
Abstract
It is remarkable that human tumour tissues can be grown for months, or even years, as serially transplantable, patient-derived xenografts (PDXs) in immunocompromised mice. The grafting technique used has been refined over the last few decades, so it is now possible to successfully engraft most tumour types, albeit with varying take rates. This review focuses on the methodological requirements to establish successful PDXs. The first step is selecting viable tumour tissue from surgical resections of local or metastatic disease, ascites, pleural effusions, biopsies, circulating tumour cells, rapid autopsies or even organoids. Once grafts are prepared, sometimes with Matrigel or stroma, their likelihood of growing is affected by the strain of immunocompromised host mice and the graft site, which may be subcutaneous, subrenal capsule or orthotopic. Finally, once PDXs are established, authentication assays can be used to rule out possible misidentification or cross-contamination following serial passaging. By carefully optimising all of these steps, PDXs can be grown as efficiently as possible, providing invaluable models for preclinical cancer research.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Patient-derived xenograft
- Tumour graft
- Metastasis
- Subcutaneous
- Renal capsule
- Orthotopic
- Immunocompromised mice
- Prostate cancer
- Breast cancer
Introduction
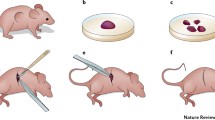
For decades, cancer researchers have attempted to grow samples of patient tumours as xenografts. Initially, there was limited success, and only a small number of patient-derived xenografts (PDXs) could be established, mostly from particularly aggressive tumours. Nevertheless, through a combination of serendipity and careful trial and error, the methods for establishing successful PDXs have gradually improved. Most tumour types can now be grown as PDXs, and large consortia are developing extensive collections of PDXs [1,2,3]. Yet, there is still room for improvement. Some tumour types still have low engraftment rates and are under-represented in PDX collections. The difficulty in establishing PDXs also means that they are beyond the resources of many laboratories, which may limit the use of PDXs in preclinical cancer research and narrow the spectrum of tumours represented by PDX models. Therefore, this chapter will focus on the methods for establishing PDXs of solid tumours. In particular, it will address four critical aspects of PDX protocols: collection of viable patient tissue, preparation of tissue for xenografting, choice of host mice and authentication of established grafts (Fig. 2.1). Continued optimisation of each of these steps will maximise the likelihood of establishing successful xenografts from patient specimens.

Overview of the main steps in establishing PDXs. Step 1: High-quality patient specimens are obtained from various stages of cancer progression, from benign tissue to metastases, using a variety of collection methods. Step 2: Patient specimens are carefully prepared for grafting as either whole pieces of tissue, tissue slices, digested cells or sorted cells, with or without the addition of Matrigel or stroma. Step 3: Specimens are engrafted into the chosen strain of immunocompromised mice at the subcutaneous, subrenal capsule or orthotopic site. Hormone implants are used for PDXs of hormone-dependent cancers. Step 4: PDXs are validated to confirm that they are not contaminated with lymphoma cells and match the original patient specimen
Primary Versus Serially Transplantable PDXs
Xenografts can be derived from various sources of cancer cells; however, this chapter will specifically focus on patient-derived xenografts, sometimes also referred to as tumour grafts or patient-derived tumour xenografts. We define PDXs as those that are established from patient tissue but not from immortalised cell lines. We will also discuss xenografts from near-patient samples, such as organoids. PDXs can be further divided into primary and serially transplantable models, which have different advantages and disadvantages.
Primary PDXs, also known as first-generation PDXs, are clinical specimens that are grown in host mice for only one generation [4,5,6,7]. Depending on the experiment, this generation can last a few months, which is sufficient time for preclinical testing of candidate therapeutics [4]. Primary PDXs have high take rates, because most tissues grow in vivo for at least one generation, assuming that the samples are of high quality and the xenografting conditions are optimal [5]. Thus, primary PDXs provide an opportunity to maximise the utility of specimens that may not produce serially transplantable PDXs, which is particularly important for rare tumours [8]. Another advantage of primary PDXs is that they maintain the complex pathology of the original samples. Benign, premalignant and malignant cells can co-exist within a single graft, just as they do in patient tissue [7, 9, 10]. Other cell types that are retained include fibroblasts, smooth muscle and endothelial cells [7, 11]. These cell types are gradually overtaken by cancer cells and recruited mouse stroma after serial transplantation [12]. Therefore, there are several advantages to only growing patient tissues in host mice for one generation as primary PDXs.
The disadvantages of primary PDXs are offset by the benefits of serially transplantable PDXs. Primary PDX experiments require ongoing access to fresh patient specimens, whereas serially transplantable PDXs are actively growing tumours that can be regrafted into new host mice for multiple generations [13]. Thus, they provide a continuing source of tissue for numerous experiments. Over several generations, cancer cells become the most prevalent cell type within serially transplantable PDXs, so their pathology becomes more similar to metastatic than localised tumours. Nevertheless, the grafts are populated by mouse stroma, so serially transplantable PDXs are more complex models compared to in vitro monocultures of cancer cells. Furthermore, like in vitro cell cultures, serially transplantable PDXs can be cryopreserved and shared between laboratories [1, 12, 13]. Collectively, these features make serially transplantable PDXs valuable preclinical models to study tumour biology and test novel therapeutics.
One of the main limitations of serially transplantable PDXs is that some tumour types are difficult to grow. Some cancers, such as melanoma, readily establish serially transplantable PDXs, while others, such as prostate and oestrogen receptor-positive breast cancer, have much lower rates of success [7, 14,15,16]. There are several interrelated explanations for why some tumours are easier to establish as serially transplantable PDXs than others. One factor is the origin and availability of tumour tissue. The surgical procedures used to remove tumours can affect the quality and viability of samples as well as the time taken to transport them to the laboratory [8]. Moreover, different patterns of early diagnosis and clinical practice between tumour types mean that samples may be available from different stages of cancer progression. Another factor influencing PDX success rates is the ability of each tumour to adapt to growing in the mouse host. Some tumours may be more sensitive to xenografting conditions, including the methods used to prepare the grafts and the choice of mouse strain. Finally, the success rate of establishing PDXs might simply reflect the aggressiveness of the cancer type and the individual patient specimen [17,18,19]. This particular variable is beyond a researcher’s control; however, many other factors can be optimised to maximise the likelihood that tumours will produce serially transplantable PDXs. Therefore, the following sections will discuss sources of tumour tissue, preparation of grafts and choice of mouse strain, because the methods of establishing PDXs may underpin their eventual success.
Methods for Generating Patient-Derived Xenografts
Sources of Tissue for Patient-Derived Xenografts
Xenografting is a challenging technique from the very first step of the process—collecting high-quality patient specimens. The sources of patient tissue determine the take rate of PDXs and the scientific questions they can be used to investigate. This section will address the benefits, limitations and applications of different sources of tissue spanning disease progression, from non-malignant samples to metastatic cancer specimens.
Non-Malignant Tissue
Non-malignant tissues are often overlooked as samples for establishing PDXs. However, they can be used to optimise xenografting techniques, study angiogenesis and the interactions between epithelium and stroma, investigate the normal physiological responses of tissues to treatment, compare the features of patient-matched benign and malignant tissue and identify cancer cells of origin [6, 10, 20,21,22,23,24]. Non-malignant tissue is often dissected from the same surgical specimen as the tumour by sampling regions that are distant from known tumour foci [10, 23, 25]. Other possible sources of non-malignant tissue include prophylactic surgeries, such as mastectomies and oophorectomies from women with pathogenic BRCA1 or BRCA2 mutations, or procedures for benign conditions, such as transurethral resection of the prostate (TURP) for treating benign prostatic hyperplasia [8, 24, 26].
Depending on the experiment and tumour type, thorough histopathology may be required to confirm that these specimens are truly non-malignant. Furthermore, some cancers are thought to exert a field effect on surrounding tissue, so non-malignant samples may be best defined as “benign” or “morphologically normal”, rather than “normal” [27, 28]. Notwithstanding this limitation, non-malignant samples are still useful because they often have high take rates as primary PDXs [10, 25]. Anecdotally, benign epithelial cells often persist in primary PDXs, even when cancer cells fail to grow. However, unlike tumours, non-malignant tissues do not produce serially transplantable PDXs. Collectively, this means that non-malignant specimens are a convenient source of tissue for short-term PDX experiments as long as their histopathology is carefully reviewed.
Localised Tumour Tissue
Localised tumours are a common source of tissue for PDXs, because surgery with curative intent is standard practice for treating many cancers. These specimens can provide large amounts of tumour tissue from each patient, and it is sometimes possible to obtain locally advanced cancer from surrounding lymph nodes. Regions of tumour tissue can be dissected from surgical specimens with a scalpel or biopsy needle, ideally avoiding benign or necrotic tissue [5, 29]. Whichever method is used, it is essential that the overall architecture of the specimen is preserved for routine pathology reporting of surgical margins, tumour differentiation and histology [5]. PDXs can also be established from primary tumours after patients have received neoadjuvant treatments [30, 31]. In cases where surgery is not performed, tissue can be obtained from biopsies, including fine needle aspirates [10, 13, 32, 33]. Thus, for many cancers localised tumours are a widely available source of tissue for xenografting.
PDXs of primary tumour have many applications. Large biobanks of PDXs have been established from primary tumours of numerous cancers [1,2,3, 34]. Genomic analyses of these large cohorts have shown that they approximate the inter-patient diversity of tumours in the clinic [1, 2]. Thus, they provide comprehensive preclinical platforms for drug screening. Serially transplantable PDXs of primary cancers are also useful for studying tumour biology. For example, some laboratories have grown PDXs under selective pressure to create models of therapy resistance [13], while others have established PDXs from matched localised and metastatic tumours from the same patient [1, 32]. Therefore, even though PDXs of primary tumours usually do not represent lethal disease, they are invaluable models for cancer research.
Metastatic Tumour Tissue
PDXs of metastatic tumours provide models of the most aggressive stages of cancer progression, including therapy resistance [14, 35]. This makes PDXs of metastatic tumours ideal for studying mechanisms of drug resistance and for testing the efficacy of novel therapeutics. For many cancers, however, there is limited access to metastatic tumours compared to localised disease. Patients with some cancers rarely undergo surgical resection of metastases and are instead treated with radiotherapy or systemic therapies like chemotherapy. Nevertheless, once the logistical and ethical challenges are overcome, it is still possible to obtain metastatic samples from patients during treatment or after death.
PDXs can be established from numerous sources of metastatic tumour cells from patients who are still undergoing treatment. For example, several laboratories have generated PDXs from surgically resected liver metastases of colon cancer [34, 36,37,38]. Surgery is less commonly performed on metastases of many other cancers; however, other types of samples are sometimes available. This includes malignant ascites or pleural effusions, from which cancer cells can be isolated and injected or grafted into mice [17, 39,40,41,42]. Metastatic tissue can also be obtained from palliative surgeries, which are sometimes used to alleviate pain or repair fractures due to bone or spinal metastases [43,44,45]. Biopsies are another common source of metastatic tumour tissue [46, 47]. They are usually performed during the course of patient treatment but are sometimes undertaken specifically to obtain tissue for research or clinical trials, which carries a small risk of complications to the patient [48,49,50]. This means that it is sometimes possible to plan the timing of biopsies and even use them to obtain serial samples from patients [49]. However, the limitations of biopsies include the small amounts of tissue they provide and the inability to sample some metastatic sites. Nevertheless, along with surgical resections, ascites and pleural effusions, biopsies are an essential source of metastatic tumour cells for establishing PDXs.
Certain limitations of collecting samples from living patients are overcome with rapid autopsy programmes. Rapid autopsy, also known as warm or immediate autopsy, involves the collection of tumour tissue within a few hours of a patient’s death [51]. The speed of this process is important for maintaining high-quality, viable tumour tissue before autolysis occurs. This creates logistic challenges, so rapid autopsy programmes typically involve team members from clinical care, palliative care, funeral services, forensic medicine, tissue banking and cancer research [14, 52]. Most rapid autopsy protocols use imaging and clinical notes to identify the locations of metastases. These sites are then reviewed macroscopically during dissection to avoid any necrotic tissue [14, 52]. Despite concerns about the viability of rapid autopsy samples, serially transplantable PDXs have been established from melanoma, rhabdomyosarcoma and breast, pancreatic, prostate and ovarian cancers [14, 35, 51,52,53]. The reported success rate for generating PDXs varies between tumour types, from 5% for prostate cancer to 100% for melanoma [14, 52].
There are benefits and limitations to rapid autopsy programmes and the samples they provide. One of the benefits of rapid autopsy is that it enables extensive sampling of multiple metastatic sites, including those that cannot be accessed before death [14]. This is particularly useful for studying intra-patient tumour heterogeneity [54,55,56]. Furthermore, compared to biopsies, rapid autopsies can provide greater amounts of tissue from more sites and without patient discomfort or the risk of complications. Yet, rapid autopsies are not routine. This makes them a low-throughput source of metastatic tissue for xenografting, especially compared to biopsies. Therefore, rapid autopsy programmes are often used for detailed studies of carefully selected patient cohorts, where samples are gradually accumulated over time. Overall, this means that rapid autopsies and other sources of metastatic tissue are complementary methods of collecting patient samples for xenografting.
Circulating Tumour Cells
Circulating tumour cells (CTCs) are an emerging source of cancer cells for PDXs. Successful PDXs have been established from breast, prostate and small-cell lung cancer CTCs directly implanted into immunocompromised host mice [57,58,59]. PDXs have also been generated from CTCs that were cultured as organoids before engraftment (see Section “Sorted or Cultured Cancer Cells”) [60,61,62]. CTCs have yielded tumours when directly injected into subcutaneous and bone sites, which is often cited as evidence that CTCs contain a subpopulation of metastasis-initiating cells [57, 58, 63]. Not surprisingly, samples with greater numbers of CTCs have higher take rates when xenografted [57, 58]. This could be due to the increased likelihood that some of the CTCs within a sample will be tumourigenic, as well as the association between high CTC counts and aggressive tumours. Since the process of collecting CTCs from blood or “liquid biopsies” is non-invasive, it might be possible to obtain serial samples to establish PDXs from different stages of disease progression, such as before and after therapeutic resistance. Therefore, CTCs are likely to become increasingly popular samples for establishing PDXs.
Sorted or Cultured Cancer Cells
PDXs are usually established from samples of intact or digested patient tissue; however, they can also be grown from tumour cells that are preselected using sorting or primary culture. Cell sorting is used to enrich defined populations of cancer cells based on their expression of cell surface antigens or phenotype. Common techniques include flow cytometry, magnetic bead separation and differential attachment to coated plates. Xenografts of sorted cells have primarily been used to study cancer-repopulating cells [37, 64, 65]. The frequency of cancer-repopulating cells can be calculated by decreasing numbers of sorted cells to establish a tumour [66]. The other common method of preselecting cancer cells, primary cell culture, is also based on the premise that only a subpopulation of cancer cells may have the potential to form tumours. In this way, in vitro cell culture can be used to select patient specimens that are more likely to establish successful PDXs. These primary cultures are increasingly being established as organoids rather than adherent or suspension cultures [60,61,62, 67]. This method is particularly useful for samples with low take rates, because it is easier to monitor their growth in vitro and then subsequently graft the cells into host mice. This also provides matched in vitro and in vivo models to study tumour biology and drug responses in various contexts. An unresolved question is whether there are differences in PDXs established from pieces of tissue compared to cultured cells, other than the obvious lack of human stroma in early generations. Nevertheless, preselecting cancer cells through sorting or primary culture provides researchers with a way to control the success rate of PDXs.
Preparation of Fresh Tissue for Xenografting
After obtaining high-quality patient specimens, the next important step in establishing successful PDXs is to carefully prepare the tissues for xenografting. The standard procedure for many tumours is to either graft whole pieces of tissue or enzymatically digested tissue as quickly as possible into the host mouse [68]. However, additional processing steps may improve the take rate of tissues that are difficult to grow as PDXs. One approach is to use a tissue slicer to cut thin and precise samples, typically about 300 μm thick [69, 70]. Once grafted, these thin slices may be more highly oxygenated than larger pieces of tissue, potentially increasing their survival during the time it takes them to become vascularised. Slicing is also useful for specimens with a heterogeneous composition of cancer and benign regions, because it is possible to compare paired slices and assess the tumour content in fixed slices [69].
Other methods of preparing tissue aim to enrich the graft microenvironment. For example, grafts are often embedded in Matrigel, which provides growth factors and extracellular matrix to encourage the growth of the patient tissue and host vasculature [17, 23, 71]. Matrigel is also useful for binding together dispersed cells or fragile pieces of tissues. The development of biomimetic scaffolds by tissue engineers may provide alternatives to Matrigel where the composition and stiffness of the matrix is customised to match the patient tissue [72]. Another way of providing a supportive microenvironment is to add stroma to grafts; however, the source of stroma is critical. The take rate of primary prostate PDXs is increased when they are recombined with mesenchyme from embryonic or neonatal mice [5, 7, 9, 73]. Similarly, the growth and vascularisation of primary breast cancer PDXs is improved by co-implanting mesenchymal stromal cells [17]. In contrast, immortalised human fibroblasts from normal breast tissue had no effect on the take rate of primary breast cancer PDXs and actually decreased their serial transplantability [42]. These studies emphasise that the graft microenvironment, and the method of preparing tissue in general, can be critical in establishing PDXs.
Influence of the Mouse Host on Patient-Derived Xenografts
Some of the most dramatic improvements in PDX protocols have been due to changes in the mouse host. This section will discuss the importance of systemic features of host mice, in particular their immunocompetence and circulating steroid levels, as well as the local features of the graft site.
Systemic Features of the Mouse Host: Strains and Steroids
It is essential to use immunocompromised host mice to avoid rejection of PDXs. Early methods of supressing the host immune system included X-ray irradiation and thymectomy [74, 75]. The subsequent discovery of athymic nude mice (nu/nu), which lack functional T cells, foreshadowed the use of increasingly immunocompromised mouse strains for xenografting [76]. Many laboratories then shifted to using severe combined immune-deficient (SCID) mice, which are deficient in both T and B cells [77]. Non-obese diabetic SCID (NOD-SCID) mice then became more popular for xenografting, because they avoid the leaky phenotype of SCID mice and also have impaired natural killer (NK) cell function [78, 79]. To further abrogate the host immune response in these strains, some laboratories pretreated mice with etoposide, an immunosuppressant, a few days prior to grafting [39, 80]. It was subsequently shown, however, that etoposide decreased the take rate and growth of breast cancer PDXs in thoracic fat pads [39].
Most recently, NOD-SCID interleukin-2 receptor gamma chain null (NSG) mice have become a common strain used for xenografting. NSG mice are highly immunocompromised because they lack functional T, B and NK cells [81, 82]. It is often assumed that using highly immunocompromised host mice can improve the take rate of PDXs; however, it seems to depend on the patient specimens. Small numbers of melanoma cells have dramatically higher take rates in NSG versus NOD-SCID mice, whereas there is no difference in the engraftment rate of breast cancer tissue in SCID versus NSG mice or prostate cancer tissue in nude versus NOD-SCID mice [11, 42, 65]. Nevertheless, many laboratories now routinely use NOD-SCID or NSG mice for xenografting given that the take rate is greater, or at least equal, to that obtained with other strains. The trade-off for high engraftment rates with immunocompromised mice is the inability to study the interactions between tumour and immune cells, leading to increasing interest in humanised PDX models [83].
In addition to the immune system, steroid hormone levels are another systemic feature of host mice that affects the engraftment and growth of hormone-dependent cancers. Breast cancer PDXs are often established in female mice implanted with oestrogen pellets. Oestrogen supplementation increases the engraftment of both oestrogen receptor-positive and receptor-negative subtypes, presumably due to paracrine signalling from the stroma [17, 42, 84, 85]. Unfortunately, higher doses of exogenous oestrogen can cause side effects in host mice, such as urine retention and hydronephrosis, leading to the development of alternative protocols with lower oestrogen levels [26, 86, 87]. Similar to breast samples, prostate tissue is grafted into male mice with testosterone implants. Higher testosterone levels are required to maintain the differentiation of benign and malignant prostate glands in PDXs, because androgen levels in adult male mice are only equivalent to hypogonadal adult men [10, 24, 88, 89]. Therefore, supplementing steroid hormone levels in host mice is a simple way to maximise the success of PDXs of hormone-dependent cancers.
Local Features of the Mouse Host: Graft Site
The graft site is an important consideration when establishing PDXs, because local features of the mouse host affect the take rate, fidelity and practicality of PDXs. Specimens are often engrafted heterotopically, that is, at a different site compared to their tissue of origin. Subcutaneous grafting into the shoulder or flank of host mice is particularly common. The advantages of subcutaneous grafting include the speed and technical simplicity of the method, the ability to graft large specimens and the ease of monitoring tumour growth [90]. Thus, it is ideal for tumours with high take rates or for serially transplantable PDXs that have already been established at other graft sites. Tumours with low take rates as subcutaneous grafts, such as oesophageal, prostate and low-grade ovarian cancer, often grow more successfully as subrenal capsule or intramuscular grafts [10, 11, 91, 92]. This is thought to be due to the greater blood supply at these sites. Subrenal grafting requires more expertise than subcutaneous grafting; however, once the grafts are implanted, they are held in place by the renal capsule, a thin membrane surrounding the kidney [5, 10, 89, 93]. Another limitation of subrenal capsule grafting is that it is more difficult to estimate tumour growth by palpating the grafts. Therefore, for heterotopic PDXs, the choice of graft site depends on the balance between optimal take rate and ease of grafting and monitoring of tumour growth.
PDXs can also be established orthotopically [10, 17, 28, 42, 90, 94,95,96,97]. It can be more difficult and time-consuming to establish orthotopic grafts, and they may need to be monitored using specialist imaging equipment [98]. Nevertheless, orthotopic grafting ensures that the host microenvironment mimics the patient tissue of origin as closely as possible. This is reflected in the high take rates of orthotopic grafts for many tumour types, compared to subcutaneous grafts [10, 17]. The graft site can also affect the phenotype of PDXs. For example, orthotopic PDXs may be more likely to metastasise [95, 99, 100] and more closely reflect patients’ responses to therapy than heterotopic PDXs [101]. Yet, most PDXs closely recapitulate the original patient tumour [2], so the degree to which orthotopic grafting improves fidelity is unclear and might depend on the tumour type. The precise anatomical site used for orthotopic grafting can also be important. For example, breast cancer PDXs exhibit better engraftment and growth in abdominal compared to thoracic fat pads [39]. Furthermore, injecting oestrogen receptor α-positive tumours into milk ducts instead of fad pads produces PDXs that more closely resemble the histopathological features of patient tumours [26]. Collectively, these studies emphasise that the local microenvironment can affect the take rate and phenotype of PDXs.
Authentication of Patient-Derived Xenografts
Once PDXs are established, it is important to verify that they recapitulate the original patient specimens. There is a risk that PDXs can become contaminated, especially with lymphomas, which can rapidly overtake grafts. Lymphomas can originate from the host mouse, particularly in ageing NOD-SCID mice [18, 102, 103]. They can also arise from human B cells within the patient specimen that are transformed by Epstein-Barr virus. PDXs from a diverse range of tumour types have been contaminated with human lymphomas [18, 92, 103,104,105,106]. Simple tests can be used to rapidly identify contaminated PDXs, allowing them to be rescued. Contaminating mouse cells can be identified using species-specific analyses of telomeres, Alu repeats and human mitochondrial antigens [26, 92, 107]. Human lymphomas can be detected, and if necessary depleted, based on CD45 expression [105].
PDXs can also be cross-contaminated with one another, so there is a growing need to authenticate that they match the correct patient. It is becoming a routine practice to authenticate immortalised cell lines to ensure that they are derived from the intended source, but this is not yet the case for PDXs. This led the International Cell Line Authentication Committee to recommend that guidelines and protocols should be developed for rigorously characterising PDXs [108]. The identity of PDXs is sometimes confirmed in the process of genomic studies, but other targeted approaches can be used for routine authentication. For example, short tandem repeat (STR) analysis is commonly used for authenticating cell lines, and it has also been used in some PDX studies [42, 109]. An alternative approach is to analyse the pattern of single nucleotide polymorphisms (SNPs), which provides similar accuracy to STRs in identifying different patient samples. An automated SNP-based PDX Authentication System (PAS) with 32 SNPs was recently validated with PDXs from paediatric acute lymphoblastic leukaemia, Ewing’s sarcoma and prostate cancer [110]. This system identifies as little as 3% cross-contamination between PDXs, providing an effective method for authenticating PDXs. Therefore, given the time and expense of PDX experiments, rapid and inexpensive approaches for authenticating the identity of PDXs will become important tools for confirming the validity of the results.
Summary and Conclusions
Over the last few decades, the availability of PDXs in laboratories around the world has increased dramatically. This has been driven by numerous methodological improvements and the realisation that PDXs are invaluable tools in preclinical cancer research. Yet, it is important to note that there is no “best practice” in establishing PDXs. Methods vary depending on the tumour type, and, in many instances, tumour take rates have not been formally compared between various protocols. Instead, a bit like a cottage industry, many individual laboratories have gradually optimised the way they establish PDXs for particular tumour types. This is changing, however, as laboratories publish detailed protocols, and national and international consortia develop large repositories of PDXs. The scale and scope of these PDX platforms is important, because multiple PDXs of each tumour type and each stage of disease progression are required to replicate the diversity of tumours in the clinic. Nevertheless, these PDX platforms depend on the expertise of cancer researchers to grow patient specimens as PDXs. Through further innovation and collaboration, this process is likely to become increasingly successful in the years ahead.
Abbreviations
- NOD-SCID:
-
Non-obese diabetic severe combined immunodeficient mice
- CTC:
-
Circulating tumour cell
- NK cell:
-
Natural killer cell
- NSG:
-
NOD-SCID interleukin-2 receptor gamma chain null mice
- PAS:
-
PDX Authentication System
- PDX:
-
Patient-derived xenograft
- SCID:
-
Severe combined immunodeficient mice
- SNP:
-
Single nucleotide polymorphism
- STR:
-
Short tandem repeat
- TURP:
-
Transurethral resection of the prostate
References
Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell. 2016;167(1):260–274. e22. doi:10.1016/j.cell.2016.08.041.
Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318–25. doi:10.1038/nm.3954.
Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4(9):998–1013. doi:10.1158/2159-8290.CD-14-0001.
Dong X, Guan J, English JC, Flint J, Yee J, Evans K, et al. Patient-derived first generation xenografts of non-small cell lung cancers: promising tools for predicting drug responses for personalized chemotherapy. Clin Cancer Res. 2010;16(5):1442–51. doi:10.1158/1078-0432.CCR-09-2878.
Lawrence MG, Taylor RA, Toivanen R, Pedersen J, Norden S, Pook DW, et al. A preclinical xenograft model of prostate cancer using human tumors. Nat Protoc. 2013;8(5):836–48. doi:10.1038/nprot.2013.043.
Presnell SC, Werdin ES, Maygarden S, Mohler JL, Smith GJ. Establishment of short-term primary human prostate xenografts for the study of prostate biology and cancer. Am J Pathol. 2001;159(3):855–60. doi:10.1016/S0002-9440(10)61761-0.
Toivanen R, Berman DM, Wang H, Pedersen J, Frydenberg M, Meeker AK, et al. Brief report: a bioassay to identify primary human prostate cancer repopulating cells. Stem Cells. 2011;29(8):1310–4. doi:10.1002/stem.668.
Lawrence MG, Pook DW, Wang H, Porter LH, Frydenberg M, Kourambas J, et al. Establishment of primary patient-derived xenografts of palliative TURP specimens to study castrate-resistant prostate cancer. Prostate. 2015;75(13):1475–83. doi:10.1002/pros.23039.
Risbridger GP, Taylor RA, Clouston D, Sliwinski A, Thorne H, Hunter S, et al. Patient-derived xenografts reveal that intraductal carcinoma of the prostate is a prominent pathology in BRCA2 mutation carriers with prostate cancer and correlates with poor prognosis. Eur Urol. 2015;67(3):496–503. doi:10.1016/j.eururo.2014.08.007.
Wang Y, Revelo M, Sudilovsky D, Cao M, Chen W, Goetz L, et al. Development and characterization of efficient xenograft models for benign and malignant human prostate tissue. Prostate. 2005;64(2):149–59.
Priolo C, Agostini M, Vena N, Ligon AH, Fiorentino M, Shin E, et al. Establishment and genomic characterization of mouse xenografts of human primary prostate tumors. Am J Pathol. 2010;176(4):1901–13. doi:10.2353/ajpath.2010.090873.
Alkema NG, Tomar T, Duiker EW, Jan Meersma G, Klip H, van der Zee AG, et al. Biobanking of patient and patient-derived xenograft ovarian tumour tissue: efficient preservation with low and high fetal calf serum based methods. Sci Rep. 2015;5:14495. doi:10.1038/srep14495.
Lin D, Wyatt AW, Xue H, Wang Y, Dong X, Haegert A, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74(4):1272–83. doi:10.1158/0008-5472.CAN-13-2921-T.
Alsop K, Thorne H, Sandhu S, Hamilton A, Mintoff C, Christie E, et al. A community-based model of rapid autopsy in end-stage cancer patients. Nat Biotechnol. 2016;34(10):1010–4. doi:10.1038/nbt.3674.
Van Weerden WM, Romijn JC. Use of nude mouse xenograft models in prostate cancer research. Prostate. 2000;43(4):263–71. doi:10.1002/1097-0045(20000601)43:4<263::AID-PROS5>3.0.CO;2-I.
Whittle JR, Lewis MT, Lindeman GJ, Visvader JE. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015;17:17. doi:10.1186/s13058-015-0523-1.
DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17(11):1514–20. doi:10.1038/nm.2454.
Ilie M, Nunes M, Blot L, Hofman V, Long-Mira E, Butori C, et al. Setting up a wide panel of patient-derived tumor xenografts of non-small cell lung cancer by improving the preanalytical steps. Cancer Med. 2015;4(2):201–11. doi:10.1002/cam4.357.
Sivanand S, Pena-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med. 2012;4(137):137ra75. doi:10.1126/scitranslmed.3003643.
Blance RN, Sims AH, Anderson E, Howell A, Clarke RB. Normal breast tissue implanted into athymic nude mice identifies biomarkers of the effects of human pregnancy levels of estrogen. Cancer Prev Res (Phila). 2009;2(3):257–64. doi:10.1158/1940-6207.CAPR-08-0161.
Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329(5991):568–71. doi:10.1126/science.1189992.
Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A. 2004;101(14):4966–71. doi:10.1073/pnas.0401064101.
Montecinos VP, Godoy A, Hinklin J, Vethanayagam RR, Smith GJ. Primary xenografts of human prostate tissue as a model to study angiogenesis induced by reactive stroma. PLoS One. 2012;7(1):e29623. doi:10.1371/journal.pone.0029623.
Staack A, Kassis AP, Olshen A, Wang Y, Wu D, Carroll PR, et al. Quantitation of apoptotic activity following castration in human prostatic tissue in vivo. Prostate. 2003;54(3):212–9.
Gray DR, Huss WJ, Yau JM, Durham LE, Werdin ES, Funkhouser Jr WK, et al. Short-term human prostate primary xenografts: an in vivo model of human prostate cancer vasculature and angiogenesis. Cancer Res. 2004;64(5):1712–21.
Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, et al. A preclinical model for eralpha-positive breast cancer points to the epithelial microenvironment as determinant of luminal phenotype and hormone response. Cancer Cell. 2016;29(3):407–22. doi:10.1016/j.ccell.2016.02.002.
Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, Alexandrov LB, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet. 2015;47(4):367–72. doi:10.1038/ng.3221.
Dotto GP. Multifocal epithelial tumors and field cancerization: stroma as a primary determinant. J Clin Invest. 2014;124(4):1446–53. doi:10.1172/JCI72589.
DeRose YS, Gligorich KM, Wang G, Georgelas A, Bowman P, Courdy SJ, et al. Patient-derived models of human breast cancer: protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr Protoc Pharmacol. 2013;60(14.23):14.23.1–14.23.43. doi:10.1002/0471141755.ph1423s60.
Kim MP, Truty MJ, Choi W, Kang Y, Chopin-Lally X, Gallick GE, et al. Molecular profiling of direct xenograft tumors established from human pancreatic adenocarcinoma after neoadjuvant therapy. Ann Surg Oncol. 2012;19 Suppl 3:S395–403. doi:10.1245/s10434-011-1839-4.
McAuliffe PF, Evans KW, Akcakanat A, Chen K, Zheng X, Zhao H, et al. Ability to generate patient-derived breast cancer xenografts is enhanced in chemoresistant disease and predicts poor patient outcomes. PLoS One. 2015;10(9):e0136851. doi:10.1371/journal.pone.0136851.
Allaway RJ, Fischer DA, de Abreu FB, Gardner TB, Gordon SR, Barth RJ, et al. Genomic characterization of patient-derived xenograft models established from fine needle aspirate biopsies of a primary pancreatic ductal adenocarcinoma and from patient-matched metastatic sites. Oncotarget. 2016;7(13):17087–102. doi:10.18632/oncotarget.7718.
Zhu Y, Tian T, Li Z, Tang Z, Wang L, Wu J, et al. Establishment and characterization of patient-derived tumor xenograft using gastroscopic biopsies in gastric cancer. Sci Rep. 2015;5:8542. doi:10.1038/srep08542.
Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goere D, Mariani P, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18(19):5314–28. doi:10.1158/1078-0432.CCR-12-0372.
Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature. 2015;518(7538):240–4. doi:10.1038/nature13948.
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1(6):508–23. doi:10.1158/2159-8290.CD-11-0109.
O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–10. doi:10.1038/nature05372.
Rashidi B, Sun FX, Jiang P, An Z, Gamagami R, Moossa AR, et al. A nude mouse model of massive liver and lymph node metastasis of human colon cancer. Anticancer Res. 2000;20(2A):715–22.
Fleming JM, Miller TC, Meyer MJ, Ginsburg E, Vonderhaar BK. Local regulation of human breast xenograft models. J Cell Physiol. 2010;224(3):795–806. doi:10.1002/jcp.22190.
Liu JF, Palakurthi S, Zeng Q, Zhou S, Ivanova E, Huang W, et al. Establishment of patient-derived tumor xenograft models of epithelial ovarian cancer for pre-clinical evaluation of novel therapeutics. Clin Cancer Res. 2016. doi:10.1158/1078-0432.CCR-16-1237.
Verschraegen CF, Hu W, Du Y, Mendoza J, Early J, Deavers M, et al. Establishment and characterization of cancer cell cultures and xenografts derived from primary or metastatic Mullerian cancers. Clin Cancer Res. 2003;9(2):845–52.
Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73(15):4885–97. doi:10.1158/0008-5472.CAN-12-4081.
Craft N, Chhor C, Tran C, Belldegrun A, DeKernion J, Witte ON, et al. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res. 1999;59(19):5030–6.
Li ZG, Mathew P, Yang J, Starbuck MW, Zurita AJ, Liu J, et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J Clin Invest. 2008;118(8):2697–710. doi:10.1172/JCI33093.
McCulloch DR, Opeskin K, Thompson EW, Williams ED. BM18: a novel androgen-dependent human prostate cancer xenograft model derived from a bone metastasis. Prostate. 2005;65(1):35–43.
Goncalves A, Bertucci F, Guille A, Garnier S, Adelaide J, Carbuccia N, et al. Targeted NGS, array-CGH, and patient-derived tumor xenografts for precision medicine in advanced breast cancer: a single-center prospective study. Oncotarget. 2016. doi:10.18632/oncotarget.12714.
Klein KA, Reiter RE, Redula J, Moradi H, Zhu XL, Brothman AR, et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat Med. 1997;3(4):402–8.
Hong MK, Sapre N, Phal PM, Macintyre G, Chin X, Pedersen JS, et al. Percutaneous image-guided biopsy of prostate cancer metastases yields samples suitable for genomics and personalised oncology. Clin Exp Metastasis. 2014;31(2):159–67. doi:10.1007/s10585-013-9617-2.
Kohli M, Wang L, Xie F, Sicotte H, Yin P, Dehm SM, et al. Mutational landscapes of sequential prostate metastases and matched patient derived xenografts during enzalutamide therapy. PLoS One. 2015;10(12):e0145176. doi:10.1371/journal.pone.0145176.
Rimondi E, Rossi G, Bartalena T, Ciminari R, Alberghini M, Ruggieri P, et al. Percutaneous CT-guided biopsy of the musculoskeletal system: results of 2027 cases. Eur J Radiol. 2011;77(1):34–42. doi:10.1016/j.ejrad.2010.06.055.
Hooper JE, Cantor EL, Ehlen MS, Banerjee A, Malempati S, Stenzel P, et al. A patient-derived xenograft model of parameningeal embryonal rhabdomyosarcoma for preclinical studies. Sarcoma. 2015;2015:826124. doi:10.1155/2015/826124.
Rubin MA, Putzi M, Mucci N, Smith DC, Wojno K, Korenchuk S, et al. Rapid (“warm”) autopsy study for procurement of metastatic prostate cancer. Clin Cancer Res. 2000;6(3):1038–45.
Xie T, Musteanu M, Lopez-Casas PP, Shields DJ, Olson P, Rejto PA, et al. Whole exome sequencing of rapid autopsy tumors and xenograft models reveals possible driver mutations underlying tumor progression. PLoS One. 2015;10(11):e0142631. doi:10.1371/journal.pone.0142631.
Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353–7. doi:10.1038/nature14347.
Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22(4):369–78. doi:10.1038/nm.4053.
Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489–94. doi:10.1038/nature14410.
Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013;31(6):539–44. doi:10.1038/nbt.2576.
Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014;20(8):897–903. doi:10.1038/nm.3600.
Williams ES, Rodriguez-Bravo V, Chippada-Venkata U, De Ia Iglesia-Vicente J, Gong Y, Galsky M, et al. Generation of prostate cancer patient derived xenograft models from circulating tumor cells. J Vis Exp. 2015;(105):53182. doi:10.3791/53182.
Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–87. doi:10.1016/j.cell.2014.08.016.
Grillet F, Bayet E, Villeronce O, Zappia L, Lagerqvist EL, Lunke S, et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut. 2016. doi:10.1136/gutjnl-2016-311447.
Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345(6193):216–20. doi:10.1126/science.1253533.
Rossi E, Rugge M, Facchinetti A, Pizzi M, Nardo G, Barbieri V, et al. Retaining the long-survive capacity of circulating tumor cells (CTCs) followed by xeno-transplantation: not only from metastatic cancer of the breast but also of prostate cancer patients. Oncoscience. 2014;1(1):49–56. doi:10.18632/oncoscience.8.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8.
Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456(7222):593–8. doi:10.1038/nature07567.
Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1–2):70–8. doi:10.1016/j.jim.2009.06.008.
Cayrefourcq L, Mazard T, Joosse S, Solassol J, Ramos J, Assenat E, et al. Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res. 2015;75(5):892–901. doi:10.1158/0008-5472.CAN-14-2613.
Morton CL, Houghton PJ. Establishment of human tumor xenografts in immunodeficient mice. Nat Protoc. 2007;2(2):247–50. doi:10.1038/nprot.2007.25.
Zhao H, Nolley R, Chen Z, Peehl DM. Tissue slice grafts: an in vivo model of human prostate androgen signaling. Am J Pathol. 2010;177(1):229–39. doi:10.2353/ajpath.2010.090821.
Zhao H, Thong A, Nolley R, Reese SW, Santos J, Ingels A, et al. Patient-derived tissue slice grafts accurately depict response of high-risk primary prostate cancer to androgen deprivation therapy. J Transl Med. 2013;11:199. doi:10.1186/1479-5876-11-199.
Bernardo C, Costa C, Sousa N, Amado F, Santos L. Patient-derived bladder cancer xenografts: a systematic review. Transl Res. 2015;166(4):324–31. doi:10.1016/j.trsl.2015.02.001.
Hutmacher DW, Holzapfel BM, De-Juan-Pardo EM, Pereira BA, Ellem SJ, Loessner D, et al. Convergence of regenerative medicine and synthetic biology to develop standardized and validated models of human diseases with clinical relevance. Curr Opin Biotechnol. 2015;35:127–32. doi:10.1016/j.copbio.2015.06.001.
Toivanen R, Frydenberg M, Murphy D, Pedersen J, Ryan A, Pook D, et al. A preclinical xenograft model identifies castration-tolerant cancer-repopulating cells in localized prostate tumors. Sci Transl Med. 2013;5(187):187ra71. doi:10.1126/scitranslmed.3005688.
Clemmesen J. On transplantation of tumor cells to normal and pre-irradiated heterologous organisms. Cancer Res. 1937;29(2):313–32.
Grogan JB, Hardy JD. Increased survival of xenogeneic tumor in thymectomized hosts. J Surg Res. 1968;8(1):7–9.
Rygaard J, Povlsen CO. Heterotransplantation of a human malignant tumour to “Nude” mice. Acta Pathol Microbiol Scand. 1969;77(4):758–60.
Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301(5900):527–30.
Bosma MJ. B and T cell leakiness in the scid mouse mutant. Immunodefic Rev. 1992;3(4):261–76.
Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154(1):180–91.
Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, Lin J, et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci U S A. 2010;107(42):18115–20. doi:10.1073/pnas.1006732107.
Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100(9):3175–82.
Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477–89.
Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7(2):118–30. doi:10.1038/nri2017.
Gupta PB, Proia D, Cingoz O, Weremowicz J, Naber SP, Weinberg RA, et al. Systemic stromal effects of estrogen promote the growth of estrogen receptor-negative cancers. Cancer Res. 2007;67(5):2062–71. doi:10.1158/0008-5472.CAN-06-3895.
Kabos P, Finlay-Schultz J, Li C, Kline E, Finlayson C, Wisell J, et al. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res Treat. 2012;135(2):415–32. doi:10.1007/s10549-012-2164-8.
Dall G, Vieusseux J, Unsworth A, Anderson R, Britt K. Low dose, low cost estradiol pellets can support MCF-7 tumour growth in nude mice without bladder symptoms. J Cancer. 2015;6(12):1331–6. doi:10.7150/jca.10890.
Gakhar G, Wight-Carter M, Andrews G, Olson S, Nguyen TA. Hydronephrosis and urine retention in estrogen-implanted athymic nude mice. Vet Pathol. 2009;46(3):505–8. doi:10.1354/vp.08-VP-0180-N-BC.
Michiel Sedelaar JP, Dalrymple SS, Isaacs JT. Of mice and men-warning: intact versus castrated adult male mice as xenograft hosts are equivalent to hypogonadal versus abiraterone treated aging human males, respectively. Prostate. 2013;73(12):1316–25. doi:10.1002/pros.22677.
Nicholson TM, Uchtmann KS, Valdez CD, Theberge AB, Miralem T, Ricke WA. Renal capsule xenografting and subcutaneous pellet implantation for the evaluation of prostate carcinogenesis and benign prostatic hyperplasia. J Vis Exp. 2013;(78):e50574. doi:10.3791/50574.
Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc. 2009;4(11):1670–80. doi:10.1038/nprot.2009.171.
Lee CH, Xue H, Sutcliffe M, Gout PW, Huntsman DG, Miller DM, et al. Establishment of subrenal capsule xenografts of primary human ovarian tumors in SCID mice: potential models. Gynecol Oncol. 2005;96(1):48–55. doi:10.1016/j.ygyno.2004.09.025.
Read M, Liu D, Duong CP, Cullinane C, Murray WK, Fennell CM, et al. Intramuscular transplantation improves engraftment rates for esophageal patient-derived tumor xenografts. Ann Surg Oncol. 2016;23(1):305–11. doi:10.1245/s10434-015-4425-3.
Shultz LD, Goodwin N, Ishikawa F, Hosur V, Lyons BL, Greiner DL. Subcapsular transplantation of tissue in the kidney. Cold Spring Harb Protoc. 2014;2014(7):737–40. doi:10.1101/pdb.prot078089.
Fu E, Nelson KE, Ramsey SA, Foley JO, Helton K, Yager P. Modeling of a competitive microfluidic heterogeneous immunoassay: sensitivity of the assay response to varying system parameters. Anal Chem. 2009;81(9):3407–13. doi:10.1021/ac802672v.
Fu XY, Theodorescu D, Kerbel RS, Hoffman RM. Extensive multi-organ metastasis following orthotopic onplantation of histologically-intact human bladder carcinoma tissue in nude mice. Int J Cancer. 1991;49(6):938–9.
Saar M, Korbel C, Linxweiler J, Jung V, Kamradt J, Hasenfus A, et al. Orthotopic tumorgrafts in nude mice: a new method to study human prostate cancer. Prostate. 2015;75(14):1526–37. doi:10.1002/pros.23027.
Topp MD, Hartley L, Cook M, Heong V, Boehm E, McShane L, et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol Oncol. 2014;8(3):656–68. doi:10.1016/j.molonc.2014.01.008.
Kraaij R, van Weerden WM, de Ridder CM, Gussenhoven EJ, Honkoop J, Nasu Y, et al. Validation of transrectal ultrasonographic volumetry for orthotopic prostate tumours in mice. Lab Anim. 2002;36(2):165–72.
Fu X, Guadagni F, Hoffman RM. A metastatic nude-mouse model of human pancreatic cancer constructed orthotopically with histologically intact patient specimens. Proc Natl Acad Sci U S A. 1992;89(12):5645–9.
Lin D, Watahiki A, Bayani J, Zhang F, Liu L, Ling V, et al. ASAP1, a gene at 8q24, is associated with prostate cancer metastasis. Cancer Res. 2008;68(11):4352–9. doi:10.1158/0008-5472.CAN-07-5237.
Garrido-Laguna I, Uson M, Rajeshkumar NV, Tan AC, de Oliveira E, Karikari C, et al. Tumor engraftment in nude mice and enrichment in stroma-related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin Cancer Res. 2011;17(17):5793–800. doi:10.1158/1078-0432.CCR-11-0341.
Kato C, Fujii E, Chen YJ, Endaya BB, Matsubara K, Suzuki M, et al. Spontaneous thymic lymphomas in the non-obese diabetic/Shi-scid, IL-2R gamma (null) mouse. Lab Anim. 2009;43(4):402–4. doi:10.1258/la.2009.009012.
Wetterauer C, Vlajnic T, Schuler J, Gsponer JR, Thalmann GN, Cecchini M, et al. Early development of human lymphomas in a prostate cancer xenograft program using triple knock-out immunocompromised mice. Prostate. 2015;75(6):585–92. doi:10.1002/pros.22939.
Bondarenko G, Ugolkov A, Rohan S, Kulesza P, Dubrovskyi O, Gursel D, et al. Patient-derived tumor xenografts are susceptible to formation of human lymphocytic tumors. Neoplasia. 2015;17(9):735–41. doi:10.1016/j.neo.2015.09.004.
Chen K, Ahmed S, Adeyi O, Dick JE, Ghanekar A. Human solid tumor xenografts in immunodeficient mice are vulnerable to lymphomagenesis associated with Epstein–Barr virus. PLoS One. 2012;7(6):e39294. doi:10.1371/journal.pone.0039294.
John T, Yanagawa N, Kohler D, Craddock KJ, Bandarchi-Chamkhaleh B, Pintilie M, et al. Characterization of lymphomas developing in immunodeficient mice implanted with primary human non-small cell lung cancer. J Thorac Oncol. 2012;7(7):1101–8. doi:10.1097/JTO.0b013e3182519d4d.
Vander Griend DJ, Konishi Y, De Marzo AM, Isaacs JT, Meeker AK. Dual-label centromere and telomere FISH identifies human, rat, and mouse cell contribution to Multispecies recombinant urogenital sinus xenografts. Prostate. 2009;69(14):1557–64. doi:10.1002/pros.21001.
Nardone RM, MacLeod RA, Capes-Davis A. Cancer: authenticate new xenograft models. Nature. 2016;532(7599):313.
Mattie M, Christensen A, Chang MS, Yeh W, Said S, Shostak Y, et al. Molecular characterization of patient-derived human pancreatic tumor xenograft models for preclinical and translational development of cancer therapeutics. Neoplasia. 2013;15(10):1138–50.
El-Hoss J, Jing D, Evans K, Toscan C, Xie J, Lee H, et al. A single nucleotide polymorphism genotyping platform for the authentication of patient derived xenografts. Oncotarget. 2016. doi:10.18632/oncotarget.11125.
Acknowledgements
We thank Laura Porter for her help preparing the manuscript. The Monash University Prostate Cancer Research Program is supported by funding from the Peter and Lyndy White Foundation, the EJ Whitten Foundation and TissuPath Pathology. GPR is supported by a fellowship from the National Health and Medical Research Council (1102752).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Risbridger, G.P., Lawrence, M.G. (2017). Towards Best Practice in Establishing Patient-Derived Xenografts. In: Wang, Y., Lin, D., Gout, P. (eds) Patient-Derived Xenograft Models of Human Cancer . Molecular and Translational Medicine. Humana Press, Cham. https://doi.org/10.1007/978-3-319-55825-7_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-55825-7_2
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-55824-0
Online ISBN: 978-3-319-55825-7
eBook Packages: MedicineMedicine (R0)