
Abstract
Hereditary tyrosinemia type I (HTI) is a rare autosomal recessive disorder caused by a fumarylacetoacetate hydrolase (FAH) deficiency. If untreated, its acute form is characterized by hepatic failure, renal dysfunction and neurological crisis, and may lead to death. Due to a genetic founder effect in the French-Canadian population, the prevalence of HTI is increased in the province of Quebec (1/19 819), with the IVS12 + 5G>A (1062 + 5G>A) splice site mutation responsible for more than 90% of mutated alleles. Universal newborn screening for (HT1) was thus established in 1970, and close to four million infants have been tested so far, allowing to identify 185 of the 190 affected newborns. During the 1970–1997 period, 2,249,000 newborns were screened at 3–7 days of life on dried filter paper blood spots by tyrosine (Tyr) concentration followed by indirect colorimetric semi-quantitative and quantitative (Q) succinylacetone (SA) testing (red blood cells δ-aminolevulinate dehydratase inhibition), with immunoreactive FAH as the confirmatory test. This approach allowed to identify 118 of 123 affected newborns. In 1998, owing to earlier hospital discharge and increased rate of breastfeeding, four cases were missed within the same year as the discriminating power of blood Tyr became inadequate. Thus, the screening algorithm was modified: indirect semi-quantitative SA measurement became the first-tier test between 1998 and 2014, and direct SA measurement by tandem mass spectrometry (MS/MS) was implemented in 2014, followed by indirect quantitative SA measurement as second tier test. Confirmation is performed by plasmatic amino acid profile and molecular testing. During the 1998–2016 period, more than 1,5 million neonates have been tested (90% sampled between 24 and 48 h of life): 67 of the 67 HTI cases were identified. Both indirect and direct SA measurement as the initial HTI screening test proved to be highly sensitive and specific, with positive and negative predicting value of 79% and 100% respectively.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Screening for HT1 in Quebec: Background
Hereditary tyrosinemia type I (HT1, McKusick 27,670), also called hepatorenal tyrosinemia, is a rare autosomal recessive inborn error of metabolism caused by a deficiency of fumarylacetoacetate hydrolase (FAH), in the last step of tyrosine catabolic pathway (Fig. 13.1). FAH deficiency leads to an accumulation of fumarylacetoacetate (FAA), maleylacetoacetate (MAA), and succinylacetone (SA), which are responsible for the clinical symptoms of the disorder. The worldwide incidence of HTI is estimated to be 1:100,000–125,000 (Mitchell et al. 2001; Halvorsen 1980). Close to 100 different mutations in the FAH gene have been identified so far, many of which have specific ethno-geographic distributions (Angileri et al. 2015).

Catabolic pathway of tyrosine
NTBC: 2-[2-nitro-4-(trifluoromethyl) benzoyl] cyclohexane-1,3-dione
The French-Canadian population of Quebec, representing about 6,5 of the 8,1 million inhabitants, descents from about 8500 French settlers from a few villages in northern France who arrived in Nouvelle-France between 1608 and 1759. The migration of those settlers and their descendants led to genetic founder effects for various disorders, including HT1, which are reflected in the geographical distribution of genetic diseases in the province (Laberge et al. 2005). Indeed, due to a complex founder effect (De Braekeleer and Larochelle 1990; Laberge 1969), the frequency of HT1 is much higher in the province of Quebec where it has been previously estimated at 1:16,786, with the highest frequency of 1:1846 in the Saguenay Lac-Saint-Jean region (SLSJ) in the northeastern part of Quebec, and an estimated carrier rate between 1:20 and 1:31 (De Braekeleer and Larochelle 1990; Laberge and Dallaire 1967). In the SLSJ region of Quebec, owing to the founder effect, about 95% of mutated alleles are due to a splice site mutation (IVS12 + 5G–>A; 1062 + 5G>A) in the FAH gene (Grompe et al. 1994; Poudrier et al. 1996).
HTI may present either as an acute or a chronic form. In the acute form, the prenatal and perinatal periods are unremarkable, but symptoms appear in the neonatal period. It is characterized by acute liver failure, peripheral porphyria-like neurological crises and coagulation disorders. In the absence of treatment, neonates affected by the acute form usually present with hepatic decompensation and porphyria-like neurological crises, caused by the accumulation of 5-aminolevulinic acid (ALA) due to ALA dehydratase inhibition by SA (Scott 2006), which is often precipitated by a catabolic state. In the chronic form, the child presents with failure to thrive and develops chronic liver disease, renal tubular dysfunction and rickets due to renal Fanconi syndrome. Untreated patients are at high risk of developing liver cirrhosis and hepatocellular carcinoma (Scott 2006). The acute form is referred to as the “French Canadian” type, while the chronic form is referred to as the “Scandinavian type”, but this classification may be misleading as both forms may coexist.
As affected neonates are usually asymptomatic in the first month of life and usually present with unspecific symptoms, in the absence of newborn screening, diagnosis may go unrecognized and will often be delayed, leading to serious complications and poor prognosis. Indeed, in severely affected neonates that were not screened and identified asymptomatically, the age of onset of symptoms is an important prognostic factor: before inception of treatment by 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3 cyclohexanedione (NTBC) in the mid-90’s, 1-year mortality was 60% in neonates who became symptomatic before 2 months of age, compared to 4% in children presenting signs and symptoms after 6 months of age (van Spronsen et al. 1994).
Treatment of HT1 has evolved greatly over the last decades. Before the 1980s, the only treatment proposed was a diet restricted in both phenylalanine and tyrosine, with the hope of obtaining successful therapy, in line with the success of early dietary treatment against phenylketonuria. However, dietary treatment alone has been disappointing in preventing complications of HT1, and in the 1980s, liver transplant started to be offered, improving the prognosis of affected children, but not without significant morbidity. More recently (mid 1990s), NTBC, an herbicide inhibiting the 4-hydroxyphenylpyruvic acid dioxygenase, an enzyme proximal to the FAH, which results in decreasing the levels of FAA, MAA and SA (Fig. 13.1), has revolutionized the treatment and prognosis of HTI (see Chaps. 18 and 19 for details on treatment of tyrosinemia in Québec). Since pregnancies carrying a foetus affected by HT1 usually evolve normally and are generally asymptomatic, these neonates may greatly benefit from newborn screening for HT1, especially with the advent of early treatment of HT1 by NTBC, allowing to greatly improve prognosis.
HT1 fulfils the Wilson and Jungner classical criteria for universal screening developed by the WHO in 1968 (Wilson et al. 1968), which were revised in 2011 (Andermann et al. 2011): it is a serious disorder, the natural history of the condition is understood, there is a an acceptable screening test and a treatment option (see Chap. 10 for details of classical screening criteria).
2 Screening for HT1 in Quebec: Historical Perspective 1970–1997
With the above considerations in mind, in the context of an increased frequency of HTI in the province of Quebec, and following the public health success of newborn screening for PKU (which began in October 1969 in Quebec), universal newborn screening for HT1 was implemented as a pilot project in the fall of 1970 and offered without interruption in the province since then. This represents close to four million newborns screened for HT1 over the last 45 years.
During the 1970–1997 period, 2,249,000 newborns were screened for HT1 at 3–7 days of life by tyrosine (tyr) concentration from dried filter paper blood spots as the first tier test, followed by α-fetoprotein (AFP) performed by a fluorimetric method as the second tier test until 1980 when first tier testing by tyr was abnormal (Grenier et al. 1976), very high levels of AFP most probably reflecting liver injury in utero. In 1980, AFP as the second tier test was replaced by indirect semi-quantitative (semi-Q) and quantitative (Q) succinylacetone (SA) measurements (by red blood cells δ-aminolevulinate dehydratase (ALA) inhibition) as the second and third tier tests before referral of screened-positive neonates to one of our clinical referral centers. In brief, the principle of indirect SA measurement is based on the inhibition of red blood cells (RBC) ALA by SA, results being compared to a standard curve with known amounts of porphobilinogen (measured at lambda = 550 nm) (Fig. 13.2). The semi-Q approach measures ALA inhibition of the neonate’s RBC, while the Q-SA measurement, performed as a validation of initial semi-Q screen-positive neonates, is based on ALA inhibition of RBCs from normal controls in the presence of eluent from the screened-positive newborn’s blood spots. Immunoreactive FAH (by enzyme-linked immunosorbent assay, ELISA) was performed as the confirmatory test on the initial blood spot, as it was shown to be associated with an absence of signal when performed in affected neonates with the IVS12 + 5G->A FAH mutation (Laberge et al. 1990).

Principle of blood spot indirect succinylacetone measurement by the inhibition of red blood cells δ-aminolevulinate dehydratase from succinlyacetone
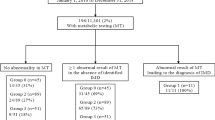
The high specificity of SA measurements as the second and third tier screening tests, although indirect through ALA dehydratase inhibition, led to very rare false-positive results, while allowing to identify neonates affected with HT (Table 13.1). However, owing to earlier hospital discharge of postpartum mothers and their babies and increasing rates of breastfeeding, even if the tyr cut-off levels to identify screen-positive neonates were gradually lowered from 414 μmol/L in 1970 to 200 μmol/L in 1997, the sensitivity, specificity and discriminating power of first tier tyr testing became insufficient. In 1997, four cases of HTI with tyr < 200 μmol/L (tyr levels of 198, 193, 168, and 166 mol/L) were missed within a year, as they did not proceed to second tier indirect semi-Q and Q-SA measurement (but showed later to have both increased semi-Q and Q-SA levels). Global figures for the 1970–1997 period are shown in Table 13.1.
Thus, if the presence of increased levels of tyr from blood spots may suggest HTI, tyr measurement in the first 24–48 h of life is neither a sensitive, nor a specific screening marker of HT1, and normal screening tyr level did rule out a diagnosis of HT1 in the new context of early discharge of mothers and infants in the mid-1990s.
3 Screening for HT1 in Quebec: Historical Perspective 1998-…
Following early discharge of neonates after birth, tyr did not qualify anymore as an adequate first tier screening test for HT1. In addition, elevated tyr is also observed in tyrosinemia type II and III, transient tyrosinemia of the newborn, prematurity and hepatocellular dysfunction of almost any etiology. Already in 1996–1997, an adapted version of the semi-Q SA measurement (Grenier and Laberge 1996) to be performed in 96-well plates was underway, allowing the screening algorithm to be modified in 1998: indirect semi-Q SA measurement became the first-tier test (normal >0.20 O.D.; optic density), followed by second tier indirect Q-SA testing (normal <2.5 μmol/L equivalent) when first tier semi-Q SA was abnormal, immunoreactive FAH testing still serving as the confirmatory test. Tyr level became an auxiliary test.
Between 1998 and April 2014, 1,366,294 newborns have been screened for HTI using indirect semi-Q SA testing as the first tier; over 90% of blood spots were sampled between 24 and 48 h of age. During this period, only 152 (0.0001%) neonates showed abnormal results after indirect semi-Q SA, 85 of them normalized after indirect Q-SA and FAH measurement, while 53 of the remaining 67 with abnormal by indirect Q-SA and FAH referred to a specialized clinic were confirmed to be affected by HTI (positive predictive value = 79,1%; incidence of HT1 = 1:25,779). No cases of acute form of HT1 were missed. Of note, between 1998 and September 2011, while tyr was still measured by fluorimetry (before we implemented tandem mass spectrometery (MS/MS) measurement), the average blood spot tyr level of confirmed HT1 cases (n = 44) was 189 ± 67 μmol/L compared with 151 ± 51 μmol/L in normal newborns (n = 1,150,888), showing significant overlap, confirming poor discriminating power of tyr.
In 2011, the newborn screening program was enhanced by the acquisition of tandem mass spectrometers. Between 2012 and 2014, direct SA measurement using MS/MS was performed using hydrazine monohydrate as per the PerkinElmer non-derivatized Neobase kit® in parallel with the first tier indirect semi Q-SA methods. In April 2014, both the indirect semi-Q and FAH measurement were removed from the screening algorithm. Since then, HT1 is screened by direct SA measurement by MS/MS as the first tier test (normal <0.7 μmol/L), followed by indirect Q-SA measurement when SA is positive by MS/MS. In case of discordance, a repeat sample is requested. During the following 20-month period (April 2014–January 2016), 150,223 neonates were screened for HT1 with the new screening algorithm, and 14/19 (positive predictive value = 73,6%) screen-positive neonates were diagnosed with HT1 (incidence of 1:10,730). Overall, during the 17-year interval since SA measurement has been used as the first tier test (either through indirect or direct SA measurement), 1,516,517 newborns have been screened for HT1: 86 screen-positive newborns were referred to a specialized referral center at about 10–15 days of life, while 67 were confirmed to be affected with the disorder, resulting to a positive predictive value = 79,1% (incidence of TYR1: 1:22,634). Since 1998, SA measurement as first tier screening test, either indirect through ALA dehydratase inhibition or by direct MS/MS measurement, proved to be highly sensitive and specific, and to our knowledge no cases were missed (negative predictive value = 100%). Data for the April 1998–January 1st 2016 period and global figures since 1970 are shown in Table 13.1.
4 Conclusion
HT1 is a rare disease presenting with non-specific signs and symptoms and may be overlooked in the investigation of an affected child. Owing to a founder effect, there is an increased incidence of HTI in the province of Quebec. Since inception of universal newborn screening for HT1 in the province in the fall of 1970, the program screened close to four million infants and identified 185 of the 190 cases of HT1 over the last 45 years, with a global frequency of HTI of 1:19,819. Interestingly, this represents more affected neonates and a higher frequency compared to classical phenylketonuria (Fig. 13.3).

Comparison of the number of newborns tested, cases confirmed and frequency of disorder between tyrosinemia type 1 (HT1) and classical phenylketonuria (PKU) by the Quebec Newborn screening program from October 1969 to January 1st 2016
Over the years, the newborn screening program had to adapt to changes in the management of the postpartum period of mothers and their babies in the health care system: when earlier hospital discharge of mothers and their newborns was instituted, tyrosine did not have sufficient sensitivity and specificity as a first tier screening test and was replaced by SA measurement.
For decades, the province of Quebec was the only jurisdiction offering HT1 universal newborn screening. More recently, the introduction of NTBC as an effective treatment, combined with the implementation of MS/MS technology in newborn screening, led to the addition of HT1, using SA as a sensitive and specific primary screening marker of the disease, to the screening panel of many jurisdictions worldwide.
References
Andermann A, Blancquaert I, Beauchamp S, Costea I (2011) Guiding policy decisions for genetic screening: developing a systematic and transparent approach. Public Health Genomics 14(1):9–16. doi:10.1159/000272898
Angileri F, Bergeron A, Morrow G, Lettre F, Gray G, Hutchin T, Ball S, Tanguay RM (2015) Geographical and ethnic distribution of mutations of the fumarylacetoacetate hydrolase gene in hereditary tyrosinemia type 1. JIMD Rep 19:43–58. doi:10.1007/8904_2014_363
De Braekeleer M, Larochelle J (1990) Genetic epidemiology of hereditary tyrosinemia in Quebec and in Saguenay-Lac-St-Jean. Am J Hum Genet 47(2):302–307
Grenier A, Laberge C (1996) Neonatal screening for tyrosinemia type I and early sampling. In: Proceedings of the Third International Society for Neonatal Screening, Boston, Oct 20–23
Grenier A, Belanger L, Laberge C (1976) alpha1-Fetoprotein measurement in blood spotted on paper: discriminating test for hereditary tyrosinemia in neonatal mass screening. Clin Chem 22(7):1001–1004
Grompe M, St-Louis M, Demers SI, al-Dhalimy M, Leclerc B, Tanguay RM (1994) A single mutation of the fumarylacetoacetate hydrolase gene in French Canadians with hereditary tyrosinemia type I. N Engl J Med 331(6):353–357. doi:10.1056/NEJM199408113310603
Halvorsen S (1980) Screening for disorders of tyrosine metabolism. In: Bickel H, Guthrie R, Hammerson G (eds) Neonatal screenings for inborn errors of metabolism. Springer, New York
JMG W, Jungner G, World Health Organization (1968) Principles and practice of screening for disease, Public Health Papers, vol 34. World Health Organization, Geneva
Laberge C (1969) Hereditary tyrosinemia in a French Canadian isolate. Am J Hum Genet 21(1):36–45
Laberge C, Dallaire L (1967) Genetic aspects of tyrosinemia in the Chicoutimi region. Can Med Assoc J 97(18):1099–1101
Laberge C, Grenier A, Valet JP, Morissette J (1990) Fumarylacetoacetase measurement as a mass-screening procedure for hereditary tyrosinemia type I. Am J Hum Genet 47(2):325–328
Laberge AM, Michaud J, Richter A, Lemyre E, Lambert M, Brais B, Mitchell GA (2005) Population history and its impact on medical genetics in Quebec. Clin Genet 68(4):287–301. doi:10.1111/j.1399-0004.2005.00497.x
Mitchell GA, Grompe M, Lambert M, Tanguay RM (2001) Hypertyrosinemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw Hill, New York, pp 1777–1806
Poudrier J, St-Louis M, Lettre F, Gibson K, Prevost C, Larochelle J, Tanguay RM (1996) Frequency of the IVS12 + 5G–>A splice mutation of the fumarylacetoacetate hydrolase gene in carriers of hereditary tyrosinaemia in the French Canadian population of Saguenay-Lac-St-Jean. Prenat Diagn 16(1):59–64. doi:10.1002/(SICI)1097-0223(199601)16:1<59::AID-PD810>3.0.CO;2-D
Scott CR (2006) The genetic tyrosinemias. Am J Med Genet C: Semin Med Genet 142C(2):121–126. doi:10.1002/ajmg.c.30092
van Spronsen FJ, Thomasse Y, Smit GP, Leonard JV, Clayton PT, Fidler V, Berger R, Heymans HS (1994) Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology 20(5):1187–1191
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Giguère, Y., Berthier, MT. (2017). Newborn Screening for Hereditary Tyrosinemia Type I in Québec: Update. In: Tanguay, R. (eds) Hereditary Tyrosinemia. Advances in Experimental Medicine and Biology, vol 959. Springer, Cham. https://doi.org/10.1007/978-3-319-55780-9_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-55780-9_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55779-3
Online ISBN: 978-3-319-55780-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)