
Abstract
The liver GSDs comprise GSD I, the hepatic presentations of GSD III, GSD IV, GSD VI, the liver forms of GSD IX, and GSD 0. GSD I, -III, -VI, and -IX present with hypoglycaemia, marked hepatomegaly, and retarded growth [6]. GSD I is the most severe of these four conditions, because both glycogenolysis and gluconeogenesis are impaired. Patients with GSD III have a syndrome that includes hepatopathy, myopathy and often cardiomyopathy. Unlike the other hepatic forms of GSD, GSD IV usually manifests in infancy or childhood as hepatic failure with cirrhosis leading to end-stage liver disease. GSD VI and the hepatic forms of GSD IX are classically the mildest forms, but there is recent evidence that optimised therapy decreases the rate of liver pathology and improves growth.
Access provided by Autonomous University of Puebla. Download to read the full chapter text
Chapter PDF
Similar content being viewed by others


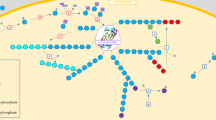
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
References
Roach PJ (2002) Glycogen and its metabolism. Curr Mol Med 2:101–120
Chen Y-T (2001) Glycogen storage diseases. In: Scriver C, Beaudet A, Sly W et al. (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 1521–1555
Wolfsdorf JI, Holm IA, Weinstein DA (1999) Glycogen storage diseases: phenotypic, genetic, and biochemical characteristics, and therapy. Endocrinol Metab Clin North Am 28:801–823
DiMauro S (2007) Muscle glycogenoses: an overview. Acta Myol 26:35–41
Benarroch EE (2010) Glycogen metabolism: metabolic coupling between astrocytes and neurons. Neurology 74:919–923
Wolfsdorf JI, Weinstein DA (2003) Glycogen storage diseases. Rev Endocr Metab Disord 4:95–102
Chou JY, Jun HS, Mansfield BC (2010) Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat Rev Endocrinol 6:676–688
Rake JP, Visser G, Labrune P et al. (2002) Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 161 [Suppl 1]:S20-S34
Elpeleg ON (1999) The molecular background of glycogen metabolism disorders. J Pediatr Endocrinol Metab 12:363–379
Visser G, Rake JP, Labrune P et al. (2002) Consensus guidelines for management of glycogen storage disease type 1b – European Study on Glycogen Storage Disease Type 1. Eur J Pediatr 161 [Suppl 1]:S120-S123
Kuijpers TW, Maianski NA, Tool AT et al. (2003) Apoptotic neutrophils in the circulation of patients with glycogen storage disease type 1b (GSD1b). Blood 101:5021–5024
Visser G, Rake JP, Fernandes J et al. (2000) Neutropenia, neutrophil dysfunction, and inflammatory bowel disease in glycogen storage disease type Ib: results of the European Study on Glycogen Storage Disease type I. J Pediatr 137:187–191
Melis D, Parenti G, Della Casa R et al. (2003) Crohn’s-like ileo-colitis in patients affected by glycogen storage disease Ib: two years’ follow-up of patients with a wide spectrum of gastrointestinal signs. Acta Paediatr 92:1415–1421
Waddell ID, Burchell A (1993) Identification, purification and genetic deficiencies of the glucose-6-phosphatase system transport proteins. Eur J Pediatr 152:S14-S17
Veiga-da-Cunha M, Gerin I, Chen YT et al. (1999) The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a. Eur J Hum Genet 7: 717–723
Burchell A (1998) A reevaluation of GLUT 7. Biochem J 331:973
Shieh JJ, Pan CJ, Mansfield BC, Chou JY (2003) A glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycaemia in glycogen storage disease type Ia. J Biol Chem 278:47098–47103
Fernandes J (1974) The effect of disaccharides on the hyperlactacidaemia of glucose-6-phosphatase-deficient children. Acta Paediatr Scand 63: 695–698
Greene HL, Swift LL, Knapp HR (1991) Hyperlipidemia and fatty acid composition in patients treated for type IA glycogen storage disease. J Pediatr 119:398–403
Alaupovic P, Fernandes J (1985) The serum apolipoprotein profile of patients with glucose-6-phosphatase deficiency. Pediatr Res 1985;19:380–384
Bandsma RH, Prinsen BH, Velden v d Mde S et al. (2008) Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatr Res 63:702–707
Binkiewicz A, Senior B (1973) Decreased ketogenesis in Von Gierke’s disease (type 1 glycogenosis). J Pediatr 83:973–978
Greene HL, Wilson FA, Hefferan P et al. (1978) ATP depletion, a possible role in the pathogenesis of hyperuricemia in glycogen storage disease type I. J Clin Invest 62:321–328
Cohen JL, Vinik A, Faller J, Fox IH (1985) Hyperuricemia in glycogen storage disease type I. Contributions by hypoglycemia and hyperglucagonemia to increased urate production. J Clin Invest 75:251–257
Matern D, Seydewitz HH, Bali D, Lang C, Chen YT (2002) Glycogen storage disease type I: diagnosis and phenotype/genotype correlation. Eur J Pediatr 161 [Suppl 1]:S10-S19
Ekstein J, Rubin BY, Anderson SL et al. (2004) Mutation frequencies for glycogen storage disease Ia in the Ashkenazi Jewish population. Am J Med Genet 129A:162–164
Lin B, Hiraiwa H, Pan CJ, Nordlie RC, Chou JY (1999) Type-1c glycogen storage disease is not caused by mutations in the glucose-6-phosphate transporter gene. Hum Genet 105:515–517
Chen SY, Pan CJ, Nandigama K et al. (2008) The glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type Ib and Ic. FASEB J 22:2206–2213
Burr IM, O’Neill JA, Karzon DT, Howard LJ, Greene HL (1974) Comparison of the effects of total parenteral nutrition, continuous intragastric feeding, and portacaval shunt on a patient with type I glycogen storage disease. J Pediatr 85:792–795
Rake JP, Visser G, Labrune P et al. (2002) Guidelines for management of glycogen storage disease type I – European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 161 [Suppl 1]:S112-S119
Chen YT, Cornblath M, Sidbury JB (1984) Cornstarch therapy in type I glycogen-storage disease. N Engl J Med 310:171–175
Weinstein DA, Wolfsdorf JI (2002) Effect of continuous glucose therapy with uncooked cornstarch on the long-term clinical course of type 1a glycogen storage disease. Eur J Pediatr 161 [Suppl 1]:S35–39
Bhattacharya K, Orton RC, Qi X et al. (2007) A novel starch for the treatment of glycogen storage diseases. J Inherit Metab Dis 30:350–357
Correia CE, Bhattacharya K, Lee PJ et al. (2008) Use of modified cornstarch therapy to extend fasting in glycogen storage disease types Ia and Ib. Am J Clin Nutr 88:1272–1276
Daublin G, Schwahn B, Wendel U (2002) Type I glycogen storage disease: favourable outcome on a strict management regimen avoiding increased lactate production during childhood and adolescence. Eur J Pediatr 161 [Suppl 1]:S40-S45
Weinstein DA, Roy CN, Fleming MD et al. (2002) Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood 100:3776–3781
Banugaria SG, Austin SL, Boney A, Weber TJ, Kishnani PS (2010) Hypovitaminosis D in glycogen storage disease type I. Mol Genet Metab 99:434–437
Das AM, Lücke T, Meyer U, Hartmann H, Illsinger S (2010) Glycogen storage disease type 1: impact of medium-chain triglycerides on metabolic control and growth. Ann Nutr Metab 56:225–232
Weinstein DA., Somers MJ, Wolfsdorf JI (2001) Decreased urinary citrate excretion in type 1a glycogen storage disease. J Pediatr 138:378–382
Wittenstein B, Klein M, Finckh B, Ullrich K, Kohlschutter A (2002) Radical trapping in glycogen storage disease 1a. Eur J Pediatr 161 [Suppl 1]:S70-S74
Lee PJ, Dalton RN, Shah V, Hindmarsh PC, Leonard JV (1995) Glomerular and tubular function in glycogen storage disease. Pediatr Nephrol 9:705–710
Chen YT, Coleman RA, Scheinman JI, Kolbeck PC, Sidbury JB (1988) Renal disease in type I glycogen storage disease. N Engl J Med 318:7–11
Martens DH, Rake JP, Navis G et al. (2009) Renal function in glycogen storage disease type I, natural course, and renopreservative effects of ACE inhibition. Clin J Am Soc Nephrol 4:1741–1746
Bandsma RH, Smit GP, Kuipers F (2002) Disturbed lipid metabolism in glycogen storage disease type 1. Eur J Pediatr 161 [Suppl 1]:S65–69
Lee PJ, Celermajer DS, Robinson J et al. (1994) Hyperlipidaemia does not impair vascular endothelial function in glycogen storage disease type 1a. Atherosclerosis 110:95–100
Nguyen AD, Pan CJ, Weinstein DA, Chou JY (2006) Increased scavenger receptor class B type I-mediated cellular cholesterol efflux and antioxidant capacity in the sera of glycogen storage disease type Ia patients. Mol Genet Metab 89:233–238
Bernier AV, Correia CE, Haller MJ et al. (2009) Vascular dysfunction in glycogen storage disease type I. J Pediatr 154:588–591
Mundy HR, Hindmarsh PC, Matthews DR, Leonard JV, Lee PJ (2003) The regulation of growth in glycogen storage disease type 1. Clin Endocrinol (Oxf) 58:332–339
Mairovitz V, Labrune P, Fernandez H, Audibert F, Frydman R (2002) Contraception and pregnancy in women affected by glycogen storage diseases. Eur J Pediatr 161 [Suppl 1]:S97–101
Kerr KG (1999) The prophylaxis of bacterial infections in neutropenic patients. J Antimicrob Chemother 44:587–591
Visser G, Rake JP, Labrune P et al. (2002) Granulocyte colonystimulating factor in glycogen storage disease type 1b. Results of the European Study on Glycogen Storage Disease Type 1. Eur J Pediatr 161 [Suppl 1]:S83-S87
Simmons PS, Smithson WA, Gronert GA, Haymond MW (1984) Acute myelogenous leukemia and malignant hyperthermia in a patient with type 1b glycogen storage disease. J Pediatr 105:428–431
Donadieu J, Barkaoui M, Bezard F et al. (2000) Renal carcinoma in a patient with glycogen storage disease Ib receiving long-term granulocyte colony-stimulating factor therapy. J Pediatr Hematol Oncol 22:188–189
Storch E, Keeley M, Merlo L et al. (2008) Psychosocial functioning in youth with glycogen storage disease type I. J Pediatr Psychol 33:728–738
Talente GM, Coleman RA, Alter C et al. (1994) Glycogen storage disease in adults. Ann Intern Med 120:218–226
Baker L, Dahlem S, Goldfarb S et al. (1989) Hyperfiltration and renal disease in glycogen storage disease, type I. Kidney Int 35:1345–1350
Chen YT, Scheinman JI, Park HK, Coleman RA, Roe CR (1990) Amelioration of proximal renal tubular dysfunction in type I glycogen storage disease with dietary therapy. N Engl J Med 323:590–593
Restaino I, Kaplan BS, Stanley C, Baker L (1993) Nephrolithiasis, hypocitraturia, and a distal renal tubular acidification defect in type 1 glycogen storage disease. J Pediatr 122:392–396
Wolfsdorf JI, Laffel LM, Crigler JF Jr (1997) Metabolic control and renal dysfunction in type I glycogen storage disease. J Inherit Metab Dis 20: 559–568
Labrune P, Trioche P, Duvaltier I, Chevalier P, Odievre M (1997) Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr 24:276–279
Bianchi L (1993) Glycogen storage disease I and hepatocellular tumours. Eur J Pediatr 152:S63-S70
Lee PJ (2002) Glycogen storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr 161 [Suppl 1]:S46-S49
Wang DQ, Fiske LM, Carreras CT, Weinstein DA (2011) Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J Pediatr Apr 8 [Epub ahead of print]; PMID: 21481415
Franco LM, Krishnamurthy V, Bali D et al. (2005) Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J Inherit Metab Dis 28:153–162
Kim YI, Chung JW, Park JH (2007) Feasibility of transcatheter arterial chemoembolization for hepatic adenoma. J Vasc Interv Radiol 18:862–867
Reddy SK, Kishnani PS, Sullivan JA et al. (2007) Resection of hepatocellular adenoma in patients with glycogen storage disease type Ia. J Hepatol 47:658–663
Labrune P (2002) Glycogen storage disease type I: indications for liver and/or kidney transplantation. Eur J Pediatr 161 [Suppl 1]:S53-S55
Davis MK, Weinstein DA (2008) Liver transplantation in children with glycogen storage disease: controversies and evaluation of the risk/benefit of this procedure. Pediatr Transplant 12:137–145
Lee PJ, Patel JS, Fewtrell M, Leonard JV, Bishop NJ (1995) Bone mineralisation in type 1 glycogen storage disease. Eur J Pediatr 154:483–487
Rake JP, Visser G, Huismans D et al. (2003) Bone mineral density in children, adolescents and adults with glycogen storage disease type Ia: a cross-sectional and longitudinal study. J Inherit Metab Dis 26:371–384
Semrin G, Fishman DS, Bousvaros et al. (2006) Impaired intestinal iron absorption in Crohn’s disease correlates with disease activity and markers of inflammation. Inflamm Bowel Dis 12:1101–1106
Lee PJ, Patel A, Hindmarsh PC, Mowat AP, Leonard JV (1995) The prevalence of polycystic ovaries in the hepatic glycogen storage diseases: its association with hyperinsulinism. Clin Endocrinol (Oxf) 42:601–606
Corby DG, Putnam CW, Greene HL (1974) Impaired platelet function in glucose-6-phosphatase deficiency. J Pediatr 85:71–76
Trioche P, Francoual J, Capel L et al. (2000) Apolipoprotein E polymorphism and serum concentrations in patients with glycogen storage disease type Ia. J Inherit Metab Dis 23:107–112
Humbert M, Labrune P, Simonneau G (2002) Severe pulmonary arterial hypertension in type 1 glycogen storage disease. Eur J Pediatr 161 [Suppl 1]:S93-S96
Martens DH, Rake JP Schwarz M et al. (2008) Pregnancies in glycogen storage disease type Ia. Am J Obstet Gynecol 198:646.e1–7
Dagli AI, Lee PJ, Correia CE et al. (2010) Pregnancy in glycogen storage disease type Ib: gestational care and report of first successful deliveries. J Inherit Metab Dis [Epub ahead of print Apr 13]; [Epub ahead of print] PubMed PMID: 20386986
Saunders AC, Feldman HA, Correia CE, Weinstein DA (2005) Clinical evaluation of a portable lactate meter in type I glycogen storage disease. J Inherit Metab Dis 28:695–701
DiMauro S, Hays AP, Tsujino S (2004) Nonlysosomal glycogenosis. In: Engel AG, Franzini-Amstrong C (eds) Myology: basic and clinical. McGraw-Hill, New York, pp 1535–1558
Mormoi T, Sano H, Yamanaka C, Sasaki H, Mikawa H (1992) Glycogen storage disease type III with muscle involvement: reappraisal of phenotypic variability and prognosis. Am J Med Genet 42:696–699
DiMauro S, Hartwig GB, Hays A et al (1979) Debrancher deficiency: neuromuscular disorder in 5 adults. Ann Neurol 5:422–436
Hobson-Webb LD, Austin SL, Bali DS, Kishnani PS (2010) The electrodiagnostic characteristics of glycogen storage disease type III. Genet Med 12:440–445
Bernier AV, Sentner CP, Correia CE et al. (2008) Hyperlipidemia in glycogen storage disease type III: effect of age and metabolic control. J Inherit Metab Dis 31:729–732
Goldstein JL, Austin SL, Boyette K et al. (2010) Molecular analysis of the AGL gene: identification of 25 novel mutations and evidence of genetic heterogeneity in patients with glycogen storage disease type III. Genet Med 12:424–430
Crushell E, Treacy EP, Dawe J, Durkie M, Beauchamp NJ (2010) Glycogen storage disease type III in the Irish population. J Inherit Metab Dis [Epub ahead of print 2010 May 20].
Lucchiari S, Fogh I, Prelle A et al (2002) Clinical and genetic variability in glycogen storage disease type IIIa: seven novel AGL gene mutations in the Mediterranean area. Am J Med Genet 109:183–190
Kishnani PS, Austin SL, Arn P et al. (2010) Glycogen storage disease type III diagnosis and management guidelines. Gene Med 12:446–463
Dagli AI, Zori RT, McCune H et al. (2009) Reversal of glycogen storage disease type IIIa-related cardiomyopathy with modification of diet. J Inherit Metab Dis [Published on line 26 Mar 2009] DOI: 10.1007/s10545-009-1088-x
Demo E, Frush D, Gottfried M et al. (2007) Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? J Hepatol 46:492–498
Matern D, Starzl TE, Arnaout W et al. (1999) Liver transplantation for glycogen storage disease types I, III, and IV. Eur J Pediatr 158 [Suppl 2]:S43-S48
Lee PJ, Deanfield JE, Burch M et al. (1997) Comparison of the functional significance of left ventricular hypertrophy in hypertrophic cardiomyopathy and glycogenosis type III. Am J Cardiol 79:834–838
Vertilus SM, Austin SL, Foster KS et al. (2010) Echocardiographic manifestations of glycogen storage disease type III: increase in wall thickness and left ventricular mass over time. Genet Med 12:413–423
Lee P (1999) Successful pregnancy in a patient with type III glycogen storage disease managed with cornstarch supplements. Br J Obstet Gynaecol 106:181–182
Moses SW, Parvari R (2002) The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med 2:177–188
De Moor RA, Schweizer JJ, Hoek v B et al. (2000) Hepatocellular carcinoma in glycogen storage disease type IV. Arch Dis Child 82:479–480
Selby R, Starzl TE, Yunis E et al. (1993) Liver transplantation for type I and type IV glycogen storage disease. Eur J Pediatr 152 [Suppl 1]:S71-S76
Lossos A, Meiner Z, Barash V et al. (1998) Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. Ann Neurol 44:867–872
Tay SK, Akman HO, Chung WK et al. (2004) Fatal infantile neuromuscular presentation of glycogen storage disease type IV. Neuromusc Disord 14:253–260
Bao Y, Kishnani P, Wu JY, Chen HT (1996) Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J Clin Invest 97:941–948
Dagli AI, Weinstein DA (2009) Glycogen storage disease type VI. In: Pagon RA, Bird TD, Dolan CR, Stephens K (eds) GeneReviews, Seattle:Universityof Washington [Internet]
Beauchamp NJ, Taybert J, Champion MP et al. (2007) High frequency of missense mutations in glycogen storage disease type VI. J Inherit Metab Dis 30:722–734
Hendrickx J, Willems PJ (1996) Genetic deficiencies of the glycogen phosphorylase system. Hum Genet 97:551–556
Chang S, Rosenberg MJ, Morton H, Francomano CA, Biesecker LG (1998) Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI. Hum Mol Genet 7:865–870
Nakai A, Shigematsu Y, Takano T, Kikawa Y, Sudo M (1994) Uncooked cornstarch treatment for hepatic phosphorylase kinase deficiency. Eur J Pediatr 153:581–583
Huijing F, Fernandes J (1969) X-Chromosomal inheritance of liver glycogenosis with phosphorylase kinase deficiency. Am J Hum Genet 21:275–284
Shin YS (2006) Glycogen storage disease: clinical, biochemical, and molecular heterogeneity. Semin Pediatr Neurol 13:115–120
Regalado JJ, Rodriguez MM, Ferrer PL (1999) Infantile hypertrophic cardiomyopathy of glycogenosis type IX: isolated cardiac phosphorylase kinase deficiency. Pediatr Cardiol 20:304–307
Willems PJ, Gerver WJ, Berger R, Fernandes J (1990) The natural history of liver glycogenosis due to phosphorylase kinase deficiency: a longitudinal study of 41 patients. Eur J Pediatr 149:268–271
Burwinkel B, Hu B, Schroers A et al. (2003) Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet 11:516–526
Carriere C, Jonic S, Mornon JP, Callebaut I (2008) 3D mapping of glycogenosis-causing mutations in the large regulatory alpha subunit of phosphorylase kinase. Biochim Biophys Acta 1782:664–670
Berg v d IE, van Beurden EA, de Klerk JB et al. (1997) Autosomal recessive phosphorylase kinase deficiency in liver caused by mutations in the gene encoding the beta subunit (PHKB). Am J Hum Genet 61:539–546
Maichele AJ, Burwinkel B, Maire I, Sǿvik O, Kilimann MW (1996) Mutations in the testis/liver isoform of the phosphorylase kinase gamma subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans. Nat Genet 14:337–340
Hendrickx J, Lee P, Keating JP et al. (1999) Complete genomic structure and mutational spectrum of PHKA2 in patients with X-linked liver glycogenosis type I and II. Am J Hum Genet 64:1541–1549
Burwinkel B, Shiomi S, Al Zaben A, Kilimann MW (1998) Liver glycogenosis due to phosphorylase kinase deficiency: PHKG2 gene structure and mutations associated with cirrhosis. Hum Mol Genet 7:149–154
Burwinkel B, Rootwelt T, Kvittingen EA, Chakraborty PK, Kilimann MW (2003) Severe phenotype of phosphorylase kinasedeficient liver glycogenosis with mutations in the PHKG2 gene. Pediatr Res 54:834–839
Wehner M, Clemens PR, Engel AG, Kilimann MW (1994) Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit. Hum Mol Genet 3:1983–1887
Beauchamp NJ, Dalton A, Ramaswami U et al. (2007) Glycogen storage disease type IX: high variability in clinical phenotype. Mol Genet Metab 92:88–99
Aynsley-Green A, Williamson DH, Gitzelmann R (1977) Hepatic glycogen synthetase deficiency. Definition of syndrome from metabolic and enzyme studies on a 9-year-old girl. Arch Dis Child 52:573–579
Weinstein DA, Correia CE, Saunders AC, Wolfsdorf JI (2006) Hepatic glycogen synthase deficiency: an infrequently recognized cause of ketotic hypoglycemia. Mol Genet Metab 87:284–288
Bachrach BE, Weinstein DA Orho-Melander M, Burgess A, Wolfsdorf JI (2002) Glycogen synthase deficiency (glycogen storage disease type 0) presenting with hyperglycemia and glucosuria: report of three new mutations. J Pediatr 140:781–783
Gitzelmann R, Spycher MA, Feil G et al. (1996) Liver glycogen synthase deficiency: a rarely diagnosed entity. Eur J Pediatr 155:561–567
Orho M, Bosshard NU, Buist NR et al. (1998) Mutations in the liver glycogen synthase gene in children with hypoglycaemia due to glycogen storage disease type 0. J Clin Invest 102:507–515
Byrne BM, Gillmer MD, Turner RC, Aynsley-Green A (1995) Glucose homeostasis in adulthood and in pregnancy in a patient with hepatic glycogen synthetase deficiency. Br J Obstet Gynaecol 102:931–933
Tonin P, Lewis PJ, Servidei S, DiMauro S (1990) Metabolic causes of myoglobinuria. Ann Neurol 27:181–185
Nadaj-Pakleza A, Vincitorio C, Laforêt P et al. (2009) Permanent muscle weakness in McArdle disease. Muscle Nerve 40:350–357
Martin MA, Rubio JC, Wevers RA et al. (2004) Molecular analysis of myophosphorylase deficiency in Dutch patients with McArdle’s disease. Ann Hum Genet 68:17–22
Bartram C, Edwards RH, Clague J, Beynon RJ (1993) McArdle’s disease: a nonsense mutation in exon 1 of the muscle glycogen phosphorylase gene explains some but not all cases. Hum Mol Genet 2:1291–1293
El Schahawi M, Tsujino S, Shanske S, DiMauro S (1996) Diagnosis of McArdle’s disease by molecular genetic analysis of blood. Neurology 47:579–580
Tsujino S, Shanske S, Carroll JE, Sabina RL, DiMauro S (1994) Two mutations, one novel and one frequently observed, in Japanese patients with McArdle’s disease. Hum Mol Genet 3:1005–1006
Martinuzzi A, Sartori E, Fanin M et al. (2003) Phenotype modulators in myophosphorylase deficiency. Ann Neurol 53:497–502
Hogrel JY, Laforêt P, Ben Yaou R et al. (2001) A non-ischemic forearm exercise test for the screening of patients with exercise intolerance. Neurology 56:1733–1738
Vissing J, Haller RG (2003) A diagnostic cycle test for McArdle‘s disease. Ann Neurol 54:539–542
Duboc D, Jehenson P, Tran Dinh S et al. (1987) Phosphorus NMR spectroscopy study of muscle enzyme deficiencies involving glycogenolysis and glycolysis. Neurology 37:663–671
Haller RG (2000) Treatment of McArdle disease. Arch Neurol 57:923–924
Andersen ST, Haller RG, Vissing J (2008) Effect of oral sucrose shortly before exercise on work capacity in McArdle disease. Arch Neurol 65:786–789
Vissing J, Haller RG (2003) The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. N Engl J Med 349:2503–2509
Andersen ST, Vissing J. (2008) Carbohydrate- and protein-rich diets in McArdle disease: effects on exercise capacity. J Neurol Neurosurg Psychiatry 79:1359–1363
Haller RG, Vissing J (2004) No spontaneous second wind in muscle phosphofructokinase deficiency. Neurology 62:82–86
Spiegel R, Gomez EA, Akman HO et al. (1009) Myopathic form of phosphoglycerate kinase (PGK) deficiency: a new case and pathogenic considerations. Neuromusc Dis 19:207–211
Stojkovic T, Vissing J, Petit F et al. (2009) Muscle glycogenosis due to phosphogluco-mutase 1 deficiency. N Engl J Med 361:425–427
Vissing J, Schmalbruch H, Haller RG, Clausen T (1999) Muscle phosphoglycerate mutase deficiency with tubular aggregates: effect of dantrolene. Ann Neurol 46:274–277
Kreuder J, Borkhardt A, Repp R et al. (1996) Inherited metabolic myopathy and hemolysis due to a mutation in aldolase A (brief report). N Engl J Med 334:1100–1104
Comi GP, Fortunato F, Lucchiari S et al. (2001) Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Ann Neurol 50:202–207
Kanno T, Maekawa M (1995) Lactate dehydrogenase M-subunit deficiencies: clinical features, metabolic background, and genetic heterogeneities. Muscle Nerve 3:S54-S60
Ploeg v d AT, Reuser AJJ (2008) Lysosomal storage disease 2. Pompe’s disease. Lancet 372:1342–1353
Hout v d HM, Hop W, Diggelen v OP et al. (2003) The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 112:332–340
Hagemans ML, Winkel LP, Doorn v PA et al. (2005) Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 128:671–677
Makos MM, McComb RD, Hart MN, Bennett DR (1987) Alphaglucosidase deficiency and basilar artery aneurysm: report of a sibship. Ann Neurol 22:629–633
Laforêt P, Petiot P, Nicolino M et al. (2008) Dilatative arteriopathy and basilar artery dolichoectasia complicating late-onset Pompe disease. Neurology 70:2063–2066
Fukuda T, Ewan L, Bauer M et al. (2006) Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol 59:700–708
Chamoles NA, Niizawa G, Blanco M, Gaggioli D, Casentini C (2004) Glycogen storage disease type II: enzymatic screening in dried blood spots on filter paper. Clin Chim Acta 347:97–102
Laforêt P, Nicolino M, Eymard B et al. (2000) Juvenile and adultonset acid maltase deficiency in France. Genotype-Phenotype correlation. Neurology 55:11222–11228
Kishnani P, Corzo D, Nicolino M et al. (2007) Recombinant human acid α-glucosidase: major clinical benefits in patients with infantile-onset Pompe disease. Neurology 68:99–109
Kishnani PS, Corzo D, Leslie ND et al. (2009) Early treatment with alglucosidase alfa prolongs long-term survival in infants with Pompe disease. Pediatr Res 66:329–335
Ploeg v d AT, Clemens PR, Corzo D et al. (2010) A randomized Study of Alglucosidase Alfa in Late-Onset Pompe’s Disease. New Engl J Med 362:1396–1406
Schorderet DF, Cottet S, Lobrinus JA et al. (2007) Retinopathy in Danon disease. Arch Ophthalmol 125:231–236
Danon MJ, Oh SJ, DiMauro S et al. (1981) Lysosomal glycogen storage disease with normal acid maltase. Neurology 31:51–57
Nishino I, Fu J, Tanji K et al. (2000) Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406:906–910
Dworzak F, Casazza F, Mora M et al. (1994) Lysosomal glycogen storage with normal acid maltase: a familial study with successful heart transplant. Neuromusc Disord 4:243–247
Moslemi AR, Lindberg C, Nilsson J et al. (2010) Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. N Engl J Med 362:1203–1209
Kollberg G, Tulinius M, Gilljam T et al. (2007) Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N Engl J Med 15:1507–1514
Laforêt P, Richard P, Ait Said M et al. (2006) A new mutation in PRKAG2 gene causing hypertrophic cardiomyopathy with conduction system disease and muscular glycogenosis. Neuromusc Disord 16:178–182
Gollob MH, Green MS, Tang AS et al. (2001) Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med 344:1823–1831
Cheung PC, Salt IP, Davies SP et al. (2000) Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J 346:659–669
Burwinkel B, Scott JW, Bührer C et al. (2005) Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the γ2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet 76:1034–1049
Benarroch EE (2010) Glycogen metabolism. Metabolic coupling between astrocytes and neurons Neurology 74:919–923
Vilchez D, Ros S, Cifuentes D et al. (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci 10:1407–1413
Robitaille Y, Carpenter S, Karpati G, DiMauro SD (1980) A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora’s disease and normal ageing. Brain 103:315–336
Cafferty MS, Lovelace RE, Hays AP et al. (1991) Polyglucosan body disease. Muscle Nerve 14:102–107
Savage G, Ray F, Halmagyi M, Blazely A, Harper C (2007) Stable neuropsychological deficits in adult polyglucosan body disease. J Clin Neurosci 14:473–477
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Laforêt, P., Weinstein, D.A., Peter A. Smit, G. (2012). The Glycogen Storage Diseases and Related Disorders. In: Saudubray, JM., van den Berghe, G., Walter, J.H. (eds) Inborn Metabolic Diseases. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-15720-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-642-15720-2_6
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-15719-6
Online ISBN: 978-3-642-15720-2
eBook Packages: MedicineMedicine (R0)