
Abstract
For assessment of mycorrhizal diversity of soils and for identification of fungal fruiting bodies, many techniques are used. The classical fungal identification starts with collection of field notes, and laboratory processing which involves the isolation of fungal cultures from soils, fruiting bodies, and spores by using direct or indirect plating methods for microbial screening. The wet-sieving and decanting technique for the extraction of spores of arbuscular mycorrhizal fungi (AMF) and microscopic identification are other important techniques.
While the work with specimens and cultures is described, it is worthy of note that direct application of molecular methods to environmental material can detect many more. Yet often different sets of fungal taxa are involved, as for example shown for the large basidiomycete diversity in agricultural soil. The advances and cost reduction in DNA-based identification of biological material has greatly improved the catalog of methods available to soil ecologists. The modern molecular method used in fungal identification of a fungal specimen or culture is to make nucleic acid available for amplification, done by the polymerase chain reaction (PCR). Commercialized and home-made “extraction-free” methods for many biological organisms are available; a simple method is PEX extraction. It involves the unique activity of liquid polyethylene glycol 200 (PEG200) in cell disruption and DNA stabilization. Other tools such as sequencing and editing sequences, cloning, database queries and alignment, where one can BLAST multiple sequences together and can also align query sequences with similar sequences, are available. Phylogenetic placement, a phenetics-based distance method, is also used to simplify fungal identification.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Arbuscular Mycorrhizal Fungus
- Fruiting Body
- Rose Bengal
- Microscopic Identification
- Fungal Identification
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
10.1 Introduction
Symbiotic mycorrhizal fungi play a pivotal role in biological interactions and biogeochemical cycles because the carbon they obtain from their photosynthetic plant hosts is allocated through the mycorrhizal mycelium to the soil ecosystem. In addition to these interactions with their host plant's roots, the mycelia also interact with a range of organic and inorganic substrates, as well as with different organisms such as bacteria, other fungi, soil micro- and mesofauna and the roots of secondary host plants (Finlay 2005).
Progress has been made in recent decades in the knowledge of root and mycorrhizae formation and turnover and its impacts on soil ecosystems; soil biota, exudations, secretions and soil aggregation phenomena; the biology of invasive species in soils; soil biodiversity, legacies and linkages to soil processes; and ecosystem functional responses (Coleman 2008).
The advances and cost reduction in DNA-based identification of biological material has been greatly improving the catalog of methods available to soil ecologists (Anderson and Cairney 2004). While we here describe the work with specimens and cultures, it is noteworthy that direct application of molecular methods to environmental material can detect many more, yet often different, sets of fungal taxa, as for example shown for the large basidiomycete diversity in agricultural soil (Lynch and Thorn 2006).
10.2 General Characteristics of Mycorrhizae
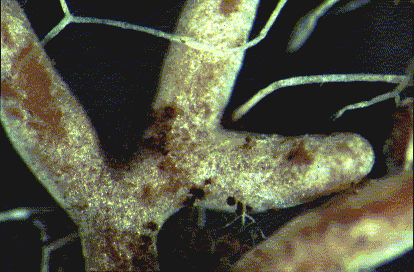
There can be no clear morphological, phylogenetic or ecological definition of soil fungi as these concepts are very difficult to implement since the geographically unbounded soil ecosystem harbors a diverse plethora of fungi with great morphological, genetic, and functional diversity. These fungi include yeasts and filamentous fungi, ascomycetes and basidiomycetes, ectomycorrhizal fungi (EMF) and arbuscular mycorrhizal fungi (AMF), anamorphic fungi and teleomorphic fungi. Mycorrhizal fungi (Fig. 10.1) have the ability to interact with the roots of more than 80% of land plants (Newman and Reddle 1987) and form symbiotic associations termed mycorrhizae. On the basis of the colonization pattern of host cells, two major types of mycorrhizae can be identified: ectomycorrhizae and arbuscular mycorrhizae. In the ectomycorrhiza the fungus does not penetrate the host cells, but forms a sheath around the roots and only traverses the cortical layers of the roots in the intercellular spaces, forming an interface called Hartig's Net. However, in arbuscular mycorrhiza the fungal hyphae penetrate cells and form intracellular structures such as coils or arbuscules. Mycorrhizal fungi provide improved access to limited soil sources such as phosphorus and nitrogen to the host plant. In exchange, mycorrhizal fungi receive carbon compounds from host plants to sustain their metabolism and complete their life cycle; they also receive protection from other microbes in the rhizosphere, and hence form a multipartite symbiotic interaction (Jeewon and Hyde 2007). A novel endophytic, root-interacting fungus, Piriformospora indica (Hymenomycetes, Basidiomycota) has been isolated and found to mimic the capabilities of a typical mycorrhizal fungus (Verma et al. 1998, Varma et al. 1999, 2001).

(a) Schematic presentation of mycorrhizosphere http://invam.caf.wvu.edu/collection/pubs/abstracts/mycorhiz.jpg (accessed 06 Sept. 2008). (b) Root hair and fungal association http://www2.warwick.ac.uk/fac/sci/whri/research/soilmicrobialdiversity/mycorrhizaldiversity_/paxillus_involutus_root.jpg (accessed 06 Sept. 2008)
10.3 Classical Fungal Processing and Identification
10.3.1 Field Notes, Processing, Fungal Identification
The field label of a specimen should include (a) field identification, (b) collector's name, (c) collection number and date, (d) detailed locality information, including coordinates and elevation (best by GPS), (e) a small description of the habitat, substratum and host. If possible, photographs should be taken on site, showing the habitat of the specimen, or on a neutral background to better show colors and details. A digital camera is of great help. Such photographic evidence can later help memorizing ephemeral characteristics required for identification and description.
The microscopic shape, size, color, manner of arrangement of spores on the fruiting bodies (sporophores), the hyphal characteristics of mycelium, as well as detailed information on fruiting bodies themselves, are the characteristics which would suggest themselves to someone somewhat experienced in the taxonomy of fungi, the class, order, family, and genus to which the particular fungus belongs. Microscopic characteristics are best examined under a compound microscope, perhaps adding various stains such as lactophenol cotton blue, toluidine blue, Giemsa stain, etc., and vital stain such as Janus green. In any case, these characteristics can be utilized to trace the fungus through published analytical, often dichotomous, keys to the genus, and perhaps finally to the species to which it belongs. Published color photograph guides and web-based keys and photographs can help further. Detailed descriptions of the known species are found in monographs of genera or in individual publications in research journals. The specimen is often kept moist for a few days to promote spore development for the production of spore print on paper or a glass slide, e.g., for help in microscopic identification or inclusion in the herbarium (see below). Alternatively, the fungus may be isolated as a culture from soil, root, sporophore or germinating spores (see below) and grown on artificial medium and identified on the basis of spores produced on the media. For some fungi, special nutrient media have been developed that allow selective growth only of the particular fungus.
The advent of molecular techniques, particularly the polymerase chain reaction (PCR), quick and inexpensive sequencing of DNA, and the accumulation of large databanks of DNA sequences have revolutionized fungal systematics and ecology (Agrios 2005).
10.3.2 Herbarium Facilities
Herbaria serve as conservatories for voucher specimens. Preservation of vouchers in publicly accessible herbaria is extremely important for further scientific studies (Agerer et al. 2000), not just because of the type requirements of nomenclature codes (McNeill et al. 2006). Herbarium specimens and their accompanying field notes document the existence of a fungus at a given place and time and provide the raw data from which taxonomic concepts are constructed. The examination of voucher specimens also provides a reliable way of verifying or correcting the identity of organisms recorded in cytological, ecological, populational, morphological, phylogenetic, and molecular studies. Scientific specimens are also a source of DNA and other compounds for phylogenetic, ecological, and other studies. Spore prints, if there are any, should be placed carefully in packets and stored with the specimens. They are also a source of DNA, identification of fungal characteristics, growing new cultures, etc.
For scientific studies specimens collected must be accompanied with informative, reproducible details:
-
A loose label and/or field tag with the collector's initial and collection number should be associated with the specimen at all times to facilitate their retrieval and identification.
-
Ideally, color photographs should be deposited with the specimen. In addition the collector should provide detailed notes on the appearance of the specimen when fresh, including pertinent information about the color (e.g., using a color guide, Kornerup and Wanscher 1978, http://www.bio.utk.edu/mycology/Color/color-intro.htm), stature, shape and general ecology.
-
Field notes should be written on archival-quality paper using permanent ink. Some collectors enter field data directly into a computer, in which case back-up files should be kept separately from the computer, and a hard copy should be printed as soon as possible. Copies generated using a laser printer and archival-quality paper is adequate for long-term preservation.
-
A specimen container (e.g., jeweler's box or folded paper, with or without attached herbarium sheet) should be large enough to accommodate fungal specimens of various shapes and sizes to provide appropriate protection. Unpacked specimens should never be mounted directly on herbarium sheets.
-
Specimens may originate from staff fieldwork or as gifts, exchanges, or loans. All incoming specimens, but especially those coming directly from the field, should be checked on arrival to determine if they are completely dry. Damp specimens must be dried thoroughly. Incoming specimens must also be disinfected (to kill insects) before they are stored in herbarium. Disinfestation can be achieved by deep freezing. Packages of specimens are wrapped in plastic bags and placed in a freezer at or below −20°C for 7 days. Bagging is essential for preventing condensation of moisture on a specimen during the process. Double plastic bags may be used to prevent air leakage. After freezing specimens should remain in their plastic bags until the whole package regains room temperature.
-
Taxonomic and sometimes other information about a specimen increases as the specimen is studied over time. Such information enhances the value of the specimen and should be recorded by the investigators on an annotation label provided by the herbarium. An annotation label should include: (a) the identification number of the specimen (i.e., collector and/or herbarium number) in case the label becomes separated from the specimen, (b) annotations for any scientific name newly applied to the organism, (c) authorship of the scientific name, (d) relevant literature citation(s), (e) the name of the annotator, (f) annotation date, and (g) other observations. Annotation labels should also be made of archival-quality paper and written with permanent ink. Policies regarding attachment of annotation labels vary among herbaria. It is recommended that investigators place annotation labels inside the packet, box, or mounting sheets with a paper clip or rubber band. A herbarium curator or technician is then notified of their existence and can attach them more securely, if necessary (Wu et al. 2004).
-
Phalloid, clathroid and other delicate fungi can be fixed and stored in liquid preservatives so that the forms and fragile textures of the fungi are not distorted. However, chemical fixation alters the DNA of an organism, so specimens fixed in that manner are not suitable for molecular studies. Consequently, warm-air drying is the preferred method of preserving fungal specimens. Freshly collected material is submerged in a fixative for 2–7 days and then transferred to storage solutions such as 70% ethanol, or they can be more complex mixtures of chemicals. Bridson and Forman (1992) recommended the Kew mixture and Copenhagen solution for fixation and storage, respectively. Both contain glycerol, which keeps the specimen from hardening. Collections stored in ethanol must be monitored closely to observe that fluid loss through evaporation can be replaced. Specimens are stored in glass jars or vials with neoprene caps or stoppers. Caps may be sealed further with sealing wax, Parafilm, and other materials to limit evaporation. Because chemical preservatives are harmful to human health, safety procedures must be followed when fixative and storage media are handled, both in the field and the laboratory. Investigators should wear gloves and safety goggles and should avoid breathing fumes by wearing masks or working in a chemical fume hood (Wu et al. 2004).
10.4 Isolation of Fungal Cultures from Soils, Mycorrhizosphere and Sporophores
10.4.1 Preparations
Before isolation, one must gather sterilized plastic items or pre-sterilize glassware, such as Petri dishes, test tubes and pipettes, by dry heat (150-160°C for 1 h or more), autoclaving, or dipping for one minute or more in 70–80% ethanol (Agrios 2005). There are many culture media available. The media have to be prepared in advance. General fungal media are malt extract agar, potato dextrose agar or Sabouraud dextrose agar. Selective agar media are designed to isolate specific groups of fungi such as cellulose agar to isolate microbes which are able to utilize long complex carbohydrates. Czapek yeast agar is used for the isolation and culture of saprobic soil microorganisms and Rose Bengal agar is used for the selective enumeration of yeast and molds. Rose Bengal is present in the media to restrict (not inhibit) the growth of Rhizopus and Mucor sp. that often overgrow culture plates. Some media are entirely synthetic, i.e., made up of known amounts of chemicals. Some are broth or semi-liquid apart from solid media (Pacioni 1992). Solutions of culture media are prepared in flasks, which are plugged and placed in an autoclave at 121°C at 15 psi pressure for 20 min. Sterilized media are allowed to cool until touchable (∼55°C) and are subsequently poured from the flask into sterilized Petri dishes, test tubes, or other appropriate containers. Pouring of the culture medium into the containers is carried out aseptically either in a separate culture room or in a clean room free from dust and contaminants. In either case the work table must be wiped with 70% ethanol, hands should be clean, and tools such as scalpels, forceps, and needles should be dipped in alcohol and flamed to prevent introduction of contaminating microorganism. Working in a laminar airflow hood greatly helps to grow the desired fungi free of airborne contaminants.
10.4.2 Isolation from Fruiting Bodies and Spores
Fungal cultures can be obtained by placing aseptically removed internal tissue of sporophores onto media. Polyspore (thus heterokaryotic) cultures can be derived from sporophore fragments glued onto Petri dish lids and sporulate onto the medium. Cutting out individual germinating spores onto separate dishes can yield monokaryotic single spore isolates. Sometimes the use of spore prints, or plating out sterilized water in which hymenophore (the spore-producing part of sporophores) fragments were vortexed, suffices to produce cultures.
10.4.3 Indirect Methods for Screening
Sporulating fungi are isolated by indirect techniques. The soil-dilution plate technique is the most common method (Fig. 10.2). In serial dilution methods, the spread plate technique is advantageous over the pour plate technique as the latter may eliminate some heat-sensitive fungi; also, some fungal spores do not germinate if submerged. Techniques such as serial root washings, soil washings, sedimentation and sieving are known for good recovery, as these techniques involve agitation in soil aggregates, and thus the spores can be easily released.

Serial dilution method: Calculation: Number of colonies on plate X reciprocal of dilution of sample = number of colony-forming units CFU/ml. For example: 32 colonies are on a plate of 1/10,000 dilution, then the count is 32 × 10,000 = 320,000/g in given soil sample. The colonies obtained in Petri plates can be further subcultured and identified
10.4.4 Soil Dilution Technique for Enumeration of Fungi
-
1.
To enumerate the total population of fungi, serial dilution and subsequent plating of the rhizosphere soil is done on the specified medium. This will estimate the number of viable fungal propagules present per gram of soil capable of growing in the specified medium, as shown in Fig. 10.2.
-
2.
Suspend 1 g of soil in 9 ml of sterile dilution blank made up of distilled water, saline, or phosphate buffer; this produces a 1:10 dilution.
-
3.
From the first dilution after proper mixing, take a further 1 ml and transfer to a fresh dilution blank, hence getting a 1:100 dilution. Continue the serial dilution until the desired dilution is obtained.
-
4.
Each suspension is shaken by hand for a few seconds and is drawn into a pipette and pour-plated or spread-plated (Fig. 10.3). 1 ml of the desired dilution is transferred to the Petri dish by micropipette or glass pipette. 15–20 ml medium is added into the Petri plate, cooled just above solidifying temperatures. The Petri dishes are swirled clockwise and anti-clockwise in order to disperse diluted soil sample in medium.
Fig. 10.3 
Schematic presentation showing pour plate method and direct plate method for isolation of fungi
-
5.
In case of spread plating for isolating surface-growing fungi 0.1–0.5 ml of the desired dilution is transferred onto the solidified agar plates. The suspension is spread over the surface of agar with help of a spreader or sterilized 2–3 mm diameter glass beads.
-
6.
Incubate at 24–30°C for 6–14 days. The colonies are counted on colony counter and average number of colonies per dish is multiplied by dilution factor to obtain the number of microorganisms per gram of the original soil sample (Mukerji et al. 1998).

10.4.5 Direct Plate Technique
For direct observation a soil profile is created in the rhizosphere zone and for other techniques, such as soil plate and soil dilution techniques, soils are collected at different depths by using a soil auger.
Direct methods are useful for quick scanning of soil fungi and are more appropriate for isolating fungi that do not sporulate and exist as mycelium attached to soil humus. Plants under study can also be uprooted carefully and either total rhizosphere soil or soil from different root zones is collected. Isolation is done by preparing a number of replicate series and after the required period of incubation. Other direct observation methods include immersion technique and soil box, with removable microscopic slides for zonal sampling at desired depth. A recent and more appropriate method to observe the rhizosphere effect is using a rhizotron, a root observation and sampling chamber (James et al. 1985).
To isolate specific fungal groups, sample pretreatment and the use of selective media have to be considered. Re-treatment involves heat shock by steaming and subsequent immersion in ethanol. Selective medium is added with enrichment compounds and certain growth retardants such as Rose Bengal and streptomycin, etc., inhibiting Actinobacteria, other bacteria, and fast-growing molds. These are used to curtail the interference and to facilitate the slow-growing fungi (Ranganayaki et al. 2006).
10.4.6 Wet–Sieving and Decanting Technique for the Extraction of Spores of AMF (Fig. 10.4)
-
1.
An optimal ratio of soil/water was found to be 1/10, that is 100 ml of soil in 1 l of water.
Fig. 10.4 
Steps in the wet-sieving technique: (a) metallic sieve, (b) decanting through a sieve series, (c) removal of debris
-
2.
The suspension should be stirred with a magnetic stirrer or by hand using a rod. Stirring time varies according to the nature of soil; usually 10 min is sufficient.
-
3.
To avoid foam that could retain debris, add an antifoam agent, for example Tween 80, 0.1–0.5%.
-
4.
Pass the suspension through the sieve with a 1 mm-wide mesh, keeping the filtrate.
-
5.
If the soil contains clay that yields a suspension blocking the sieve, precipitate the particles in 0.1 M sodium pyrophosphate.
-
6.
Debris retained by this sieve must be re-suspended into the kept suspension, stirred again, or washed under a stream of water, saving the suspension.
-
7.
Decant all suspensions through a sieve series ranging from 1 mm to 40 μm.
-
8.
With the aid of a jet of water directed at both sides of the sieve, the content of each sieve is transferred to a Petri dish.
-
9.
The shallow suspension can be observed with a stereo-dissecting microscope, and spores and microsporocarps picked up with a flattened needle or a Pasteur pipette.
-
10.
Small concave watch glasses or slides can be used for further observations.

{kind=link}
{kind=link}
Further modification of the filtering apparatus can be introduced particularly when working with either humic or clay soils and where the efficiency of the filter is lowered by the presence of foam or suspended particles. The removal of spores from metallic sieves can be difficult, particularly when working with soil where predominantly rare, single spores occur:
-
Nylon filters with standard meshes of the kind used in palynology or with cellular cultures for the separation of protoplasts can be used.
-
A series of filters, decreasing in pore size (1 mm–40 μm), are attached to each other by plastic tubing of 20 cm diameter, cut in lengths of 20 cm.
-
Once filled, the filters are placed onto a transparent Plexiglas grid-lined sheet and placed under a stereoscope; the spores are supported by the filter meshes where they can be counted accurately and manipulated easily. Using a transmitting light stereoscope, the transparency of the supports (nylon filter and Plexiglas sheet) allows easy observation of spores.
10.4.7 Techniques for Large Volumes of Soil
The techniques mentioned above are suitable for small-scale studies, involving up to 100 g of soil at a time. If kilogram quantities of soil are to be examined, a drum, for example a gasoline barrel without cover but with a lateral overflow pipe, can be utilized. The filtering apparatus is made up of a metallic sieve with meshes of 1 mm for retaining larger debris and a nylon filter bag held by strings or tapes. The size of the mesh is selected with the dimensions of the spores in mind:
-
1.
Place a plastic pipe or insert a fixed water source at the bottom of the drum.
-
2.
Place the filtering apparatus to catch the water suspension.
-
3.
Fill the drum, suspend the soil, stir with a rod, treat as necessary and then turn on the water, leaving the suspension to flow through the filtering apparatus.
10.5 Preservation and Maintenance
10.5.1 Culture Collections
Primary methods of culture preservation are continuous growth, drying, and freezing. Continuous growth methods, in which cultures are grown on agar, typically are used for short-term storage. Such cultures are stored at temperatures of 5–20°C, or they may be frozen to increase the interval between subculturing. The methods are simple and inexpensive because specialized equipment is not required. Freezing methods, including cryopreservation, are versatile and widely applicable. Most fungi can be preserved, with or without cryoprotectants, in liquid nitrogen or in standard home freezers. With freeze-drying, or lyophilization, the fungal cultures are frozen and subsequently dried under vacuum. The method is highly successful with cultures that produce mitospores. Freeze-drying and freezing below −135°C are excellent methods for permanent preservation, and we highly recommend them. However, both methods require specialized and expensive equipment. Permanent preservation is essential for strains with critically important characteristics and for type specimens. Cultures that are permanently preserved in metabolically inactive states can serve as type specimens (Nakasone et al. 2004).
In long-term preservation, sclerotization is an important method. Some fungi develop sclerotia or other long-term survival propagules in culture as well as in nature. Preserving such structures, usually at 3–5°C, is a good way of maintaining fungal strains. Two important methods are the oil overlay method or storage in distilled, sterilized water (Burdsall and Dorworth 1994; Nakasone et al. 2004). They are low-cost and low-maintenance methods for preserving cultures, either growing on agar slants and covered with mineral oil, or placed on cut-out pieces of solid medium in vials of sterile water. These cultures can be kept for several years or, in exceptional cases, up to 32 years at room temperature or 15−20°C. This method is especially appropriate for mycelial or nonsporulating cultures that are not amenable to freezing or freeze-drying, or when lacking suitable lab facilities (Nakasone et al. 2004).
Traditional methods for assessing fungal diversity in the soil environment rely mainly on the dilution-plating technique (coupled with use of selective media) and microscopy to identify sporulating fruiting bodies. The traditional methods tend to overestimate species that sporulate in soil, while those in mycelial state or those that have slow growth in culture are largely overlooked. In addition, most of these methods result in isolation of only the most common and abundant fungi (often referred to as generalists) such as the asexual ascomycetous molds Fusarium, Penicillium and Trichoderma, Zygomycota (especially Mucor), and the Oomycota (e.g., Pythium). These cultivated microorganisms are those that can utilize the energy source under the physical and chemical limitations of the growth medium. This clearly indicates that many other fungi do not respond readily to cultural techniques. Therefore the diversity data cannot be considered as accurate. Altered and optimized growth media, coupled with advanced molecular biology techniques, have demonstrated that a larger proportion of uncultured fungi belonging to novel fungal lineages could be isolated and identified (Jeewon and Hyde 2007).
Public culture collections are repositories of often published or of economically critical fungi. Some of the larger culture collections are ATCC (http://www.atcc.org), CBS (http://www.cbs.knaw.nl), BCCM-MUCL http://bccm.belspo.be), DSMZ (http://www.dsmz.de/), INVAM (http://invam.caf.wvu.edu), and IFO (http://www.nbrc.nite.go.jp).
10.5.2 Cultures in Herbaria
The choice of preservation methods depends on the species of concern, the resources available, and the goal of the project. Therefore it can be a long-term or short-term preservation. Fungal cultures can be preserved as voucher specimens by drying. The mature fungal colony, in its Petri plate, is placed in a frost-free freezer. The freezer must be frost-free or the preparation will not dry properly. Data are written on the plate bottom. After 4–6 weeks the colony should be sufficiently dry so that it is loose in the plate. The colony is then transferred either to an archival-paper envelope or to a thin (e.g., 8 mm) Petri plate and placed in a herbarium packet, and a label with all accompanying data is prepared. The old plate and lid are discarded. The paper envelopes can be used for most pyrenomycetes, many discomycetes and all loculomycetes. Delicate taxa such as zygomycetes, hyphomyctes, coelomycetes and some ascomycetes are best stored in thin Petri plates. The material is then placed in a 100% cotton rag packet to which the label is attached with acid-free glue (Wu et al. 2004). Drying is the most useful method of preservation for cultures that produce spores or other resting structures.
10.6 Modern Molecular Methods Used in Fungal Identification
The initial task towards molecular identification of a fungal specimen or culture is to make nucleic acid available for amplification by the PCR (Saiki et al. 1985). While there is a plethora of modifications and personal preferences, all at some step must include the leaking of nucleic acid into a buffer that preserves it but allows further purification to remove it from PCR-interfering cell wall components and compounds. A workflow of steps possible in molecular identification is given with Figs. 10.5–10.8.

Workflow of bioinformatic processes in identifying an unknown fungal object (process step query DNA sequence) Part 1. Process step a: example Xerocomus badius (http://www.mtsn.tn.it/bresadola/gallery.asp?code = 31) specimens, or b: Xerocomus badius mycorrhiza on Pinus roots (http://www.ektomykorrhiza.de/mycorh1.gif) serve in the production of a c: culture on malt extract agar. The biological material is extracted, resulting DNA cleaned, amplified, and sequenced, yielding d: electropherogram (e.g., FinchTV, Geospiza, Inc, Seattle, WA, USA) — note the potential need to edit the data, as e.g., there are “dye blobs” that are introduced by the sequence cleaning method with ethanol precipitation
{kind=link}



Workflow of bioinformatic processes in identifying an unknown fungal object (process step query DNA sequence) Part 2. Process Step e: graphical result of BLAST-n search on the website of the DNA Databank of Japan (DDBJ), allowing query and anchoring sequences to be sent to ClustalW for alignment. Process step f: the program QAlign 2 (Panta Rhei) aligns sequences and can also compute simple phylogenetic trees, here a jackknife resampling, T-Coffee alignment with associated Kimura 2-parameter Neighbour-Joining tree. g: graphical representation of highly similar (top lines) and dissimilar (bottom lines) areas of alignment in POAVIZ (Grasso et al 2003)



Workflow of bioinformatic processes in identifying an unknown fungal object (process step query DNA sequence) Part 3. Process step h: visualization of the FASTA formatted alignment as a PDF output from SeaView (Galtier et al 1996). Process step i: using the software Seqverter (GeneStudio, Inc., Suwanee, GA, USA) data can be converted, e.g., from FASTA to the NEXUS format j as for example used in PAUP*, Mesquite (Maddison & Maddison 2007a), MacClade (Maddison & Maddison 2007b), or MrBayes



(continued)
While for some time the direct amplification from bacteria, especially Escherichia coli but also others, such as Actinobacteria (e.g., Ishikawa et al. 2000) has been routine in molecular biology labs, commercialized and home-made “extraction-free” methods for other biological organisms have become available and can be inquired with your local sales representative. To ease the identification of soil fungi, we suggest starting with such a simple method here. It involves the unique activity of liquid polyethylene glycol 200 (PEG200) in cell disruption and DNA stabilization (Chomczynski and Rymaszewski 2006). In order to allow adjustment to the equipment available, we are intentionally vague in the protocol given below.
10.6.1 “Extraction-Free” Preparation of PCR-Ready Material
-
1.
Prepare 2 ml screw-cap microtubes by adding 300 μl PEG200-KOH (adjusted with KOH to pH 13.3–13.5 and autoclaved) and a sterile glass bead of 2–3 mm diameter.
-
2.
Take a loop full of yeast culture or a few mm3 of fungal fruiting body or a small amount of fungal aerial hyphae from a Petri dish and mix into the PEG200. Freeze if not used right away.
-
3.
Before going to PCR, vortex (the glass bead here supports homogenization) and spin down.
The use of sterile sand, a commercial grinding resin, or a microtube pestle to help disrupt the material is optional, e.g., in place of the glass bead.
10.6.2 PEX Extraction
For tough fungal herbarium specimens (Krüger 2002) or from mycorrhizal root tips (Martin and Rygiewicz 2005) the xanthogenate (PEX) protocol is a useful and time-saving method. This can be implemented as an alternative to the above PEG200 protocol.
Buffers needed:
-
(a)
100 ml TEx buffer stock:
-
10 mM Tris-HCl (starting from 0.5 ml 2 M Tris-HCl pH 7.4)
-
1 mM EDTA (starting from 0.2 ml 0.5 M EDTA pH 8.0)
-
0.5 M CaCl2 (5.5 g)
-
Autoclave
-
-
(b)
100 ml XT buffer stock:
-
100 mM Tris-HCl (starting from 5 ml 2 M Tris-HCl pH 7.4)
-
20 mM EDTA (starting from 4 ml of 0.5 M pH 8.0)
-
5 ml Tween 20
-
800 mM sodium acetate (starting from 6.16 g or 16 ml 5 M stock)
-
Autoclave
-
-
(c)
0.1X TE pH 8.0:
-
=1 mM Tris-HCl, 0.1 mM EDTA, autoclaved.
-
Prior to extraction, for each sample dissolve 7.5 mg potassium ethyl xanthogenate (O-ethyl xanthic acid, PEX) in 0.75 ml XT buffer. Possibly heat it slightly. Do not inhale vapours:
-
1.
Place the fungal material, some sterile sand, and 50 μl phosphate buffered saline–Tween 20 (10%) or 1% sodium pyrophosphate with 10% Tween 20 in a 1.5 ml microtube. Mesh the material with a sterile microtube pestle. Add 50 μl TEx. Prepare a heating block to 70°C.
-
2.
Add 750 μl XT buffer with added PEX and vortex vigorously.
-
3.
Incubate 60 min at 70°C, shaking occasionally.
-
4.
Incubate 30 min on ice.
-
5.
Centrifuge 10 min at full speed.
-
6.
Mix the supernatant in a new microtube with 80% cold isopropyl alcohol. Potentially use a coloured co-precipitant from a commercial vendor – it will improve visibility of small pellets as a precaution against loss. Prepare heating block to 90°C.
-
7.
Centrifuge 10 min at ca. 10,000 rpm. Remove the alcohol.
-
8.
Wash the pellet with 100 μl cold ethanol (100%), scraping the tube on a rack to allow the alcohol to reach all sides.
-
9.
Centrifuge 10 min at full speed.
-
10.
Pipette off alcohol, then incubate 1 min at 90°C. Air dry the pellet.
-
11.
Dissolve the pellet in 50 μl 0.1X TE, placing it 1 min at 90°C.
-
12.
Add 0.5 μl RNAse A and spin down. Store at −20°C.
If PCR is inhibited by too much pigment or carbohydrates, clean/re-extract further with additional chloroform-isoamyl alcohol extraction, a commercial kit, polyvinylpyrrolidone (Berthelet et al. 1996, Young et al. 1993), polyethylene glycol 8,000 (Howeler et al. 2003), glassmilk and guanidine (Saltikov and Olson 2002), or hydroxyapatite (Purdy et al. 1996).
10.6.3 Choice of PCR Target
Because the fungal ITS rDNA barcoding region is the most commonly used genetic marker deposited in databases, usually exhibiting high variability descriptive at the species level, this is the target of choice. In general, the highly repetitive nature of the ribosomal DNA genes (Fig. 10.9) by itself makes them easier amplification loci, offering a higher amount of template than single-copy genes. However, it is possible that not all loci are created equal, and also noteworthy is that a single gene may not accurately represent the phylogenetic background of the organism (Rokas and Carroll 2005). Using ribosomal DNA genes complicates this matter because it codes for RNA and not protein, and thus the standard models of sequence evolution may not be adequate (Gesell and von Haeseler 2006). However, for molecular identification and measuring diversity, precise branching order in phylogenetic trees is not troublesome, but rather of interest to hardcore molecular systematists. Cloning PCR products even from heterogeneous copies of rDNA from pure cultures or specimens is no more a necessity than if amplifying from polygenomic extractions. PCR errors and natural sequence heterogeneity within an organism will play no major role in further analyses explained below.

Scheme of the tandemly arrayed ribosomal RNA gene cluster (rDNA). IGS Intergenic Spacer; ETS External Transcribed Spacer; ITS Internal Transcribed Spacer (ITS1, ITS2). 18S rDNA is also called the SSU (small subunit) gene, 28S rDNA the LSU (large subunit) gene. Note the unfortunate naming of standard primers, e.g., ITS5 and ITS4 (yes, there are primers ITS1 and ITS2 as well)
For the PCR amplification we recommend using a ready mastermix that already contains the nucleotides, Taq polymerase, and loading dye for subsequent electrophoresis. More and more companies provide such solutions which help decrease cost, workload, and contamination potential. With one such mastermix, we use the following standard protocol with primers ITS5 and ITS4 (White et al. 1990) (see Table 10.1). Ideally, some sterile environment and gamma-irradiated reaction tubes will also decrease chances of contamination.
For ITS5 (GGAAGTAAAAGTCGTAACAAGG) and ITS4 (TCCTCCGCTT ATTGATATGC) we use the following cycling conditions: initial denaturation 94°C/10 min, 32 cycles of denaturation 94°C/40 s, primer-annealing 54°C/30 s, extension 72°C/40 s, followed by final extension 72°C/4 min, and a hold at 10°C. Generally, the annealing temperature is to be about 5°C below the melting temperature of the lower one of a primer pair. The melting temperature is given by the manufacturer of the primers, or can be calculated at their website. Other primer information is available from, e.g., http://www.biology.duke.edu/fungi/mycolab/primers.htm.
10.6.4 PCR Troubleshoot
There are many variants of PCR, but for extractions from pure cultures it should normally be possible to use standard settings. Sometimes it is necessary to dilute the samples, increase primer concentrations, or to optimize the program. Of good use is a temperature-gradient thermocycler to judge susceptibility to annealing temperature variation. PCR additives can facilitate the success of PCR, yet this needs to be optimized (Rådström et al. 2004). Besides choosing a commercial, proprietary product, freely available enhancer compounds include
-
0.2–0.6 M trehalose (Spiess et al. 2004)
-
The alcohols glycerol at 5–20% (Nagai et al. 1998) or polyethylene glycol (PEG) of various molecular weights at 0.75–15% (Giordano et al. 2001; Rådström et al. 2004; Lareu et al. 2007)
-
Polyamines such as 0.1–2 M spermidine (Wan and Wilkins 1993; Ahokas and Erkkila 1993)
-
Amides such as 0.4 M 2 pyrrolidone (Chakrabarti and Schutt 2001a)
-
Methylammonia such as 1 M monohydrate of betaine (carboxymethyl trimethylammonium) (Baskaran et al. 1996; Rees et al. 1993), tetramethylammonium chloride (TMAC, Chevet et al. 1995), tetramethylammonium oxalate (Kovárová and Dráber 2000)
-
0.5% non-ionic detergents Triton X-100, Tween 20 or Nonidet P-40 (Demeke and Adams 1992)
-
Solvents such as 1–5% formamide (Sarkar et al. 1990), 0.15–0.4% tetramethylene sulfone (Chakrabarti and Schutt 2001b), 2–10% dimethyl sulfoxide (DMSO, Bookstein et al. 1990)
-
Proteins 0.1-0.8 μg μl−1 BSA or T4 gp32 (Kreader et al. 1996), 0.01–1% gelatin (Ohler and Rose 1992)
-
10–20 mM ammonium sulfate
10.6.5 Sequencing and Editing Sequences
In the modern molecular biology lab, the dye-dideoxy variant of the enzymatic method by Sanger (Sanger and Coulson 1975) is the standard. Here, a special PCR reaction containing only one primer synthesizes the DNA from the template, the prior PCR product. A certain number of deoxy nucleotides are modified to interrupt chain elongation upon being randomly incorporated in the growing DNA strands. These also carry different fluorescence labels for each of the four different bases. Hence, all lengths are produced, and the final nucleotide will be known when the cycle-sequencing product is electrophoresed on the sequencer. There, it is separated on either a polyacrylamide gel or a capillary. It is necessary to clean the PCR product prior to cycle sequencing, and after. The former can be achieved by enzymatic means (exonuclease/shrimp alkaline phosphatase), or centrifugation through commercially available columns. After cycle sequencing, one uses columns, alcohol precipitation (perhaps modified with magnetic beads or coprecipitants), or centrifugation through self-prepared Sephadex columns.
Sequence output files (electropherograms) can be visualized and edited in a variety of free software programs, such as Chromas Lite (Technelysium Pty Ltd, Australia), Staden Package trev.exe (https://sourceforge.net/projects/staden), Ridom TraceEdit (Ridom GmbH, Würzburg, Germany), FinchTV (Geospiza, Inc., Seattle, WA, USA), BioEdit (Hall 1999; Tippmann 2004) or one of the commercial products. It is necessary to decide on nucleotide composition where there are artefacts such as “dye blobs” caused by alcohol precipitation. Unreliable ends need to be trimmed off. In addition, it might be necessary to repeat the sequencing, sequence with another primer, or clone the PCR products if they appear to be mixed, rather then having occasional point errors or single nucleotide polymorphisms (SNPs). DNA analysis software such as used for phylogenetics, as well as BLAST-n (Altschul et al. 1997) can deal with the ambiguity codes for nucleotides (see Table 10.2, Cornish-Bowden 1985).
10.6.6 Cloning
If no good sequence can be obtained, e.g., because the specimen or culture is impure or the ribosomal DNA is heterogeneous, cloning is necessary. It is simplest to use a T/A or U/A cloning kit (Mead et al. 1991) where the PCR products, which should have A overhangs due to the peculiarity of Taq polymerase, are ligated into a special cloning vector for the bacterium E. coli. In that case, one can then directly add a little pipette tip full of E. coli single colony material into a PCR mix such as above (barely touch individual colony on plate!), but using primers anchored in the vector, such as recommended by the cloning kit manufacturer. The sequencing of the colony PCR product also has the advantage of getting a clean sequence from the start, as the otherwise usually “messy” start and the region where dye blobs would appear are in the vector sequence area. It is necessary to find out in what region of the DNA sequence is the vector sequence, which can be done with the aid of commercial tools, as well as by special BLAST (e.g., EMVEC database query, http://www.ebi.ac.uk/blastall/vectors.html).
The edited DNA data must also be oriented in the correct way, which can be gauged by BLAST search. Sequences can then be “reverse complemented.” Such manipulations can be done within the program BioEdit or with the BCM Search Launcher (http://searchlauncher.bcm.tmc.edu).
10.6.7 Database Queries and Alignment
A good way to BLAST is to use the site of the Japanese data repository DDBJ (http://www.ddbj.nig.ac.jp/). Here, it is possible to BLAST multiple sequences together, and also to align query sequences with similar sequences. BLAST outputs contain two numerical values, one score correlating with similarity of query and database entry, and the other being the E-value that reflects the probability of finding matches with equal scores. Hence, one looks for high scores and low E-values. More information can be found in Korf et al. (2003). The data maintained at DDBJ are synchronized with the American NCBI GenBank and the European EMBL database. Alignment can be performed with the ClustalW algorithm (Thompson et al. 1994), which in DDBJ can also be used to obtain sequences for anchoring and new sequences for rooting the tree with an outgroup. ClustalW is also available within BioEdit, or comes with its own graphical interface that can even produce simple phylogenetic trees (Thompson et al. 1997). There are other, alternative alignment programs such as Dialign-T (Morgenstern 2004, Subramanian et al. 2005), Divide-and-Conquer (Stoye et al. 1997), Align-m (Van walle et al. 2004), MAVID (Bray and Pachter 2004), MUSCLE (Edgar 2004), POA (Lee et al. 2002), MAFFT (Katoh et al. 2005), multalign (Corpet 1988), T-Coffee (Notredame et al. 2000), or combinations in the program QAlign (Sammeth et al. 2003, 2006). A variety of these alignment programs is available for online use at EMBL (http://www.ebi.ac.uk/Tools/sequence.html). Local databases of sequence data can be well-managed with the programs ARB (Ludwig et al. 2004), BioEdit, GeneDoc (Nicholas et al. 1997), or the sequence editor of MEGA (Tamura et al. 2007).
It is also possible to align sequences to the specialized database mor (Hibbett et al. 2005), which contains the curated data of a large fungal phylogenetic project (AFTOL, Lutzoni et al. 2004), or to use seeded alignments against the SILVA database, a progression of ARB and the European Small Subunit Ribosomal RNA database (http://www.arb-silva.de/ Pruesse et al. 2007). A specialized database for ectomycorrhizal fungal sequences exists in UNITE (http://www.unite.ut.ee). The molecular approach to systematics has even led people to propose new classifications entirely based on cladistic principles (PhyloCode, Donoghue and Gauthier 2004).
Alignment, which also decides on positional homology, is a prerequisite for phylogenetic analysis showing the putative evolutionary relationship between the culture or specimen to be identified and database comparison sequences. For the ITS spacers, alignment can be very difficult beyond closely related taxa. Thus, for uncertain homology, stretches of sequence should be excluded in the phylogenetic analysis, although of course the complete data without potential vector sequence would be deposited in the public repositories upon publication. SequIn (http://www.ncbi.nlm.nih.gov/Sequin/) at NCBI GenBank can be used when publishing own sequences. Secondary structure of the ribosomal RNA transcript can sometimes help in alignment, yet when recoding data for structure features information content is lost (Krüger and Gargas 2004).
10.6.8 Phylogenetic Placement
To simply identify fungi, a phenetics-based distance method such as neighbor-joining NJ (Kimura 1980) or minimal-evolution ME (Rzhetsky and Nei 1993) suffices. These are implemented in the package PHYLIP (Felsenstein 2005), or in MEGA, and rudimentary in ARB, ClustalX and QAlign. Some of these programs allow specification of a model of DNA sequence evolution. For simple identification, this may not be so important. Otherwise, the use of the popular program PAUP* (Swofford 2002) together with modeltest (Posada and Crandall 1998) is needed to decide on the model of sequence evolution. More sophisticated phylogenetic analysis based on different search strategies, e.g., on the principles of maximum-parsimony MP (available for example in MEGA and PAUP*), maximum-likelihood ML (programs PUZZLE, PHYML, or Treefinder; Schmidt et al. 2002, Guindon and Gascuel 2003, Jobb et al. 2004 respectively), or Bayesian inference (program MrBayes, Ronquist and Huelsenbeck 2003), is often not needed. One is advised to read Hall (2004) for instructions. Conflicting signals in phylogenetic data can be visualized as reticulogram (e.g., Makarenkov 2001, Huson and Bryant 2006). For likelihood approaches, including Bayesian, more computing power than for NJ is advised. Commonly, a random resampling/reanalysis strategy is used to gauge consistency of a dataset and the support for the branching order. The two most common forms of frequentist support of data-to-tree topology are the jackknife and the bootstrap over characters (Krüger and Gargas 2006). For example, a clade on a phylogenetic tree with 67% bootstrap support was found in 67 of 100 resampled datasets. Branch lengths in phylograms can variably depict the distance, likelihood, or steps of mutations (the latter in parsimony). A consensus tree usually collapses all branches with less than 50% support, though perhaps 90% support is considered as significant in the relationship of the branches. The root of the tree is usually a sequence or group of sequences a priori known to be closely related to yet outside the sequences of interest. The programs TreeView (Page 1996), PhyloDraw (Choi et al. 2000) and Dendroscope (Huson et al. 2007) are three of many for visualization of phylogenetic trees. Because file types are not always compatible, a conversion tool such as ForCon (Raes and Van de Peer 1999) is useful. A website that contains several programs to cover various steps in phylogenetic analysis is in development in France (http://www.phylogeny.fr/) and hopefully will be further expanded.
10.7 Conclusions
Mycorrhizal fungi have the ability to interact with roots and form symbiotic associations termed mycorrhizae. For fungal identification, field notes, drawings, and photographs are essential tools. A duplicate set of field notes should be kept separately although original field notes should be placed with the specimen and, if available, spore print, both deposited in a publicly accessible herbarium. There are several traditional methods such as the isolation of fungi from soil samples by serial dilution, culture plating, microscopic identifications, etc. which form the preliminary part of fungal identification. Cultures should be kept at a publicly accessible culture collection. A variety of extraction methods for fungal material yields the DNA template identification by sequencing, subsequent database comparison, alignment, and phylogenetic placement of the fruiting bodies and cultures.
References
Agerer R, Ammirati J, Blanz P, Courtecuisse R, Desjardin ED, Gams W, Hallenberg N, Halling R, Hawksworth LD, Horak E, Korf K, Mueller MG, Oberwinkler F, Rambold G, Summerbell CR, Triebel D, Watling R (2000) Open letter to the scientific community of Mycologists. Can J Bot 78:981–983
Agrios NG (2005) Plant Pathology, 5th edn. Academic Press, New York
Ahokas H, Erkkila MJ (1993) Interference of PCR amplification by the polyamines, spermine and spermidine. PCR Methods Appl 3:65–68
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Anderson IC, Cairney JW (2004) Diversity and ecology of soil fungal communities: increased understanding through the application of molecular techniques. Environ Microbiol 6:769–779
Baskaran N, Kandpal RP, Bhargava AK, Glynn MW, Bale A, Weissman SM (1996) Uniform amplification of a mixture of deoxyribonucleic acids with varying GC content. Genome Res 6:633–638
Berthelet M, Whyte LG, Greer CW (1996) Rapid, direct extraction of DNA from soils for PCR analysis using polyvinylpolypyrrolidone spin columns. FEMS Microbiol Lett 138:17–22
Bookstein R, Lai CC, To H, Lee WH (1990) PCR-based detection of a polymorphic BamHI site in intron 1 of the human retinoblastoma (RB) gene. Nucleic Acids Res 18:1666
Bray N, Pachter L (2004) MAVID: Constrained ancestral alignment of multiple sequences. Genome Res 14:693–699
Bridson DM, Forman L (1992) The Herbarium Handbook, 2nd edn. Royal Botanic Gardens, Kew
Burdsall HH Jr, Dorworth EB (1994) Preserving cultures of wood-decaying Basidiomycotina using sterile distilled water in cryovials. Mycologia 86:275–280
Candrian U, Furrer B, Höfelein C, Lüthy J (1991) Use of inosine-containing oligonucleotide primers for enzymatic amplification of different alleles of the gene coding for heat-stable toxin type I of enterotoxigenic Escherichia coli. Appl Environ Microbiol 57:955–961
Chakrabarti R, Schutt CE (2001a) The enhancement of PCR amplification by low molecular weight amides. Nucleic Acids Res 29:2377–2381
Chakrabarti R, Schutt CE (2001b) The enhancement of PCR amplification by low molecular weight sulfones. Gene 274:293–298
Chevenet F, Brun C, Bañuls AL, Jacq B, Christen R (2006) TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics 7:439
Chevet E, Lemaftre G, Katinka MD (1995) Low concentrations of tetramethylammonium chloride increase yield and specificity of PCR. Nucleic Acids Res 23:3343–3344
Choi JH, Jung HY, Kim HS, Cho HG (2000) PhyloDraw: a phylogenetic tree drawing system. Bioinformatics 16:1056–1058
Chomczynski P, Rymaszewski M (2006) Alkaline PEG-based method for direct PCR from Bacteria, Eukaryotic tissue samples, and whole blood. Biotechniques 40:454–458
Coleman DC (2008) From peds to paradoxes: linkages between soil biota and their influences on ecological processes. Soil Biol Biochem 40:271–289
Cornish-Bowden A (1985) Nomenclature for incompletely specified bases in nucleic acid sequences: recommendations 1984. Nucleic Acids Res 13:3021–3030
Corpet F (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890
Demeke T, Adams RP (1992) The effects of plant polysaccharides and buffer additives on PCR. Biotechniques 12:332–334
Donoghue MJ, Gauthier JA (2004) Implementing the PhyloCode. Trends Ecol Evol 19:281–282
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113
Felsenstein J (2005) PHYLIP (phylogeny inference package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle
Finlay DR (2005) Mycorrhizal symbiosis: myths, misconceptions, new perspectives and further research priorities. Mycologist 19:90–95
Galtier N, Gouy M, Gautier C (1996) SeaView and Phylo_win, two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci 12:543–548
Gesell T, von Haeseler A (2006) In silico sequence evolution with site-specific interactions along phylogenetic trees. Bioinformatics 22:716–722
Giordano BC, Ferrance J, Swedberg S, Hühmer AF, Landers JP (2001) Polymerase chain reaction in polymeric microchips: DNA amplification in less than 240 seconds. Anal Biochem 291:124–132
Grasso C, Quist M, Ke K, Lee C (2003) POAVIZ: a partial order multiple sequence alignment visualizer. Bioinformatics 19:1446–1448
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hall B (2004) Phylogenetic trees made easy: a how-to manual, 2nd edn. Sinauer Associates, Sunderland, MA
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hibbett DS, Nilsson RH, Snyder M, Fonseca M, Costanzo J, Shonfeld M (2005) Automated phylogenetic taxonomy: an example in the Homobasidiomycetes (mushroom-forming Fungi). Syst Biol 54:660–668
Howeler M, Ghiorse WC, Walker LP (2003) A quantitative analysis of DNA extraction and purification from compost. J Microbiol Meth 54:37–45
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R (2007) Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics 8:460
Ishikawa J, Tsuchizaki N, Yoshida M, Ishiyama D, Hotta K (2000) Colony PCR for detection of specific DNA sequences in Actinomycetes. Actinomycetologica 14:1–5
James RB, Bartlett RJ, Amadon JF (1985) A root observation and sampling chamber (rhizotron) for pot studies. Plant Soil 85:291–293
Jeewon R, Hyde KD (2007) Detection and diversity of fungi from environmental samples: traditional versus molecular approaches. In: Varma A, Oelmüller R (eds) Advanced Techniques in Soil Microbiology. Springer, Heidelberg, pp 1–3
Jobb G, von Haeseler A, Strimmer K (2004) TREEFINDER: A powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol 4:18
Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy multiple sequence alignment. Nucleic Acids Res 33:511–518
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Korf I, Yandell M, Joseph Bedell J (2003) BLAST. Basic local alignment search tool. O'Reilly Media, Sebastopol, CA
Kornerup A,Wanscher JH (1978) Handbook of colour, 3rd revised edn. Eyre Methuen, London
Kovárová M, Dráber P (2000) New specificity and yield enhancer of polymerase chain reactions. Nucleic Acids Res 28:E70–E70
Kreader CA (1996) Relief of amplification inhibition of PCR with bovine serum albumin or T4 gene 32 protein. Appl Environ Microbiol 62:1102–1106
Krüger D (2002) Monographic studies in the genus Polyporus (Basidiomycotina) [doctoral thesis]. University of Tennessee, Knoxville. Available from: http://idserver.utk.edu/?id = 200300000001704
Krüger D, Gargas A (2004) The basidiomycete genus Polyporus — an emendation based on phylogeny and putative secondary structure of ribosomal RNA molecules. Feddes Repertorium 115:530–546
Krüger D, Gargas A (2006) New measures of topological stability in phylogenetic trees — Taking taxon composition into account. Bioinformation 1:327–330
Lareu RR, Harve KS, Raghunath M (2007) Emulating a crowded intracellular environment in vitro dramatically improves RT-PCR performance. Biochem Biophys Res Commun 363:171–177
Lee C, Grasso C, Sharlow MF (2002) Multiple sequence alignment using partial order graphs. Bioinformatics 18:452–464
Ludwig W et al (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371
Lutzoni F et al (2004) Assembling the fungal tree of life: progress, classification, and evolution of subcellular traits. Am J Bot 91:1446–1480
Lynch MD, Thorn RG (2006) Diversity of basidiomycetes in Michigan agricultural soils. Appl Environ Microbiol 72:7050–7056
Maddison WP, Maddison DR (2007a) Mesquite: a modular system for evolutionary analysis. Version 2.01. Available from: http://mesquiteproject.org/
Maddison WP, Maddison DR (2007b) MacClade 4. Sinauer Associates, MA
Makarenkov V (2001) T-REX: reconstructing and visualizing phylogenetic trees and reticulation networks. Bioinformatics 17:664–668
Martin KJ, Rygiewicz PT (2005) Fungal-specific primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiology 5:28
McNeill J, Barrie FR, Burdet HM, Demoulin V, Hawksworth DL, Marhold K, Nicolson DH, Prado J, Silva PC, Skog JE, Wiersema JH, Turland NJ (2006) International code of botanical nomenclature (Vienna code). Regnum Vegetabile 146. A.R.G. Gantner Verlag, Ruggell, Liechtenstein
Mead DA, Pey NK, Herrnstadt C, Marcil RA, Smith LM (1991) A universal method for the direct cloning of PCR amplified nucleic acid. Biotechnology 9:657–663
Morgenstern B (2004) DIALIGN: multiple DNA and protein sequence alignment at BiBiServ. Nucleic Acids Res 32:W33–W36
Mukerji KG, Mandeep, Varma A (1998) Mycorrhizosphere microorganisms: screening and evaluation. In: Varma A (ed) Mycorrhiza Manual. Springer, Heidelberg, pp 85–96
Nagai M, Yoshida A, Sato N (1998) Additive effects of bovine serum albumin, dithiothreitol, and glycerol on PCR. Biochem Mol Biol Int 44:157–163
Nakasone KK, Peterson WS, Jong C-S (2004) Preservation and distribution of fungal cultures. In: Mueller GM, Bills FG, Foster SM (eds) Biodiversity of fungi: inventory and monitoring methods. Elsevier, Amsterdam, pp 37–47
Newman EI, Reddel P (1987) The distribution of mycorrhizas among families of vascular plants. New Phytol 106:745–751
Nicholas KB, Nicholas HB Jr, Deerfield DW II (1997) GeneDoc: analysis and visualization of genetic variation. EMBNET.news 4:1–4
Notredame C, Higgins D, Heringa J (2000) T-Coffee: a novel method for multiple sequence alignments. J Mol Biol 302:205–217
Ohler LD, Rose EA (1992) Optimization of long-distance PCR using a transposon-based model system. PCR Methods Appl 2:51–59
Pacioni G (1992) Wet-Sieving and decanting techniques for the extraction of spores of vesicular–arbuscular fungi. In: Norris JR, Read DJ, Varma AK (eds) Methods in Microbiology. Springer, Heidelberg, pp 317–322
Page RD (1996) TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 124:357–358
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Pruesse E, Quast C, Knittel K, Fuchs B, Ludwig W, Peplies J, Glöckner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucl Acids Res 35:7188–7196
Purdy KJ, Embley TM, Takii S, Nedwell DB (1996) Rapid extraction of DNA and rRNA from sediments by a novel hydroxyapatite spin-column method. Appl Environ Microbiol 62:3905–3907
Rådström P, Knutsson R, Wolffs P, Lövenklev M, Löfström C (2004) Pre-PCR processing: strategies to generate PCR-compatible samples. Mol Biotechnol 26:133–146
Raes J, Van de Peer Y (1999) ForCon: a software tool for the conversion of sequence alignments. EMBnet.news 6. Distributed for free at: http://bioinformatics.psb.ugent.be/webtools/ForCon/
Ranganayaki N, Tilak VBRK, Manoharachary C, Mukerji GK (2006) Methods and techniques for isolation, enumeration and characterization of rhizosphere microorganism. In: Mukerji KG, Manoharachary C, Singh J (eds) Microbial activity in the rhizosphere. Springer, Heidelberg, pp 17–25
Rees WA, Yager TD, Korte J, von Hippel PH (1993) Betaine can eliminate the base pair composition dependence of DNA melting. Biochemistry 32:137–144
Rokas A, Carroll SB (2005) More genes or more taxa? The relative contribution of gene number and taxon number to phylogenetic accuracy. Mol Biol Evol 22:1337–1344
Ronquist F, Huelsenbeck JP (2003) MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rzhetsky A, Nei M (1993) Theoretical foundation of the minimum evolution. Method of phylogenetic inference. Mol Biol Evol 10:1073–1095
Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N (1985) Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230:1350–1354
Saltikov CW, Olson BH (2002) Homology of Escherichia coli R773 arsA, arsB, and arsC genes in arsenic-resistant bacteria isolated from raw sewage and arsenic-enriched creek waters. Appl Environ Microbiol 68:280–288
Sammeth M, Griebel T, Tille F, Stoye J (2006) Panta rhei (QAlign2): an open graphical environment for sequence analysis. Bioinformatics 22:889–890
Sammeth M, Rothgänger J, Esser W, Albert J, Stoye J, Harmsen D (2003) QAlign: quality-based multiple alignments with dynamic phylogenetic analysis. Bioinformatics 19:1592–1593
Sanger F, Coulson AR (1975) A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 94:441–448
Sarkar G, Kapelner S, Sommer SS (1990) Formamide can dramatically improve the specificity of PCR. Nucleic Acids Res 18:7465
Schmidt HA, Strimmer K, Vingron M, von Haeseler A (2002) TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18:502–504
Spiess A-N, Mueller N, Ivell R (2004) Trehalose is a potent PCR enhancer: lowering of DNA melting temperature and thermal stabilization of Taq Polymerase by the disaccharide trehalose. Clin Chem 50:1256–1259
Stoye J, Moulton V, Dress AW (1997) DCA: an efficient implementation of the divide-and-conquer approach to simultaneous multiple sequence alignment. Comput Appl Biosci 13:625–626
Subramanian AR, Weyer-Menkhoff J, Kaufmann M, Morgenstern B (2005) DIALIGN-T: An improved algorithm for segment-based multiple sequence alignment. Bioinformatics 6:66
Swofford DL (2002) PAUP*: phylogenetic analysis using parsimony (and other methods), Version 4.0 Beta. Sinauer Associates, Sunderland, MA
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tippmann FH (2004) Analysis for free: comparing programs for sequence analysis. Brief Bioinform 5:82–87
Van Walle I, Lasters I, Wyns L (2004) Align-m — a new algorithm for multiple alignment of highly divergent sequences. Bioinformatics 20:1428–1435
Varma A, Verma S, Sudha, Sahay N, Britta B, Franken P (1999) Piriformospora indica — a cultivable plant growth promoting root endophyte with similarities to arbuscular mycorrhizal fungi. Appl Environ Microbiol 65:2741–2744
Varma A, Singh A, Sudha, Sahay N, Sharma J, Roy A, Kumari M, Rana D, Thakran S, Deka D, Bharti K, Franken P, Hurek T, Blechert O, Rexer K-H, Kost G, Hahn A, Hock B, Maier W, Walter M, Strack D, Kranner I (2001) Piriformospora indica: A cultivable mycorrhiza-like endosymbiotic fungus. In: Esser K (eds) Mycota IX. Springer, Heidelberg, pp 123–150
Verma S, Varma A, Rexer KH, Hassel A, Kost G, Sarbhoy A, Bisen P, Buetehorn P, Franken P (1998) Priformospora indica gen. et sp. nov., a new root-colonizing fungus. Mycologia 90:895–909
Wan CY, Wilkins TA (1993) Spermidine facilitates PCR amplification of target DNA. PCR Methods Appl 3:208–210
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic, London, pp 315–322
Wu Q, Thiers MB, Pfister HD (2004) Preparation, preservation and use of fungal specimen in herbaria. In: Mueller GM, Bills FG, Foster SM (eds) Biodiversity of fungi: inventory and monitoring methods. Academic, London, pp 23–28
Young CC, Burghoff RL, Keim LG, Minak-Berners V, Lute JR, Hinton SM (1993) Polyvinylpyrrolidone-agarose gel electrophoresis purification of polymerase chain reaction amplifiable DNA from soils. Appl Environ Microbiol 59:1972–1974
Acknowledgments
DK thanks Stefan Hempel and Stephan König for critical reading of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Krüger, D., Sharma, M., Varma, A. (2009). Assessing the Mycorrhizal Diversity of Soils and Identification of Fungus Fruiting Bodies and Axenic Cultures. In: Varma, A., Kharkwal, A.C. (eds) Symbiotic Fungi. Soil Biology, vol 18. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-95894-9_10
Download citation
DOI: https://doi.org/10.1007/978-3-540-95894-9_10
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-95893-2
Online ISBN: 978-3-540-95894-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)