
Abstract
Spinocerebellar ataxia type 2 (SCA2) is autosomal dominantly inherited and caused by CAG repeat expansion in the ATXN2 gene. Because the CAG repeat expansion is localized to an encoded region of ATXN2, the result is an expanded polyglutamine (polyQ) tract in the ATXN2 protein. SCA2 is characterized by progressive ataxia, and slow saccades. No treatment for SCA2 exists. ATXN2 mutation causes gains of new or toxic functions for the ATXN2 protein, resulting in abnormally slow Purkinje cell (PC) firing frequency and ultimately PC loss. This chapter describes the characteristics of SCA2 patients briefly, and reviews ATXN2 molecular features and progress toward the identification of a treatment for SCA2.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
8.1 SCA2 Clinical Characteristics
While patients with SCA2 possess many of the core clinical characteristics that define the SCAs as a group of neurodegenerative disorders, SCA2 is a clinically distinct. Considered a hallmark characteristic of any SCA, the most noticeable symptom of onset is gait ataxia. In SCA2, onset also frequently, although not always, coincides with muscle cramping. Ataxia onset is then followed by multiple other symptoms characteristic of cerebellar degeneration . For SCA2 these symptoms include appendicular ataxia with instability of stance, dysarthria, and ocular signs including nystagmus , and ocular dysmetria. The signs and symptoms of SCA2 are almost entirely of cerebellar origin, with clearly defined involvement of cerebellar regions and associated cerebellar circuits. However, one predominant ocular feature typical of SCA2, slow or absent saccades, arises from degeneration of neurons of the oculomotor brainstem. Dystonia and myoclonus are also frequent in patients with SCA2, as well as neuropathy, muscle spasticity and frontal-executive dysfunction [1–3].
SCA2 phenotype is characterized by gait ataxia in most SCA2 patients, however variant phenotypes have been defined. These variant phenotypes reside outside of the cerebellar spectrum and include L-DOPA responsive parkinsonism and amyotrophic lateral sclerosis (ALS) [3, 4]. Patients with these variant phenotypes present with idiopathic forms of parkinsonism or ALS. While ATXN2-associated parkinsonism and ALS present with no symptoms of cerebellar ataxia , more imaging data are necessary to define whether these variant phenotypes are accompanied with cerebellar atrophy.
8.1.1 SCA2 and ALS
ATXN2 CAG repeat expansions are also associated with ALS-like motor phenotypes. For ATXN2 CAG repeats in the normal range for SCA2, between 27 and 33 repeats in length, a statistically significant increased risk for ALS has been defined [4, 5]. A meta-analysis of ATXN2 alleles drawing on worldwide reporting of ALS, however, showed that ALS-risk only increased significantly for CAG repeats ≥31. In these patients, the phenotype is indistinguishable from other idiopathic forms of ALS. The causes of the ALS-like phenotypes in patients with ATXN2 expansions are not well described but may associate with the ATXN2 interacting proteins TDP-43 and FUS [4, 6], since mutations in the genes encoding these proteins can cause ALS. ALS and intermediately expanded ATXN2 connects functionally to the action of C9ORF72, since aggregates partially depleted of C9ORF72, including intermediately expanded ATXN2, were neurotoxic due to impaired autophagy [7]. However, it remains to be determined why intermediate expanded ATXN2 increases risk of ALS in the absence of C9ORF72 mutations. In our meta-analysis an 11-fold increased risk was observed for ATXN2 repeats of 32 [5]. Thus the rarest ATXN2 alleles represent the highest risk for ALS. However, SCA2 patients with longer ATXN2 mutations can also present with ALS-like phenotypes [5, 8].
8.2 Discovery of the ATXN2 Gene
SCA2 was first described in India with the discovery of nine patient families [9]. Nearly two decades later a large population of SCA2 patients was discovered in eastern Cuba [9]. Both discoveries noted that the affected families were characterized by ataxia and other cerebellar signs as well as slow saccades; clinical features that are now known to typify patients with SCA2. In Cuba it was later noted that the prevalence of SCA2 was especially high compared to other regions in the world, attributed to a founder population in the eastern part of the island where 4 out of every 10,000 inhabitants of Holguin province has SCA2 [10, 11]. The existence of the large SCA2 populations in Cuba and elsewhere has aided the identification of the ATXN2 gene. Varying age of onset (AO) in SCA2 pedigrees helped to establish anticipation in SCA2 of 14.4 ± 7.9 years per generation strongly hinting that the ATXN2 mutation was likely a repeat expansion mutation [12]. Mapping studies ultimately localized ATXN2 to Chr 12q24.12 following initial mapping to chromosome 12 [13], and fine mapping to 12q24 [12]. The ATXN2 gene was identified in 1996 demonstrating the causative mutation as a CAG repeat expansion in the coding region of ATXN2 resulting in a polyglutamine expansion in the ATXN2 protein [14–16].
8.3 Molecular Genetics of SCA2
Most commonly, the ATXN2 gene has 22 CAG repeats while ≥33 CAG repeats causes SCA2 [17]. SCA2 is characterized by anticipation with strong correlation between age of onset and CAG repeat length (Fig. 8.1). The CAG repeat expansion is dynamic and unstable during meiosis with a strong propensity for expansion.

Anticipation in SCA2. SCA2 age of onset is negatively correlated with ATXN2 CAG repeat length. Note that the variability in AO for any CAG repeat length is partly associated with CACNA1A repeat length (but no other polyglutamine disease genes) and likely also to other genetic and environmental factors [10]
The ATXN2 gene consists of 25 exons and spans a total of 147 megabase pairs (147,463 bp). The ATXN2 transcript is 4699 bp long with relatively small untranslated regions (162 bp 5′-UTR, 601 bp 3′-UTR). There are two in-frame start codons at the 5′-end of the sequence with the second one located four codons upstream of the CAG repeat. Transcriptional studies have only partly described which of these are utilized in translation . The predicted molecular weight for ATXN2, when made from the first start codon, is 144 kDa and ATXN2 made from the second start codon is 17 kDa smaller. While western blot analyses typically produce a single ATXN2 protein band consistent for use of the further-most upstream ATG, artificial luciferase tagged ATXN2 promoter constructs lacking the second ATG fail to express proteins [18]. Note that a smaller, approximately 42 kDa, fragment of ATXN2 was observed in brain extracts from SCA2 patients and SCA2 mice [19–21]. Huynh et al. [20] identified a consensus aa sequence for caspase-3 cleavage at ATXN2 aa 396–399 that could explain the origin of this band. ATXN2 is a cytoplasmic protein that also localizes to the trans-Golgi network [22, 23], and is a phosphorylated protein with half-life of ≥21 h [22]. ATXN2 transcription is also regulated by the ETS1 transcription factor [18], and might be altered by CAG repeat expansion since the ATXN2 CAG repeat is located inside a CpG island [24]. The molecular features of the ATXN2 gene and the encoded protein are summarized in Table 8.1.
8.3.1 Macromolecules Interacting with ATXN2
The ATXN2 interacting proteins provided clues on the functions controlled by ATXN2. ATXN2 interacts with multiple RNA binding proteins (RBPs) suggesting that ATXN2 has a role in RNA metabolism. ATXN2 also interacts with staufen, which controls stress granule formation and itself interacts with RBPs. ATXN2 interactions with IP3R and with the RGS8 mRNA transcript support the ATXN2 roles in calcium homeostasis. ATXN2 also interacts with endophilins and CIN85 indicating a function for ATXN2 in synaptic vesicle endocytosis. The ATXN2 interacting proteins are summarized in Table 8.2, and a graphic representation of the binding sites within ATXN2 is presented in Fig. 8.2.

ATXN2 domain structure and sites of interactions. The diagram depicts the amino acids of the ATXN2 protein with locations of known domains indicated. Lines represent the minimal regions experimentally tested for interaction. The domains and their locations are as follows: Polyglutamine tract (PolyQ) (aa 166–187), SRC homology 3 (SH3) domain binding motifs 1 (SBM1) (aa 117–126) and 2 (SBM2) (aa 587–596), Like sm domain (Lsm) (aa 254–345), Lsm associated domain (LsmAD) (aa 353–475), PABP interacting motif 2 (PAM2) (aa 908–925). In addition, ATXN2 has an acidic domain (aa 256–405), a predicted clathrin-mediated sorting signal (aa 414–416), and a predicted site for caspase cleavage (aa 396–399) [20]
8.3.2 A2BP1/RBFOX1
A2BP1/RBFOX1 is a regulator of RNA alternative splicing . We discovered A2BP1 as an ATXN2 interacting protein by yeast two-hybrid screening. Secondary two-hybrid assays using protein fragments determined that A2BP1 interacted with the C-terminal half of ATXN2 residues 760–1313 and that the full-length A2BP1 protein interacted stronger than did the C-terminal A2BP1 fragment while an N-terminal A2BP1 fragment did not interact with ATXN2 [25]. A2BP1 labeled in granules present in SCA2 patient dentate neurons and Purkinje neurons. A2BP1 functions in RNA splicing suggested that ATXN2 may regulate alternative splicing in a tissue-specific manner or of a subset of RNAs. A2BP1 was the first RNA binding protein discovered to interact with ATXN2, and in simultaneous work in our research group, this led to our first attention on the Lsm and LsmAD domains located in the N-terminal region of ATXN2 [19] that are common in spliceosomal small nuclear ribonucleoproteins (snRNPs) and function in RNA binding and protein-protein interactions [33].
8.3.3 PABP & DDX6
Poly(A)-binding protein (PABP) interacts with the polyA end of mRNAs in the initiation of protein translation [26]. Other interactions made by PABP are facilitated by the 12 amino acid PABP-interacting motif 2 (PAM2) domain. A survey of multiple PAM2 proteins demonstrated that a PAM2 domain in ATXN2 and a high level of conservation of the PAM2 domain among the proteins [34]. A physical interaction between PABP and ATXN2 was demonstrated by yeast two hybrid testing and co-immunoprecipitation [26]. PABP is a component of mammalian stress granules, and ATXN2 and PABP colocalized in stress granules in heat-shock treated COS1 cells [26]. The study was the first to demonstrate the localization of ATXN2 to stress granules. The same research group further investigated ATXN2 in stress granules by characterizing its interaction with the DEAD/H-box RNA helicase (DDX6) [35]. DDX6 is a stress granule protein that like PABP is localized to stress granules as well as processing bodies (p -bodies). ATXN2 was shown to directly interact with DDX6 by way of the Lsm and LsmAD domains in ATXN2 by yeast two-hybrid interaction testing [27]. Upon identifying DDX6 as an ATXN2 interacting protein, the investigators further demonstrated that ATXN2 localized to both stress granules and processing bodies (p -bodies). ATXN2 also interacts with polyribosomes which are also known to be regulated by RNA granule formation [35].
Interaction between ATXN2 and PABP appears to connect functionally to the control of translation by ATXN2 involving mTOR signaling. Increased ATXN2 mRNA was observed in SH-SY5Y cells stressed by serum starvation [36]. The authors also showed sequestration of PABP and proteins of the cap-binding complex with ATXN2 in stress granules in mouse embryo fibroblasts (MEFs) stressed with arsenite [36]. A connection between ATXN2 and mTOR signaling was further confirmed by demonstrating increased phosphorylation of S6 and 4EBP1 in MEFs null for ATXN2, as well as elevated ATXN2 mRNA abundance in SH-SY5Y cells treated with the mTOR inhibitor rapamycin but not the PI3-kinase inhibitor LY294002 [36], suggesting the presence of a compensatory feed-back mechanism activating ATXN2 when mTOR is inhibited.
8.3.4 TDP-43 & FUS
Both TDP-43 and FUS are RBDs that are mutated in amyotrophic lateral sclerosis (ALS). The identification of ATXN2 as an interactor with TDP-43 was presented along with the discovery that moderate expansions in the ATXN2 gene CAG repeat are associated with increased risk for ALS [4]. More on ATXN2 and ALS is discussed below. The interaction between TDP-43 and ATXN2 was demonstrated by yeast two-hybrid interaction testing, and in HEK293 cells by coimmunoprecipitation (co-IP) of overexpressed GFP-TDP-43 fragments with endogenous ATXN2 and by immunofluorescent colocalization. In co-IP tests, including RNAse or including TDP-43 proteins mutated to abolish RNA binding, the TDP-43-ATXN2 interaction was abolished, demonstrating that the interaction is RNA dependent. FUS was also demonstrated to interact with ATXN2 [6]. Both TDP-43 and FUS have been characterized in RNA granules containing ATXN2 [37], suggesting a pathogenic connection for ATXN2 in increased ALS risk is associated with abnormal stress granule function.
8.3.5 Parkin
To investigate a functional connection that might explain why Parkinsonism is sometimes observed in SCA2, Huynh et al. [28] tested ATXN2 as an interacting protein with Parkin. Parkin directly interacted with the ATXN2 N-terminal domain (residues 1–396) when hemagglutinin (HA)-tagged Parkin was pulled down with an anti-HA antibody in HEK293 cells expressing GFP fused to full-length ATXN2 or N- or C-terminal fragments of ATXN2 [28]. The interaction was verified for ATXN2 proteins with Q22, as well as expanded polyglutamine tracts of Q58 and Q104. Parkin, an E3 ubiquitin ligase, ubiquitinated the full-length ATXN2 more efficiently than ATXN2 N-terminal fragments. ATXN2 ubiquitination by Parkin was more pronounced when ATXN2 was polyglutamine expanded but less efficient with Parkin-C289G mutated. The induced overexpression of Parkin in tetracycline inducible PC12 cells was associated with increased turnover of the ATXN2 protein. ATXN2 and Parkin colocalized in cytoplasmic structures of Purkinje cells from normal (non-SCA2) individuals [28]. The interaction between Parkin and ATXN2 was independently confirmed by coimmunoprecipition of polyglutamine expanded ATXN2 with Parkin from the cerebella of ATXN2-CAG42 knock-in mice [38]. Note that the latter study also demonstrated that the E3 ubiquitin ligase Fbxw8 also coimmunoprecipitated with ATXN2.
8.3.6 Staufen
Recently we demonstrated that ATXN2 interacts with Staufen1. Staufen is a key regulator of stress granule formation. We demonstrated that staufen expression is increased in SCA2 derived patient fibroblasts, lymphoblasts, iPSCs, and in the cerebella of our ATXN2-Q127 transgenic and our ATXN2-Q72 BAC mice. The result of elevated staufen expression in these systems is constitutively present stress granules. The identification of staufen as an interacting protein with ATXN2, whose expression is elevated upon ATXN2 mutation, demonstrates a functional role for ATXN2 in either staufen mediated decay, stress granule mediated mRNA processing or stress granule mediated dendritic mRNA trafficking for localized expression control.
8.3.7 RGS8 mRNA & IP3R
ATXN2 interacts with RGS8 mRNA and IP3R supporting roles for ATXN2 in calcium homeostasis. We determined that Rgs8 expression is reduced in the cerebella of SCA2 mice by transcriptome analysis, and verified RGS8 reduction in SCA2 patient lymphoblast cells. We also demonstrated that Rgs8 translation is reduced in the presence of mutant ATXN2 using rabbit reticulocyte in vitro translation assays. Thus, reduced Rgs8 could be the result of mRNA degradation, as well as RGS8 mRNA translation inhibition perhaps mediated by sequestration in stress granules. RGS8 inhibition could impact calcium levels in Purkinje cells , since RGS8 is believed to be an inhibitor of mGluR1. The role of mGlur1 in the normal functioning of Purkinje neuron and motor coordination is well described in a review by Hartmann et al. [39]. In Purkinje cells , mGluRs produce two distinct signals including a local dendritic Ca2+ signal and a slow excitatory postsynaptic potential. The dendritic Ca2+ signal originates through Ca2+ release from the ER mediated by the inositol-triphosphate receptor type 1 (IP3R). The slow excitatory postsynaptic potentials are mediated by Ca2+ influx, via the transient receptor potential cation channel 3 (TRPC3) that is gated by diacylglycerol (DAG) and IP3R [39]. IP3R is abnormally activated upon interaction by mutant ATXN2, resulting in abnormal release of Ca2+ from intracellular stores [29]. The Bezprozvanny group verified that IP3R specifically interacts with the polyglutamine expanded ATXN2 protein but not the normal ATXN2 protein [29]. The interaction was demonstrated between endogenous ATXN2 and overexpressed GST-IP3R in COS7 cells using a pull-down assay. A second assay using cerebellar homogenates from Pcp2-ATXN2-Q58 mice demonstrated that polyglutamine expanded ATXN2 co-precipitated with greater abundance of radiolabeled IP3 than did wildtype ATXN2, consistent with an interaction between mutant ATXN2 and IP3R [29]. Further experiments on modulating IP3R function for modifying SCA2 mouse phenotypes are described below.
8.3.8 Endophilins and CIN85
Endophilin and CIN85 are proteins that function along with Cbl in endocytosis of cell surface receptor tyrosine kinases [40]. The Huntington disease protein huntingtin , another polyglutamine disease protein, interacts with endophilin A3 resulting in abnormal sequestration of proteins of endocytic vesicle systems [41, 42]. Therefore, to test whether ATXN2 could interact with endophilins, Ralser et al. [31] performed two-hybrid interaction tests that demonstrated ATXN2 direct binding with both endophilin A1 and endophilin A3. The interaction was mediated by the SH3 domain binding motif 2 (SBM2) in ATXN2, and the investigators also showed that ATXN2 failed to interact with endophilin A2. The study also demonstrated competitive binding between ATXN2 and huntingtin for endophilin A3 in the yeast two-hybrid system. Another study investigated ATXN2 interactions with endophilin A1 and endophilin A3 by GST pull-down tests showing that the endophilins interacted with other ATXN2 protein regions not including the SBM2 domain [32]. Extensive cytoplasmic colocalization of ATXN2 with endophilins A1 and A3 was also demonstrated by immunofluorescent labeling of HEK293 and SH-SY5Y cells [31, 32]. Co-immunoprecipitation assays demonstrated ATXN2 exists in complexes containing endophilin A3, CIN85, Cbl, and EGF receptor (EGFR) [32]. Overexpression of ATXN2 in CHO cells inhibited EGF-stimulated EGFR internalization, demonstrating a functional role of ATXN2 in endocytosis [32]. Endocytosis controlled by the Endophilin-CIN85-Cbl complex is mediated by clathrin-coated pits. A putative site for clathrin binding in ATXN2 was described by Huynh et al. [20] at aa 414–416. However Turnbull et al. [22] could demonstrate no co-localization between ATXN2 and clathrin-coated pits or vesicles.
8.3.9 Other ATXN2 Interaction Studies
Various studies have demonstrated proteins with which ATXN2 coimmunoprecipitated without formally testing direct interactions. Discussed briefly in the PABP paragraph above, Lastres-Becker et al. [36] demonstrated that ATXN2 coimmuniprecipitated with TIA1, eIF3B, eIF4G, eIF4A1 and S6 from HEK293 cells treated with or without arsenite. Blokhuis et al. [43] characterized the ATXN2 interactome in Neuro2A cells using mass spectrometry with validations performed by coimmunoprecipitation. Key interacting proteins verified in coimmunoprecipitation experiments included Fmrp, Upf1, Caprin1, HuD, Pabpc4, and Dhx9. The investigators also produced interactomes for Fus and Tdp43 and presented interactions shared among these proteins and ATXN2 [43].
Studies of the ATXN2 yeast homolog Pbp1 suggest other proteins likely to interact with the ATXN2 protein. The PAS kinase 1 (Psk1) was shown to interact with the C-terminal half of Pbp1 resulting in Pbp1 phosphorylation proximal to the interaction [44]. Another study of Pbp1 demonstrated interactions with Lsm12 and Pbp4 in addition to the yeast homologs of PABP and DDX6 [45].
8.4 SCA2 Mouse Models
We and others have produced multiple SCA2 mouse models, including transgenic and knockout models [20, 30, 46, 47, 48, 49, 50, 51]. In this section we describe these mice. Note that a recent review comprehensively describes each of these mice [52].
8.4.1 Pcp2-ATXN2 Transgenic Mice
We have made two types of Pcp2-ATXN2 transgenic mice, including one with ATXN2-CAG58 (Q58) and another with ATXN2-CAG127 (Q127). Both of these mice have ATXN2 expressed under the control of the Purkinje cell protein 2 (Pcp2)/L7 promoter. These mice are characterized by age-dependent molecular, motor and electrophysiological phenotypes. Rotarod testing demonstrated ATXN2-dose dependent motor phenotype for ATXN2-Q58 mice first observed at six months of age, and Purkinje cells in these mice contained cytoplasmic, but not nuclear, inclusion bodies [20]. The ATXN2-Q58 mouse was also used in studies demonstrating dantrolene treatment could restore ATXN2 mouse motor phenotypes [29]. This is discussed in further detail in the section below, on calcium homeostasis. ATXN2-Q127 mice also has Purkinje cells with cytoplasmic inclusion bodies, but with the longer repeat length we have observed the motor phenotype as early as eight weeks of age [46]. The Auburger group has also created an ATXN2-CAG42 knock-in mouse by replacing the single CAG in the mouse Atxn2 gene with an expanded CAG42 repeat [50]. The ATXN2-CAG42 mouse was characterized for how mutant ATXN2 alters PABPC1 solubility and availability for functions in RNA metabolism.
8.4.2 SCA2 BAC Transgenic Mice
SCA2 BAC mice possess the entire 176 kb ATXN2 gene region including 16 kb upstream sequence and 2.5 kb downstream sequence. Presently we have two SCA2 BAC lines including alleles expressing ATXN2-Q22 normal length and ATXN2-Q72 expanded [30]. The Q22 line has no motor, transcriptomic or neurophysiological phenotype. However, the Q72 line has a progressive onset of its motor phenotype, determined using the accelerating rotarod that is mimicked by progressive reduction of the expression of various neuronal and Purkinje cell specific genes, beginning at 8 weeks of age [30]. More recently, we have identified changes in Purkinje cell firing frequencies in the SCA2-Q72 BAC mice, compared to wildtype littermates, present for mice age 6 and 12 months but not mice 4 months of age (unpublished observation). This demonstrates that the neurophysiological phenotype of the BAC-Q72 mice appears later than for the ATXN2-Q127 mice, mirroring the later motor phenotype observed in the BAC-Q72 mice. The delayed onset of SCA2 phenotypes in the BAC-Q72 mice is due to the lower expression from the native ATXN2 promoter, as well as the shorter Q72 repeat compared to the ATXN2-Q127 mouse. Cerebellar molecular phenotype changes determined by qPCR and RNA -seq were largely similar to those observed in ATXN2-Q127 mice [30, 46].
8.4.3 ATXN2-Q75 Transgenic Mice
ATXN2-Q75 mice are transgenic for the ATXN2 cDNA under transcriptional control by the native ATXN2 promoter, and include ATXN2 with 75 CAG repeats [51]. Ubiquitous transgene expression was observed, and hemizygous mice were ataxic by 12 weeks of age in rotarod tests, corresponding with abnormal Purkinje cell morphology.
8.4.4 Atxn2 Knockout Mice
Our group and the Auburger group have both produced Atxn2 knockout mice and we have demonstrated key characteristics that are common to both, which include viable mice with marked obesity and the lack of any significantly debilitating neuropathology [48, 49]. We have also demonstrated normal Purkinje cell physiology in Atxn2 knockout mice [53], but these mice are also characterized by abnormal fear-related behavior [47]. The Auburger group demonstrated that Atxn2 knockout mice had abnormally low insulin receptor expression in both the cerebellar and the liver and concluded that these molecular changes associate with the onset of obesity [49]. These investigators further evaluated their A2 knockout mouse employing microarray analysis revealing increased expression of transcription factors but overall lower translation [54]. The lack of neuropathology in these mice supports the concept for developing SCA2 therapeutics that target the total expression of ATXN2, such as we are with ATXN2 compounds and ATXN2 antisense oligonucleotides (ASOs), described below.
8.5 Transcriptome Analyses
Multiple studies on the cerebellar transcriptomes have been conducted using SCA2 mice. In our initial study we used cerebellar RNAs isolated from wildtype and BAC-ATXN2-Q72 mice at ages 1 day, 3 weeks and 6 weeks. In day 1 mice we observed ~200 transcripts that were significantly dysregulated, and more transcripts were altered in the older animals [30]. Most transcripts were reduced in abundance in SCA2 mice as compared to wildtype mice. We also performed transcriptome analysis using Pcp2-ATXN2-Q127 mice for the purpose of identifying pathways commonly modified in these mice and BAC-ATXN2-Q127 mice [30]. It was these studies that resulted in the identification of significant reductions in the RGS8 mRNA as described above. Other genes that were significantly reduced in both of these SCA2 mouse models included Pcp2, Fam107b and others. Pathways that we identified that are altered in both Pcp2-ATXN2-Q127 and BAC-ATXN2-Q127 mice include glutamate signaling, calcium signaling and others [30]. We have also begun to compare the transcriptomes of these mice with that of age-matched knockout mice. Unlike the transcriptomes in the SCA2 mice we observed that few transcripts were altered in the A txn2 knockout mice (unpublished observations). Similarly, the Auburger group compared transcriptomes of ATXN2-CAG42 transgenic mice with A txn2 knockout mice using a microarray analysis approach, demonstrating overlapping abnormalities in calcium homeostasis pathways [55].
8.6 SCA2 Therapeutics
Despite presence of many ATXN2 interactors, we still lack information on which ATXN2 functions are targetable to prevent SCA2 pathogenesis or to delay SCA2 progression. The concept of two different approaches for developing SCA2 therapeutics discussed here, closely follows that developed in a recent review on precision medicine for the spinocerebellar ataxias [56]. One approach is to develop the known functions for ATXN2 as therapeutic targets for SCA2, including glutamate signaling, calcium homeostatsis, and RNA metabolism (Fig. 8.3). Another promising approach is to target ATXN2 expression directly, because, like for other SCAs caused by mutations leading to polyglutamine expansions, SCA2 is characterized predominantly by a toxic gain of function. We are making efforts to develop SCA2 therapeutics that target ATXN2 functionally and we are also developing antisense oligonucleotides that lower overall ATXN2 expression.

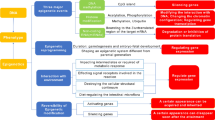
Pathogenic outcomes of ATXN2 mutation. Polyglutamine expansion in ATXN2 results in misfolding and increased staufen1 (STAU1) expression and ATXN2 aggregations leading to stress granule formation and abnormal RNA processing. Misfolded ATXN2 interacts directly with IP3R leading to abnormal IP3R channel activity followed by calcium release from internal stores. ATXN2 also directly interacts with RGS8 mRNA resulting in reduced RGS8 protein abundance. RGS8 reduction is attributed to decreased RGS8 translation which may be caused by sequestration in stress granules, and to decreased RGS8 mRNA abundance possibly also related to stress granule functions. The consequence of reduced RGS8 is overactive mGluR1 leading to increased cytoplasmic calcium. The result of is abnormally slow Purkinje cell firing
8.6.1 Therapies Targeting ATXN2-Related SCA2 Pathways
The approach that we have taken for developing therapeutics that target ATXN2-related SCA2 pathways is to target pathways leading to abnormal rise of cytoplasmic Ca2+. These efforts were initiated upon the identification that the mutant, but not the wildtype, ATXN2 protein interacted with the inositol-triphosphate receptor type 1 (ITPR1), also described above [29]. Targeting the Ca2+ pathway in SCA2 is in line with the notion that defective Ca2+ signaling underlies most neurodegenerative diseases [57]. ITPR1 mutations or haploinsufficiency are also causative for SCA15/16 [58, 59]. ITPR1 is a calcium channel located on the endoplasmic reticulum membrane controlling the release of intracellular Ca2+ stores, and is expressed highly in PCs. Mice harboring ATXN2-Q58 were characterized with increased Ca2+ release from the endoplasmic reticulum associated with molecular layer thinning and Purkinje cell loss [20, 29]. Blocking of the functionally coupled ryanodine receptor with dantrolene reduced abnormal calcium release and cell death in culture [29]. Liu et al. [29] also demonstrated that SCA2 motor phenotypes of ATXN2-Q58 mice, as well as improved Purkinje cell survival was delayed by oral treatment of ATXN2-Q58 mice with dantrolene. Tests included the beam walk and accelerating rotarod.
We have also begun to investigate mGlur1 as a therapeutic target for SCA2. As described above, we demonstrated that RGS8 expression is reduced in an age-dependent manner in SCA2 mice. RGS8 is a putative regulator of mGlur1 and its reduced expression is predicted to deregulate mGlur1 [30]. We have now used our Pcp2-ATXN2-Q127 model to replicate the in vitro findings and show that the mGlur1 agonist DHPG enhances firing frequency of Pcp2-ATXN2-Q127 mouse PCs accompanied by abnormally elevated intracellular Ca2+ at specific PC firing rates [60]. ATXN2 expression itself may be regulated by intracellular calcium and mGluR1. These data suggest that mGlur1 antagonists could be therapeutic for SCA2.
8.6.2 ATXN2 ASO Therapeutics
Antisense oligonucleotides (ASOs) represent a promising approach for treating SCA2. SCA2 is characterized by a gain of toxic function, thus we hypothesize that lowering ATXN2 expression would be therapeutic for SCA2. Reducing total expression as a therapeutic approach is supported for polyQ diseases by several observations. In SCA2 patients two copies of the mutant ATXN2 allele can be accompanied by earlier age of onset and more rapid disease progression [61]. The importance of gene dosage is further supported by studies on mice. Doubling of gene dosage in transgenic ATXN2-Q58 mice led to earlier onset of abnormal rotarod performance [20]. Studies using tetracycline-regulated promoters in HD, SCA1 or SCA3 mice have demonstrated reversibility of phenotypes even after disease onset [62,61,62,65]. Another study showed that intracerebellar injection of AAV virus encoding shRNA Ataxin-1 reduced transgene expression, improved motor coordination, restored cerebellar morphology and resolved ataxin-1 inclusions in Purkinje neurons (PNs) of SCA1 mice [66]. Recently, ASOs have proven useful for the treatment of spinal muscular atrophy and SOD-ALS [67, 68], and newer phase 1 clinical trials have been initiated using ASOs for the treatment of myotonic dystrophy (DM1) and Huntington disease.
Our ASO approach to therapeutics is being conducted collaboratively with Ionis Pharmaceuticals utilizing modified 2′-MOE-gapmer ASOs. The 2′-MOE-gapmer are 20 bp in length, are phosphorothioate throughout, and have a 2′-O-methoxyethyl group (MOE) on the terminal 5 bps at each end of the oligonucleotide [69]. These modifications prevent degradation by nucleases and the MOE chemical modifications also aid in increasing specificity of target mRNA interaction, supporting target degradation by RNase-H [70].
The SCA2 ASOs that we are developing are for non-allele-specific targeting of ATXN2 unlike the approach undertaken for Huntington’s disease . In Huntington’s disease ASOs are made to target SNPs in linkage with CAG repeat expanded alleles [71, 72] because Htt knockout in mice disrupts neuronal development [73]. In our approach we permit the ASO to target the mutant ATXN2 allele as well as the non-mutant allele because knockout of the Atxn2 gene in mice is well tolerated and associated with no neurodegeneration [47, 48]. However, progress on developing non-allele-specific RNAi therapeutics for HD had favorable outcomes in mice and non-human primates [74, 75].
In collaboration with Ionis Pharmaceuticals we have produced ATXN2 ASOs that lower human ATXN2 expression in both our Pcp2-ATXN2-Q127 mouse model, as well as our BAC-ATXN2-Q72 mouse model. We have observed as much as 80% ATXN2 mRNA reduction when delivered to mice by introcerebroventricular (ICV) injection to the right lateral ventricle, and we have observed no cytotoxicity indicated by following AIF1 and GFAP expression post injection. We have employed our most promising lead ASO, designated ASO7, in a blinded preclinical treatment trial using Pcp2-ATXN2-Q127 mice (Fig. 8.4). Mice treated at 8 weeks of age were tested at different treatment timepoints, on the accelerating rotarod, demonstrating delayed progression of the SCA2 motor phenotypes in both mouse models. At the endpoint we determined the cerebellar expression of ATXN2, Rgs8 and Pcp2 by qPCR and Western blotting demonstrating prolonged ATXN2 reduction and increases in Rgs8 and Pcp2 expression. Moreover, subsets of mice were tested to determine the effect of the ASO7 treatments on Purkinje cell physiology. We observed that ASO7 treatment could restore the mean PC firing frequency to that unlike observed in age matched mice. The result of our ATXN2 ASO study was recently published [76]. Finally, we also have ongoing studies to characterize the transcriptomes of SCA2 mice treated with or without ATXN2 ASO7. Information resulting from the latter work might indicate new pathways that can be exploited for the treatment of ALS associated with ATXN2. This is supported by a recent finding that lowering Atxn2 expression either genetically or by ASO therapy improved survival of TDP-43 ALS mice [77].

Reduction of ATXN2 expression improved SCA2 mouse phenotypes. a Compared with saline-treated animals, spinocerebellar ataxia type 2 mice (Pcp2-ATXN2-Q127) show significantly reduced progression of motor disability. Mean latency to fall in 3 trials on day 3 of rotarod testing is shown. b Cerebellar endogenous mouse and human transgenic ataxin-2 mRNA (ATXN2) are reduced by ASO treatment 14 weeks after intracerebroventricular (ICV) injection compared with saline. NS, not significant
8.7 Conclusions
SCA2 is a debilitating disorder for which there is no treatment. Research by numerous teams on SCA2 has resulted in the identification of multiple interacting proteins that have given rise to clues about ATXN2 function. Additionally, multiple SCA2 transgenic mouse models and Atxn2 knockout models have been studied giving rise to understanding on pathways dysregulated in SCA2 mice. Collectively, these studies have aided understanding on ATXN2 function and have indicated possible pathways that can be targeted in order to delay SCA2 progression. Progress toward developing drugs targeting calcium signaling and related pathways is accumulating. But even with this increased knowledge of ATXN2 function, the opportunity to target ATXN2 directly using antisense oligonucleotides remains a primary goal of our research group for treating SCA2, garnered by positive preliminary data in SCA2 mice and recent data demonstrating that ASOs are tolerated in humans and effective for SMA and SOD ALS. Preliminary data demonstrating that SCA2 ASOs lower ATXN2 expression in mouse spinal cord also lend hope toward developing ATXN2 ASOs for SCA2 ALS and perhaps ALS in a more generalized manner.
References
Geschwind DH, Perlman S, Figueroa CP, Treiman LJ, Pulst SM (1997) The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am J Hum Genet 60:842–850
Ashizawa T, Figueroa KP, Perlman SL, Gomez CM, Wilmot GR, Schmahmann JD et al (2013) Clinical characteristics of patients with spinocerebellar ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet J Rare Dis 8:177
Gwinn-Hardy K, Chen JY, Liu HC, Liu TY, Boss M, Seltzer W et al (2000) Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology 55:800–805
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X et al (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466:1069–1075
Neuenschwander AG, Thai KK, Figueroa KP, Pulst SM (2014) Amyotrophic lateral sclerosis risk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis. JAMA Neurol 71:1529–1534
Farg MA, Soo KY, Warraich ST, Sundaramoorthy V, Blair IP, Atkin JD (2013) Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum Mol Genet 22:717–728
Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M et al (2016) Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J
Fischbeck KH, Pulst SM (2011) Amyotrophic lateral sclerosis and spinocerebellar ataxia 2. Neurology 76:2050–2051
Wadia NH, Swami RK (1971) A new form of heredo-familial spinocerebellar degeneration with slow eye movements (nine families). Brain 94:359–374
Pulst SM, Santos N, Wang D, Yang H, Huynh D, Velazquez L et al (2005) Spinocerebellar ataxia type 2: polyQ repeat variation in the CACNA1A calcium channel modifies age of onset. Brain 128:2297–2303
Velazquez-Perez L, Rodriguez-Labrada R, Canales-Ochoa N, Montero JM, Sanchez-Cruz G, Aguilera-Rodriguez R et al (2014) Progression of early features of spinocerebellar ataxia type 2 in individuals at risk: a longitudinal study. Lancet Neurol 13:482–489
Pulst SM, Nechiporuk A, Starkman S (1993) Anticipation in spinocerebellar ataxia type 2. Nat Genet 5:8–10
Gispert S, Twells R, Orozco G, Brice A, Weber J, Heredero L et al (1993) Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1. Nat Genet 4:295–299
Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I et al (1996) Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 14:269–276
Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H et al (1996) Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 14:277–284
Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM et al (1996) Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 14:285–291
Fernandez M, McClain ME, Martinez RA, Snow K, Lipe H, Ravits J et al (2000) Late-onset SCA2: 33 CAG repeats are sufficient to cause disease. Neurology 55:569–572
Scoles DR, Pflieger LT, Thai KK, Hansen ST, Dansithong W, Pulst SM (2012) ETS1 regulates the expression of ATXN2. Hum Mol Genet 21:5048–5065
Huynh DP, Del Bigio MR, Ho DH, Pulst SM (1999) Expression of ataxin-2 in brains from normal individuals and patients with Alzheimer’s disease and spinocerebellar ataxia 2. Ann Neurol 45:232–241
Huynh DP, Figueroa K, Hoang N, Pulst SM (2000) Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat Genet 26:44–50
Koyano S, Uchihara T, Fujigasaki H, Nakamura A, Yagishita S, Iwabuchi K (1999) Neuronal intranuclear inclusions in spinocerebellar ataxia type 2: triple-labeling immunofluorescent study. Neurosci Lett 273:117–120
Turnbull VJ, Storey E, Tarlac V, Walsh R, Stefani D, Clark R et al (2004) Different ataxin-2 antibodies display different immunoreactive profiles. Brain Res 1027:103–116
Huynh DP, Yang HT, Vakharia H, Nguyen D, Pulst SM (2003) Expansion of the polyQ repeat in ataxin-2 alters its Golgi localization, disrupts the Golgi complex and causes cell death. Hum Mol Genet 12:1485–1496
Aguiar J, Santurlidis S, Nowok J, Alexander C, Rudnicki D, Gispert S et al (1999) Identification of the physiological promoter for spinocerebellar ataxia 2 gene reveals a CpG island for promoter activity situated into the exon 1 of this gene and provides data about the origin of the nonmethylated state of these types of islands. Biochem Biophys Res Commun 254:315–318
Shibata H, Huynh DP, Pulst SM (2000) A novel protein with RNA-binding motifs interacts with ataxin-2. Hum Mol Genet 9:1303–1313
Ralser M, Albrecht M, Nonhoff U, Lengauer T, Lehrach H, Krobitsch S (2005) An integrative approach to gain insights into the cellular function of human ataxin-2. J Mol Biol 346:203–214
Nonhoff U, Ralser M, Welzel F, Piccini I, Balzereit D, Yaspo ML et al (2007) Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell 18:1385–1396
Huynh DP, Nguyen DT, Pulst-Korenberg JB, Brice A, Pulst SM (2007) Parkin is an E3 ubiquitin-ligase for normal and mutant ataxin-2 and prevents ataxin-2-induced cell death. Exp Neurol 203:531–541
Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP et al (2009) Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci 29:9148–9162
Dansithong W, Paul S, Figueroa KP, Rinehart MD, Wiest S, Pflieger LT et al (2015) Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet 11:e1005182
Ralser M, Nonhoff U, Albrecht M, Lengauer T, Wanker EE, Lehrach H et al (2005) Ataxin-2 and huntingtin interact with endophilin-A complexes to function in plastin-associated pathways. Hum Mol Genet 14:2893–2909
Nonis D, Schmidt MH, van de Loo S, Eich F, Dikic I, Nowock J et al (2008) Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell Signal 20:1725–1739
Hermann H, Fabrizio P, Raker VA, Foulaki K, Hornig H, Brahms H et al (1995) snRNP Sm proteins share two evolutionarily conserved sequence motifs which are involved in Sm protein-protein interactions. EMBO J 14:2076–2088
Albrecht M, Lengauer T (2004) Survey on the PABC recognition motif PAM2. Biochem Biophys Res Commun 316:129–138
Satterfield TF, Pallanck LJ (2006) Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet 15:2523–2532
Lastres-Becker I, Nonis D, Eich F, Klinkenberg M, Gorospe M, Kotter P et al (2016) Mammalian ataxin-2 modulates translation control at the pre-initiation complex via PI3 K/mTOR and is induced by starvation. Biochim Biophys Acta 1862:1558–1569
Nihei Y, Ito D, Suzuki N (2012) Roles of ataxin-2 in pathological cascades mediated by TAR DNA-binding protein 43 (TDP-43) and Fused in Sarcoma (FUS). J Biol Chem 287:41310–41323
Halbach MV, Stehning T, Damrath E, Jendrach M, Sen NE, Basak AN et al (2015) Both ubiquitin ligases FBXW8 and PARK2 are sequestrated into insolubility by ATXN2 PolyQ expansions, but only FBXW8 expression is dysregulated. PLoS ONE 10:e0121089
Hartmann J, Henning HA, Konnerth A (2011) mGluR1/TRPC3-mediated synaptic transmission and calcium signaling in mammalian central neurons. Cold Spring Harb Perspect Biol 3
Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I (2002) Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 416:183–187
Sittler A, Walter S, Wedemeyer N, Hasenbank R, Scherzinger E, Eickhoff H et al (1998) SH3GL3 associates with the Huntingtin exon 1 protein and promotes the formation of polygln-containing protein aggregates. Mol Cell 2:427–436
Qin ZH, Wang Y, Sapp E, Cuiffo B, Wanker E, Hayden MR et al (2004) Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J Neurosci 24:269–281
Blokhuis AM, Koppers M, Groen EJ, van den Heuvel DM, Dini Modigliani S, Anink JJ et al (2016) Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta Neuropathol 132:175–196
DeMille D, Badal BD, Evans JB, Mathis AD, Anderson JF, Grose JH (2015) PAS kinase is activated by direct SNF1-dependent phosphorylation and mediates inhibition of TORC1 through the phosphorylation and activation of Pbp1. Mol Biol Cell 26:569–582
Swisher KD, Parker R (2010) Localization to, and effects of Pbp1, Pbp4, Lsm12, Dhh1, and Pab1 on stress granules in Saccharomyces cerevisiae. PLoS ONE 5:e10006
Hansen ST, Meera P, Otis TS, Pulst SM (2013) Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet 22:271–283
Huynh DP, Maalouf M, Silva AJ, Schweizer FE, Pulst SM (2009) Dissociated fear and spatial learning in mice with deficiency of ataxin-2. PLoS ONE 4:e6235
Kiehl TR, Nechiporuk A, Figueroa KP, Keating MT, Huynh DP, Pulst SM (2006) Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem Biophys Res Commun 339:17–24
Lastres-Becker I, Brodesser S, Lutjohann D, Azizov M, Buchmann J, Hintermann E et al (2008) Insulin receptor and lipid metabolism pathology in ataxin-2 knock-out mice. Hum Mol Genet 17:1465–1481
Damrath E, Heck MV, Gispert S, Azizov M, Nowock J, Seifried C et al (2012) ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet 8:e1002920
Aguiar J, Fernandez J, Aguilar A, Mendoza Y, Vazquez M, Suarez J et al (2006) Ubiquitous expression of human SCA2 gene under the regulation of the SCA2 self promoter cause specific Purkinje cell degeneration in transgenic mice. Neurosci Lett 392:202–206
Alves-Cruzeiro JM, Mendonca L, Pereira de Almeida L, Nobrega C (2016) Motor dysfunctions and neuropathology in mouse models of spinocerebellar ataxia type 2: a comprehensive review. Front Neurosci 10:572
Pflieger LT, Dansithong W, Paul S, Scoles DR, Figueroa KP, Meera P, Otis TS, Facelli JC, Pulst SM (2017) Gene co-expression network analysis for identifying modules and functionally enriched pathways in SCA2. Hum Mol Genet 26(16):3069–3080
Fittschen M, Lastres-Becker I, Halbach MV, Damrath E, Gispert S, Azizov M et al (2015) Genetic ablation of ataxin-2 increases several global translation factors in their transcript abundance but decreases translation rate. Neurogenetics 16:181–192
Halbach MV, Gispert S, Stehning T, Damrath E, Walter M, Auburger G (2016) ATXN2 Knockout and CAG42-Knock-in cerebellum shows similarly dysregulated expression in calcium homeostasis pathway. Cerebellum
Bushart DD, Murphy GG, Shakkottai VG (2016) Precision medicine in spinocerebellar ataxias: treatment based on common mechanisms of disease. Ann Transl Med 4:25
Bezprozvanny IB (2010) Calcium signaling and neurodegeneration. Acta Naturae 2:72–82
van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR et al (2007) Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet 3:e108
Iwaki A, Kawano Y, Miura S, Shibata H, Matsuse D, Li W et al (2008) Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J Med Genet 45:32–35
Meera P, Pulst S, Otis T (2017) A positive feedback loop linking enhanced mGluR function and basal calcium in spinocerebellar ataxia type 2. Elife 6. pii:e26377
Ragothaman M, Muthane U (2008) Homozygous SCA 2 mutations changes phenotype and hastens progression. Mov Disord 23:770–771
Yamamoto A, Lucas JJ, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 101:57–66
Boy J, Schmidt T, Wolburg H, Mack A, Nuber S, Bottcher M et al (2009) Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3. Hum Mol Genet 18:4282–4295
Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB et al (2004) Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci 24:8853–8861
Oz G, Vollmers ML, Nelson CD, Shanley R, Eberly LE, Orr HT et al (2011) In vivo monitoring of recovery from neurodegeneration in conditional transgenic SCA1 mice. Exp Neurol 232:290–298
Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT et al (2004) RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med 10:816–820
Hache M, Swoboda KJ, Sethna N, Farrow-Gillespie A, Khandji A, Xia S et al (2016) Intrathecal injections in children with spinal muscular atrophy: nusinersen clinical trial experience. J Child Neurol
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH et al (2013) An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 12:435–442
Rigo F, Seth PP, Bennett CF (2014) Antisense oligonucleotide-based therapies for diseases caused by pre-mRNA processing defects. Adv Exp Med Biol 825:303–352
Bennett CF, Swayze EE (2010) RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Ann Rev Pharmacol Toxicol 50:259–293
Skotte NH, Southwell AL, Ostergaard ME, Carroll JB, Warby SC, Doty CN et al (2014) Allele-specific suppression of mutant Huntingtin using antisense oligonucleotides: providing a therapeutic option for all Huntington disease patients. PLoS ONE 9:e107434
Ostergaard ME, Southwell AL, Kordasiewicz H, Watt AT, Skotte NH, Doty CN et al (2013) Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic Acids Res 41:9634–9650
White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL et al (1997) Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet 17:404–410
McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR et al (2011) Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther 19:2152–2162
Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ et al (2009) Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther 17:1053–1063
Scoles DR, Meera P, Schneider MD, Paul S, Dansithong W, Figueroa KP, Hung G, Rigo F, Bennett CF, Otis TS, Pulst SM (2017) Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 544(7650):362–366
Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, Pulst SM, Taylor JP, Rigo F, Gitler AD (2017) Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544(7650):367–371
Acknowledgements
We thank Duong P. Huynh and Sharan Paul for editing the chapter. We thank Darren Ames and Lance Pflieger for assistance with transcriptome analysis computations.
Funding
This work was supported by the Carmen and Louis Warschaw Endowment Fund, the National Ataxia Foundation, grants R01NS33123, R56NS33123 and R37NS033123 from the National Institutes of Neurological Disorders and Stroke to SMP, the Noorda foundation to SMP, and grants RC4NS073009 and R21NS081182 to DRS and SMP. SMP received grant support from the Target ALS Foundation.
Conflict of Interest Statement
SMP is a consultant for Progenitor Life Sciences.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Scoles, D.R., Pulst, S.M. (2018). Spinocerebellar Ataxia Type 2. In: Nóbrega, C., Pereira de Almeida, L. (eds) Polyglutamine Disorders. Advances in Experimental Medicine and Biology, vol 1049. Springer, Cham. https://doi.org/10.1007/978-3-319-71779-1_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-71779-1_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-71778-4
Online ISBN: 978-3-319-71779-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)