
Abstract
The field of tumor immunology and immunotherapy has undergone a renaissance in the past decade do in large part to a better understanding of the tumor immune microenvironment. After suffering countless successes and setbacks in the twentieth century, immunotherapy has now come to the forefront of cancer research and is recognized as an important tool in the anti-tumor armamentarium. The goal of therapy is to aid the immune system in recognition and destruction of tumor cells by enhancing its ability to react to tumor antigens. This traditionally has been accomplished by induction of adaptive immunity through vaccination or through passive delivery of immunologic effectors as in the case of adoptive cell transfer. The recent discovery of immune “checkpoints” whose purpose is to suppress immune activity and prevent auto-immunity has created a new angle by which reactivity to tumors can be enhanced. Blockers of these checkpoints have yielded impressive clinical results and have recently been approved for use in a wide variety of malignancies. With data showing increasing rates of not only treatment response, but complete remissions, immunotherapy is poised to become an increasingly utilized therapy in the treatment of cancer.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Introduction
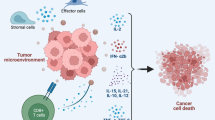
In the past decade, immunotherapy has undergone a metamorphosis, transforming from complex experimental protocols to “off-the-shelf” first line therapy for many previously untreatable malignancies. Unlike traditional chemotherapeutics which target biologic processes of cancer cells, immunotherapy seeks to boost the bodies natural immunologic defense against cancer [1, 2]. This is accomplished by either training resident immune cells to recognize and eliminate cells bearing tumor specific antigens, providing external stimuli to enhance immune mediated tumor cell lysis or abrogating signals directed by tumor cells to dampen immune responsiveness. Both cellular and molecular components of the tumor microenvironment (TME) can serve to impair the efficacy of immunotherapy and strategies to abrogate this are the source of on-going research [3].
The principle components of immune based tumor ablation are T-lymphocytes (T-cells) and natural killer cells (NK cells), which target cells by antigen specific and non-specific means, respectively. The process of antigen specific immune recognition is a complex one that involves orchestrated steps from both the cell presenting the antigen and the lymphocyte which recognizes it. Self and foreign proteins are digested by proteasomes in the cytoplasm to form 8 to 9 amino acid peptides which are transported to the endoplasmic reticulum. They are loaded by chaperones onto major histocompatibility complex (MHC) class I proteins and the pair is transported to the cell surface for display. Once on the surface, a T-cell bearing a receptor specific for a given MHC class I-peptide complex can bind and with the help of co-stimulatory molecules, trigger activation of the immune cell. This physiologic process is often dysfunctional within the TME as cells lose key components of antigen breakdown and processing rendering cancers effectively hidden to immune cells [4]. An example of this is the defect in MHC surface expression and antigen display on dendritic cells in the TME of head and neck cancers and other malignancies [5, 6].
In addition to impaired processing and display, production of antigen themselves can be down-regulated by epigenetic silencing in the TME [7]. Alterations in DNA promoter methylation and histone modifications have been implicated in repression of expression of key tumor specific antigens including cancer-testes antigens [8, 9]. Efforts to enhance immunotherapy by altering epigenetic pathways with the goal of enhancing antigen expression have been met with variable success [10,11,12].
The theory of immune surveillance posits that the body is composed of near countless numbers of T-cells with a vast array of receptors capable of recognizing a wide variety of infectious and cancerous antigens and initiating immune activation [13, 14]. When activated by either a primary transformed cell or via an intermediary cell such as an antigen-presenting cell (APC) within the TME, T-cells begin a cascade of signaling events that results in recruitment of cellular and non-cellular immune components, clonal expansion of antigen specific T-cells and release of stimulatory cytokines. The end result is a local accumulation of pro-inflammatory cells and destruction of the cancerous or infected cell. We now know, however, that despite this complex network of cellular signaling, tumor cells often evade immune detection leading to growth and eventual spread [15,16,17,18,19,20,21,22].
Failure of immune-editing can be attributed to one of five phenomena: (1) lack of recognition by T-cell receptors (TCR) [23, 24], (2) lack of sufficient activation in response to T-cell recognition, (3) failure of clonal expansion of antigen specific T-cells [25], (4) suppression of immune activation by tumor bearing inhibitors of co-stimulation [26], and (5) repression of activation by inhibitory immune cells within the TME [27, 28]. In this chapter we will discuss strategies developed to overcome these failures with the aim of subverting immunosuppression and enhancing the immunologic destruction of cancer cells [29,30,31,32].
4.2 History
The birth of modern day immunotherapy traces back to renowned New York surgeon William B. Coley in the early 1890s [33, 34]. He inoculated the unresectable sarcoma of a young man with cultures of erysipelas and noted a dramatic reduction in tumor size [35]. Subsequent to this, he created a mixture of filtered bacteria and lysates composed primarily of Streptococcus pyogenes and Bacillus prodigiosus which he termed “Coley’s Toxins.” In 1893, he published a report of 10 patients treated with his concoction, many of whom experienced tumor reduction [36]. An interesting observation was that, anecdotally, the severity of infectious symptoms seemed to correlate with degree of response. Over the next four decades, Dr. Coley treated close to 1000 patients with his toxin and reported a 10% complete response rate [37]. While his results were unprecedented, they were met with significant skepticism. Many in the scientific and medical communities derided his lack of supporting mechanistic data and noted his therapy was associated with significant toxicity and results difficult to reproduce. These same criticisms would plague immunotherapy research over the next century. Despite this, Dr. Coley is considered by most to be the father of modern immunotherapy [38].
The field of tumor immunology stalled over the next three decades as scientists failed to consistently demonstrate immune specific rejection of transplantable tumors. This led to the statement of Dr. William Woglom in 1929 that “it would be as difficult to reject the right ear and leave the left ear intact as it is to immunize against cancer.” [39] This concept was fortified by the work of Frank Burnet who in 1948 published his theory of self-tolerance and thymic deletion. In it, he described how lymphocytes that were capable of recognizing self antigens were deleted in prenatal life during immunologic development [40].
The field of cancer immunotherapy, which seemed defeated at this point, underwent a resurrection in the 1950s with the discovery that carcinogen induced tumors could effectively immunize mice against re-challenge with the same syngeneic tumor [41, 42]. In a classic experiment, Prehn and colleagues induced formation of sarcoma in mice by treatment with the carcinogen methyl-cholanthrene (MCA). Tumors were then removed and after recovery, the same tumor cells transplanted back to the mice. Tumors failed to establish in those mice that had previously harbored malignancy. Researchers suggested that there must be antigens present on tumor cells that are not expressed by the host [42]. This ushered in the concept of tumor specific or associated antigens (TAA) that could be recognized by the host immune system.
In the late 1950s, theories emerged that the immune system is not only involved in tumor rejection, but that one of the principle roles of lymphocytes is to troll through the microenvironment deleting transformed cells [43]. This theory of “immune-surveillance” was met by harsh criticism and essentially dismissed, as many pointed to obvious flaws such as the observation that immunodeficient mice were no more prone to develop tumors than their immunocompetent counterparts [44, 45] and emerging data that previously reported tumor immunity may have been virally mediated [46]. It wasn’t until the 1980s that that the field experienced a re-birth with the discovery of auto-reactive T-cells in the periphery which had evaded thymic deletion and technologic advancements allowed for the discovery of scores of tumor specific antigens [47, 48]. Thus the modern era of immunotherapy was born with a focus on identifying ways to heighten the capabilities of dormant immune cells to eradicate tumors.
4.3 Active Immunotherapy
Active immunity is defined as immunologic recognition and protection using the body’s resident antibodies or lymphocytes. It comes following exposure to antigen and typically takes days to weeks to develop, but lasts a lifetime. Natural active immunity is achieved during exposure over the course of ones life to antigens such as viral proteins and confers protection against future infection. For example, once infected with the hepatitis B virus, the active immune response to surface and core antigens allows for clearance of the virus and lifelong immunity to re-infection [49, 50]. Contrary to this, acquired active immunity is accomplished by forced exposure to typically non-infective or minimally infective antigens such as the hepatitis B vaccine. After repeated exposure to portions of the hepatitis B surface antigens, active immunity is achieved without the need for systemic infection [50].
There are many factors present in the TME which serve to counteract active immunity [51]. Both natural and acquired immunity can be derailed if “primed” immune cells fail to reach their target or are suppressed by tolerizing cells or molecules. While certain immunogenic tumors such as melanoma and renal cell carcinoma express high levels of lymphocytic homing chemokines such as CCL2 [52] others release signals into the TME which actively suppress immune cell infiltration rendering active immunotherapy futile [53].
If educated antigen specific cytotoxic lymphocytes are able to infiltrate tumors, they face yet another hurdle in the form of immunosuppressive cells within the TME. Whereas some tumors secrete molecules to block lymphocytic infiltration, others attract a specific subset, T regulatory cells, which function to suppress cytotoxicity [20, 54]. Disproportionally high quantities of T-regs with the TME have been identified in multiple tumor types including breast [55], melanoma [56], and ovarian cancer [20]. Regulatory T-cells are directly implicated in suppressing the effects of active immunotherapy [57, 58] and efforts to remove them from the local microenvironment have shown promise in improving the efficacy of treatment [59].
4.3.1 Cancer Vaccines
The principle forms of acquired active immunotherapy for cancer treatment that are currently used or under development are vaccine-based. The premise behind vaccination is that T-cells specific for any one tumor antigen are present in such low numbers within the body that they are unlikely to encounter tumor cells and trigger an immune response [60, 61]. By providing an antigen in high quantity or more importantly with high affinity for receptor binding, immune activation and expansion can be artificially triggered [62]. Vaccines come in various platforms and can be categorized as peptides, DNA-based or dendritic cell/APC derived. Although they use different mechanisms for T-cell activation, their central premise is display of a TAA which can vary from tumor specific overexpressed self-antigens, mutated antigens or cancer-testis antigens [62].
Peptide vaccines are perhaps the simplest form of active immunization and have been studied extensively since the mid-1990s [63,64,65]. MHC class 1 restricted peptides are delivered with the hope that they will be displayed on cell surfaces and encountered by cognate TCRs triggering activation. Peptide vaccines are often administered with immune stimulatory compounds such as Freund’s adjuvant or cytokines to heighten the immune response [66]. The benefit of this approach is that they are relatively inexpensive to produce in large scale, pose little biologic risk to patients and can be administered with other peptide vaccines increasing likelihood of immune activation. While animal studies showed this strategy to be efficacious, multiple human trials have failed to reveal significant treatment effect [67, 68]. Despite documented ability to expand antigen specific T-cells, peptide vaccination alone leads to only a 2–4% objective response in patients with metastatic melanoma [69, 70]. This phenomenon highlights the complexity of tumor-immune cell interaction and was the impetus for strategies to enhance activation after antigen recognition.
An obvious shortcoming of peptide based vaccine therapy is the assumption that the chosen amino acid sequence is the optimal one for immune activation. DNA vaccines correct this by introducing a plasmid encoding the entire TAA into APCs [71]. The plasmids consist of the gene for the antigen of interest as well as a mammalian promoter that drives its expression. After the plasmid is injected subcutaneously or intramuscularly, it is taken up by resident APCs and the gene is transcribed in the nucleus [72]. The standard cellular machinery then processes the resultant protein and all relevant peptides are displayed on the cell surface for recognition by passing immune surveyors. Despite its ability to process and display multiple epitopes from a single antigen, DNA vaccines suffer from low transduction efficiency and a relative lack of immunogenicity in large mammals such as humans [73].
Use of APCs, particularly dendritic cells (DC), as a vehicle to present antigen has many theoretical advantages. As “professional” antigen presenting cells, they possess the machinery and more importantly the co-stimulatory molecules to produce profound activation of the immune system. DCs can be generated ex vivo from bone marrow stem cells or monocytes using a cocktail of cytokines, pulsed with tumor specific antigens and then re-infused [74]. They can also be transfected with TAA expressing plasmids or tumor genomic DNA to allow for expression of a wider variety of epitopes and antigens. A trial in metastatic prostate cancer demonstrated that pulsation of DCs with a prostatic acid phosphatase-GM-CSF fusion protein followed by re-infusion resulted in a significantly improved 3 year survival, leading to its approval by the FDA in 2010 [75]. Despite its theoretical advantage, most trials of DC based vaccination have been met with disappointing results with few reporting better than 15% overall response rates [76, 77].
Despite countless trials and preparations, there has been only marginal success with active immunotherapy with only three vaccines currently approved by the FDA [78]. Two of these are prophylactic treatments of viruses linked to cancer formation. Vaccination against the hepatitis B virus prevents chronic infection which subsequently reduces the risk of hepatocellular carcinoma [79]. A polyvalent vaccine against the human papilloma virus, has been shown to prevent infection with the most carcinogenic forms of the virus thereby reducing long-term development of cervical cancer [80]. The third, Sipuleucel-T for the treatment of prostate cancer as mentioned above, is the sole vaccine approved for the treatment, not prevention, of cancer [75].
4.3.2 Interleukin-2
The ability of vaccines to produce tumor specific T-cells without significant tumor reduction led many to believe that it was not the lack of recognition, but insufficient activation that was at the heart of failed immuno-editing. Discovered in 1976, interleukin-2 (IL-2) is capable of inducing growth and activation of bone marrow derived T-cells [81]. In the early 1980s, IL-2 was found to be capable of promoting cytotoxic T-cell expansion in vivo and enhancing their tumor lytic abilities transforming dormant cells into lymphokine activated killer cells (LAK) [82, 83]. In a sentinel paper, Rosenberg et al. demonstrated that delivery of high dose IL-2 to mice with metastatic sarcoma to the lungs resulted in profound tumor regression [84]. Immunohistochemical analysis of lung sections 6 days after treatment revealed a massive infiltration of cytotoxic T-cells in the pulmonary interstitium. These promising in vivo findings led to the first clinical trial of IL-2 for the treatment of metastatic cancer. Published in 1986, Lotze and colleagues at the National Cancer Institute treated 10 patients with metastases from various tumor types with increasing doses of IL-2 [85]. Of the ten participants, only those with metastatic melanoma (n = 6) showed tumor response with 50% of patients demonstrating clinically significant tumor reduction. The toxicity of the therapy was dramatic with many patients experiencing renal and respiratory failure, infection and mental status changes harking back to times of Coley’s toxin [85]. Like the murine models, biopsies of tumors revealed a profound infiltration with cytotoxic lymphocytes and active tumor necrosis. In follow-up studies of patients with metastatic melanoma and renal cell carcinoma, overall and complete response rates were 17% and 7% and 20% and 7%, respectively. These studies resulted in approval by the FDA of IL-2 for the treatment of renal cell carcinoma in 1992 and melanoma in 1998 introducing the era of non-specific stimulation based immunotherapy [86].
While it remains impossible to predict which patients will respond to IL-2 therapy, important differences observed within the TME after treatment shed light on the potential mechanisms of action of IL-2. Tumors with high prevalence of infiltrating immune cells within the tumor tend to respond better to therapy than those without [87]. It has also been suggested that the presence of T-reg cells within the TME may predict failure of IL-2 therapy [88], but this requires more dedicated research.
4.4 Passive Immunotherapy
While active immunity entails training the body’s natural defenses to better recognize pathogens and transformed cells, passive immunity simply delivers the end effectors in the form of antibodies (humoral passive immunity) or cytotoxic cells (adoptive cell transfer). The theoretical advantage of passive immunity is that it avoids potential shortcomings innately present in antigen processing and immune cell recognition to achieve the desired effect. In the case of adoptive cell transfer (ACT) is also allows for ex vivo cellular manipulation and stimulation, decreasing the systemic toxicities such as those experienced following IL-2 administration. The disadvantage of passive immunotherapy is that the effectors are often short-lived limiting their ability to provide long-term remission.
4.4.1 Humoral Immunotherapy
The passive transfer of antibodies for the treatment of disease has existed for over a century with the discovery that “anti-toxins” to diphtheria and tetanus could considerably ameliorate symptoms of the infection. The serum of immunized horses was injected into patients with tetanus, neutralizing the toxin and preventing disease dissemination [89]. Antibody based treatment for cancer can be divided into unconjugated and conjugated groups.
Unconjugated or “naked” antibodies function by binding to cancer cells and either alerting the immune system or interfering with cell signaling. An example of the former is alemtuzumab, a monoclonal antibody used to treat chronic lymphocytic leukemia (CLL) by binding to CD52 present on lymphocytes targeting them for immune clearance [90]. Trastuzumab is a monoclonal antibody which binds the overactive HER2/neu receptor decreasing its signaling and subsequently cell growth. Use of this antibody in overexpressing HER2/neu breast and gastric cancers improves both overall and disease specific survival [91, 92].
Conjugated antibodies utilize the specificity of the variable region to deliver toxic cargo to cancer cells. Ibritumomab-tiuxetan, used to treat non-Hodgkin’s lymphoma, is a radio-labeled antibody which binds to the B-lymphocyte specific marker CD20 delivering its radioactive payload, inducing cell death [93]. Other antibody-based strategies involve fusing chemotherapeutics to antibodies, better directing their delivery and increasing efficacy while limiting off-target side effects. Ado-trastuzumab emtansine fuses the same anti HER2/neu monoclonal antibody mentioned previously to the cytotoxic chemotherapy DM1 which, upon binding, enters the cell disrupting tubulin and promoting cell death [94].
4.4.2 Adoptive Cell Transfer (ACT)
The fundamental principle of ACT is removal of cytotoxic lymphocytes from the body to allow for ex vivo expansion and activation followed by re-infusion [95]. It accomplishes the goals of T-cell immunization without relying on unpredictable factors such as antigen processing and presentation and T-cell recognition and activation. Cells can be manipulated with either cytokines or genetic modification without the need for systemic administration and exposure, thereby limiting off target effects [96].
First proposed in the mid 1950s, Mitchison and colleagues demonstrated in mouse models that “adoptive immunity” could be transferred from one animal to another by transplant of tumor draining lymph node fragments [97]. Until the mid-1970s it was difficult to culture or expand T-cells in vitro, limiting the potential of this finding clinically. The discovery of IL-2 allowed not only for the activation and expansion of lymphocytes ex vivo, but also conferred T-cells with greater cytotoxicity [81, 83]. Murine models in the early 1980s harvested splenocytes from non-tumor bearing mice and activated them by co-culture with IL-2. Reinfusion of these LAK cells in mice with established pulmonary metastases resulted in a dramatic decrease in disease burden and improved overall survival [98]. Unfortunately, these results were not as impressive when translated into human trials. A prospective randomized trial of high-dose IL-2 alone or in conjunction with LAK cells demonstrated only a non-statistically significant trend towards improved survival in patients receiving ACT [85]. Impressively, however, there was a 12% complete response rate in patients receiving LAK therapy, a result that was unprecedented up to that point and sparked considerable interest.
An obvious shortcoming of ACT with LAK cells is that the reactivity is non-specific relying on expansion and activation of lymphocytes indiscriminate of antigen specificity. Over the next decade, strategies to improve ACT were sought by harvesting lymphocytes from resected tumors [95]. This was based in part on the recognition that patients bearing tumors with higher infiltration of cytotoxic cells have improved overall outcomes [99,100,101]. In theory, tumor-infiltrating lymphocytes (TILs) should inherently possess the chemokine receptors and antigen specific TCRs necessary to hone to and destroy tumors. Animal data reveals that TILs are 50–100 times more potent then LAK cells when adoptively transferred [102]. The cloning and development of techniques to produce therapeutic grade IL-2 allowed lymphocytes to be grown from resected tumors making clinical use of TIL possible.
The procedure for TIL harvest begins with surgical resection of tumors followed by fragmentation and culture in lymphocyte sustaining media supplemented with high dose IL-2 [103]. Over the subsequent days to weeks, non-IL-2 dependent tumor and stromal cells die off leaving only a culture of purified lymphocytes. Classically, these various fragment cultures are assayed for reactivity against autologous tumor and/or established tumor cell lines. Reactive culture wells are then separated and rapidly expanded using radiated autologous antigen presenting cells as feeders. After one to two rapid expansions, sufficient quantities of cells are present for re-infusion [103].
It was identified through in vivo experiments, that administration of TIL alone was not sufficient for tumor reduction and that preparative chemotherapy with cyclophosphamide was required [104]. This created a state of relative immuno-depletion allowing transferred cells less competition for resources such as nutrients and pro-inflammatory cytokines. Because of these findings, patients receive some form of non-myeloablative therapy prior to adoptive cell transfer. In 1988, Rosenberg and colleagues published a report on 20 patients with metastatic melanoma treated with ACT of TIL followed by high-dose IL-2 and noted a 50% objective response rate and lower toxicity then prior trials of IL-2 alone [105]. An important finding of early trials was that persistence of transferred cells was associated with improved magnitude and duration of treatment response. Fueled by animal data linking persistence to increased levels of lymphodepletion, increasing degrees of preparative immune ablation were studied including the addition of fludarabine and whole body irradiation. Over the next decade, clinical trials revealed that increased intensity of radiation resulted in improved lymphodepletion and cellular persistence leading to improved treatment response, with an objective response rate of 72% in patients with refractory melanoma [106]. This increased intensity was not, however, without consequence as patients occasionally suffered from long-term renal insufficiency secondary to radiation induced thrombotic microangiopathy [107].
There were many translational correlates that emerged from early clinical trials of ACT, which were later studied to improve efficacy of therapy. One such finding was the association of telomere length of the infused TIL with cancer regression signifying that “younger” lymphocytes may be more potent inducers of treatment response [108]. Based on these findings as well as animal data suggesting that naive lymphocytes may be better effectors, a trial was undertaken using minimally cultured TIL [109]. Unlike prior studies, harvested lymphocytes were not tested for reactivity prior to infusion and cells underwent a shorter rapid expansion. Theoretical benefits of this approach include administration of less exhausted lymphocytes as well as simplifying the pre-ACT protocol allowing for more rapid delivery of TIL and wider acceptance into clinical practice. Two trials utilizing young TIL showed similar efficacy to prior approaches with significantly improved ease in cell preparation [110, 111].
There are several important shortcomings to TIL therapy that have limited its widespread acceptance into clinical practice. First, the therapy requires surgical resection of a metastatic lesion, which can often mean a major operation in patients already debilitated by widespread disease. Second, ACT requires ex vivo expansion of lymphocytes which is labor intensive and unpredictable. Finally, and most importantly, until recently, ACT with TIL has been limited to the treatment of melanoma as multiple attempts at harvesting and expanding reactive lymphocytes from other malignancies have failed. The one exception to this is the recent report by Tran et al. of successful treatment of a patient with cholangiocarcinoma using TIL reactive to a mutated cancer specific protein [112].
A recent strategy to overcome these shortcomings has been genetic modification of peripheral lymphocytes to confer tumor reactivity [113]. To accomplish this, TCR genes from lymphocyte clones isolated from TIL which are reactive to shared TAAs are cloned. These genes are then inserted into pheresed non-reactive peripheral lymphocytes via retroviral or lentiviral transduction allowing for expression of the transplanted TCR [114]. Culture of these genetically modified lymphocytes with cell lines expressing the shared tumor antigen confirm transferred reactivity [115]. This technique avoids the need for surgical intervention, produces reliably reactive lymphocytes for infusion and creates an “off-the-self” reagent that could improve accessibility to this therapy. ACT with genetically modified PBL occurs in a similar manner to traditional TIL with plasma pheresis, viral mediated gene transfer to PBL, preparative chemotherapy and then cellular infusion. The first trial using ACT with genetically modified PBL by Morgan et al. treated metastatic melanoma patients with lymphocytes engineered to express a TCR specific for the melanoma-associated antigen (MAA) MART-1 [114]. They noted significant tumor reduction in two patients but unfortunately no response in a vast majority. Believing the poor efficacy was due to the relative low-affinity of the MART-1 receptor, a second trial was undertaken using PBL transduced to express a much higher affinity receptor to MART-1 or a receptor to the MAA gp-100 [116, 117]. These higher-affinity receptors proved more efficacious, but created significant off target effects attacking melanin-expressing cells in the skin, eyes, and ears [117].
Use of genetically modified lymphocytes has allowed expansion of ACT outside of the realm of melanoma. TCRs specific for multiple cancer testes antigens (CTA) have been cloned conferring lymphocyte specificity for antigens expressed on a wide variety of tumor types. A trial utilizing PBL transduced with a TCR specific for the CTA NY-ESO-1 demonstrated the ability of this therapy to treat sarcomas which have proven refractory to standard treatment [118]. A second trial using a TCR specific for the CTA MAGE-A3 enrolled patients with melanoma, sarcoma as well as esophageal cancer [119].
While genetically engineered PBL show promise, the restriction of TCRs to specific MHC subtypes limit their widespread utility. For example, most TCRs in development are specific for the HLA-A2 haplotype which is expressed by only 50% of Caucasians and 35% of African-Americans [120]. To expand the potential treatment population, chimeric antigen receptors (CAR) have been developed which utilize the antibody binding region joined by a linker to the TCR intracellular signaling domain [121]. Benefits of this strategy are a vast expansion of potential targets and the lack of MHC restriction. Antibodies, however, lack the specificity of TCRs making off target toxicity a concern.
One of the first CARs developed targeted CD19, which is widely expressed in B-cell lymphoma and lymphoblastic leukemia [122]. Patients with medically refractory disease underwent lymphocyte harvest followed by transduction of cells with a CAR composed of an antibody to CD19 and the CD28/CD3ζ intracellular signaling domains. Treatment induced rapid remission in 50% of patients with B-cell lymphomas and up to 100% of patients with acute lymphoblastic leukemia resulting in its approval by the FDA [123,124,125].
While CAR directed therapy is promising, it is not without its drawbacks. As previously stated, antibodies lack the specificity of TCRs and off target toxicity can be common. A dramatic example of this is the case report by Morgan et al. of a patient receiving a CAR directed to the TAA ERBB2 [126]. Within hours of ACT, the patient suffered multi-organ failure and eventually death due to cytokine storm and pulmonary congestion. Analysis of the CAR transduced lymphocytes showed activity to primary lung tissue lines leading authors to conclude that activation of the cells during first-pass in the lungs led to the subsequent outcome. While results from ACT continue to improve, the complexity of therapy and potential toxicity still limit its use to highly specialized centers.
Despite the numerous advancements in ACT over the past decades, it remains effective in only a small subset of patients. To better determine why the therapy is often ineffective researchers have turned their attention to other components of the TME. There are three principle factors that appear to impair function of transferred cytotoxic T-cells: (1) immunosuppressive cellular elements, (2) local secreted factors, and (3) immune checkpoints.
Tumor infiltrating lymphocyte function can be blocked by various immune cells including T-regs (as discussed in the active immunotherapy section), myeloid derived suppressor cells (MDSC) and type-2 macrophages [127,128,129]. While difficult to remove from the TME, strategies aimed at suppressing their function are currently being investigated with the goal of enhancing ACT efficacy. Delivery of a cyclooxygenase inhibitor can prevent differentiation of MDSCs and enhance immunotherapy in mesothelioma [130]. Type-2 macrophages, which function primarily by releasing arginase have been targeted by attempts to reprogram them to the more tumor destroying type-1 macrophage [131] and inhibition of arginase activity [132].
The cytokine milieu of the TME can tip the balance towards immune mediated destruction or protection. High levels of IL-10 and TFG-B suppress cytotoxic T-cell function and promote expansion of suppressive cellular elements [129]. Contrary to this, cytokines such as IL-12 lead to accumulation of T-cells and enhance efficacy of ACT. Attempts to utilize these pro-inflammatory cytokines clinically has been met with some difficulty as their potency can often lead to undesirable off target effects [133].
The final suppressor of ACT efficacy within the microenvironment is up-regulation of immune regulatory receptors and ligands. As immune excitation occurs, a proportional increase in immunosuppressive signals occurs to prevent the reaction from spiraling out of control. Cells that are expanded for ACT often express high levels of inhibitory receptors and attempts to block these have resulted in increased effector activity [134, 135]. More details regarding the function of checkpoint blockade will be discussed in the following section.
4.5 Checkpoint Blockade
Arguably the greatest advancement in immunotherapy over the past decades has been discovery [31] and clinical introduction of checkpoint inhibitors [26]. As previously stated, lymphocyte mediated immune destruction requires recognition via the TCR and co-stimulation via a variety of cell surface molecules. While these co-stimulatory proteins serve the heighten lymphocyte response to antigen, an assortment of inhibitory cell surface proteins within the TME serve to quell the reaction. This balance ensures adequate immune response without over-activation. Two notable inhibitory proteins cytotoxic T-lymphocyte protein 4 (CTLA-4) [136] and programmed cell-death protein 1 (PD-1) [137] have come to the forefront as pharmacologic strategies to block their activity has yielded impressive anti-tumor response.
4.5.1 CTLA-4
Originally described in the late 1980s and early 1990s, CTLA-4 is a member of the immunoglobulin superfamily and binds to B7-1 and B7-2 on antigen presenting cells. It is similar in structure to the co-stimulatory protein CD28 and functions to suppress T-cell activation [136]. Leach et al. determined that antibody blockade of CTLA-4 resulted in enhanced tumor immunity [138]. In 2010, Hodi et al. published a clinical trial of anti-CTLA-4 in the treatment of refractory metastatic melanoma [139]. Authors demonstrated a response rate of 25% and a long-term disease control rate of 15%. The drug was well tolerated with the principle side effects consisting of autoimmune colitis and hypophysitis. Results led to FDA approval of anti-CTLA-4 (ipilimumab) for the treatment of melanoma in 2011. Follow-up studies using ipilimumab in patients with less heavily treated disease and in combination with other immune modifying agents have led to improved results [140].
4.5.2 PD-1 and PD-L1
Like CTLA-4, PD-1 is present on lymphocytes and serves to inhibit antigen-mediated reactivity preventing autoimmunity. The principle ligand of PD-1, PD-L1 (B7H1), is often over expressed by tumor cells and APCs [19, 31], and used as a mechanism to evade immune destruction [137]. Blockade of the PD-1 -PD-L1 axis leads to improved tumor recognition and destruction. Unlike ipilimumab, the efficacy of PD-1 and PD-L1 inhibitors extends beyond melanoma and has been successfully used to treat non-small cell lung cancer (NSCLC), renal cell carcinoma and ovarian cancer [141]. These promising studies have led to the approval of the PD-1 blockers pembrolizumab and nivolumab by the FDA [142]. As the expression of PD-L1 and PD-1 is largely in the tumor microenvironment [19, 31], the side effects are less severe and highly manageable.
Tumor immunology and immunotherapy has undergone a renaissance of late. After suffering countless successes and setbacks in the twentieth century, it has now come to the forefront of cancer research and is recognized as an important tool in the anti-tumor armamentarium. Most exciting, is the potential for immunotherapy to not just result in tumor response, but complete and long-term remission. As our understanding of the intricate interactions between T-cells and the tumor microenvironment improves, so to will strategies aimed at derailing tumor mediate immune suppression. While the current focus is on T-cell mediated immunotherapy, emerging literature is suggesting an important role for myeloid derived cells such as macrophages and myeloid derived suppressor cells [143, 144] within the TME. It may be that someday a cocktail of different immune modulators is required to destroy established tumors and achieve cure. Achievements in the last decade have made this dream closer to a reality with more advancements soon to come.
References
Gravitz L. Cancer immunotherapy. Nature. 2013;504(7480):S1.
Rosenberg SA, Dudley ME, Restifo NP. Cancer immunotherapy. N Engl J Med. 2008;359(10):1072.
Gajewski TF, Woo SR, Zha Y, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25(2):268–76.
Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today. 2000;21(9):455–64.
Whiteside TL, Stanson J, Shurin MR, et al. Antigen-processing machinery in human dendritic cells: up-regulation by maturation and down-regulation by tumor cells. J Immunol. 2004;173(3):1526–34.
Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27(45):5904–12.
Heninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol. 2015;6:29.
Yu J, Ni M, Xu J, et al. Methylation profiling of twenty promoter-CpG islands of genes which may contribute to hepatocellular carcinogenesis. BMC Cancer. 2002;2:29.
Rao M, Chinnasamy N, Hong JA, et al. Inhibition of histone lysine methylation enhances cancer-testis antigen expression in lung cancer cells: implications for adoptive immunotherapy of cancer. Cancer Res. 2011;71(12):4192–204.
Simova J, Pollakova V, Indrova M, et al. Immunotherapy augments the effect of 5-azacytidine on HPV16-associated tumours with different MHC class I-expression status. Br J Cancer. 2011;105(10):1533–41.
Wang LX, Mei ZY, Zhou JH, et al. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS One. 2013;8(5):e62924.
West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology. 2014;3(1):e27414.
Iannello A, Raulet DH. Immune surveillance of unhealthy cells by natural killer cells. Cold Spring Harb Symp Quant Biol. 2013;78:249–57.
Chow MT, Moller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol. 2012;22(1):23–32.
Groth A, Kloss S, von Strandmann EP, et al. Mechanisms of tumor and viral immune escape from natural killer cell-mediated surveillance. J Innate Immun. 2011;3(4):344–54.
Melchionda F, McKirdy MK, Medeiros F, et al. Escape from immune surveillance does not result in tolerance to tumor-associated antigens. J Immunother. 2004;27(5):329–38.
Al-Tameemi M, Chaplain M, d’Onofrio A. Evasion of tumours from the control of the immune system: consequences of brief encounters. Biol Direct. 2012;7:31.
Zou W, Machelon V, Coulomb-L’Hermin A, et al. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat Med. 2001;7(12):1339–46.
Curiel TJ, Wei S, Dong H, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9(5):562–7.
Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9.
Kryczek I, Zou L, Rodriguez P, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203(4):871–81.
Cui TX, Kryczek I, Zhao L, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. 2013;39(3):611–21.
Bai XF, Liu J, Li O, et al. Antigenic drift as a mechanism for tumor evasion of destruction by cytolytic T lymphocytes. J Clin Invest. 2003;111(10):1487–96.
Bubenik J. MHC class I down-regulation: tumour escape from immune surveillance? (review). Int J Oncol. 2004;25(2):487–91.
Topfer K, Kempe S, Muller N, et al. Tumor evasion from T cell surveillance. J Biomed Biotechnol. 2011;2011:918471.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Singh S, Ross SR, Acena M, et al. Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. J Exp Med. 1992;175(1):139–46.
Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res. 2007;13(18 Pt 1):5256–61.
Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–74.
Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6(4):295–307.
Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8(6):467–77.
Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010;10(4):248–56.
Parish CR. Cancer immunotherapy: the past, the present and the future. Immunol Cell Biol. 2003;81(2):106–13.
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–9.
Coley WB II. Contribution to the knowledge of sarcoma. Ann Surg. 1891;14(3):199–220.
Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. Clin Orthop Relat Res. 1893;1991(262):3–11.
Coley WB. The treatment of inoperable sarcoma by bacterial toxins (the mixed toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med. 1910;3(Surg Sect):1–48.
American Association for Cancer Research. The 2013 William B. Coley award for distinguished research in basic and tumor immunology. Cancer Immunol Res. 2013;1(6):362–4.
Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999;10(3):281–7.
Burnet FM, Fenner F. Genetics and immunology. Heredity. 1948;2(Pt. 3):289–324.
Foley EJ. Antigenic properties of methylcholanthrene-induced tumors in mice of the strain of origin. Cancer Res. 1953;13(12):835–7.
Prehn RT, Main JM. Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst. 1957;18(6):769–78.
Burnet M. Cancer; a biological approach. I. The processes of control. Br Med J. 1957;1(5022):779–86.
Outzen HC, Custer RP, Eaton GJ, et al. Spontaneous and induced tumor incidence in germfree "nude" mice. J Reticuloendothel Soc. 1975;17(1):1–9.
Stutman O. Immunodepression and malignancy. Adv Cancer Res. 1975;22:261–422.
Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121(1):1–14.
Herberman RB, Holden HT. Natural cell-mediated immunity. Adv Cancer Res. 1978;27:305–77.
Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–11.
Gerlich WH. Medical virology of hepatitis B: how it began and where we are now. Virol J. 2013;10:239.
Shepard CW, Simard EP, Finelli L, et al. Hepatitis B virus infection: epidemiology and vaccination. Epidemiol Rev. 2006;28:112–25.
Gajewski TF, Meng Y, Blank C, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–45.
Zhang T, Somasundaram R, Berencsi K, et al. Migration of cytotoxic T lymphocytes toward melanoma cells in three-dimensional organotypic culture is dependent on CCL2 and CCR4. Eur J Immunol. 2006;36(2):457–67.
Buckanovich RJ, Facciabene A, Kim S, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36.
Turk MJ, Guevara-Patino JA, Rizzuto GA, et al. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200(6):771–82.
Liyanage UK, Moore TT, Joo HG, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169(5):2756–61.
Viguier M, Lemaitre F, Verola O, et al. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol. 2004;173(2):1444–53.
Casares N, Arribillaga L, Sarobe P, et al. CD4+/CD25+ regulatory cells inhibit activation of tumor-primed CD4+ T cells with IFN-gamma-dependent antiangiogenic activity, as well as long-lasting tumor immunity elicited by peptide vaccination. J Immunol. 2003;171(11):5931–9.
Antony PA, Piccirillo CA, Akpinarli A, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174(5):2591–601.
Viehl CT, Moore TT, Liyanage UK, et al. Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann Surg Oncol. 2006;13(9):1252–8.
Wang RF, Rosenberg SA. Human tumor antigens for cancer vaccine development. Immunol Rev. 1999;170:85–100.
Chan AD, Morton DL. Active immunotherapy with allogeneic tumor cell vaccines: present status. Semin Oncol. 1998;25(6):611–22.
Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today. 1997;18(4):175–82.
Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4(3):321–7.
Maeurer MJ, Storkus WJ, Kirkwood JM, et al. New treatment options for patients with melanoma: review of melanoma-derived T-cell epitope-based peptide vaccines. Melanoma Res. 1996;6(1):11–24.
Berinstein NL. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol. 2002;20(8):2197–207.
Wang F, Bade E, Kuniyoshi C, et al. Phase I trial of a MART-1 peptide vaccine with incomplete Freund’s adjuvant for resected high-risk melanoma. Clin Cancer Res. 1999;5(10):2756–65.
Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–27.
Eggermont AM. Immunotherapy: vaccine trials in melanoma—time for reflection. Nat Rev Clin Oncol. 2009;6(5):256–8.
Rosenberg SA, Sherry RM, Morton KE, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175(9):6169–76.
Phan GQ, Touloukian CE, Yang JC, et al. Immunization of patients with metastatic melanoma using both class I- and class II-restricted peptides from melanoma-associated antigens. J Immunother. 2003;26(4):349–56.
Liu MA. DNA vaccines: a review. J Intern Med. 2003;253(4):402–10.
Kutzler MA, Weiner DB. Developing DNA vaccines that call to dendritic cells. J Clin Invest. 2004;114(9):1241–4.
Lu S, Wang S, Grimes-Serrano JM. Current progress of DNA vaccine studies in humans. Expert Rev Vaccines. 2008;7(2):175–91.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–77.
Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22.
Engell-Noerregaard L, Hansen TH, Andersen MH, et al. Review of clinical studies on dendritic cell-based vaccination of patients with malignant melanoma: assessment of correlation between clinical response and vaccine parameters. Cancer Immunol Immunother. 2009;58(1):1–14.
Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5(4):296–306.
Bodey B, Bodey B Jr, Siegel SE, et al. Failure of cancer vaccines: the significant limitations of this approach to immunotherapy. Anticancer Res. 2000;20(4):2665–76.
Lai CL, Yuen MF. Prevention of hepatitis B virus-related hepatocellular carcinoma with antiviral therapy. Hepatology. 2013;57(1):399–408.
Paavonen J, Naud P, Salmeron J, et al. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet. 2009;374(9686):301–14.
Welte K, Wang CY, Mertelsmann R, et al. Purification of human interleukin 2 to apparent homogeneity and its molecular heterogeneity. J Exp Med. 1982;156(2):454–64.
Donohue JH, Rosenstein M, Chang AE, et al. The systemic administration of purified interleukin 2 enhances the ability of sensitized murine lymphocytes to cure a disseminated syngeneic lymphoma. J Immunol. 1984;132(4):2123–8.
Cheever MA, Greenberg PD, Fefer A, et al. Augmentation of the anti-tumor therapeutic efficacy of long-term cultured T lymphocytes by in vivo administration of purified interleukin 2. J Exp Med. 1982;155(4):968–80.
Rosenberg SA, Mule JJ, Spiess PJ, et al. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J Exp Med. 1985;161(5):1169–88.
Lotze MT, Rosenberg SA. Results of clinical trials with the administration of interleukin 2 and adoptive immunotherapy with activated cells in patients with cancer. Immunobiology. 1986;172(3–5):420–37.
Gaffen SL, Liu KD. Overview of interleukin-2 function, production and clinical applications. Cytokine. 2004;28(3):109–23.
Phan GQ, Attia P, Steinberg SM, et al. Factors associated with response to high-dose interleukin-2 in patients with metastatic melanoma. J Clin Oncol. 2001;19(15):3477–82.
Antony PA, Restifo NP. CD4+CD25+ T regulatory cells, immunotherapy of cancer, and interleukin-2. J Immunother. 2005;28(2):120–8.
Plotkin S. History of vaccination. Proc Natl Acad Sci U S A. 2014;111(34):12283–7.
Fraser G, Smith CA, Imrie K, et al. Alemtuzumab in chronic lymphocytic leukemia. Curr Oncol. 2007;14(3):96–109.
Hortobagyi GN. Trastuzumab in the treatment of breast cancer. N Engl J Med. 2005;353(16):1734–6.
Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365(14):1273–83.
Witzig TE. Yttrium-90-ibritumomab tiuxetan radioimmunotherapy: a new treatment approach for B-cell non-Hodgkin’s lymphoma. Drugs Today (Barc). 2004;40(2):111–9.
Amiri-Kordestani L, Blumenthal GM, Xu QC, et al. FDA approval: ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin Cancer Res. 2014;20(17):4436–41.
Rosenberg SA, Restifo NP, Yang JC, et al. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308.
Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39(1):49–60.
Mitchison NA. Adoptive transfer of immune reactions by cells. J Cell Physiol Suppl. 1957;50(Suppl 1):247–64.
Mazumder A, Rosenberg SA. Successful immunotherapy of natural killer-resistant established pulmonary melanoma metastases by the intravenous adoptive transfer of syngeneic lymphocytes activated in vitro by interleukin 2. J Exp Med. 1984;159(2):495–507.
Murray D, Hreno A, Dutton J, et al. Prognosis in colon cancer: a pathologic reassessment. Arch Surg. 1975;110(8):908–13.
Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–43.
Naito Y, Saito K, Shiiba K, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58(16):3491–4.
Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233(4770):1318–21.
Dudley ME, Wunderlich JR, Shelton TE, et al. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26(4):332–42.
North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155(4):1063–74.
Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–80.
Dudley ME, Yang JC, Sherry R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–9.
Tseng J, Citrin DE, Waldman M, et al. Thrombotic microangiopathy in metastatic melanoma patients treated with adoptive cell therapy and total body irradiation. Cancer. 2014;120(9):1426–32.
Zhou J, Shen X, Huang J, et al. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175(10):7046–52.
Tran KQ, Zhou J, Durflinger KH, et al. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother. 2008;31(8):742–51.
Itzhaki O, Hovav E, Ziporen Y, et al. Establishment and large-scale expansion of minimally cultured "young" tumor infiltrating lymphocytes for adoptive transfer therapy. J Immunother. 2011;34(2):212–20.
Dudley ME, Gross CA, Langhan MM, et al. CD8+ enriched "young" tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. 2010;16(24):6122–31.
Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–5.
Rossig C, Brenner MK. Genetic modification of T lymphocytes for adoptive immunotherapy. Mol Ther. 2004;10(1):5–18.
Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9.
Frankel TL, Burns WR, Peng PD, et al. Both CD4 and CD8 T cells mediate equally effective in vivo tumor treatment when engineered with a highly avid TCR targeting tyrosinase. J Immunol. 2010;184(11):5988–98.
Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–46.
Seaman BJ, Guardiani EA, Brewer CC, et al. Audiovestibular dysfunction associated with adoptive cell immunotherapy for melanoma. Otolaryngol Head Neck Surg. 2012;147(4):744–9.
Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–24.
Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–51.
Ellis JM, Henson V, Slack R, et al. Frequencies of HLA-A2 alleles in five U.S. population groups. Predominance of A*02011 and identification of HLA-A*0231. Hum Immunol. 2000;61(3):334–40.
Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21(2):215–23.
Kochenderfer JN, Feldman SA, Zhao Y, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32(7):689–702.
Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–9.
Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38.
Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18.
Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–51.
Marigo I, Dolcetti L, Serafini P, et al. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–79.
Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30(5):636–45.
Devaud C, John LB, Westwood JA, et al. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology. 2013;2(8):e25961.
Veltman JD, Lambers ME, van Nimwegen M, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. 2010;10:464.
Buhtoiarov IN, Sondel PM, Wigginton JM, et al. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology. 2011;132(2):226–39.
Serafini P, Meckel K, Kelso M, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203(12):2691–702.
Gately MK, Renzetti LM, Magram J, et al. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495–521.
Blake SJ, Ching AL, Kenna TJ, et al. Blockade of PD-1/PD-L1 promotes adoptive T-cell immunotherapy in a tolerogenic environment. PLoS One. 2015;10(3):e0119483.
Mahvi DA, Meyers JV, Tatar AJ, et al. Ctla-4 blockade plus adoptive T-cell transfer promotes optimal melanoma immunity in mice. J Immunother. 2015;38(2):54–61.
Chambers CA, Kuhns MS, Egen JG, et al. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565–94.
Keir ME, Butte MJ, Freeman GJ, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6.
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–33.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Barbee MS, Ogunniyi A, Horvat TZ, et al. Current status and future directions of the immune checkpoint inhibitors ipilimumab, pembrolizumab, and nivolumab in oncology. Ann Pharmacother. 2015;49(8):907–37.
Panni RZ, Linehan DC, DeNardo DG. Targeting tumor-infiltrating macrophages to combat cancer. Immunotherapy. 2013;5(10):1075–87.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Frankel, T., Lanfranca, M.P., Zou, W. (2017). The Role of Tumor Microenvironment in Cancer Immunotherapy. In: Kalinski, P. (eds) Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy. Advances in Experimental Medicine and Biology, vol 1036. Springer, Cham. https://doi.org/10.1007/978-3-319-67577-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-67577-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-67575-6
Online ISBN: 978-3-319-67577-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)