heart Circ Physiol 282:H1778–H1786
Fernandes R, Dreher G, Schenkel P, Fernandes T, Ribeiro M, Araujo A, Belló‐Klein A (2011) Redox status and pro‐survival/pro‐apoptotic protein expression in the early cardiac hypertrophy induced by experimental hyperthyroidism. Cell Biochem Funct 29:617–623
Feuerstein GZ, Young PR (2000) Apoptosis in cardiac diseases: stress- and mitogen-activated signaling pathways. Cardiovasc Res 45:560–569
Fiebeler A, Schmidt F, Müller DN, Park JK, Dechend R, Bieringer M, Shagdarsuren E, Breu V, Haller H, Luft FC (2001) Mineralocorticoid receptor affects AP-1 and nuclear factor-κB activation in angiotensin II–induced cardiac injury. Hypertension 37:787–793
Finkel T (2011) Signal transduction by reactive oxygen species. J Cell Biol 194:7–15
Force T, Hajjar R, Del Monte F, Rosenzweig A, Choukroun G (1999) Signaling pathways mediating the response to hypertrophic stress in the heart. Gene Expr 7:337–348
Francis GS (2001) Pathophysiology of chronic heart failure. Am J Med 110:37S–46S
Frantz S, Fraccarollo D, Wagner H, Behr TM, Jung P, Angermann CE, Ertl G, Bauersachs J (2003) Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res 57:749–756
Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J (2006) Absence of NF-κB subunit p50 improves heart failure after myocardial infarction. FASEB J 20:1918–1920
Frantz S, Stoerk S, Ok S, Wagner H, Angermann CE, Ertl G, Bauersachs J (2004) Effect of chronic heart failure on nuclear factor kappa B in peripheral leukocytes. Am J Cardiol 94:671–673
Fu YC, Yin SC, Chi CS, Hwang B, Hsu SL (2006) Norepinephrine induces apoptosis in neonatal rat endothelial cells via a ROS-dependent JNK activation pathway. Apoptosis 11:2053–2063
Gaitanaki C, Papatriantafyllou M, Stathopoulou K, Beis I (2006) Effects of various oxidants and antioxidants on the p38-MAPK signalling pathway in the perfused amphibian heart. Mol Cell Biochem 291:107–117
Gao X, Zhang H, Belmadani S, Wu J, Xu X, Elford H, Potter BJ, Zhang C (2008) Role of TNF-α-induced reactive oxygen species in endothelial dysfunction during reperfusion injury. Am J Physiol Heart Circ Physiol 295:H2242–H2249
Garlie JB, Hamid T, Gu Y, Ismahil MA, Chandrasekar B, Prabhu SD (2011) Tumor necrosis factor receptor 2 signaling limits β-adrenergic receptor-mediated cardiac hypertrophy in vivo. Basic Res Cardiol 106:1193–1205
Gaspar-Pereira S, Fullard N, Townsend PA, Banks PS, Ellis EL, Fox C, Maxwell AG, Murphy LB, Kirk A, Bauer R (2012) The NF-κB subunit c-Rel stimulates cardiac hypertrophy and fibrosis. Am J Pathol 180:929–939
Gavin A, Struthers A (2005) Allopurinol reduces B-type natriuretic peptide concentrations and haemoglobin but does not alter exercise capacity in chronic heart failure. Heart 91:749–753
Ghatak A, Brar MJS, Agarwal A, Goel N, Rastogi AK, Vaish AK, Sircar AR, Chandra M (1996) Oxy free radical system in heart failure and therapeutic role of oral vitamin E. Int J Cardiol 57:119–127
Gill JS, McKenna WJ, Camm AJ (1995) Free radicals irreversibly decrease Ca2 currents in isolated guinea-pig ventricular myocytes. Eur J Pharmacol 292:337–340
Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, Ruiz-Lozano P, Gu Y, Nath AK, Huang Y, Hickey R (2001) A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci USA 98:5780–5785
Giordano F (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 115:500–508
GISSI-3 (1994) GISSI-3: effects of lisinopril and transdermal glyceryl trinitrate singly and together on 6-week mortality and ventricular function after acute myocardial infarction. Gruppo Italiano per lo Studio della Sopravvivenza nell'infarto Miocardico. Lancet 343:1115–1122
Gordon JW, Shaw JA, Kirshenbaum LA (2011) Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res 108:1122–1132
Gradman AH, Alfayoumi F (2006) From left ventricular hypertrophy to congestive heart failure: management of hypertensive heart disease. Prog Cardiovasc Dis 48:326–341
Grewal IS, Flavell RA (1997) The CD40 ligand. At the center of the immune universe? Immunol Res 16:59–70
Griendling KK, Sorescu D, Ushio-Fukai M (2000) NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 86:494–501
Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, Shah AM (2006) Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J Am Coll Cardiol 47:817–826
Gupta MK, Neelakantan T, Sanghamitra M, Tyagi RK, Dinda A, Maulik S, Mukhopadhyay CK, Goswami SK (2006) An assessment of the role of reactive oxygen species and redox signaling in norepinephrine-induced apoptosis and hypertrophy of H9c2 cardiac myoblasts. Antioxid Redox Signal 8:1081–1093
Gupta S, Young D, Maitra RK, Gupta A, Popovic ZB, Yong SL, Mahajan A, Wang Q, Sen S (2008) Prevention of cardiac hypertrophy and heart failure by silencing of NF-κB. J Mol Biol 375:637–649
Gupta S, Young D, Sen S (2005) Inhibition of NF-κB induces regression of cardiac hypertrophy, independent of blood pressure control, in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 289:H20–H29
Haller ST, Kennedy DJ, Shidyak A, Budny GV, Malhotra D, Fedorova OV, Shapiro JI, Bagrov AY (2012) Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am J Hypertens 25:690–696
Hamblin M, Smith HM, Hill MF (2007) Dietary supplementation with vitamin E ameliorates cardiac failure in type I diabetic cardiomyopathy by suppressing myocardial generation of 8-iso-prostaglandin F2α and oxidized glutathione. J Card Fail 13:884–892
Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD (2011) Cardiomyocyte NF-κB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res 89:129–138
Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, Prabhu SD (2009) Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-κB and inflammatory activation. Circulation 119:1386–1397
Hampton MB, Kettle AJ, Winterbourn CC (1998) Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92:3007–3017
Häuselmann SP, Rosc-Schlüter BI, Lorenz V, Plaisance I, Brink M, Pfister O, Kuster GM (2011) β1-Integrin is up-regulated via Rac1-dependent reactive oxygen species as part of the hypertrophic cardiomyocyte response. Free Radic Biol Med 51:609–618
Hawkins BJ, Solt LA, Chowdhury I, Kazi AS, Abid MR, Aird WC, May MJ, Foskett JK, Madesh M (2007) G protein-coupled receptor Ca2 -linked mitochondrial reactive oxygen species are essential for endothelial/leukocyte adherence. Mol Cellular Biol 27:7582–7593
Hayashi H, Kobara M, Abe M, Tanaka N, Gouda E, Toba H, Yamada H, Tatsumi T, Nakata T, Matsubara H (2008) Aldosterone nongenomically produces NADPH oxidase-dependent reactive oxygen species and induces myocyte apoptosis. Hypertens Res 31:363–375
Heart Protection Study Collaborative Group (2002) MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 360:23–33
Heineke J, Auger-Messier M, Xu J, Oka T, Sargent MA, York A, Klevitsky R, Vaikunth S, Duncan SA, Aronow BJ (2007) Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J Clin Invest 117:3198–3210
Herzig TC, Jobe SM, Aoki H, Molkentin JD, Cowley AW, Izumo S, Markham BE (1997) Angiotensin II type1a receptor gene expression in the heart: AP-1 and GATA-4 participate in the response to pressure overload. Proc Natl Acad Sci USA 94:7543–7548
Hess J, Angel P, Schorpp-Kistner M (2004) AP-1 subunits: quarrel and harmony among siblings. J Cell Sci 117:5965–5973
Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen-Tervaert JW, Drexler H, Filippatos G, Felix SB, Gullestad L (2009) Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 11:119–129
Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nübe O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P (1999) Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 5:1135–1142
Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM (2003) Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 41:2164–2171
Hidalgo C, Donoso P (2008) Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid Redox Signal 10:1275–1312
Higuchi M, Manna SK, Sasaki R, Aggarwal BB (2002) Regulation of the activation of nuclear factor κB by mitochondrial respiratory function: Evidence for the reactive oxygen species-dependent and-independent pathways. Antioxid Redox Signal 4:945–955
Hikoso S, Yamaguchi O, Nakano Y, Takeda T, Omiya S, Mizote I, Taneike M, Oka T, Tamai T, Oyabu J (2009) The IκB kinase β/nuclear factor κB signaling pathway protects the heart from hemodynamic stress mediated by the regulation of manganese superoxide dismutase expression. Circ Res 105:70–79
Hilfiker-Kleiner D, Hilfiker A, Castellazzi M, Wollert KC, Trautwein C, Schunkert H, Drexler H (2006) JunD attenuates phenylephrine-mediated cardiomyocyte hypertrophy by negatively regulating AP-1 transcriptional activity. Cardiovasc Res 71:108–117
Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E (2004) Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res 95:187–195
Hill MF, Singal PK (1996) Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol 148:291–300
Hingtgen SD, Li Z, Kutschke W, Tian X, Sharma RV, Davisson RL (2010) Superoxide scavenging and Akt inhibition in myocardium ameliorate pressure overload-induced NF-κB activation and cardiac hypertrophy. Physiol Genomics 41:127–136
Hingtgen SD, Tian X, Yang J, Dunlay SM, Peek AS, Wu Y, Sharma RV, Engelhardt JF, Davisson RL (2006) Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics 26:180–191
Hirotani S, Otsu K, Nishida K, Higuchi Y, Morita T, Nakayama H, Yamaguchi O, Mano T, Matsumura Y, Ueno H, Tada M, Hori M (2002) Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation 105:509–515
Hu CM, Chen YH, Chiang MT, Chau LY (2004) Heme oxygenase-1 inhibits angiotensin II-induced cardiac hypertrophy in vitro and in vivo. Circulation 110:309–316
Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A (2001) Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 88:529–535
Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K, Utsumi H, Machida Y, Egashira K, Takeshita A (2000) Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res 86:152–157
Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A (1999) Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res 85:357–363
Irukayama-Tomobe Y, Miyauchi T, Sakai S, Kasuya Y, Ogata T, Takanashi M, Iemitsu M, Sudo T, Goto K, Yamaguchi I (2004) Endothelin-1-induced cardiac hypertrophy is inhibited by activation of peroxisome proliferator-activated receptor-α partly via blockade of c-Jun NH2-terminal kinase pathway. Circulation 109:904–910
Ismahil MA, Hamid T, Haberzettl P (2011) Chronic oral exposure to the aldehyde pollutant acrolein induces dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 301:H2050–H2060
Jankowska EA, von Haehling S, Czarny A, Zaczynska E, Kus A, Anker SD, Banasiak W, Ponikowski P (2005) Activation of the NF-κB system in peripheral blood leukocytes from patients with chronic heart failure. Eur J Heart Fail 7:984–990
Jessup M, Brozena S (2003) Heart failure. N Engl J Med 348:2007–2018
Jindal E, Goswami SK (2011) In cardiac myoblasts, cellular redox regulates FosB and Fra-1 through multiple cis-regulatory modules. Free Radic Biol Med 51:1512–1521
Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM (2006) Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J 20:1546–1548
Kaludercic N, Carpi A, Menabò R, Di Lisa F, Paolocci N (2011) Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta 1813:1323–1332
Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, Chen K, Gabrielson KL, Blakely RD, Shih JC (2010) Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res 106:193–202
Kang PM, Izumo S (2000) Apoptosis and heart failure: a critical review of the literature. Circ Res 86:1107–1113
Karin M (1995) The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem 270:16483–16486
Kawakami M, Okabe E (1998) Superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum through ryanodine receptor Ca2+ channel. Mol Pharmacol 53:497–503
Kawano S, Kubota T, Monden Y, Tsutsumi T, Inoue T, Kawamura N, Tsutsui H, Sunagawa K (2006) Blockade of NF-κB improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol 291:H1337–H1344
Kietzmann T, Gorlach A (2005) Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol 16:474–486
Kim NN, Villegas S, Summerour SR, Villarreal FJ (1999) Regulation of cardiac fibroblast extracellular matrix production by bradykinin and nitric oxide. J Mol Cell Cardiol 31:457–466
Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y (2005) Mitochondria-derived reactive oxygen species and vascular MAP kinases; comparison of angiotensin II and diazoxide. Hypertension 45:438–444
Kinugawa S, Tsutsui H, Hayashidani S, Ide T, Suematsu N, Satoh S, Utsumi H, Takeshita A (2000) Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res 87:392–398
Klebanoff SJ (2005) Myeloperoxidase: friend and foe. J Leukoc Biol 77:598–625
Kong SW, Bodyak N, Yue P, Liu Z, Brown J, Izumo S, Kang PM (2005) Genetic expression profiles during physiological and pathological cardiac hypertrophy and heart failure in rats. Physiol Genomics 21:34–42
Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT (2002) Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol 282:L1324–L1329
Kunsch C, Medford RM (1999) Oxidative stress as a regulator of gene expression in the vasculature. Circ Res 85:753–766
Kurrelmeyer K, Kalra D, Bozjurt B, Wang F, Dibbs Z, Seta Y, Baumgarten G, Engle D, Sivasubramanian N, Mann DL (1998) Cardiac remodeling as a consequence and cause of progressive heart failure. Clin Cardiol 21:I14–I19
Kuster GM, Lancel S, Zhang J, Communal C, Trucillo MP, Lim CC, Pfister O, Weinberg EO, Cohen RA, Liao R, Siwik DA, Colucci WS (2010) Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic Biol Med 48:1182–1187
Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS (2003) H2O2 regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol 35:615–621
Lakshminarayanan V, Lewallen M, Frangogiannis NG, Evans AJ, Wedin KE, Michael LH, Entman ML (2001) Reactive oxygen intermediates induce monocyte chemotactic protein-1 in vascular endothelium after brief ischemia. Am J Pathol 159:1301–1311
Lambeth JD (2004) NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4:181–189
Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ, Kang YJ, Siwik DA, Cohen RA, Colucci WS (2010) Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ Res 107:228–232
Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG (2002) Role of p47phox in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40:511–515
Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG (2003) Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111:1201–1210
Latchman DS (1997) Transcription factors: an overview. Int J Biochem Cell Biol 29:1305–1312
Lee SB, Bae IH, Bae YS, Um HD (2006) Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem 281:36228–36235
Lee IM, Cook NR, Gaziano JM, Gordon D, Ridker PM, Manson JE, Hennekens CH, Buring JE (2005) Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women's Health Study: a randomized controlled trial. JAMA 294:56–65
Leyva F, Anker S, Godsland I, Teixeira M, Hellewell P, Kox W, Poole-Wilson P, Coats A (1998) Uric acid in chronic heart failure: a marker of chronic inflammation. Eur Heart J 19:1814–1822
Li HL, Wang AB, Huang Y, Liu DP, Wei C, Williams GM, Zhang CN, Liu G, Liu YQ, Hao DL (2005) Isorhapontigenin, a new resveratrol analog, attenuates cardiac hypertrophy via blocking signaling transduction pathways. Free Radic Biol Med 38:243–257
Li JM, Gall NP, Grieve DJ, Chen M, Shah AM (2002) Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 40:477–484
Li S, Li X, Zheng H, Xie B, Bidasee KR, Rozanski GJ (2008) Pro-oxidant effect of transforming growth factor-β1 mediates contractile dysfunction in rat ventricular myocytes. Cardiovasc Res 77:107–117
Li Y, Ha T, Gao X, Kelley J, Williams DL, Browder IW, Kao RL, Li C (2004) NF-κB activation is required for the development of cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol 287:H1712–H1720
Li YY, Feldman AM, Sun Y, McTiernan CF (1998) Differential expression of tissue inhibitors of metalloproteinases in the failing human heart. Circulation 98:1728–1734
Litwin SE, Grossman W (1993) Diastolic dysfunction as a cause of heart failure. J Am Coll Cardiol 22:A49–A55
Liu Q, Chen Y, Auger-Messier M, Molkentin JD (2012) Interaction Between NFκB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res 110:1077–1086
Lonn E, Yusuf S, Hoogwerf B, Pogue J, Yi Q, Zinman B, Bosch J, Dagenais G, Mann JF, Gerstein HC (2002) Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes. Diabetes Care 25:1919–1927
Looi YH, Grieve DJ, Siva A, Walker SJ, Anilkumar N, Cave AC, Marber M, Monaghan MJ, Shah AM (2008) Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 51:319–325
Lu Z, Xu X, Hu X, Zhu G, Zhang P, Van Deel ED, French JP, Fassett JT, Oury TD, Bache RJ (2008) Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension 51:19–25
Maalouf RM, Eid AA, Gorin YC, Block K, Escobar GP, Bailey S, Abboud HE (2012) Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. Am J Physiol Cell Physiol 302:C597–C604
MacCarthy PA, Grieve DJ, Li JM, Dunster C, Kelly FJ, Shah AM (2001) Impaired endothelial regulation of ventricular relaxation in cardiac hypertrophy: role of reactive oxygen species and NADPH oxidase. Circulation 104:2967–2974
Machida Y, Kubota T, Kawamura N, Funakoshi H, Ide T, Utsumi H, Li YY, Feldman AM, Tsutsui H, Shimokawa H (2003) Overexpression of tumor necrosis factor-α increases production of hydroxyl radical in murine myocardium. Am J Physiol Heart Circ Physiol 284:H449–H455
Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A (1998) Elevated levels of 8-iso-prostaglandin F2α in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 97:1536–1539
Mann DL (1999) Mechanisms and models in heart failure: a combinatorial approach. Circulation 100:999–1008
Mann DL, McMurray JJV, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L (2004) Targeted anticytokine therapy in patients with chronic heart failure. Circulation 109:1594–1602
Marchioli R, Levantesi G, Macchia A, Marfisi RM, Nicolosi GL, Tavazzi L, Tognoni G, Valagussa F (2006) Vitamin E increases the risk of developing heart failure after myocardial infarction: results from the GISSI-Prevenzione trial. J Cardiovasc Med (Hagerstown) 7:347–350
Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG (2006) Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18:69–82
Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, Kinugawa S, Tsutsui H (2006) Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension 47:711–717
Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara T (2006) Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 113:1779–1786
Maulik N, Goswami S, Galang N, Das DK (1999) Differential regulation of Bcl-2, AP-1 and NF-κB on cardiomyocyte apoptosis during myocardial ischemic stress adaptation. FEBS Lett 443:331–336
Maulik N, Sasaki H, Addya S, Das D (2000) Regulation of cardiomyocyte apoptosis by redox-sensitive transcription factors. FEBS Lett 485:7–12
McMurray J, Chopra M, Abdullah I, Smith W, Dargie H (1993a) Evidence of oxidative stress in chronic heart failure in humans. Eur Heart J 14:1493–1498
McMurray J, McDonagh T, Morrison C, Dargie H (1993b) Trends in hospitalization for heart failure in Scotland 1980–1990. Eur Heart J 14:1158–1162
McNally JS, Davis ME, Giddens DP, Saha A, Hwang J, Dikalov S, Jo H, Harrison DG (2003) Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol 285:H2290–H2297
Mercuro G, Cadeddu C, Piras A, Dessi M, Madeddu C, Deidda M, Serpe R, Massa E, Mantovani G (2007) Early epirubicin-induced myocardial dysfunction revealed by serial tissue Doppler echocardiography: correlation with inflammatory and oxidative stress markers. Oncologist 12:1124–1133
Minhas KM, Saraiva RM, Schuleri KH, Lehrke S, Zheng M, Saliaris AP, Berry CE, Vandegaer KM, Li D, Hare JM (2006) Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res 98:271–279
Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, Ketner EA, Majmudar M, Gabrielson K, Halushka MK (2008) Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation 117:2626–2636
Monti J, Fischer J, Paskas S, Heinig M, Schulz H, Gösele C, Heuser A, Fischer R, Schmidt C, Schirdewan A (2008) Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat Genet 40:529–537
Morita H, Khanal S, Rastogi S, Suzuki G, Imai M, Todor A, Sharov VG, Goldstein S, O'Neill TP, Sabbah HN (2006) Selective matrix metalloproteinase inhibition attenuates progression of left ventricular dysfunction and remodeling in dogs with chronic heart failure. Am J Physiol Heart Circ Physiol 290:H2522–H2527
Muller DN, Dechend R, Mervaala E, Park JK, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H (2000) NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 35:193–201
Murata H, Ihara Y, Nakamura H, Yodoi J, Sumikawa K, Kondo T (2003) Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem 278:50226–50233
Nahrendorf M, Sosnovik D, Chen JW, Panizzi P, Figueiredo JL, Aikawa E, Libby P, Swirski FK, Weissleder R (2008) Activatable magnetic resonance imaging agent reports myeloperoxidase activity in healing infarcts and noninvasively detects the antiinflammatory effects of atorvastatin on ischemia-reperfusion injury. Circulation 117:1153–1160
Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, Namba M (1998) Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-α and angiotensin II. Circulation 98:794–799
Nava E, Noll G, Lüscher TF (1995) Increased activity of constitutive nitric oxide synthase in cardiac endothelium in spontaneous hypertension. Circulation 91:2310–2313
Nordberg J, Arner ESJ (2001) Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 31:1287–1312
Oikonomou E, Tousoulis D, Siasos G, Zaromitidou M, Papavassiliou AG, Stefanadis C (2011) The role of inflammation in heart failure: new therapeutic approaches. Hellenic J Cardiol 52:30–40
Ojaimi C, Qanud K, Hintze TH, Recchia FA (2007) Altered expression of a limited number of genes contributes to cardiac decompensation during chronic ventricular tachypacing in dogs. Physiol Genomics 29:76–83
Onai Y, Suzuki J, Maejima Y, Haraguchi G, Muto S, Itai A, Isobe M (2007) Inhibition of NF-κB improves left ventricular remodeling and cardiac dysfunction after myocardial infarction. Am J Physiol Heart Circ Physiol 292:H530–H538
Pacher P, Nivorozhkin A, Szabó C (2006) Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev 58:87–114
Park YM, Park MY, Suh YL, Park JB (2004) NAD(P)H oxidase inhibitor prevents blood pressure elevation and cardiovascular hypertrophy in aldosterone-infused rats. Biochem Biophys Res Commun 313:812–817
Pchejetski D, Kunduzova O, Dayon A, Calise D, Seguelas MH, Leducq N, Seif I, Parini A, Cuvillier O (2007) Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ Res 100:41–49
Peng M, Huang L, Xie ZJ, Huang WH, Askari A (1995) Oxidant-induced activations of nuclear factor-kappa B and activator protein-1 in cardiac myocytes. Cell Mol Biol Res 41:189–197
Peterson JT, Hallak H, Johnson L, Li H, O'Brien PM, Sliskovic DR, Bocan T, Coker ML, Etoh T, Spinale FG (2001) Matrix metalloproteinase inhibition attenuates left ventricular remodeling and dysfunction in a rat model of progressive heart failure. Circulation 103:2303–2309
Pimentel DR, Amin JK, Xiao L, Miller T, Viereck J, Oliver-Krasinski J, Baliga R, Wang J, Siwik DA, Singh K (2001) Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res 89:453–460
Planavila A, Rodríguez-Calvo R, Palomer X, Coll T, Sánchez RM, Merlos M, Laguna JC, Vázquez-Carrera M (2008) Atorvastatin inhibits GSK-3β phosphorylation by cardiac hypertrophic stimuli. Biochim Biophys Acta 1781:26–35
Qin F, Lennon-Edwards S, Lancel S, Biolo A, Siwik DA, Pimentel DR, Dorn GW, Kang YJ, Colucci WS (2010) Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and-independent phases of myocardial remodeling and prevents the progression to overt heart failure in Gαq-overexpressing transgenic mice. Circ Heart Fail 3:306–313
Qin F, Patel R, Yan C, Liu W (2006) NADPH oxidase is involved in angiotensin II-induced apoptosis in H9C2 cardiac muscle cells: effects of apocynin. Free Radic Biol Med 40:236–246
Qin F, Simeone M, Patel R (2007) Inhibition of NADPH oxidase reduces myocardial oxidative stress and apoptosis and improves cardiac function in heart failure after myocardial infarction. Free Radic Biol Med 43:271–281
Rajesh M, Mukhopadhyay P, Bátkai S, Patel V, Saito K, Matsumoto S, Kashiwaya Y, Horváth B, Mukhopadhyay B, Becker L (2010) Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol 56:2115–2125
Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C, Niebauer J, Hooper J, Volk HD, Coats AJ (2000) Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 102:3060–3067
Reddy VS, Prabhu SD, Mummidi S, Valente AJ, Venkatesan B, Shanmugam P, Delafontaine P, Chandrasekar B (2010) Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP-9 in part via EMMPRIN and through AP-1 and NF-κB activation. Am J Physiol Heart Circ Physiol 299:H1242–H1254
Ritchie RH, Schiebinger RJ, Lapointe MC, Marsh JD (1998) Angiotensin II-induced hypertrophy of adult rat cardiomyocytes is blocked by nitric oxide. Am J Physiol Heart Circ Physiol 275:H1370–H1374
Robinson MJ, Cobb MH (1997) Mitogen-activated protein kinase pathways. Curr Opin Cell Biol 9:180–186
Rose BA, Force T, Wang Y (2010) Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev 90:1507–1546
Rössig L, Hoffmann J, Hugel B, Mallat Z, Haase A, Freyssinet JM, Tedgui A, Aicher A, Zeiher AM, Dimmeler S (2001) Vitamin C inhibits endothelial cell apoptosis in congestive heart failure. Circulation 104:2182–2187
Rude MK, Duhaney TA, Kuster GM, Judge S, Heo J, Colucci WS, Siwik DA, Sam F (2005) Aldosterone stimulates matrix metalloproteinases and reactive oxygen species in adult rat ventricular cardiomyocytes. Hypertension 46:555–561
Rueckschloss U, Villmow M, Klöckner U (2010) NADPH oxidase-derived superoxide impairs calcium transients and contraction in aged murine ventricular myocytes. Exp Gerontol 45:788–796
Ruetten H, Dimmeler S, Gehring D, Ihling C, Zeiher AM (2005) Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc Res 66:444–453
Sakai S, Miyauchi T, Irukayama-Tomobe Y, Ogata T, Goto K, Yamaguchi I (2002) Peroxisome proliferator-activated receptor-g activators inhibit endothelin-1-related cardiac hypertrophy in rats. Clin Sci 103:16S–20S
Saliaris AP, Amado LC, Minhas KM, Schuleri KH, Lehrke S, John MS, Fitton T, Barreiro C, Berry C, Zheng M (2007) Chronic allopurinol administration ameliorates maladaptive alterations in Ca2+ cycling proteins and β-adrenergic hyporesponsiveness in heart failure. Am J Physiol Heart Circ Physiol 292:H1328–H1335
Sam F, Kerstetter DL, Pimental DR, Mulukutla S, Tabaee A, Bristow MR, Colucci WS, Sawyer DB (2005) Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Cardiac Fail 11:473–480
Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, Koyasu S, Matsui H, Yamauchi-Takihara K, Harada M (2001) ERK and p38 MAPK, but not NF-κB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res 89:661–669
Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M (2007) p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446:444–448
Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM (2011) Redox signaling in cardiac myocytes. Free Radic Biol Med 50:777–793
Santos D, Resende M, Mill J, Mansur A, Krieger J, Pereira A (2010) Nuclear Factor (NF) κB polymorphism is associated with heart function in patients with heart failure. BMC Med Genet 11:89
Sauer H, Rahimi G, Hescheler J, Wartenberg M (2000) Role of reactive oxygen species and phosphatidylinositol 3-kinase in cardiomyocyte differentiation of embryonic stem cells. FEBS Lett 476:218–223
Sawyer DB, Colucci WS (2000) Mitochondrial oxidative stress in heart failure: “oxygen wastage” revisited. Circ Res 86:119–120
Schäfer M, Schäfer C, Ewald N, Piper H, Noll T (2003) Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ Res 92:1010–1015
Schenkel PC, Tavares AM, Fernandes RO, Diniz GP, Bertagnolli M, da Rosa Araujo AS, Barreto-Chaves ML, Ribeiro MFM, Clausell N, Belló-Klein A (2010) Redox-sensitive prosurvival and proapoptotic protein expression in the myocardial remodeling post-infarction in rats. Mol Cell Biochem 341:1–8
Scherrer-Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, Lindsey ML, Vancon AC, Huang PL, Lee RT (2001) Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation 104:1286–1291
Schreck R, Rieber P, Baeuerle PA (1991) Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J 10:2247–2258
Seko Y, Takahashi N, Ishiyama S, Nishikawa T, Kasajima T, Hiroe M, Suzuki S, Ishiwata S, Kawai S, Azuma M, Yagita H, Okumura K, Yazaki Y (1998) Expression of costimulatory molecules B7-1, B7-2, and CD40 in the heart of patients with acute myocarditis and dilated cardiomyopathy. Circulation 97:637–639
Shaulian E, Karin M (2002) AP-1 as a regulator of cell life and death. Nat Cell Biol 4:E131–E136
Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K (2005) Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115:2108–2118
Sia YT, Parker TG, Liu P, Tsoporis JN, Adam A, Rouleau JL (2002) Improved post-myocardial infarction survival with probucol in rats: effects on left ventricular function, morphology, cardiac oxidative stress and cytokine expression. J Am Coll Cardiol 39:148–156
Sies H (1997) Oxidative stress: oxidants and antioxidants. Exp Physiol 82:291–295
Silberman GA, Fan THM, Liu H, Jiao Z, Xiao HD, Lovelock JD, Boulden BM, Widder J, Fredd S, Bernstein KE (2010) Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 121:519–528
Sinning C, Schnabel R, Peacock WF (2008) Blankenberg S (2008) Up-and-coming markers: myeloperoxidase, a novel biomarker test for heart failure and acute coronary syndrome application? Congest Heart Fail 14(4 Suppl 1):46–48
Siwik DA, Pagano PJ, Colucci WS (2001) Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol 280:C53–C60
Siwik DA, Tzortzis JD, Pimental DR, Chang DL, Pagano PJ, Singh K, Sawyer DB, Colucci WS (1999) Inhibition of copper-zinc superoxide dismutase induces cell growth, hypertrophic phenotype, and apoptosis in neonatal rat cardiac myocytes in vitro. Circ Res 85:147–153
Sorescu D, Griendling KK (2002) Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail 8:132–140
Spallarossa P, Altieri P, Garibaldi S, Ghigliotti G, Barisione C, Manca V, Fabbi P, Ballestrero A, Brunelli C, Barsotti A (2006) Matrix metalloproteinase-2 and-9 are induced differently by doxorubicin in H9c2 cells: The role of MAP kinases and NAD (P) H oxidase. Cardiovasc Res 69:736–745
Spinale FG (2002) Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res 90:520–530
Spinale FG, Coker ML, Bond BR, Zellner JL (2000) Myocardial matrix degradation and metalloproteinase activation in the failing heart: a potential therapeutic target. Cardiovasc Res 46:225–238
Srivastava S, Chandrasekar B, Bhatnagar A, Prabhu SD (2002) Lipid peroxidation-derived aldehydes and oxidative stress in the failing heart: role of aldose reductase. Am J Physiol Heart Circ Physiol 283:H2612–H2619
Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K, Mitchinson MJ (1996) Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 347:781–786
Stuehr D, Pou S, Rosen GM (2001) Oxygen reduction by nitric-oxide synthases. J Biol Chem 276:14533–14536
Stull LB, Leppo MK, Szweda L, Gao WD, Marbán E (2004) Chronic treatment with allopurinol boosts survival and cardiac contractility in murine postischemic cardiomyopathy. Circ Res 95:1005–1011
Sud N, Wells SM, Sharma S, Wiseman DA, Wilham J, Black SM (2008) Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterial endothelial cells: role of mitochondrial dysfunction. Am J Physiol Cell Physiol 294:C1407–C1418
Sugden PH, Clerk A (2006) Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid Redox Signal 8:2111–2124
Sun Y, Weber KT (2000) Infarct scar: a dynamic tissue. Cardiovasc Res 46:250–256
Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT (2002) Aldosterone-induced inflammation in the rat heart: role of oxidative stress. Am J Pathol 161:1773–1781
Swynghedauw B (1999) Molecular mechanisms of myocardial remodeling. Physiol Rev 79:215–262
Taimor G, Rakow A, Piper HM (2001) Transcription activator protein 1 (AP-1) mediates NO-induced apoptosis of adult cardiomyocytes. FASEB J 15:2518–2520
Takac I, Schröder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP (2011) The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem 286:13304–13313
Takemoto Y, Yoshiyama M, Takeuchi K, Omura T, Komatsu R, Izumi Y, Kim S, Yoshikawa J (1999) Increased JNK, AP-1 and NF-κB DNA binding activities in isoproterenol-induced cardiac remodeling. J Mol Cell Cardiol 31:2017–2030
Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y (2005) Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest 115:1221–1231
Tanaka K, Honda M, Takabatake T (2001) Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J Am Coll Cardiol 37:676–685
Tang WH, Wu Y, Nicholls SJ, Hazen SL (2011) Plasma myeloperoxidase predicts incident cardiovascular risks in stable patients undergoing medical management for coronary artery disease. Clin Chem 57:33–39
Taniyama Y, Walsh K (2002) Elevated myocardial Akt signaling ameliorates doxorubicin-induced congestive heart failure and promotes heart growth. J Mol Cell Cardiol 34:1241–1247
Thakur S, Du J, Hourani S, Ledent C, Li JM (2010) Inactivation of adenosine A2A receptor attenuates basal and angiotensin II-induced ROS production by Nox2 in endothelial cells. J Biol Chem 285:40104–40113
Tian J, Liu J, Garlid KD, Shapiro JI, Xie Z (2003) Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol Cell Biochem 242:181–187
Treasure CB, Klein JL, Vita JA, Manoukian SV, Renwick GH, Selwyn AP, Ganz P, Alexander RW (1993) Hypertension and left ventricular hypertrophy are associated with impaired endothelium-mediated relaxation in human coronary resistance vessels. Circulation 87:86–93
Tsutsui H, Ide T, Shiomi T, Kang D, Hayashidani S, Suematsu N, Wen J, Utsumi H, Hamasaki N, Takeshita A (2001) 8-oxo-dGTPase, which prevents oxidative stress-induced DNA damage, increases in the mitochondria from failing hearts. Circulation 104:2883–2885
Turan B, Saini HK, Zhang M, Prajapati D, Elimban V, Dhalla NS (2005) Selenium improves cardiac function by attenuating the activation of NF-κB due to ischemia-reperfusion injury. Antioxid Redox Signal 7:1388–1397
Turner NA, Xia F, Azhar G, Zhang X, Liu L, Wei JY (1998) Oxidative stress induces DNA fragmentation and caspase activation via the c-Jun NH2-terminal kinase pathway in H9c2 cardiac muscle cells. J Mol Cell Cardiol 30:1789–1801
Ueland T, Aukrust P, Yndestad A, Otterdal K, Frøland SS, Dickstein K, Kjekshus J, Gullestad L, Damås JK (2005) Soluble CD40 ligand in acute and chronic heart failure. Eur Heart J 26:1101–1107
Ukai T, Cheng CP, Tachibana H, Igawa A, Zhang ZS, Cheng HJ, Little WC (2001) Allopurinol enhances the contractile response to dobutamine and exercise in dogs with pacing-induced heart failure. Circulation 103:750–755
Ushio-Fukai M, Alexander RW, Akers M, Griendling KK (1998) p38 mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. J Biol Chem 273:15022–15029
Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW (2002) Novel role of gp91phox-containing NAD (P) H oxidase in vascular endothelial growth factor–induced signaling and angiogenesis. Circ Res 91:1160–1167
Van der Heiden K, Cuhlmann S, le Luong L, Zakkar M, Evans P (2010) Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci (Lond) 118:593–605
van Empel VP, Bertrand AT, Hofstra L, Crijns HJ, Doevendans PA, De Windt LJ (2005) Myocyte apoptosis in heart failure. Cardiovasc Res 67:21–29
Vasilyev N, Williams T, Brennan ML, Unzek S, Zhou X, Heinecke JW, Spitz DR, Topol EJ, Hazen SL, Penn MS (2005) Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 112:2812–2820
Viedt C, Soto U, Krieger-Brauer HI, Fei J, Elsing C, Kubler W, Kreuzer J (2000) Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol 20:940–948
von Harsdorf R, Li PF, Dietz R (1999) Signaling pathways in reactive oxygen species–induced cardiomyocyte apoptosis. Circulation 99:2934–2941
Walsh K (2006) Akt signaling and growth of the heart. Circulation 113:2032–2034
Wang ZH, Chen YX, Zhang CM, Wu L, Yu Z, Cai XL, Guan Y, Zhou ZN, Yang HT (2011) Intermittent hypobaric hypoxia improves postischemic recovery of myocardial contractile function via redox signaling during early reperfusion. Am J Physiol Heart Circ Physiol 301:H1695–H1705
Watson PA, Birdsey N, Huggins GS, Svensson E, Heppe D, Knaub L (2010) Cardiac-specific overexpression of dominant-negative CREB leads to increased mortality and mitochondrial dysfunction in female mice. Am J Physiol Heart Circ Physiol 299:H2056–H2068
Weber K, Sun Y, Guarda E (1994) Structural remodeling in hypertensive heart disease and the role of hormones. Hypertension 23:869–877
Wei L, Fahey T, Struthers A, MacDonald T (2009) Association between allopurinol and mortality in heart failure patients: a long‐term follow‐up study. Int J Clin Pract 63:1327–1333
Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN (2003) A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 111:1497–1504
Wenzel S, Taimor G, Piper HM, Schlüter KD (2001) Redox-sensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-ß expression in adult ventricular cardiomyocytes. FASEB J 15:2291–2293
Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A (2011) Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4:44–52
Westermann D, Van Linthout S, Dhayat S, Dhayat N, Schmidt A, Noutsias M, Song XY, Spillmann F, Riad A, Schultheiss HP (2007) Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol 102:500–507
Widder JD, Fraccarollo D, Galuppo P, Hansen JM, Jones DP, Ertl G, Bauersachs J (2009) Attenuation of angiotensin II–induced vascular dysfunction and hypertension by overexpression of thioredoxin 2. Hypertension 54:338–344
Wong JL, Créton R, Wessel GM (2004) The oxidative burst at fertilization is dependent upon activation of the dual oxidase Udx1. Dev Cell 7:801–814
Wosniak J Jr, Santos CX, Kowaltowski AJ, Laurindo FR (2009) Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal 11:1265–1278
Wu S, Gao J, Ohlemeyer C, Roos D, Niessen H, Köttgen E, Geβner R (2005) Activation of AP-1 through reactive oxygen species by angiotensin II in rat cardiomyocytes. Free Radic Biol Med 39:1601–1610
Xia M, Li G, Ma J, Ling W (2010) Phosphoinositide 3-kinase mediates CD40 ligand-induced oxidative stress and endothelial dysfunction via Rac1 and NADPH oxidase 2. J Thromb Haemost 8:397–406
Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, Sawyer DB (2002) Role of reactive oxygen species and NAD(P)H oxidase in α1-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol 282:C926–C934
Xie S, Nie R, Wang J, Li F, Yuan W (2009) Transcription factor decoys for activator protein-1 (AP-1) inhibit oxidative stress-induced proliferation and matrix metalloproteinases in rat cardiac fibroblasts. Transl Res 153:17–23
Xie Z, Kometiani P, Liu J, Li J, Shapiro JI, Askari A (1999) Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J Biol Chem 274:19323–19328
Xie Z, Xie J (2005) The Na/K-ATPase-mediated signal transduction as a target for new drug development. Front Biosci 10:3100–3109
Xu KY, Zweier JL, Becker LC (1997) Hydroxyl radical inhibits sarcoplasmic reticulum Ca2 -ATPase function by direct attack on the ATP binding site. Circ Res 80:76–81
Xu Q, Dalic A, Fang L, Kiriazis H, Ritchie R, Sim K, Gao XM, Drummond G, Sarwar M, Zhang YY (2011) Myocardial oxidative stress contributes to transgenic β2‐adrenoceptor activation‐induced cardiomyopathy and heart failure. Br J Pharmacol 162:1012–1028
Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, Wagner T, Vatner SF, Sadoshima J (2003) Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest 112:1395–1406
Yang HY, Liu JC, Chen YL, Chen CH, Lin H, Lin JW, Chiu WT, Chen JJ, Cheng TH (2005) Inhibitory effect of trilinolein on endothelin-1-induced c-fos gene expression in cultured neonatal rat cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol 372:160–167
Yang J, Marden JJ, Fan C, Sanlioglu S, Weiss RM, Ritchie TC, Davisson RL, Engelhardt JF (2003) Genetic redox preconditioning differentially modulates AP-1 and NFκB responses following cardiac ischemia/reperfusion injury and protects against necrosis and apoptosis. Mol Ther 7:341–353
Yasunari K, Maeda K, Watanabe T, Nakamura M, Yoshikawa J, Asada A (2004) Comparative effects of valsartan versus amlodipine on left ventricular mass and reactive oxygen species formation by monocytes in hypertensive patients with left ventricular hypertrophy. J Am Coll Cardiol 43:2116–2123
Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, Epstein PN (2004) Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 53:1336–1343
Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P (2000) Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 342:154–160
Zelarayan L, Renger A, Noack C, Zafiriou MP, Gehrke C, Van Der Nagel R, Dietz R, De Windt L, Bergmann MW (2009) NF-κB activation is required for adaptive cardiac hypertrophy. Cardiovasc Res 84:416–424
Zhang C, Hein TW, Wang W, Ren Y, Shipley RD, Kuo L (2006) Activation of JNK and xanthine oxidase by TNF-α impairs nitric oxide-mediated dilation of coronary arterioles. J Mol Cellular Cardiol 40:247–257
Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, Yao L, Nagai Y, Fujisawa Y, Miyatake A, Abe Y (2005) Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovasc Res 65:230–238
Zhang GX, Lu XM, Kimura S, Nishiyama A (2007) Role of mitochondria in angiotensin II-induced reactive oxygen species and mitogen-activated protein kinase activation. Cardiovasc Res 76:204–212
Zhang HJ, Zhao W, Venkataraman S, Robbins MEC, Buettner GR, Kregel KC, Oberley LW (2002) Activation of matrix metalloproteinase-2 by overexpression of manganese superoxide dismutase in human breast cancer MCF-7 cells involves reactive oxygen species. J Biol Chem 277:20919–20926
Zhang L, Jiang H, Gao X, Zou Y, Liu M, Liang Y, Yu Y, Zhu W, Chen H, Ge J (2011a) Heat shock transcription factor-1 inhibits H2O2-induced apoptosis via down-regulation of reactive oxygen species in cardiac myocytes. Mol Cell Biochem 347:21–28
Zhang Y, Kang YM, Tian C, Zeng Y, Jia LX, Ma X, Du J, Li HH (2011b) Overexpression of Nrdp1 in the heart exacerbates doxorubicin-induced cardiac dysfunction in mice. PLoS One 6:e21104
Zhang M, Brewer AC, Schröder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM (2010) NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci USA 107:18121–18126
Zhang M, Looi YH, Brewer AC, Shah AM (2009) Cardiomyocyte-specific overexpression of Nox4 attenuates adverse cardiac remodeling after myocardial infarction. Circulation 120:S1097
Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, McDermott BJ, Grieve DJ (2010) Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with doxorubicin chemotherapy. Cancer Res 70:9287–9297
Zhou L, Azfer A, Niu J, Graham S, Choudhury M, Adamski FM, Younce C, Binkley PF, Kolattukudy PE (2006) Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ Res 98:1177–1185
Zhu Y, Shi YP, Wu D, Ji YJ, Wang X, Chen HL, Wu SS, Huang DJ, Jiang W (2011) Salidroside protects against hydrogen peroxide-induced injury in cardiac H9c2 cells via PI3K-Akt dependent pathway. DNA Cell Biol 30:809–819
Zima AV, Blatter LA (2006) Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71:310–321
Zima AV, Copello JA, Blatter LA (2004) Effects of cytosolic NADH/NAD levels on sarcoplasmic reticulum Ca2+ release in permeabilized rat ventricular myocytes. J Physiol 555:727–741
Zinkevich NS, Gutterman DD (2011) ROS-induced ROS release in vascular biology: redox-redox signaling. Am J Physiol Heart Circ Physiol 301:H647–H653
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this entry
Cite this entry
Harvey, A.P., Grieve, D.J. (2014). Reactive Oxygen Species (ROS) Signaling in Cardiac Remodeling and Failure. In: Laher, I. (eds) Systems Biology of Free Radicals and Antioxidants. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-30018-9_50
Download citation
DOI: https://doi.org/10.1007/978-3-642-30018-9_50
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-30017-2
Online ISBN: 978-3-642-30018-9
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences