
Abstract
Hemophilia B or Christmas disease is an X-linked recessive bleeding disorder with a prevalence of ~1 in 30,000 males worldwide that is clearly less common than hemophilia A. Patients with hemophilia B suffer from recurrent joint bleeds, ecchymosis, epistaxis, and post-dental extraction bleeding. Nevertheless women who are carriers of this abnormality are asymptomatic. Timely diagnosis of disorder is made based on family history, clinical manifestations, and appropriate laboratory studies. Replacement therapies with intravenous injection of plasma-derived factor IX (FIX) and recombinant FIX (rFIX) are current therapeutic choices that have significantly improved life expectancy and quality of life in these patients; but inhibitor formation occurring in ~1% of patients remains as a challenge of replacement therapy that can cause infused FIX concentrate to be less efficient. Gene therapy is the only definitive curative option, but some time will pass before it becomes available as a routine treatment choice.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Blood coagulation factor IX (FIX) is a vitamin K-dependent glycoprotein which consists of 415 amino acid residues with a molecular weight of 57 kDa [1]. This coagulation factor has a vital role in coagulation cascade. FIX is mainly activated to FIXa by tissue factor-FVII (TF-FVII) complex and also by FXIa. FIXa with FVIIIa, phospholipid and calcium, makes an activating complex, called tenase, which converts FX to FXa. This reaction has a crucial role in the propagation phase of the coagulation cascade (please refer to Chap. 1) [2].
Hemophilia B is a congenital bleeding disorder caused by mutation in the F9 gene, which is located in chromosome Xq27.1 [3]. Although hemophilia B is an inherited sex-linked recessive disorder, in ~30% of patients, mutations are sporadic, with no preceding family history of the disease [1]. In addition to genetically determined defect, FIX deficiency can be an acquired deficiency due to vitamin K deficiency, in which all vitamin K-dependent coagulation factors including FII, FVII, FIX, and FX and proteins C, S, and Z are decreased [4].
There is a strong correlation between severity of the disorder and residual FIX activity in the bloodstream in hemophilia B. Hemophilia B is classified according to residual plasma FIX activity into severe (<1%), moderate (1–5%), and mild (>5–30%) deficiency [1]. Hemarthrosis, ecchymosis, epistaxis, and dental-related bleeding are common clinical presentations of patients with hemophilia B. Rarely, patients with severe deficiency may present with life-threatening bleeds such as intracranial hemorrhage (ICH) [5].
In initial laboratory examination, patients with hemophilia B have prolonged activated partial thromboplastin time (aPTT) and normal prothrombin time (PT), thrombin time (TT), and platelet count. Factor assay should be performed to confirm the diagnosis. In addition, molecular studies can contribute to detection of underling mutation, carrier detection, and prenatal diagnosis (PND) [5].
Fresh frozen plasma (FFP) and prothrombin complex concentrate (PCC) were therapeutic choices, but these possessed risks of blood-borne disease transmission [2]. Todays, plasma-derived FIX (pdFIX) and recombinant FIX (rFIX) are treatments of choice. With timely diagnosis and appropriate management, burden of the disorder can be decreased, and the patients’ quality of life can be improved [6].
2 Factor IX Structure and Function
FIX is a vitamin K-dependent single-chain glycoprotein which contains 17% carbohydrates and is produced by hepatocytes in the liver. Plasma half-life of FIX is ~18 h, and its average plasma concentrate is 2.5–5 μg/ml [7].
The complete sequence of F9 gene was determined in 1985 [7]. The gene is located in the long arm of the X chromosome in the chromosomal location Xq27.1 and spans 33 kb. F9 gene has eight exons that transcribe to a 2.8 kb mRNA. Of this, 1.4 kb mRNA translate to a precursor protein with 461 amino acids, from which the signal peptide (28 amino acid residues) and propeptide (18 amino acid residues) are removed by proteolytic cleavage. As a result, mature FIX zymogen of 415 amino acids and 57 kDa of molecular weight are released to bloodstream (Fig. 5.1) [8].

F9 gene located in Xq27.1. It has eight exons and spans 33 kilobase (kb) pairs. F9 gene transcribed to an mRNA with 2.8 kb, from which 1.4 kb translated to a precursor FIX with 461 amino acids. Then two domains of FIX, including signal peptide and propeptide domains, are removed by proteolytic cleavage. The remained zymogene FIX has 415 amino acids with 57 kDa molecular weight. UTR untranslated region, aa amino acid, FIX factor IX, sig.p signal peptide, pro.p propeptide
Blood coagulation FIXa glycoprotein has several domains including γ-carboxylation domain (Gla domain), short hydrophobic stack, epidermal growth factor-1 (EGF-1)-like domain, EGF-2, activation peptide (AP), and serine protease (SP) domain (Fig. 5.2) [9, 10].

Precursor FIX which synthesized in the liver has several domains including Gla domain, EGF1 and EGF2 domains, activation peptide, and serine protease domain. Proteolytic cleavages remove signal peptide and propeptide domains. Thus initial FIX converts to FIX zymogene. Then FIX zymogene is released to blood circulation. Cleavage of activated peptide by activated FXI (FXIa) or tissue factor-FVII (TF-FVII) complex leads to conversion of FIX to activated FIX (FIXa). FIXa comprises of a light chain (Gla, EGF-1, EGF-2) and a heavy chain (serine protease domain) which bonded together by a single disulfide bridge. L linker, Gla glutamic acid, EGF epidermal grow factor, FIX factor IX, AP activated peptide, sig.p signal peptide, Pro.p propeptide, TF-FVIIa, tissue factor-activated factor VII
Gla domain has 11 glutamic acids which post-translationally is modified to γ-carboxy-glutamate. These modified residues are necessary for binding to calcium ions. These calciums with positive charge make feasible binding of Gla domain to membrane phospholipids with negative charge. Regions with high affinity for connecting to calcium were reported in SP and EGF-1 domains [8].
Both EGF domains are related to EGF superfamily and have six cysteine residues, which form disulfide bridges. AP is removed to convert FIX zymogen to FIXa. The catalytic activity of FIXa results from a catalytic triad of Ser411, Asp315, and His267 in SP domain [1].
However, crystal structure of full-length human FIX has not been determined, but a structure model could be constructed by considering porcine FIXa (because of 84% identity) and determined three partial structures of human FIXa, Gla, EGF-1, and EGF2-SP domains (Fig. 5.3). According to these findings, FIXa comprises of a light chain with three domains (Gla, EGF-1, and EFG-2) and a heavy chain with one domain (SP domain), which linked together by a single disulfide bond (Cys178-Cys335) [10]. Light chain of FIX which includes Gla, EGF-1, and EGF-2 domains creates a stalk so that Gla domain connects to the phospholipid membrane. Serine protease which is catalytic domain places above the structure [8, 10]. FVIIIa as cofactor of FIX, in order to playing its important cofactor role, makes interaction with catalytic domain and critical residues in EGF-1 and EGF-2 domains either [11, 12].

(a) Epidermal growth factor (EGF)-like domain from human factor IX. B chain and C chain are shown in purple and blue, respectively. Calcium ions are illustrated in gray residues. (b) Porcine FIX domains. The blue color shows light chain which includes Gla domain and EGF-1 and EGF-2 domains. The serine protease (SP) domain which is the catalytic region of the coagulation factor is shown in purple color
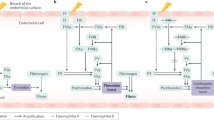
Activation of FIX either via extrinsic pathway by TF-FVII complex, or via intrinsic pathway by FXI, occurs upon double cleavages of peptide bonds after Arg192 and Arg226 residues. This phenomenon leads to removal of AP and release of FIXa to the plasma [13]. When FIXa binds to FVIIIa as its cofactor, its function is dramatically increased by 50,000 fold. In addition, its binding to membrane phospholipid increases its catalytic activity. FIXa as a component of intrinsic complex which includes FVIIIa, calcium, and phospholipid cleaves an arginine-isoleucine bond to convert FX to FXa (Fig. 5.4). Then, FXa initiates prothrombin to thrombin conversion and ultimately results in clot formation. Therefore, FIX has a vital role in the coagulation cascade. Hence mutations in F9 gene causing FIX quantitative deficiencies (type I) or/and qualitative defects (type II) lead to hemophilia B—a bleeding disease [10].

Factor IX (FIX) which activated by tissue factor-activated FVIIa (FVIIa) complex or factor FXIa leads to formation of intrinsic tenase complex that converts FX to FXa. Ca calcium, PL phospholipid, TF tissue factor, FIX factor IX, FIXa activated factor IX, FVIIIa activated factor VIII, FX factor X
3 Hemophilia B
Hemophilia B or Christmas disease is a congenital X chromosome-linked bleeding disorder, which arises from different mutations in F9 gene located in Xq27.1 [13, 14]. The overall incidence of hemophilia B is 1 per 30,000 male live births, whereas hemophilia A has a prevalence of 1 per 5000 male infants [15]. Although most cases of hemophilia B are hereditary, 30% of cases are due to sporadic mutations. In the case of hereditary deficiency, because of X-linked recessive manner of inheritance, a male with one mutant X chromosome and a female with two mutants X chromosome (although scarce) have hemophilia. Females by one mutant X chromosome are carriers of this genetic abnormality. In the case of a hemophilic father and a normal mother, all sons would be non-hemophilic, while daughters would be carriers of hemophilia B. On the other hand, when father has none mutant X chromosome and mother is carrier, 50% of sons would have mutant allele, and 50% of their daughters would be carrier (Fig. 5.5) [1].

Inheritance pattern in hemophilia B. (a) In a family with hemophilic father and non-hemophilic mother, all daughters are genetic carrier, and all sons are non-hemophilic. (b) In the case of a non-hemophilic father and carrier mother with one mutant X chromosome, 50% of sons are hemophilic, and 50% of daughters are hemophilic carrier. Black, hemophilia patient; white, nonhemophilia; gray, carrier
The most prevalent genetic abnormalities in patients with hemophilia B with a frequency of 64% are point mutations. Overall, mutations are categorized in two main groups: type I mutations which cause quantitative deficiency of FIX and type II mutations which manifest as functional defects [1, 10]. In rare cases, hemophilia B can be an acquired phenomenon due to antibody against FIX [16].
Patients with hemophilia B present with recurrent joint bleeding, soft tissue hematoma, and rarely in severely affected cases, life-threatening manifestations such as ICH. Recurrent joint bleeding may lead to disabling arthropathy [5]. Diagnosis of disorder can be made based on clinical presentations, family history, and appropriate laboratory approach. Routine coagulation tests such as PT and aPTT accompany with FIX activity assay, and in well-equipped laboratories, molecular analysis can be used for diagnosis of the disorder [5]. The main drawback with replacement therapy via injection of plasma-derived protein and rFIX is producing of inhibitors against exogenous factor [17].
4 Molecular Basis
F9 gene is located on Xq27.1 and comprises of eight exons. Approximately 1000 distinct mutations have been reported in F9 gene, up to now [3]. The miscellaneous genetic abnormalities were observed within F9 gene including missense/nonsense mutations (64%), small insertion/deletion (indels) (18%), splicing (9%), large indels (6%), regulatory (2%), and complex rearrangement (1%) (Fig. 5.6) [1]. Therefore, unlike hemophilia A, the most common abnormalities in hemophilia B are missense mutations which most of them are combined with normal FIX antigen level (type II mutations) [15].

This pie illustrates percentage of genetic abnormalities in patients with hemophilia B. The most prevalent genetic abnormalities are point mutations (64%). Prevalence of other abnormalities reported within F9 gene are small indels (insertion and deletion) (18%), splicing (9%), large indels (6%), regulatory (2%), and rearrangement (1%)
There are some mutational hotspots within F9 gene that are more prone to occurrence of mutation. For instance, the high rate of mutations in regions with high frequency of guanine and cytosine, typically near the initiation transcript region, revealed that they are mutational hotspots. The loci of mutation can affect production and function of coagulation FIX and therefore can influence disease severity. Point mutations can manifest as missense, premature stop codon, or splice site mutations. F9 gene mutations are classified in two main types, type I and type II, which lead to quantitative deficiencies and qualitative defects, respectively. Type I mutations often result in reduced transcription and FIX secretion to the bloodstream and can cause severe hemophilia B [1].
A signal peptide (residues 1–28) directs intracellular trafficking of the protein and has no effect on the protein function; its mutations lead to impaired FIX secretion and therefore lead to type I mutation. An example of functional deficit in F9 gene is mutation in triad residues of serine protease domain which produces protein defective in catalytic activity. This results in severe hemophilia B and associated with increased susceptibility to inhibitor formation. In addition, mutations in propeptide (residues 29–46) which mediates interaction with the vitamin K-dependent γ-carboxylase lead to impaired phospholipid-binding capacity that presents as mild or moderate phenotype [10].
Generally, nonsense mutations are associated with severe type I defect, without any protein production. Nevertheless, when a nonsense mutation occurs in coding gene of C-terminal region of protein, results in producing nonfunctional FIX and type II defect. However a splicing defect creates a variable range of disease severity; a single point mutation that impairs a splice donor site or occurs in the sequence of an intron which creates an alternative splice acceptor site results in a nonsense mRNA and severe hemophilia B. In north America, England, and Ireland, some recurrent mutations due to founder effect were reported. Founder effect illustrates how certain genetic abnormalities occur more frequently in a specific geographically restricted hemophilia B population due to identity by descent. These are usually mild or moderate disease causing mutations for the reason that a severe defect reduces reproductive fitness of affected males (please refer to Chap. 2) [1].
There are some mutations in the F9 gene promoter that cause permanent low level of FIX throughout patient’s life. However in hemophilia B Leyden with more than 20 different identified mutations in proximal F9 gene promoter, a severe FIX deficiency presents at birth. The FIX level starts to increase in the second decade of life at the onset of puberty, and finally reaches to near normal level in the third decade of life, that then remains stable throughout life. It is apparent that the defect in the promoter makes FIX transcription dependent on testosterone level, and hence the severity of disease is reduced after puberty [18].
Small insertion or deletion with a prevalence of 18%, generally occurs in association with runs of dinucleotide repeats. This abnormality usually occurs within introns without any effect on FIX protein. However, occasionally these indels can occur in regulatory regions or lead to frameshifts which result in a severe hemophilia B. As inhibitor formation is influenced by genetic abnormalities, a null allele which means complete deletion of F9 gene has the most probability for inhibitor formation [19]. Although the overall prevalence of inhibitor formation against therapeutic products is 1–3%, this rate is increased to nearly 20% in cases with complete deletion of the F9 gene [10].
5 Factor IX Propeptide Mutation-Associated Hypersensitivity to Coumarin Therapy
There are two missense mutations in FIX propeptide which lead to major bleeding after coumarin therapy. These mutations occur in Ala-10 locus which is a high protected site in vitamin K-dependent clotting factors (FII, FVII, FIX, and FX). These two point mutations, Ala-10(GCC) → Val(GTC) and Ala-10(GCC) → Thr(ACC) in exon 2 of F9 gene result in a propeptide to which carboxylase enzyme has low affinity and therefore exhibit extremely low FIX coagulant activity (FIX:C) after coumarin therapy. It is considered that these patients have normal phenotype and coagulation screening test without any bleeding episodes in their life. They only show a defect once they undergo oral anticoagulation therapy with warfarin. Upon receipt of anticoagulant, they would show extreme fall in FIX:C to <1–3% and subsequently abnormal increase in aPTT in therapeutic range of PT and INR. In other people usually after coumarin therapy FIX:C decreases to 15–30% as do other vitamin K-dependent factors [20,21,22].
Prothrombin complex factors (FII, FVII, FIX and FX) for connection to membrane phospholipid need γ-carboxylation in NH2 terminal of mature protein. The propeptide is a carboxylase recognition site, and their junction together is a key step of the reaction (Fig. 5.7). In cases with propeptide mutations, although the affinity of hydroxylase enzyme is reduced, its activity is normal in absence of coumarin therapy. So it could be the probable cause of normal FIX:C and lack of bleeding before treatment with oral anticoagulants as warfarin. After coumarin therapy, in addition of low affinity, the activity of enzyme falls down because of inhibition of vitamin K reductase and lack of KH2 as cofactor (Table 5.1). Therefore, the result is FIX:C less than 1–3%, abnormally increased aPTT, and severe bleeding in these patients after treatment with coumarin [20,21,22].

The mechanism of vitamin K-dependent clotting factor carboxylation. The propeptide is carboxylase recognition site, and after connection, KH2 is used as enzyme cofactor. Generated KO coverts to KH2 through epoxide reductase function. It is goal enzyme for inhibition by warfarin. Pro.p propeptide, FIX factor IX, KH2 dihydro vitamin K, KO epoxide vitamin K
Propeptide mutations in other vitamin K-dependent clotting factors show less coumarin sensitivity due to their autosomal recessive inheritance (unlike to F9 gene which is located on X chromosome). Consequently, the effect of these mutations should be observed greatly in males [21, 22]. In conclusion, in cases with unusual bleeding pattern after treatment with oral anticoagulant which are vitamin K antagonist, aPTT and FIX:C should be evaluated, even in therapeutic range of PT and INR [22].
6 Clinical Manifestations
Patients with hemophilia B present various clinical symptoms including hemarthrosis, epistaxis, ecchymosis, and post-dental extraction bleeding. The most common site of spontaneous bleeding is the joints [5]. Life-threatening symptoms such as central nervous system (CNS) bleeding and umbilical cord bleeding are rare and usually only seen in severely affected individuals [5]. Over the long period, the main complications of recurrent joint bleeding and soft tissue hematomas are extensive arthropathy, muscle contractures, and pseudotumors that result in disability and chronic pain [15].
According to FIX:C, hemophilia B is classified into three types: severe (FIX:C <1% or <0/01 IU ml−1), moderate (FIX:C of 1–5% or 0/01–0/05 IU ml−1), and mild (FIX:C of 5–30% or >0/05–<0/30 IU ml−1) deficiency. The main presentation of patients with severe hemophilia B is spontaneous bleeding, while patients with moderat FIX deficiency has seldom spontaneous bleeding and patient with mild FIX deficiency is lack of this clinical feature. Most commen clinical presentations in patients with moderate hemophilia B is post-traumatic hemorrhage, while post-surgical bleeding, post-dental extraction hemorrhage and bleeding after major injories are more common in patients with mild phenotype (Table 5.2) [3, 10, 14].
In overall, severity of disease influenced by type of mutation and some of mutations lead to severe hemophilia B. Frameshifts, nonsense mutations, large deletions, and splicing mutations are the most common genetic abnormalities that lead to severe hemophilia B. However, small deletions and missense mutations cause severe, moderate, or mild deficiency, depending on their location on genome (Table 5.3) [23].
Generally, hemophilia B is a less severe bleeding disease with lower bleeding frequency and better long-term outcomes than hemophilia A [14]. Approximately one-third of heterozygous women are classified as symptomatic carriers because their FIX activity is between 40% and 60%. Their clinical manifestations are similar to mild hemophilia B. In addition, they often have more prolonged menstrual bleedings [1].
7 Diagnosis
The usual age of patients with hemophilia B at diagnosis relies on the severity of disorder. In the case of severe hemophilia, usual age at diagnosis is ≤2 years. Moderate hemophilia typically is diagnosed at age of 2–5 years, while mild deficiency is often diagnosed later in the life. In the cases with sporadic mutations or in the cases without family history, the time of diagnosis is related to outward symptoms of patients [1].
Generally, diagnosis of disorder is based on clinical presentations, family history, and appropriate laboratory approach. As recurrent bleeding episodes are relatively similar in various coagulopathies, proper laboratory assessment is necessary for correct and timely diagnosis of the disorder [5]. Complete blood count (CBC) is normal in hemophilia B; however a reduction in hemoglobin and RBC might be seen due to prolonged hemorrhages. In screening coagulation tests, aPTT is prolonged up to 2.5 times, while PT is normal [1].
Diagnosis of hemophilia B should be differentiated from vitamin K deficiency, heparin consumption, von Willebrand disease (VWD), and hemophilia A which all presented with increased aPTT (Table 5.4) [24].
Factor assay could be used for determining each factor activity level. In hemophilia B, FIX level is decreased [5]. There are two methods for FIX activity assay including one-stage clot assay which is based on aPTT and chromogenic assay. The former is traditional common method, whereas the later rarely is used for hemophilia B. Chromogenic assay is used more commonly in hemophilia A than hemophilia B. According to recent reports from seven countries, chromogenic assay is used in 68% of laboratories for hemophilia A, while only 11% of laboratories used this method for hemophilia B [25].
For FIX:C, blood should be collected in citrated tube and immediately be centrifuged in 2000g for 20 min. Then plasma should be separated and be freezed in −70 °C [25]. After melting, FIX:C could be performed with one of two methods: one-stage assay or chromogenic assay [26]. The one-stage assay is an aPTT-based method and the most routinely used method. Patient plasma and FIX-deficient plasma preincubate with aPTT reagent for 3–5 min. Then, after addition of calcium, the clotting time should be recorded. The result of patient’s plasma clotting time is compared with a standard curve that is generated from plasma samples with determined FIX activity. Each patient’s plasma at least should be measured in three different dilutions for analyzing of parallelism between standard dilutions and patient plasma dilutions. Two lines should be parallel, unless there is an inhibitor [24].
Chromogenic assay include two stages: at first stage, a reactive mixture consisting of FXIa, thrombin, phospholipid, and calcium chloride adding to patient’s plasma with unknown FIX activity. It is assumed that amount of generated FXa is proportional with the amount of residual plasma FIX. In the second stage, a specific peptide of FXa, nitroanilin substrate, is measured through a peptide cleavage. Generated p-nitroaniline is analyzed photometrically in absorbance of 405 nm. The created color is directly proportional with the amount of functional FIX in plasma according to a standard curve (Fig. 5.8) [24, 25].

Principle of factor IX (FIX) chromogenic assay. A reactive mixture (FXIa, Thr, Ph, and Ca) adding to patient plasma with unknown FIX activity. In the first stage, generated FX is directly proportional to FIX activity in patient’s plasma. In the second stage, a peptide cleavage of chromogenic FXa substrate leading to generation of p-nitroaniline and therefore analyzing of this color in absorbance of 405 nm. FXIa activated factor XI, Thr thrombin, Ph phospholipid, Ca calcium, FX factor X, FXa activated factor X, FIX factor IX
According to a study on patients with hemophilia B, discrepancy between two activities assay methods is not observed in patients with severe hemophilia B, while in non-severe hemophilia B, discrepancy between one-stage assay and chromogenic assay is observed. This study illustrated that those patients with non-severe hemophilia B and mutation in N-terminal site of activation peptide and propeptide domains of FIX have twofold more differentiate between results of one-stage and chromogenic stage assays (higher activity in chromogenic assay) [25].
Different aPTT reagents don’t have any impact on discrepancy between these assays. In fact, each method has some advantages and disadvantages (Table 5.5). Furthermore, the use of both assays is contributory for favorable diagnosis and classification of hemophilia B (Table 5.6) [25].
To assess inhibitor development in response to factor replacement therapy or when presence of inhibitor is suspected, a mixing test is used, in which normal pooled plasma as a source of FIX is mixed with equal parts of patient’s plasma. The assay is based on incubation of this mixture for 1 h at 37° followed by aPTT assay. A prolonged repeated aPTT should be followed by Nijmegen assay to determine the inhibitor titer. One unit of inhibitor is the amount of inhibitor that will inactivate 50% or 0.5 unit of FIX activity over 10 min at 37°. This assessment helps to make a correct decision for management of patients with hemophilia B with inhibitor [5, 24].
In the case of a known carrier mother, chorionic villus sampling (CVS) and amniocentesis can be performed for screening of the fetus. After birth, coagulation tests for identifying clotting factor level should be performed for such cases. It is considerable that although low level of FVIII is pointing to hemophilia A, results for FIX are not so decisive. FIX takes approximately 6 months after age to reach normal level, so in interpretation of FIX assay at birth, this issue should be considered. On the other hand, if there is mild decrease in FIX level at birth, it is not representative of hemophilia B, while an extensive reduction of FIX level is suggestive of the disorder [1].
Molecular analysis can be used for confirmation of the disorder and can assist to achieving valuable data about carrier detection, prenatal diagnosis (PND), and prediction of inhibitor formation and therefore management of disorder [5]. Additionally determination of eligibility for gene therapy in future will benefit from molecular analysis. Since there is no common reported mutation within F9 gene, for molecular diagnosis full F9 gene sequencing is required. The DNA amplification by polymerase chain reaction (PCR) and then direct sequencing is a well-standardized manner for molecular analysis; however large deletions or other gross abnormalities due to the existence of other normal allele could not be detectable by this method. Multiplex ligation-dependent probe amplification (MLPA) and multiplex amplification and probe hybridization (MAPH) could be used to reduce percent of F9 gene mutations in patients with hemophilia B [27, 28].
8 Treatment
In the past, initial treatment of patients with hemophilia B was injection of fresh frozen plasma (FFP) or prothrombin complex concentrate (PCC) which, in addition to FIX, contains additional coagulation factors. The problems associated with these products were extreme activation of coagulation cascade, volume enhancement, and risk of blood-borne diseases especially in using of FFP [1, 2].
Next achievement for hemophilia treatment was advent of plasma-derived FIX (pdFIX) products and recombinant FIX (rFIX) concentrates. pdFIX and FVIII are available since 1970 which lead to self-infusion and home therapy. rFIX which are available since 1999 eradicate risk of animal and human infectious agent transmission. In addition, rFIX is safe and effective and with lower rate of allergic and thrombosis reactions and inhibitor formation [29].
The purpose of replacement therapy is achievement of plasma FIX level to 60–80% and 20–40% for major and minor bleeding, respectively. There are several plasma-derived and recombinant FIX products for treatment of hemophilia B (Table 5.7) [30]. Over past years, the safety and purity of these products were improved, so the current treatment of choice for hemophilia B is intravenous injection of these products. However, the fear, pain, and annoyance related to needle-based injection of these products are causes of low patient requisition [5].
In 2014, for the first time, Food and Drug Administration (FDA) confirmed a long-lasting rFIX Fc fusion protein (rFIXFc) which required a lower frequency of injections to maintain hemostasis. This product meets the current goal of producing a FIX with extended circulation time which can decrease injection frequency and improve quality of life. rFIXFc has an extended half-life up to 48 h, compared to standard rFIX with half-life of approximately 18 h, in animal models of hemophilia B. Alprolix which is a rFIXFc comprises of a single-chain FIX that is recombinantly connected to constant region (Fc) of immunoglobulin IgG. Fc domain protects protein from catabolism via binding to Fc receptors (FcRn). These receptors protect IgG from degradation [30].
GlycoPEGylated rFIX is another rFIX concentrate which has twofolds recovery time and fivefolds longer half-life than standard FIX. PEG covalently connects to activation peptide, so PEGylation of rFIX in this special site, saves its biological activation and increases its circulation time Therefore patients requier lower frequency of injections. Once per week administration of 40 IU/kg of PEG-rFIX is required for hemophilia B treatment [30,31,32].
Another type of fusion protein is albumin-rFIX which has five times increased half-life compared with standard rFIX. However, more studies are required for establishing safety and efficacy of newer rFIX products [30, 33].
Replacement therapy could be administrated as prophylaxis or on-demand regimens [30]. Primary prophylaxis therapy is replacement protein injection in absence of bleeding as a protective procedure. This trend has improved life expectancy and quality of life in patients with hemophilia B. On-demand therapy is treatment when bleeding is occurred [29]. Recommended dosage of FIX concentrate for hemophilia B treatment is shown in Table 5.8 [30, 34]. Prophylaxis is recommended care for patients with severe hemophilia B; it prevents joint bleeding and hemophilic arthropathy and improves patient’s quality of life [30]. Common prophylaxis is injection of FIX concentrate twice per week, but emergence of extended half-life of FIX products leads to once per week injection. In a study, it has been shown that 100 IU/kg injection of rFIX nonacog alfa once per week is as effective as 50 IU/kg injection of it twice per week in 50 patients [35].
9 Inhibitor Formation a Challenge of Replacement Therapy
The most important complication of replacement therapy in hemophilia B is inhibitor development which leads to increasing risk of bleeding-related morbidity such as nephrosis and mortality [36]. The FIX inhibitor is commonly IgG4 subclass with affinity to Gla domain and SP domain in light and heavy chains, respectively. This phenomenon not only reduces efficacy of replacement therapy but also uses lots of economic resources [17, 37]. Although the prevalence of inhibitor in hemophilia B is less than 5%, in severely affected patients with major gene defects, this increases to 9–23% (Fig. 5.9) [19, 29, 36].

Crystal structure of human factor IX GLA domain in complex with an inhibitory antibody. Heavy chain and light chain of inhibitory antibody are shown in blue and purple, respectively. Gla domain is shown in brown color. The grey residues are calcium ions. Ab antibody
Approximately 50% of patients with FIX inhibitor might experience life-threatening anaphylactic reactions. The inhibitor development results from genetic and non-genetic reasons which the mutation type is the major risk factor. In HB, null mutations in which the genetic abnormality such as large deletions and nonsense mutations prevents synthesizes of coagulation factor result in inhibitor development. In contrast, in milder molecular defects such as small deletions, insertions, splice site, and missense mutations which result in synthesis of coagulation factor but loss of function, inhibitory development risk is less [5, 17, 36, 37]. Since the missense mutations are the most common molecular defects in hemophilia B, it might be the reason of low prevalence of inhibitor antibody in these patients than hemophilia A [5, 36]. The other risk factors, for instance, non-genetic reasons, are less studied and analyzed in hemophilia B because of rarity of inhibitor development in these patients [19].
The treatment strategy for these patients depends on the inhibitor levels. When the inhibitor titer is low (<5 Bethesda unit (BU)), the treatment approach is injection of FIX in high level of standard dose. In contrast, the high titer of inhibitor (>5 BU) requires more rigorous therapeutic options. In these cases, management of patients is divided into two parts: prevention of further hemorrhage and eradication of inhibitor. Bypassing agents, recombinant activated FVII (rFVIIa), and activated prothrombin complex concentrate (APCC) are main options for management of bleeding episodes. Immune tolerance induction (ITI), requiring high-dose injection of FIX for months to years, is used for inhibitor eradication. In cases of ITI-resistant, immunosuppressive drugs such as rituximab are the alternative choice for inhibitor elimination (Fig. 5.10). ITI is less effective in patients with hemophilia B in comparison with cases with hemophilia A (<50% and 60–80% for hemophilia B and hemophilia A, respectively) [17, 36]. Since rFVIIa as a bypassing agent doesn’t include FIX, it’s a suitable choice for hemophilia B patients who developed the inhibitor and manifest anaphylactic reactions to FIX injections [38].

The treatment approach for patients with hemophilia B and inhibitor antibody. HB hemophilia B, U/B unite/Bethesda, FIX factor IX, ITI immune tolerance induction, rFVIIa recombinant activated factor VII, APCC activated prothrombin complex concentrate
10 Gene Therapy
Gene therapy is one of the curative options for hemophilia B. It can lead to long-stay synthesis of FIX. Additionally, it can eliminate spontaneous bleeding and requiring for frequently injections. In fact gene therapy treats hemophilia completely. Adeno-associated virus (AAV)-mediated gene therapy is full of promise because this vector could induce long term FIX level [39, 40].
Current therapy using pdFIX products and rFIX concentrates has increased life expectancy and quality of life in patients with hemophilia B. However, these treatments require high-cost and lifelong factor injection [41, 42].
A promising strategy for effective treatment of patients with hemophilia disease is gene therapy. This approach is especially effevtive for severe untreated patients with less than 1% factor activity accompanied with arthropathy and life-threatening bleeds. Gene therapy is a conclusive treatment using AAV vectors to transfer F9 gene through intramuscular injections or liver-targeting delivery [40]. According to a study on ten patients with severe hemophilia B, a single AAV8 vector dose injection causes long-term expression of FIX and clinical improvement [41].
AAV is a nonpathogenic virus with a single-strand genome which is 4.7 kb in length. Results of gene transferring through the AAV vector are reduction of prophylactic costs and bleeding episodes. Among various serotypes of AAV, AAV serotype 8 is more effective because it is lack of abiliy to cross react with antibodies produced against other AAV serotypes [40, 43]. Nevertheless the main complication of gene therapy is liver toxicity and aminotransferase elevation 7–10 weeks after gene transfer [41]. This phenomenon is a result of T cell-mediated immune responses to conducted hepatocytes. It can be simply resolved by using glucocorticoid therapy like prednisolone without loss of transgene expression. The level of increase of FIX is dose-dependent. In high-dose vector injection, FIX level reaches to 8–12% of normal levels [39].
In conclusion, gene therapy is the potential curative option for hemophilia because it induces long-term endogenous production of FIX. A small enhancement in factor level at least 1% of normal, ameliorates bleeding episodes [44]. So, despite increase in aminotransferase levels which could return to basic level through glucocorticoid therapy, without loss of gene expression, gene therapy could convert severe hemophilia to mild form or reverse it completely [39]. In fact, the future of gene therapy for management and treatment of hemophilia patients looks bright [40].
References
Horava SD, Peppas NA. Recent advances in hemophilia B therapy. Drug Deliv Transl Res. 2017;7:359–71.
DeLoughery TG. Basics of coagulation. In: Hemostasis and thrombosis. Cham: Springer; 2015. p. 1–7.
Wang Q-Y, et al. A genetic analysis of 23 Chinese patients with hemophilia B. Sci Rep. 2016;6:25024.
Eby CS. Bleeding and vitamin K deficiency. In: Management of bleeding patients. Cham: Springer; 2016. p. 145–50.
Dorgalaleh A, Dadashizadeh G, Bamedi T. Hemophilia in Iran. Hematology. 2016;21(5):300–10.
Berntorp E, Shapiro AD. Modern haemophilia care. Lancet. 2012;379(9824):1447–56.
Yoshitake S, et al. Complete nucleotide sequences of the gene for human factor IX (antihemophilic factor B). Biochemistry. 1985;24(14):3736–50.
Lee C, Berntorp E, Hoots K. Textbook of hemophilia. Chichester, West Sussex, UK. Hoboken, NJ: Wiley-Blackwell; 2010.
Schmidt AE, Bajaj SP. Structure–function relationships in factor IX and factor IXa. Trends Cardiovasc Med. 2003;13(1):39–45.
Rallapalli P, et al. An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B. J Thromb Haemost. 2013;11(7):1329–40.
Ngo JCK, et al. Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure. 2008;16(4):597–606.
Autin L, et al. Molecular models of the procoagulant factor VIIIa–factor IXa complex. J Thromb Haemost. 2005;3(9):2044–56.
Li T, et al. The CDC Hemophilia B mutation project mutation list: a new online resource. Mol Genet Genomic Med. 2013;1(4):238–45.
Li T, et al. Mutation analysis of a cohort of US patients with hemophilia B. Am J Hematol. 2014;89(4):375–9.
Mannucci P, Franchini M. Is haemophilia B less severe than haemophilia A? Haemophilia. 2013;19(4):499–502.
Jedidi I, et al. Acquired haemophilia B: a case report and literature review. Ann Biol Clin. 2011;69:685–8.
Franchini M, Mannucci PM. Inhibitors of propagation of coagulation (factors VIII, IX and XI): a review of current therapeutic practice. Br J Clin Pharmacol. 2011;72(4):553–62.
Crossley M, et al. Recovery from hemophilia B Leyden: an androgen-responsive element in the factor IX promoter. Science. 1992;257(5068):377–9.
DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br J Haematol. 2007;138(3):305–15.
Ulrich S, et al. Congenital hypersensitivity to vitamin K antagonists due to FIX propeptide mutation at locus-10: a (not so) rare cause of bleeding under oral anticoagulant therapy in Switzerland. Swiss Med Wkly. 2008;138(7–8):100–7.
Chu K, et al. A mutation in the propeptide of Factor IX leads to warfarin sensitivity by a novel mechanism. J Clin Investig. 1996;98(7):1619.
Oldenburg J, et al. Missense mutations at ALA-10 in the factor IX propeptide: an insignificant variant in normal life but a decisive cause of bleeding during oral anticoagulant therapy. Br J Haematol. 1997;98(1):240–4.
Surin V, et al. Mutational analysis of hemophilia B in Russia: molecular-genetic study. Russ J Genet. 2016;52(4):409–15.
Kitchen S, McCraw A, Echenagucia M. Diagnosis of hemophilia and other bleeding disorders. A laboratory manual. Montreal: World Federation of Hemophilia; 2000.
Kihlberg K, et al. Discrepancies between the one-stage clotting assay and the chromogenic assay in haemophilia B. Haemophilia. 2017;23(4):620–7.
Kitchen S, Signer-Romero K, Key N. Current laboratory practices in the diagnosis and management of haemophilia: a global assessment. Haemophilia. 2015;21(4):550–7.
Sellner LN, Taylor GR. MLPA and MAPH: new techniques for detection of gene deletions. Hum Mutat. 2004;23(5):413–9.
KWON MJ, et al. Identification of mutations in the F9 gene including exon deletion by multiplex ligation-dependent probe amplification in 33 unrelated Korean patients with haemophilia B. Haemophilia. 2008;14(5):1069–75.
Bolton-Maggs PH, Pasi KJ. Haemophilias a and b. Lancet. 2003;361(9371):1801–9.
Franchini M, et al. Treatment of hemophilia B: focus on recombinant factor IX. Biol Targets Ther. 2013;7:33.
Elm T, Oestergaard H, Tranholm M. Dose response and prolonged effect of 40K PEG-FIX on bleeding in hemophilia B mice. J Thromb Haemost. 2009;7:134.
Carcao M, et al. Insight into health-related quality of life of young children with haemophilia B treated with long-acting nonacog beta pegol recombinant factor IX. Haemophilia. 2017;23(3):e222–4.
Metzner HJ, et al. Genetic fusion to albumin improves the pharmacokinetic properties of factor IX. Thromb Haemost. 2009;102(4):634–44.
Brown DL. Congenital bleeding disorders. Curr Probl Pediatr Adolesc Health Care. 2005;35(2):38–62.
Nummi V, Jouppila A, Lassila R. Monitoring once-weekly recombinant factor IX prophylaxis in hemophilia B with thrombin generation assay and factor IX activity. Int J Lab Hematol. 2017;39:359–68.
Dolan G, et al. Haemophilia B: Where are we now and what does the future hold? Blood Rev. 2017;32:52–60.
Key NS. Inhibitors in congenital coagulation disorders. Br J Haematol. 2004;127(4):379–91.
White G, et al. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85(3):560.
Nathwani AC, et al. Adenovirus-associated virus vector–mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357–65.
Nienhuis AW, Nathwani AC, Davidoff AM. Gene therapy for hemophilia. Mol Ther. 2017;25:1163–7.
Nathwani AC, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;2014(371):1994–2004.
Park C-Y, et al. Genome-editing technologies for gene correction of hemophilia. Hum Genet. 2016;135(9):977–81.
Kay MA, et al. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet. 2000;24(3):257.
Herzog RW, et al. Long-term correction of canine hemophilia B by gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat Med. 1999;5(1):56.
Acknowledgment
We appreciate Professor Edward Tuddenham for his valuable comments that significantly improved the quality of this chapter.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Motlagh, H., Pezeshkpoor, B., Dorgalaleh, A. (2018). Hemophilia B. In: Dorgalaleh, A. (eds) Congenital Bleeding Disorders . Springer, Cham. https://doi.org/10.1007/978-3-319-76723-9_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-76723-9_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-76722-2
Online ISBN: 978-3-319-76723-9
eBook Packages: MedicineMedicine (R0)