
Abstract
In hematopoietic stem progenitor cell (HSPC) gene therapy (GT) applications for the treatment of genetic diseases, retroviral vectors (RVs) are used to efficiently transduce and integrate therapeutic genes in the genome of patient-derived HSPCs, which, upon reinfusion, reconstruct the entire hematopoietic system and restore the correct hematopoietic functions, or deliver the therapeutic factor to different tissues. However, in initial HSPC-GT clinical trials using early-generation γ-RVs, vector insertions near proto-oncogenes triggered their overexpression and induced leukemia in some of the transplanted patients. These unexpected adverse events have prompted the development of highly sensitive preclinical assays to test the genotoxic potential of different GT vector types and designs, and the development of powerful PCR-based techniques, combined with next generation sequencing (NGS) and bioinformatics analyses, have allowed to study integration sites (ISs) present in leukemic and dominant expanding cells, identify the genes targeted by insertions and to investigate the clonal composition of complex vector-marked cell populations. The positive safety data obtained from the testing in highly sensitive preclinical models and, successively, in clinical trials, of the more advanced lentiviral vectors (LVs) with self-inactivating (SIN) long terminal repeats (LTRs), have reduced the concerns related to insertional mutagenesis , encouraging the adoption of this vector platform in GT protocols for the treatment of many other diseases. Nevertheless, the evidences collected from genotoxicity assays and a β-thalassemia clinical trial, during which a vector-driven clonal dominance event has occurred, point to the fact that even SIN LVs insertions are not entirely neutral, and thus to the importance of a continuous effort to improve both the design of GT vectors, and the sensitivity of preclinical assays aimed at assessing their residual genotoxicity .
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Genotoxicity
- Insertional mutagenesis
- Hematopoietic stem cell gene therapy
- Viral integration
- Retroviral vectors
- Common integration sites
- Clonal dominance
Hematopoietic Stem Progenitor Cell Gene Therapy with Gamma Retroviral Vectors
Retroviruses belong to a family of enveloped viruses with single-stranded positive-sense RNA genome that, once entered the host cell cytoplasm, is reverse-transcribed into a DNA intermediate by the activity of the viral enzyme reverse transcriptase to produce DNA from its RNA genome. This new DNA is then integrated in semi-random positions of the host cell genome by the viral enzyme integrase. The stably integrated provirus will thus produce the viral proteins and RNA genomes to produce new viral particles and reinitiate the infectious cycle.
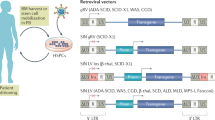
RVs have been the classical gene delivery vehicles for hematopoietic cells, including stem cells [1]. The ability of retroviruses to stably integrate in the host cell genome allows to take advantage of the cellular transcriptional machinery for the permanent genetic modification of the host cells and their progeny. Moloney murine leukemia virus (MoMLV) was in fact the first retroviral genome to be engineered to carry a foreign gene into a host cell [2]. In this initial work, MoMLV was used to transfer a copy of the herpes simplex virus thymidine kinase into murine hematopoietic stem progenitor cells (HSPCs) that were subsequently able to rescue irradiated recipient mice from lethality upon transplantation [2]. Few years later, MLV vectors were demonstrated able to mediate gene transfer also into human bone marrow stem progenitor cells [3], successfully mediating the transfer of neomycin and methotrexate resistance genes, and conferring functional drug resistance to the recipient cells. Early gene therapy clinical trials exploiting gamma (γ) Retroviral Vectors (RVs) were developed for the treatment of primary immunodeficiencies (Table 1). For these patients, HLA-matched bone marrow transplantation or T-lymphocyte infusion were the only available treatment options, and the well-known risks associated with these procedures, coupled to the lack of HLA-matched donors, pressed to find alternative curative solutions for these patients [4].
The keystone for using γ-RVs for gene therapy clinical applications was set as the first trials with these vectors revealed a tangible clinical application: in 1990 Blaese et al. [5] started a clinical trial for the treatment of adenosine deaminase (ADA) gene deficiency, a metabolic disorder characterized by the accumulation of toxic metabolites causing near to total absence of lymphocytes in the affected subjects. In this opening study, patient-derived T-lymphocytes were modified ex vivo with a γ-RV carrying the functional copy of ADA and infused back into the patients. Although this first attempt did not revert the immunodeficiency, it demonstrated the feasibility and safety of the procedure [5, 6].
Since the treatment of the first patients, improvements in gene therapy protocols made significant advances, smoothing ground for fruitful trials that followed.
Indeed, noteworthy success was achieved in a later clinical trial [7], which used improved gene transfer protocols taking advantage of hematopoietic stem/progenitors cells (HSPC) as cell target to achieve the correction of ADA-deficiency (Table 1). In this setting, gene therapy with genetically modified HSPC would warrant a constant supply of gene-corrected cell progeny, able to restore the healthy phenotype in treated patients. Combined with a non-myeloablative conditioning regimen and the withdrawal of enzyme replacement treatment, this strategy advantaged the gene-corrected cells over the enzyme-deficient ones, contributing to a first perceptible efficacy of the treatment. With a slight variation in conditioning regimen and vector constructs, similar protocols have been used by different centers for ADA treatment [8,9,10,11,12]. Overall, a total of forty-two patients with ADA-deficiency have been treated by gamma retroviral HSC gene therapy worldwide so far. In highlighting that no adverse events have been reported in any of the treated patients of these trials, thirty-one became independent from life-long pharmacological enzyme replacement therapy. The strategy of using γ-RV-based gene therapy clinical trials with HSPCs was exploited also for additional trials to cure different primary immunodeficiencies such as X-linked severe combined immunodeficiency (SCID-X1) [13, 14], X-linked chronic granulomatous disease (X-CGD) [15,16,17,18,19], and Wiskott–Aldrich syndrome (WAS) (Table 1) [20,21,22].
SCID-X1 is caused by mutations of the interleukin 2 receptor subunit gamma (IL2RG) gene, which encodes for the common gamma chain (γc) subunit shared in the interleukin (IL)-2, 4, 7, 9, 15, and 21 receptor complexes. SCID-X1 accounts for 40–50% of all SCID cases [23]. SCID-X1 patients present profound immunological defects caused by low numbers or complete absence of T and NK cells, and presence of nonfunctional B-cells [24]. The observation that spontaneous somatic reversion of the mutation in the γc-encoding IL2RG gene in lymphocyte progenitors resulted in the restoration of immunological competence in some patients [25] suggested that γ-RV-meditated gene transfer of a normal γc transgene into lymphocyte progenitors could provide a selective advantage over their non-transduced counterparts, after autologous transplantation into patients. Aimed at restoring the healthy immunological phenotype, different clinical trials have been performed, treating, from 1999 to 2006, a total of twenty-three SCID-X1 patients, of which twenty recovered immunological functions after the gene therapy treatment [13, 14, 26]. Despite the successes, as explained in detail below, leukemogenesis occurred in a number of cases as the result of vector insertions that deregulated the expression of nearby cellular oncogenes in transduced cells.
Chronic granulomatous diseases encompass a group of pathologies caused by defects in the nicotinamide dinucleotide phosphate (NADPH) oxidase complex, vital for antimicrobial activity of phagocytes. In X-CGD, this defect relies on mutations in the CYBB gene, the enzymatic center of NADPH oxidase [27, 28]. Initial clinical trials with gamma retroviral vectors for X-CGD had limited success compared to the abovementioned trials, since they reached only transitory functional correction of less than 0.5% of peripheral blood granulocytes [29,30,31], and although following trials extended restoration of functional neutrophils up to 30%, the long-term engraftment of gene-corrected cells fell short, and myelodysplastic cell clones were selected as the result of vector insertions near a specific oncogene [15,16,17,18,19].
For Wiskott–Aldrich syndrome (WAS), a severe X-linked disorder caused by mutations in the leukocyte migration involved gene WAS, a phase I/II clinical trial was initiated in 2007 [21]. Nine out of the ten patients treated in this trial had sustained engraftment and correction of WAS protein expression in platelets, lymphoid and myeloid cells, resulting in partial or complete resolution of immunodeficiency [22].
Taken together, these clinical trials demonstrate that vector-mediated stable gene transfer into hematopoietic stem and progenitor cells can provide clinical benefits to many patients.
Amidst of its advantages, vector integration, in altering the host genetic code, is an intrinsic mutagenic event that can lead to cell damage with deleterious consequences. As a result of harmful vector integration events, lymphoproliferative or myelodysplastic disorders have been reported in X-SCID, X-CGD and WAS clinical trials (Table 1) [18, 19, 21, 22, 32,33,34].
In the X-SCID trial, five successfully treated patients developed leukemia. In four out of five cases, analysis of malignant clones found integrations close to the Lim Domain Only-2 (LMO-2) proto-oncogene (Table 1) [33, 34]. These vector integrations caused increased gene expression and consequent enhanced protein production. Although the IL2Rγc-deficient background and the transgene itself have been hypothesized as potential cofactors of clonal expansion [35], the vector-mediated aberrant expression of this proto-oncogene is still considered the main cause of oncogenesis.
A similar scenario hit WAS clinical trial when the same locus was found to be targeted in patients’ expanded clones, triggering, in seven treated subjects, hematologic malignancies (Table 1). Four patients developed T-cell acute lymphoblastic leukemia (T-ALL), two primary T-ALL with secondary acute myeloid leukemia (AML) and one patient displayed Primary AML [21, 22].
Myelodysplastic disorders originated in three patients that enrolled in the X-CGD clinical trial. Aberrantly expanded clones where characterized by vector-driven upregulation of MDS-EVI1, PRDM16 and SETBP1 proto-oncogenes (Table 1) [18, 19].
Mechanisms of Insertional Mutagenesis
γ-RVs conventionally used in gene therapy applications were derived from slow transforming retroviruses, a class of oncogenic viruses capable of inducing, after a moderately long period of latency, the development of tumors in infected animals. Differently from other oncogenic viruses, carrying coding sequences for proto-oncogenes [36] or for viral proteins able to interfere with cellular tumor suppressor genes [37], the capacity of γ-retroviruses to promote malignant transformation is strictly dependent on their integration into the host’s cellular genome. This event allows the provirus to interact in various ways with the genomic elements surrounding the site of integration, potentially leading to alterations in physiological cellular gene expression, a phenomenon referred to as “insertional mutagenesis” [38, 39].
Retroviral integration can cause upregulation of cellular genes as well as their disruption by three different mechanisms, involving enhancer and promoter elements contained within the proviral long terminal repeats (LTRs), splicing signals and/or polyadenylation sites present in the vector [38,39,40].
Integrations, either upstream or downstream a cellular gene, can trigger insertional mutagenesis through a mechanism of enhancer activation [38, 39]. In this scenario, gene transcription, driven by the gene’s endogenous promoter, is augmented by the activity of the enhancers present in the nearby integrated proviral genome (Fig. 1a). This effect can be exerted not only on genes that are most proximal to the insertion site, but also on genes lying far apart on the linear genome and that are brought in spatial vicinity via the generation of chromatin loops in the nucleus [41].

Insertional mutagenesis mechanisms by retroviral insertions. a Enhancer-mediated gene activation events; b promoter insertion, transcript truncation and altered splicing mechanisms. E/P viral enhancer/promoter elements; pA viral polyadenylation signal; SD and SA splice donor and splice acceptor signals, respectively
Retroviral integrations can lead to the overexpression of genes also by the mechanism of promoter insertion (Fig. 1b), which takes place when integrations are in the same transcriptional orientation of the gene, and the proviral promoter elements within the LTR replace the cellular promoter in driving gene transcription [38, 39]. Although the enhancer/promoter elements of both LTRs have the potential to initiate gene transcription, this phenomenon is frequently driven by the 5′ LTR, since the 3′ LTR is inactivated through a phenomenon called promoter occlusion [36].
The interference of cis-acting elements between virus and host genome can lead to formation of dangerous transcription-signals that deceive the cellular transcription machinery. Aberrant splicing and read-through mechanisms, overcoming the LTR-polyadenylation site (polyA), allow generating aberrant fusion transcripts containing vector and cellular sequences (Fig. 1b). Specifically, when starting from the 5′ LTR, aberrant transcripts are fused to cellular sequences by using canonical or cryptic viral splice donor (SD) sites and cellular splice acceptors (SA) signals [38, 39].
Intragenic insertions, which are integrations landing inside the host genomic transcriptional units, can trigger insertional mutagenesis by disrupting coding domains, thus leading to gene inactivation. Given the presence in the R region of the LTR of a canonical polyA signal in the same orientation as the viral transcription, and of a cryptic polyA signal in antisense orientation, proviral intragenic integrations in both orientations can elicit the premature termination of gene transcription (Fig. 1b).
Such events may occur in concert with aberrant splicing, enhancer or promoter activation. Viral insertions may also target the 3′ UTR region of genes, resulting in the deletion of mRNA-destabilizing motifs such as miRNA target sequences or AUUUA hairpins, leading to the increase of the mRNA expression levels and consequently increased protein levels [39].
Common Integration Sites : The Hallmarks of Insertional Mutagenesis
Owing to their oncogenic potential, slow transforming retroviruses have long been exploited as mutagenic agents in forward genetic screens aimed at identifying novel cancer genes [38, 39, 42,43,44]. Such screens were originally performed either by infecting newborn mice with replication competent retroviruses, or by using constitutively infected recombinant inbred mouse strains, in which the virus is vertically transmitted [43]. In both cases, retroviral infection in early life allows the establishment of a life-long viremia during which multiple rounds of proviral integration in millions of cells and within the same cellular genome can occur. This may result in the deregulation of multiple growth-regulatory genes that can in turn confer a selective advantage to cells which, upon the acquisition of additional mutations, can become fully transformed and malignant [43].
The type of tumor developed depends on the specific tissue tropism of the virus used; for instance, the mouse mammary tumor virus (MMTV) and the Moloney murine leukemia virus (MoMLV), extensively employed in such studies, induce mammary and hematopoietic tumors, respectively [44]. Compared to other mutagens, such as chemicals and radiations, retroviruses display the feature of integration that can be advantageously used as a molecular tag, allowing the mutated genes to be easily identified by retrieving the viral integration site (IS) and mapping its sequence to the reference genome.
Genomic loci targeted by proviral insertions in multiple independent tumors, at a frequency higher than expected by chance, and being consequence of clonal selection triggered by integrations conferring a growth and proliferative advantage to the cells, are termed common insertion sites (CIS).
Targeting of genes responsible for malignant events occurred during the initial γ-RV-based GT clinical trials. Genes, such as LMO2 in the X-SCID and WAS trials, MDS1-EVI1 in the X-CGD and WAS trials and PRDM16 and SETPB1 in X-CGD trial, were found to be highly targeted by vector integrations in malignant and pre-malignant clones retrieved from different patients (Table 1). Hence, the identification and monitoring of CIS in vivo in GT patients is of great importance in clinical applications, to monitor the safety outcome of GT procedures.
Since some genomic regions can be frequently targeted as a result of retroviral integration biases, the classical methods to identify CIS based on comparison with simulated random integration distributions, founding on the assumption that retroviral integrations occur randomly into the genome, had to be revisited [39]. The total number of insertions of the analyzed dataset must be considered since the higher the number of overall integrations, the greater is the probability that these could target by chance the same genomic region. Differently, oncogenic CIS tend to be highly clustered within narrow genomic regions targeting a single gene [45]. Furthermore, the orientation of the vector integration compared to the targeted gene can be an important feature defining genotoxic CIS, since it provides important insights on the probable mechanisms of genotoxicity leading to deregulation. As HIV-1/LV integrations have the tendency to distribute over megabase-wide genomic areas, an additional analysis step to avoid the overestimation of potential cancer-associated CIS has been proposed [45]. This approach takes advantage of the Grubbs test for outliers to define if integrations within a CIS gene are significantly enriched compared to genes contained in the flanking genomic regions, in which case the identified CIS will be considered the result of a selection process; conversely, it will be considered the product of an integration bias [45].
Overall, the different analytical strategies that have been used over the years to identify CIS can be classified into two main groups: (i) whole genome scanning, in which CIS are computed by parsing all IS in the whole covered genome independently from the functional or genomic annotation, and (ii) gene-centric approach, in which all IS are associated with the closest gene and CIS are computed with respect to the corresponding gene size. The first group includes the majority of the methods, from the first approaches based on genomic sliding window [46] to Kernel convolution-based methods [47], Poisson distribution statistics [48], Monte Carlo-based methods [49], and, more recently, on scan statistics [50], whereas the Grubbs test for outliers [45, 51, 52] is classified in the gene-centric group.
State of the Art Methods for Integration Sites Retrieval
Methods
Gene therapy safety studies and retroviral integration analyses require identifying the genomic site of the integrated provirus. To this purpose, during the last decade, different techniques have been developed and optimized. All these methods rely on polymerase chain reaction (PCR) for the isolation and amplification of proviral-host genome junctions, starting amplifications from known sequences in the proviral LTR, followed by sequencing and mapping to the reference genome. Among these methods, ligation-mediated PCR (LM-PCR) [53, 54] and linear amplification-mediated PCR (LAM-PCR) [55,56,57] have been the most frequently exploited ones. In both approaches, restriction enzymes are used to fragment DNA and a common oligonucleotide sequence (a DNA-linker cassette) is ligated to the resulting fragments, followed by exponential amplification with primers complementary to LTR and linker cassette sequences.
In LM-PCR, genomic DNA is directly subjected to the above-described flow, while LAM-PCR involves a prior step of linear amplification and second strand synthesis, preceding the enzymatic restriction (Fig. 2).

Schematic representation of: ligation-mediated (LM) PCR; left panel and linear amplification-mediated (LAM) PCR; right panel. RE Restriction enzymes; gDNA genomic DNA
Identification of vector integration sites is crucial for evaluating the efficacy and the safety of gene therapy clinical trials. Moreover, in clonal tracking studies, like the ones performed for the monitoring of HSPC-GT patients, the total number of reads derived from the same sequence, called sequence count, can be used to calculate the relative abundance of clones “marked” by a specific integration within the pool of vector-marked clones [51, 52, 56,57,58,59,60]. This strategy is based on the assumption that the more one integration site is sequenced, the more the clone harboring the specific integration is abundant in the sample.
However, the use of restriction enzymes for DNA fragmentation in LM and LAM-PCR can introduce biases, which impact the sensitivity of these techniques and the abundance estimates obtained from sequence counts [61]. Specifically, the non-uniform distribution of restriction sites throughout the genome leads to the generation, upon digestion, of fragments of different lengths. During the PCR, shorter fragments can be preferentially amplified over longer ones, thus misrepresenting the relationship between the real frequency of an integration and its calculated estimate. Additionally, integration loci lacking close restriction sites may be missed, since those fragments will result being too long to be amplified by standard polymerases [61], or, even in case of successful amplification, the long products could be too long to be analyzed by specific sequencing platforms [62]. Furthermore, some of the generated fragments might be too short to be unequivocally mapped to the reference genome after sequencing [61]. To overcome these technical limitations, restriction-free LM and LAM-PCR methods have been introduced [22, 61,62,63,64,65]. The use of sonication to fragment DNA allows controlling DNA-shearing, achieving an evenly sized distribution of the fragments. This permits to take advantage of novel strategies for clonal abundance quantification, which do not rely on the sequence count, and should therefore improve the accuracy of such measurements [58, 62, 63, 65].
Although of similar size, being the sites of genomic DNA fragmentation random, the sonicated fragments will display different shearing ends. The number of different break points (called: shear sites) associated with each integration site reflects the abundance of cells containing that specific insertion. Ligating the linker cassette directly to the sonicated fragments prior to the amplification steps generates unique ligation points (LPs), which serve as molecular barcodes to identify individual cells [58, 62, 63, 65].
One limitation of this technique is that the maximum number of clones with the same integration that can be distinguished corresponds to the maximum fragment length generated by sonication, leading in some cases to underestimate the abundance of large clones [58, 65,66,67]. To overcome this, the use of adaptors that contain random barcode sequences has been proposed, so that individual cells can be identified by coupling unique shear sites to unique barcodes [65, 67].
Applications
Following HSPC gene therapy, gene-corrected stem cells are expected to give rise to a gene-corrected hematopoietic cell progeny, with a plethora of vector-marked cells harboring different integration sites, which is referred to as to as polyclonal integration pattern [4]. Vector integration studies can be exploited to follow the clonal composition of hematopoietic reconstitution over time after gene therapy and are known as clonal tracking studies [18, 22, 51, 52, 60, 66]. In allowing the detection of clonal dominance events, which might represent early steps of tumorigenesis, these studies are important safety readouts of gene therapy trials. Pioneering work has been performed by large-scale mapping analysis of retroviral insertion sites achieved in the trial for X-CGD disease [18], where the clones that became malignant were found to be the most represented among gene-corrected cells already five months after gene therapy, and before symptoms-based diagnosis of myelodysplastic syndromes and leukemia [18, 19].
Similarly, in the γ-RV-based clinical trial for WAS [22], the progressive expansion of the clones that triggered leukemia could be followed during time by IS retrieval. Therefore, longitudinal tracking studies aimed at quantifying over time the clonal abundance of cell clones harboring different integration sites are extremely important to monitor the safety of HSPC gene transfer applications.
To assess clonal diversity in each lineage over time, an index, measuring the entropy of the integration data sets, the Shannon Diversity Index, is used, and takes in account the total number of integrations and their relative contribution to the clonal output over time [22, 51, 52, 60]. Analyses of the clonal contribution of gene-marked cells to hematopoiesis in gene therapy patients in the LV-based clinical trials for WAS and MLD showed that no clonal dominance events had occurred during the time of follow-up [51, 52] with almost all cell clones marked by a specific integration accounting for only a fraction of less than 5% of the total clones, while the few clones displaying a higher percentage at a specific time point then disappeared or were strongly reduced at later time points [51, 52]. In line with this, the Shannon Diversity Index calculated for each patient at respective time points in both trials revealed that hematopoietic reconstitution after transplantation became increasingly polyclonal in all hematopoietic compartments, providing a further indication of the safety of these treatments [51, 52].
Beyond the safety assessment purposes, given the powerful information that can be earned from clonal tracking studies, such investigations can be also exploited to study the dynamics and lineages in hematopoietic reconstitution [51].
Based on results from the WAS clinical trial, Aiuti and coworkers [51] were able to propose a model in which the hematopoietic output after transplantation by long-term HSPCs occurs in a defined time window after gene therapy, generating a diverse clonal repertoire in the blood progeny. As an example, the diversity of the HSPC (CD34+ cells) compartment of patient 1 from the WAS trial progressively decreased, initially reaching its minimum at six months post gene therapy, and rebounded over time after that time point, while diversity in the other hematopoietic lineages increased stably over time. This trend is believed to result from an initial contribution of multiple progenitor cells to hematopoiesis that are subsequently supplanted by engrafted long-term HSCs upon exhaustion of short-term progenitors.
Long-term reconstituting HSCs are expected to share identical integrations among bone marrow CD34+ progenitors and multiple myeloid and lymphoid lineages persisting overtime, whereas integrations shared only among some hematopoietic lineages, but not others, might indicate lineage-restricted progenitors [51, 52, 59]. The output of short-lived mature myeloid cells in the peripheral blood long-term after HSC-GT has been exploited as a readout to estimate the number of transduced HSCs contributing to human hematopoiesis in vivo [51, 52]. Lineage tracking studies could also provide insights in the lifespan and fluctuations in lineage output of hematopoietic progenitors [59].
Assessing Retroviral Vector Genotoxicity
Despite the well-known oncogenic potential of the parental viruses, the risk of tumor development associated with gene therapy-grade retroviral vectors was considered to be remote, owing to the replication-defective nature of the vectors and to the unlikelihood that few vector integrations could activate multiple proto-oncogenes within the same cell, promoting neoplastic transformation. Furthermore, the results of preclinical studies performed on animal models did not highlight the occurrence of such adverse events [68,69,70,71,72,73,74].
However, unexpected malignant events occurred during the early γ-RV-based GT clinical trials (Table 1), and the identification of vector-induced insertional mutagenesis as their major drivers highlighted the need for a deeper understanding of the mechanisms and the intrinsic vector features underlying vector genotoxicity , which is instrumental for designing safer vectors for clinical applications.
Several models, both in vitro and in vivo, have thus been proposed over the years to address the mutagenic risk and transforming potential of integrative vectors (Fig. 3).

a In vitro immortalization assay (IVIM); b IL-3 independence growth assay with plating in limiting dilution or expansion in mice; c in vivo serial transplantation assay with lineage negative (Lin−) wt murine cells; d tumor-prone mice genotoxicity assay with transduction and transplantation of Cdkn2a −/− Lin− cells in wt recipients or direct vector injection in newborn Cdkn2a −/− mice
In Vitro Assays
Cell-based assays are a convenient means to perform rapid functional screens, by scoring for macroscopic gain of functions induced by vector treatment. Molecular analyses can then be performed, to identify the genes targeted by integrations, and the mechanisms by which they were deregulated (Figs. 3a, 4b).

Genotoxicity mechanisms induced by different vector designs: From top to bottom: active LTR lentiviral vector with strong enhancer/promoters performing promoter insertion; SIN.LV with strong enhancer/promoter activating oncogenes by enhancer-mediated mechanisms and truncation transactivation; SIN LV with moderate enhancer/promoter causing gene truncation and enhancer-mediated genotoxicity; SIN.LV with strong enhancer/promoter in internal position bracketed by chromatin insulators in LTRs induces mostly gene truncation events. SFFV spleen focus forming virus enhancer/promoter elements; GFP green fluorescent protein; PRE post-transcriptional regulatory element; PGK phospho-glycerate kinase promoter; INS chromatin insulator sequences
One of the strategies takes advantage of growth factor-dependent cell lines, such as interleukin-3 (IL-3) (Fig. 3b) [75, 76]. Cellular transduction with the vector to be tested, followed by growth factor withdrawal from the culture medium, gives rise to growth factor-independent clones generated by vector-induced insertional mutagenesis . Using this assay, the mutagenic potential of matched design γ-RVs and LVs has been compared, revealing that, while both vectors are able to generate insertional mutants at similar frequencies, they do so by different mechanisms: Moloney leukemia-virus (MLV)-derived γ-RV tested induced the overexpression of the IL-3 gene or other cancer-associated genes by enhancer insertion, whereas LV intragenic insertions lead to aberrant fusion transcripts starting at the proviral 5’-LTR encoding for the growth hormone receptor [75].
A similar strategy is employed in the in vitro immortalization (IVIM) assay based on transduction of primary hematopoietic stem and progenitor cells isolated from untreated adult mice (Fig. 3a) [77]. Cell transformation is detected by culturing transduced cells under myeloid differentiation conditions, followed by a replating step in limiting dilution that suppresses the residual self-renewal ability of the cells, unless insertional upregulation of cellular proto-oncogenes occurred. To allow direct comparison of the mutagenic potential of different vectors, an index is calculated by correcting the replating efficiency for the post-transduction vector copy number (i.e. the average number of integrated vectors per cellular genome). Comparative studies using the IVIM assay have shown that, compared to conventional γ-RVs, third-generation SIN LVs have a lower transforming potential and that improvement in vector design such as the use of moderate cellular enhancer/promoters or chromatin insulators can reduce the transforming capacity and thus the vector genotoxic potential [78,79,80,81,82,83].
In Vivo Assays
In vivo genotoxicity assays are important to investigate genotoxicity since different components like tumor development, immune response, cellular microenvironment and interactions between different cell-types cannot be readily assessed in in vitro models. Both murine and non-human primates in vivo models have been largely exploited in genotoxicity studies, in order to assess not only the mutagenicity of gene therapy vector integrations, but also their actual oncogenic potential, and their impact on the hematopoiesis of animals subjected to HSPC gene therapy-like procedures [76, 84,85,86,87,88,89,90].
Initial mouse-based studies relied on the transplantation of transduced wild-type Lineage negative (Lin−) murine HSPCs into primary recipients, followed by serial transplantation of bone marrow-derived cells from these primary recipients into secondary and tertiary animals, in order to promote potential leukemic progression (Fig. 3c). This strategy has allowed detecting and investigating the molecular bases of vector-mediated oncogenesis in a number of different studies providing experimental evidence that the overexpression of oncogenes, such as the murine Evi1, is the major driver of γ-RV-induced cellular transformation [76, 91] and that the onset of γ-RV-associated leukemia, at least in this model, necessitates the cooperation of multiple vector insertions in growth-regulatory genes within the same clone [87]. However, the predictive power of safety tests based on the use of wild-type murine models is limited by the difficulty of inducing cell transformation, which requires time to occur since the acquisition of multiple mutations is needed, so that even serial transplantation can result inefficient at promoting the development of leukemia [87]. Non-human primates have also been used as models to study the genotoxicity of retroviral vectors in the context of HSPC-GT [86, 90, 92, 93]. These models too, however, seem to lack enough sensitivity to detect the oncogenic potential of retroviral vectors , including the ones that have caused overt malignancies in humans, and to compare the genotoxic potential of vectors with different designs, since a malignant event has been reported only in one rhesus macaque that had been transplanted with γ-RV-transduced autologous HSPCs six years earlier, while the long-term follow-up of a large cohort of animals in two other studies has revealed that all of them had retained a completely normal hematopoiesis during time [86, 90, 92, 93].
To increase the sensitivity to vector genotoxicity , reducing the time required to obtain safety readouts in vivo, genotoxicity assays based on the use of mouse strains with a predisposition to tumor development have been developed (Fig. 3d) [84, 88, 89]. Specifically, these studies have taken advantage of Cdkn2a −/− mice [94], in which the knock out of the Cdkn2a locus results in the combined deficiency of the p53 and Rb pathways, rendering the mice more susceptible, compared to wild-type mice, to mutagenic insults such as those that can be delivered by a genotoxic vector. Cdkn2a −/− genotoxicity assays have been performed either by transducing and transplanting tumor-prone Lin− cells into wild-type recipients [88, 89], or by directly injecting the vector intravenously into newborn Cdkn2a −/− mice [84]. Since all animals develop hematologic malignancies, the readout for vector genotoxicity is the accelerated tumor onset in vector-treated mice compared to mock-treated animals, which increases according to the mutagenic potential of the tested vectors. The genes involved in tumorigenesis can then be identified by harvesting malignant tissues from the mice and performing integration site studies for CIS identification and molecular analyses. Studies using the Cdkn2a −/− transplantation model have established a correlation between γ-RV dosage and the risk of malignant transformation, since γ-RV-treatment triggered a significant dose-dependent acceleration of tumor onset in mice compared to Mock and provided a strong evidence of the safety of third-generation SIN LVs, which, instead, did not cause accelerated tumor onset [89]. Experiments in the same model provided a formal proof of the predominant role of active proviral LTRs in mediating vector-induced tumorigenesis, showing that the re-introduction of strong enhancer/promoter elements into LV’s LTRs renders these vectors extremely genotoxic, and validating the safety of the SIN configuration in both retroviral and lentiviral vector platforms [88]. The direct-injection-based Cdkn2a −/− genotoxicity assay has allowed detecting the residual oncogenic potential of SIN LVs, which resulted undetectable by the Cdkn2a −/− transplantation assay [84]. The sensitivity of the injection-based assay has allowed to compare and rank the genotoxic potential of LVs with different designs, revealing that both the mechanism of insertional mutagenesis and the genetic drivers of oncogenesis strongly depend on the specific features of each vector [84].
The combination of in vitro and in vivo approaches has also been exploited, to test the ability of different insulator sequences to reduce the mutagenic and oncogenic potential of γ-RVs (Fig. 3b) [95]. In this assay, IL-3-dependent cell lines were transduced either with insulated vectors, or non-insulated, match-design, vectors and, after selection, the expanded clones were transplanted into B lymphocyte-deficient C3H/HeJ mice, which were monitored for tumor development. Mice transplanted with mock-treated cells did not develop tumors, whereas the readout for the insulating activity was both the reduction in the frequency of animals developing tumors and the delay on tumor onset in mice receiving cells transduced with insulated vectors, compared to the ones receiving cells transduced with the non-insulated counterparts.
Factors Influencing Genotoxicity
Different factors may influence the success of gene therapy applications, such as the patients’ age and health-status, as well as the specific type of disease. Besides patient-related aspects, the outcome of the vector treatment can be influenced by three main vector-related factors: (a) the genomic integration profile, (b) the vector design and (c) the vector dose.
Vector Genomic Integration Profile
The integration pattern of retroviruses is not uniform throughout the host genome, and different retrovirus families display diverse integration preferences, as distinctive fingerprints of their identity [86, 96,97,98,99,100,101,102,103].
First studies on HIV integration revealed preferences of lentiviruses—and their derived vectors—to target genes with a greater percentage opposed to the theoretical of randomly targeted transcriptional units present in the human genome. For these vectors, gene-dense regions and gene-rich chromosomes are the main targets, and integrations are consistently present along the entire length of the transcriptional unit [101, 102]. Differently, MLV integrations displayed a lower preference to integrate within genes [103] and a marked bias to integrate near the transcriptional start site (TSS) of expressed genes, where a bimodal integration distribution around the transcription start was observed for γ-RVs, probably due to physical inaccessibility of specific positions within the promoter when bound by transcription factors [100]. MLV integrations cluster around CpG islands, likely due to MLV bias for TSS, and correlate with epigenetic markers for active promoters and enhancers, like H3k4me1or H3K9ac. Differently, LVs insertion pattern target actively transcribed regions marked by H3K36me3 [86, 92, 100, 101].
Overall, the integration pattern of γ-RV and LV vectors is defined as semi-random and indicates that some intrinsic cellular characteristics are particular appealing to integration of specific viral vectors [97]. The interaction of the pre-integration complex with cellular intrinsic nuclear factors, tethers the complex to the preferred integration loci. LEDGF/p75 and bromo/extraterminal-domain (BET) proteins have recently been shown to be involved in lentiviral and γ-RV tethering, respectively. Specifically, the chromatin-binding domain of LEDGF/p75 involved in interactions with chromatin marks of actively transcribed gene bodies, such as H3K36me3, is responsible for LV bias of integrations. To similar extent but with a different result, BET proteins interacting with chromatin marks enriched around the genes’ TSS, are responsible of γ-RV integrations around these genomic features [104]. The knowledge of the molecular players involved in integration sites preferences can allow engineering vectors to fuse to alternative binding domains allowing for safer integration site choice.
Vector Design
Different features present in the vector are able to deregulate host cellular transcripts (Fig. 4). Vector features, like the LTRs, the promoters, the transgene plus additional regulatory elements, can be modified or adjusted in order to improve the vector safety profile.
The vector type itself can influence genotoxicity : with the IVIM assay Baum and colleagues compared γ-RV and LV vectors with the same design and showed that the latter induced mutants with a threefold lower incidence compared to γ-RVs [80]. Moreover, in vivo assays with tumor-prone mice showed a cumulative higher genotoxic potential of RVs compared to LVs [89]. In mice carrying vector copy number (VCN)-matched integrations of γ-RV and LV with the same design, it was found that γ-RV was significantly more genotoxic than LV. It was estimated that a ten-fold higher integration load of the design-matched LV would be required to have the same oncogenic risk, meaning that the relative risk differs between these two vector types [88]. As previously described, vector LTRs can deregulate neighboring genes through enhancer-mediated effect and through aberrant transcript mechanisms (Fig. 1). γ-RVs’ transforming potential can be significantly reduced by removing the strong retroviral enhancer/promoter sequences from the LTR and placing them in single copy in internal position [80, 88] and the use of SIN LTR is able to reduce the genotoxic potential of γ-RV and LV vectors carrying strong enhancer/promoter sequences (Fig. 4).
The internal promoter in SIN vectors can be either a moderate cellular promoter or a promoter of viral-origin and with different strengths. A safe vector design should avoid strong viral promoters and support the use of moderate cellular promoters, like elongation factor 1α (EF1α) and phospho-glycerate kinase (PGK) promoters [83]. Indeed, a LV with active proviral LTRs was found to be highly genotoxic in Cdkn2a −/− mice and induced tumors by predominantly activating Braf proto-oncogene, through the promoter insertion mechanism (Fig. 4) [84]. Differently, the SIN LV carrying the same enhancer/promoter elements in internal position caused tumorigenesis mainly by activating a different proto-oncogene, Map3k8, through a combination of enhancer-mediated overexpression and transcript truncation using cryptic vector splice acceptor sites and/or the LV polyadenylation site present in the LTR (Fig. 4) [84]. The same SIN LV backbone carrying the moderate PGK promoter in internal position, while still being able to cause the enhancer-mediated overexpression of Map3k8, triggered tumorigenesis also by inactivating tumor suppressor genes like Pten, indicating that the propensity of SIN LVs to induce enhancer-mediated activation of oncogenes or to inactivate tumor suppressor genes depends on the strength of the internal enhancer/promoter used (Fig. 4) [84]. Indeed, when blunting the interaction between the internal vector enhancers and the surrounding cellular genes with chromatin insulators, inactivation of tumor suppressor genes became even more predominant (Fig. 4) [84]. Furthermore, these results highlighted that tumor suppressor disruption endured as escape genotoxicity mechanism that cannot be prevented when using integration competent retroviral vectors (Fig. 4) [84].
Vector Dose
A single mutation event is rarely able to induce neoplastic transformation, suggesting that also in vector-induced genotoxicity , the collaboration with other mutations is needed. The increase in vector load with the consequent increase of vector integrations in the cells results in an enhancement of the risk of targeting cancer-related genes in vitro and in vivo [87,88,89]. Wild-type mice transplanted with bone marrow-derived Lin− cells transduced with high or low doses of γ-RV sporadically developed leukemia only in the high dose vector treatment group [105]. Other genotoxicity studies performed by transplanting wild-type mice with vector-transduced tumor-prone Cdkn2a −/− bone marrow-derived Lin− cells showed a better correlation between vector dose and genotoxicity [88, 89]. γ-RVs or LVs with active LTRs were able to trigger a dose-dependent acceleration of tumor onset although to a different extent. The vector dose needed for detecting genotoxicity in this highly sensitive genotoxicity assay was in part dependent on the vector integration profile (dictated by the vector type). Indeed, the innate tendency of γ-RV to integrate near the TSS and growth-promoting genes resulted in 10-fold higher risk of leukemia compared to an LV with a matched design. Most importantly, the design of the vector was the most relevant factor modulating the genotoxic potential of vector integration, since SIN LTR RVs or LVs even at high dose did not accelerate tumor onset [88] highlighted that different doses of a genotoxic vector lead to different genotoxic readout. Mice that received tumor-prone cells transduced at high vector dose died significantly earlier not only compared to mock-treated mice, but also compared to mice that had received low-dose transduced cells. Moreover, by stratifying the mice according to the retrieved VCN in the tumors, it was demonstrated that the ones with high copy number (>6) died significantly earlier compared to the one with lower VCN (1–6). The same studies have revealed that LVs with active LTRs require a 10-fold higher integration load compared to γ-RVs with active LTRs to achieve the same oncogenic risk, likely reflecting the differences in the integration preferences of the two vectors, which may increase the probability of oncogene activation and, consequently, cancer development by γ-RVs as compared to LVs.
Lentiviral Vector-Based Clinical Trials
The unexpected adverse events in γ-RV gene therapy trials highlighted that clinical benefits of HSPC gene therapy were offset by limitations and risks associated with γ-RV-based gene therapy applications. On one side, the occurrence of leukemia posed major issues concerning the safety of these applications, which, together with the limiting unfeasibility to transduce non-dividing cells, promoted the use of different vectors to deliver the corrected gene copy to the diseased cells.
Lentiviral vectors allured scientists for such purposes, since they subsume important features of retroviral vectors —as the ability to stably integrate within the host genome—as well as grant advantages of reaching higher vector titers and ability to transduce non-dividing cells. Genotoxicity studies also showed that these vectors harbor a reduced genotoxic potential compared to analogous γ-RV constructs [75, 80, 89, 105]. Thus, gene therapy clinical trials using LVs as vehicles to deliver therapeutic genes expanded beyond primary immunodeficiencies, such as the LV-based clinical trial for WAS [52], toward the treatment of numerous monogenic disorders (Table 1).
A LV-based clinical trial for X-linked adrenoleukodystrophy (ALD), a severe demyelinating disease caused by ABCD1 gene mutations, showed disease correction by engineered HSPC cell progeny able to replace diseased microglia (Table 1) [106]. Along the same line, a clinical trial for the correction of a lysosomal storage disorder caused by Arylsulfatase A (ARSA) deficiency, namely metachromatic leukodystrophy (MLD) was performed (Table 1) [51]. Interestingly, MLD gene therapy patients greatly profited from gene therapy edited HSPC by means of corrected microglia replacement and cross-correction phenomenon, a mechanism by which gene-corrected monocyte-derived cells release the therapeutic enzyme, whose’ uptake from enzyme-deficient cells of the central nervous system allows restoration of enzymatic function although these cells do not directly express the therapeutic transgene [4, 51].
Beta-thalassemia, caused by mutations in the beta chains of hemoglobin leading to decreased or absent globin protein and, consequently, anemia, was also a target disease of LV-based gene therapy trials (Table 1) [66, 107]. One of the beta-thalassemia treated patients experienced a transient and benign clonal dominance event, attributed to lentiviral vector-induced overexpression of HMGA2 gene. Molecular investigations revealed LV integrations in HMGA2 engendering a chimeric transcript between the third exon of HMGA2 and a cryptic splice-site located inside the 3′ end of the vector construct causing, by the vector polyadenylation signal, premature truncation and loss of host microRNA Let-7 regulatory sequences in charge of physiological degradation of HMGA2 transcript [66]. Present in over 60% of vector-marked nucleated blood cell population, this overt clonal expansion was undermined by untransduced cells that continued to dominate on hematopoiesis so that positive clones for this insertion site represented only near to 3% of the total nucleated blood cells’ population and over time this extent reduced. Nevertheless, this patient turned independent from transfusion treatments and never displayed oncologic malignancies [66]. More recently, clinical trials have initiated for the treatment of ADA-SCID and X-CGD using LVs (Table 1) [6, 9]. In the current short time treatment follow-up interval, lack of transplantation-related side effects as well as absence of vector-related oncogenic events was reported.
Overall, no severe adverse events have been reported for any of the LV-based gene therapy trials so far, and most patients displayed hematopoietic gene modified cells reconstitution underlying clinical benefit. Comparison of γ-RV and LV-based trials for WAS bests recapitulates the safety of the different vector platforms for gene therapy applications. While γ-RV integrations next to LMO2 proto-oncogene in patients conferred growth advantage to these clones, driving leukemia occurrence, vector integration sites studies for WAS LV-based clinical trial confirmed absence of recurrent integrations targeting potential oncogenes [22, 52]. Moreover, no evidences of clonal expansion was detected, since CIS harboring cell clones were not the most abundant at any given time point during the first three years of follow-up [51, 52]. Indeed, CIS found in the LV-based trial, e.g. KDM2A or PACS1, are LV insertion hot spots likely being the result of vector integration biases at the time of transduction and not consequence of in vivo genetic selection [45, 52, 99].
Concluding Remarks
The successful results of gene therapy are embodied by the clinical benefits and positive long-term follow-up of treated patients. With gene therapy becoming a curative treatment option for many patients with severe diseases, improvements both in vector engineering and in genotoxicity assessment will help sustaining further improved therapies to safely cure patients. Ultrasensitive genotoxicity assays and powerful technologies for safety testing and clonal monitoring have shown that retroviral vector insertions are not neutral to the host genome, since they can alter the mRNAs structure and stability or expression levels of targeted genes in human and mouse HSPCs cells and even promote cancer formation [18, 19, 21, 22, 33, 34, 66, 84, 108]. Several novel vector designs and novel genetic elements are being developed to improve the safety of vector integration and tested in different genotoxicity assays. However, when more advanced vector designs with lower genotoxic potential or with low vector doses requirements will be available, even the currently most sensitive tumor-prone mouse models may not be sensitive enough to score for possible residual insertional mutagenesis events. Therefore, the development of increasingly sensitive genotoxicity assays, and the assessment of mutagenicity and oncogenicity of vector treatments is still a crucial, outstanding issue for the whole gene therapy field.
References
Karlsson S, Ooka A, Woods NB. Development of gene therapy for blood disorders by gene transfer into haematopoietic stem cells. Haemophilia: The Official Journal of the World Federation of Hemophilia. 2002;8:255–60.
Williams DA, Lemischka IR, Nathan DG, Mulligan RC. Introduction of new genetic material into pluripotent haematopoietic stem cells of the mouse. Nature. 1984;310:476–80.
Hock RA, Miller AD. Retrovirus-mediated transfer and expression of drug resistance genes in human haematopoietic progenitor cells. Nature. 1986;320:275–7.
Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. 2011;12:301–15.
Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, Shearer G, Chang L, Chiang Y, Tolstoshev P, Greenblatt JJ, Rosenberg SA, Klein H, Berger M, Mullen CA, Ramsey WJ, Muul L, Morgan RA, Anderson WF. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995;270:475–80.
Kaufmann KB, Buning H, Galy A, Schambach A, Grez M. Gene therapy on the move. EMBO molecular medicine. 2013;5:1642–61.
Aiuti A, Vai S, Mortellaro A, Casorati G, Ficara F, Andolfi G, Ferrari G, Tabucchi A, Carlucci F, Ochs HD, Notarangelo LD, Roncarolo MG, Bordignon C. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat Med. 2002;8:423–5.
Candotti F, Shaw KL, Muul L, Carbonaro D, Sokolic R, Choi C, Schurman SH, Garabedian E, Kesserwan C, Jagadeesh GJ, Fu PY, Gschweng E, Cooper A, Tisdale JF, Weinberg KI, Crooks GM, Kapoor N, Shah A, Abdel-Azim H, Yu XJ, Smogorzewska M, Wayne AS, Rosenblatt HM, Davis CM, Hanson C, Rishi RG, Wang X, Gjertson D, Yang OO, Balamurugan A, Bauer G, Ireland JA, Engel BC, Podsakoff GM, Hershfield MS, Blaese RM, Parkman R, Kohn DB. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120:3635–46.
Cicalese MP, Aiuti A. Clinical applications of gene therapy for primary immunodeficiencies. Hum Gene Ther. 2015;26:210–9.
Cicalese MP, Ferrua F, Castagnaro L, Pajno R, Barzaghi F, Giannelli S, Dionisio F, Brigida I, Bonopane M, Casiraghi M, Tabucchi A, Carlucci F, Grunebaum E, Adeli M, Bredius RG, Puck JM, Stepensky P, Tezcan I, Rolfe K, De Boever E, Reinhardt RR, Appleby J, Ciceri F, Roncarolo MG, Aiuti A. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood;2016.
Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Adams S, Howe SJ, Al Ghonaium A, Bayford J, Brown L, Davies EG, Kinnon C, Thrasher AJ. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2011;3:97ra79.
Montiel-Equihua CA, Thrasher AJ, Gaspar HB. Gene therapy for severe combined immunodeficiency due to adenosine deaminase deficiency. Curr Gene Ther. 2012;12:57–65.
Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J, Brouns G, Schmidt M, Von Kalle C, Barington T, Jakobsen MA, Christensen HO, Al Ghonaium A, White HN, Smith JL, Levinsky RJ, Ali RR, Kinnon C, Thrasher AJ. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. The Lancet. 2004;364:2181–7.
Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP, Thrasher AJ, Wulffraat N, Sorensen R, Dupuis-Girod S, Fischer A, Davies EG, Kuis W, Leiva L, Cavazzana-Calvo M. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. New Eng J Med. 2002;346:1185–93.
Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, Reichenbach J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114:2619–22.
Kang EM, Choi U, Theobald N, Linton G, Long Priel DA, Kuhns D, Malech HL. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood. 2010;115:783–91.
Kang HJ, Bartholomae CC, Paruzynski A, Arens A, Kim S, Yu SS, Hong Y, Joo CW, Yoon NK, Rhim JW, Kim JG, Von Kalle C, Schmidt M, Kim S, Ahn HS. Retroviral gene therapy for X-linked chronic granulomatous disease: results from phase I/II trial. Mol Ther: The Journal of the American Society of Gene Therapy. 2011;19:2092–101.
Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, Glimm H, Kuhlcke K, Schilz A, Kunkel H, Naundorf S, Brinkmann A, Deichmann A, Fischer M, Ball C, Pilz I, Dunbar C, Du Y, Jenkins NA, Copeland NG, Luthi U, Hassan M, Thrasher AJ, Hoelzer D, von Kalle C, Seger R, Grez M. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–9.
Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Kramer A, Schwable J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Gohring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kuhlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204.
Bosticardo M, Ghosh A, Du Y, Jenkins NA, Copeland NG, Candotti F. Self-inactivating retroviral vector-mediated gene transfer induces oncogene activation and immortalization of primary murine bone marrow cells. Mol Ther: The Journal of the American Society of Gene Therapy. 2009;17:1910–8.
Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Diez IA, Dewey RA, Bohm M, Nowrouzi A, Ball CR, Glimm H, Naundorf S, Kuhlcke K, Blasczyk R, Kondratenko I, Marodi L, Orange JS, von Kalle C, Klein C. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. New Eng J Med. 2010;363:1918–27.
Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M, Modlich U, Beier R, Gohring G, Steinemann D, Fronza R, Ball CR, Haemmerle R, Naundorf S, Kuhlcke K, Rose M, Fraser C, Mathias L, Ferrari R, Abboud MR, Al-Herz W, Kondratenko I, Marodi L, Glimm H, Schlegelberger B, Schambach A, Albert MH, Schmidt M, von Kalle C, Klein C. Gene therapy for Wiskott-Aldrich syndrome–long-term efficacy and genotoxicity. Sci Transl Med. 2014;6:227ra233.
Mukherjee S, Thrasher AJ. Gene therapy for PIDs: progress, pitfalls and prospects. Gene. 2013;525:174–81.
Fischer A, Cavazzana-Calvo M. Gene therapy of inherited diseases. Lancet. 2008;371:2044–7.
Bousso P, Wahn V, Douagi I, Horneff G, Pannetier C, Le Deist F, Zepp F, Niehues T, Kourilsky P, Fischer A, de Saint Basile G. Diversity, functionality, and stability of the T cell repertoire derived in vivo from a single human T cell precursor. Proc Natl Acad Sci USA. 2000;97:274–8.
Chinen J, Davis J, De Ravin SS, Hay BN, Hsu AP, Linton GF, Naumann N, Nomicos EY, Silvin C, Ulrick J, Whiting-Theobald NL, Malech HL, Puck JM. Gene therapy improves immune function in preadolescents with X-linked severe combined immunodeficiency. Blood. 2007;110:67–73.
Chatziandreou I, Siapati EK, Vassilopoulos G. Genetic correction of X-linked chronic granulomatous disease with novel foamy virus vectors. Exp Hematol. 2011;39:643–52.
Grez M, Reichenbach J, Schwable J, Seger R, Dinauer MC, Thrasher AJ. Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther: The Journal of the American Society of Gene Therapy. 2011;19:28–35.
Goebel WS, Dinauer MC. Gene therapy for chronic granulomatous disease. Acta Haematol. 2003;110:86–92.
Malech HL. Progress in gene therapy for chronic granulomatous disease. J Infect Dis. 1999;179(Suppl 2):S318–25.
Malech HL, Maples PB, Whiting-Theobald N, Linton GF, Sekhsaria S, Vowells SJ, Li F, Miller JA, DeCarlo E, Holland SM, Leitman SF, Carter CS, Butz RE, Read EJ, Fleisher TA, Schneiderman RD, Van Epps DE, Spratt SK, Maack CA, Rokovich JA, Cohen LK, Gallin JI. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci U S A. 1997;94:12133–8.
Candotti F. Gene transfer into hematopoietic stem cells as treatment for primary immunodeficiency diseases. Int J Hematol. 2014;99:383–92.
Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Investig. 2008;118:3132–42.
Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. New Eng J Med. 2003;348:255–6.
Woods NB, Muessig A, Schmidt M, Flygare J, Olsson K, Salmon P, Trono D, von Kalle C, Karlsson S. Lentiviral vector transduction of NOD/SCID repopulating cells results in multiple vector integrations per transduced cell: risk of insertional mutagenesis. Blood. 2003;101:1284–9.
Coffin JM, Stephen HH, Varmus HE. Retroviruses, Plainview, N.Y.; 1997.
Doorbar J. The papillomavirus life cycle. J Clin Virol: The Official Publication of the Pan American Society for Clinical Virology. 2005;32(Suppl 1):S7–15.
Jonkers J, Berns A. Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochim Biophys Acta. 1996;1287:29–57.
Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005;24:7656–72.
Coffin JM, Hughes SH, Varmus H. Retroviruses. N.Y. xv: Cold Spring Harbor Laboratory Press, Plainview; 1997 843 pp.
West AG, Fraser P. Remote control of gene transcription. Hum Mol Genet. 2005;14(Spec No 1):R101–111.
Hayward WS, Neel BG, Astrin SM. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature. 1981;290:475–80.
Kool J, Berns A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat Rev Cancer. 2009;9:389–99.
Ranzani M, Annunziato S, Adams DJ, Montini E. Cancer gene discovery: exploiting insertional mutagenesis. Mol Cancer Res: MCR. 2013;11:1141–58.
Biffi A, Bartolomae CC, Cesana D, Cartier N, Aubourg P, Ranzani M, Cesani M, Benedicenti F, Plati T, Rubagotti E, Merella S, Capotondo A, Sgualdino J, Zanetti G, von Kalle C, Schmidt M, Naldini L, Montini E. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332–9.
Suzuki M, Ketterling MG, McCarty DR. Quantitative statistical analysis of cis-regulatory sequences in ABA/VP1- and CBF/DREB1-regulated genes of Arabidopsis. Plant Physiol. 2005;139:437–47.
de Ridder J, Uren A, Kool J, Reinders M, Wessels L. Detecting statistically significant common insertion sites in retroviral insertional mutagenesis screens. PLoS Comput Biol. 2006;2:e166.
Sarver AL, Erdman J, Starr T, Largaespada DA, Silverstein KA. TAPDANCE: an automated tool to identify and annotate transposon insertion CISs and associations between CISs from next generation sequence data. BMC Bioinformatics. 2012;13:154. PMID: 22748055. doi: 10.1186/1471-2105-13-154.
Abel U, Deichmann A, Bartholomae C, Schwarzwaelder K, Glimm H, Howe S, Thrasher A, Garrigue A, Hacein-Bey-Abina S, Cavazzana-Calvo M, Fischer A, Jaeger D, von Kalle C, Schmidt M. Real-time definition of non-randomness in the distribution of genomic events. PLoS ONE. 2007;2:e570.
Berry CC, Ocwieja KE, Malani N, Bushman FD. Comparing DNA integration site clusters with scan statistics. Bioinformatics. 2014;30:1493–500.
Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, Banerjee PP, Orange JS, Galimberti S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151.
Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, Benedicenti F, Vallanti G, Biasco L, Leo S, Kabbara N, Zanetti G, Rizzo WB, Mehta NA, Cicalese MP, Casiraghi M, Boelens JJ, Del Carro U, Dow DJ, Schmidt M, Assanelli A, Neduva V, Di Serio C, Stupka E, Gardner J, von Kalle C, Bordignon C, Ciceri F, Rovelli A, Roncarolo MG, Aiuti A, Sessa M, Naldini L. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158.
Kustikova OS, Baum C, Fehse B. Retroviral integration site analysis in hematopoietic stem cells. Methods Mol Biol. 2008;430:255–67.
Mueller PR, Wold B. In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science. 1989;246:780–6.
Harkey MA, Kaul R, Jacobs MA, Kurre P, Bovee D, Levy R, Blau CA. Multiarm high-throughput integration site detection: limitations of LAM-PCR technology and optimization for clonal analysis. Stem Cells Dev. 2007;16:381–92.
Schmidt M, Schwarzwaelder K, Bartholomae C, Zaoui K, Ball C, Pilz I, Braun S, Glimm H, von Kalle C. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR). Nat Methods. 2007;4:1051–7.
Schmidt M, Zickler P, Hoffmann G, Haas S, Wissler M, Muessig A, Tisdale JF, Kuramoto K, Andrews RG, Wu T, Kiem HP, Dunbar CE, von Kalle C. Polyclonal long-term repopulating stem cell clones in a primate model. Blood. 2002;100:2737–43.
Berry CC, Gillet NA, Melamed A, Gormley N, Bangham CR, Bushman FD. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics. 2012;28:755–62.
Biasco L, Baricordi C, Aiuti A. Retroviral integrations in gene therapy trials. Mol Ther: The Journal of the American Society of Gene Therapy. 2012;20:709–16.
Wang GP, Berry CC, Malani N, Leboulch P, Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M, Bushman FD. Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood. 2010;115:4356–66.
Gabriel R, Eckenberg R, Paruzynski A, Bartholomae CC, Nowrouzi A, Arens A, Howe SJ, Recchia A, Cattoglio C, Wang W, Faber K, Schwarzwaelder K, Kirsten R, Deichmann A, Ball CR, Balaggan KS, Yanez-Munoz RJ, Ali RR, Gaspar HB, Biasco L, Aiuti A, Cesana D, Montini E, Naldini L, Cohen-Haguenauer O, Mavilio F, Thrasher AJ, Glimm H, von Kalle C, Saurin W, Schmidt M. Comprehensive genomic access to vector integration in clinical gene therapy. Nat Med. 2009;15:1431–6.
Koudijs MJ, Klijn C, van der Weyden L, Kool J, ten Hoeve J, Sie D, Prasetyanti PR, Schut E, Kas S, Whipp T, Cuppen E, Wessels L, Adams DJ, Jonkers J. High-throughput semiquantitative analysis of insertional mutations in heterogeneous tumors. Genome Res. 2011;21:2181–9.
Gillet NA, Malani N, Melamed A, Gormley N, Carter R, Bentley D, Berry C, Bushman FD, Taylor GP, Bangham CR. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood. 2011;117:3113–22.
Wu C, Jares A, Winkler T, Xie J, Metais JY, Dunbar CE. High efficiency restriction enzyme-free linear amplification-mediated polymerase chain reaction approach for tracking lentiviral integration sites does not abrogate retrieval bias. Hum Gene Ther. 2013;24:38–47.
Zhou S, Bonner MA, Wang YD, Rapp S, De Ravin SS, Malech HL, Sorrentino BP. Quantitative shearing linear amplification polymerase chain reaction: an improved method for quantifying lentiviral vector insertion sites in transplanted hematopoietic cell systems. Hum Gene Ther Methods. 2015;26:4–12.
Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, Cavallesco R, Gillet-Legrand B, Caccavelli L, Sgarra R, Maouche-Chretien L, Bernaudin F, Girot R, Dorazio R, Mulder GJ, Polack A, Bank A, Soulier J, Larghero J, Kabbara N, Dalle B, Gourmel B, Socie G, Chretien S, Cartier N, Aubourg P, Fischer A, Cornetta K, Galacteros F, Beuzard Y, Gluckman E, Bushman F, Hacein-Bey-Abina S, Leboulch P. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–22.
Firouzi S, Lopez Y, Suzuki Y, Nakai K, Sugano S, Yamochi T, Watanabe T. Development and validation of a new high-throughput method to investigate the clonality of HTLV-1-infected cells based on provirus integration sites. Genome Med. 2014;6:46.
Biffi A, De Palma M, Quattrini A, Del Carro U, Amadio S, Visigalli I, Sessa M, Fasano S, Brambilla R, Marchesini S, Bordignon C, Naldini L. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113:1118–29.
De Palma M, Montini E, Santoni de Sio FR, Benedicenti F, Gentile A, Medico E, Naldini L. Promoter trapping reveals significant differences in integration site selection between MLV and HIV vectors in primary hematopoietic cells. Blood. 2005;105:2307–15.
Dupre L, Marangoni F, Scaramuzza S, Trifari S, Hernandez RJ, Aiuti A, Naldini L, Roncarolo MG. Efficacy of gene therapy for Wiskott-Aldrich syndrome using a WAS promoter/cDNA-containing lentiviral vector and nonlethal irradiation. Hum Gene Ther. 2006;17:303–13.
Lo M, Bloom ML, Imada K, Berg M, Bollenbacher JM, Bloom ET, Kelsall BL, Leonard WJ. Restoration of lymphoid populations in a murine model of X-linked severe combined immunodeficiency by a gene-therapy approach. Blood. 1999;94:3027–36.
May C, Rivella S, Chadburn A, Sadelain M. Successful treatment of murine beta-thalassemia intermedia by transfer of the human beta-globin gene. Blood. 2002;99:1902–8.
Montini E, Cesana D. Genotoxicity assay for gene therapy vectors in tumor prone Cdkn2a(–)/(–) mice. Methods Enzymol. 2012;507:171–85.
Vigna E, Naldini L. Lentiviral vectors: excellent tools for experimental gene transfer and promising candidates for gene therapy. J Gene Med. 2000;2:308–16.
Bokhoven M, Stephen SL, Knight S, Gevers EF, Robinson IC, Takeuchi Y, Collins MK. Insertional gene activation by lentiviral and gammaretroviral vectors. J Virol. 2009;83:283–94.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Du Y, Jenkins NA, Copeland NG. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood. 2005;106:3932–9.
Arumugam PI, Higashimoto T, Urbinati F, Modlich U, Nestheide S, Xia P, Fox C, Corsinotti A, Baum C, Malik P. Genotoxic potential of lineage-specific lentivirus vectors carrying the beta-globin locus control region. Mol Ther: The Journal of the American Society of Gene Therapy. 2009;17:1929–37.
Gaussin A, Modlich U, Bauche C, Niederlander NJ, Schambach A, Duros C, Artus A, Baum C, Cohen-Haguenauer O, Mermod N. CTF/NF1 transcription factors act as potent genetic insulators for integrating gene transfer vectors. Gene Ther. 2012;19:15–24.
Modlich U, Bohne J, Schmidt M, von Kalle C, Knoss S, Schambach A, Baum C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108:2545–53.
Modlich U, Navarro S, Zychlinski D, Maetzig T, Knoess S, Brugman MH, Schambach A, Charrier S, Galy A, Thrasher AJ, Bueren J, Baum C. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther: The Journal of the American Society of Gene Therapy. 2009;17:1919–28.
Schambach A, Zychlinski D, Ehrnstroem B, Baum C. Biosafety features of lentiviral vectors. Hum Gene Ther. 2013;24:132–42.
Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E, Mishra A, Baum C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther: The Journal of the American Society of Gene Therapy. 2008;16:718–25.
Cesana D, Ranzani M, Volpin M, Bartholomae C, Duros C, Artus A, Merella S, Benedicenti F, Sergi Sergi L, Sanvito F, Brombin C, Nonis A, Serio CD, Doglioni C, von Kalle C, Schmidt M, Cohen-Haguenauer O, Naldini L, Montini E. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol Ther. 2014;22:774–85.
Cornils K, Bartholomae CC, Thielecke L, Lange C, Arens A, Glauche I, Mock U, Riecken K, Gerdes S, von Kalle C, Schmidt M, Roeder I, Fehse B. Comparative clonal analysis of reconstitution kinetics after transplantation of hematopoietic stem cells gene marked with a lentiviral SIN or a gamma-retroviral LTR vector. Exp Hematol. 2013;41(28–38):e23.
Hematti P, Hong BK, Ferguson C, Adler R, Hanawa H, Sellers S, Holt IE, Eckfeldt CE, Sharma Y, Schmidt M, von Kalle C, Persons DA, Billings EM, Verfaillie CM, Nienhuis AW, Wolfsberg TG, Dunbar CE, Calmels B. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004;2:e423.
Kustikova O, Fehse B, Modlich U, Yang M, Dullmann J, Kamino K, von Neuhoff N, Schlegelberger B, Li Z, Baum C. Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science. 2005;308:1171–4.
Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M, Benedicenti F, Sergi LS, Ambrosi A, Ponzoni M, Doglioni C, Di Serio C, von Kalle C, Naldini L. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119:964–75.
Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, Sergi Sergi L, Benedicenti F, Ambrosi A, Di Serio C, Doglioni C, von Kalle C, Naldini L. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–96.
Nienhuis AW, Dunbar CE, Sorrentino BP. Genotoxicity of retroviral integration in hematopoietic cells. Mol Ther: The Journal of the American Society of Gene Therapy. 2006;13:1031–49.
Li Z, Dullmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J, Forster M, Stocking C, Wahlers A, Frank O, Ostertag W, Kuhlcke K, Eckert HG, Fehse B, Baum C. Murine leukemia induced by retroviral gene marking. Science. 2002;296:497.
Beard BC, Dickerson D, Beebe K, Gooch C, Fletcher J, Okbinoglu T, Miller DG, Jacobs MA, Kaul R, Kiem HP, Trobridge GD. Comparison of HIV-derived lentiviral and MLV-based gammaretroviral vector integration sites in primate repopulating cells. Mol Ther: The Journal of the American Society of Gene Therapy. 2007;15:1356–65.
Kiem HP, Sellers S, Thomasson B, Morris JC, Tisdale JF, Horn PA, Hematti P, Adler R, Kuramoto K, Calmels B, Bonifacino A, Hu J, von Kalle C, Schmidt M, Sorrentino B, Nienhuis A, Blau CA, Andrews RG, Donahue RE, Dunbar CE. Long-term clinical and molecular follow-up of large animals receiving retrovirally transduced stem and progenitor cells: no progression to clonal hematopoiesis or leukemia. Mol Ther: The Journal of the American Society of Gene Therapy. 2004;9:389–95.
Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37.
Liu M, Maurano MT, Wang H, Qi H, Song CZ, Navas PA, Emery DW, Stamatoyannopoulos JA, Stamatoyannopoulos G. Genomic discovery of potent chromatin insulators for human gene therapy. Nat Biotechnol. 2015;33:198–203.
Ambrosi A, Glad IK, Pellin D, Cattoglio C, Mavilio F, Di Serio C, Frigessi A. Estimated comparative integration hotspots identify different behaviors of retroviral gene transfer vectors. PLoS Comput Biol. 2011;7:e1002292.
Bushman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, Hannenhalli S, Hoffmann C. Genome-wide analysis of retroviral DNA integration. Nat Rev Microbiol. 2005;3:848–58.
Bushman FD. Retroviral integration and human gene therapy. J Clin Investig. 2007;117:2083–6.
Cattoglio C, Facchini G, Sartori D, Antonelli A, Miccio A, Cassani B, Schmidt M, von Kalle C, Howe S, Thrasher AJ, Aiuti A, Ferrari G, Recchia A, Mavilio F. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood. 2007;110:1770–8.
Cattoglio C, Pellin D, Rizzi E, Maruggi G, Corti G, Miselli F, Sartori D, Guffanti A, Di Serio C, Ambrosi A, De Bellis G, Mavilio F. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood. 2010;116:5507–17.
Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, Ecker JR, Bushman FD. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:E234.
Schröder ARW, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–9.
Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–51.
Craigie R, Bushman FD. Host factors in retroviral integration and the selection of integration target sites. Microbiol Spectr. 2014;2.
Modlich U, Kustikova OS, Schmidt M, Rudolph C, Meyer J, Li Z, Kamino K, von Neuhoff N, Schlegelberger B, Kuehlcke K, Bunting KD, Schmidt S, Deichmann A, von Kalle C, Fehse B, Baum C. Leukemias following retroviral transfer of multidrug resistance 1 (MDR1) are driven by combinatorial insertional mutagenesis. Blood. 2005;105(11):4235–46. PMID: 15713797.
Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Bougneres P, Schmidt M, Kalle CV, Fischer A, Cavazzana-Calvo M, Aubourg P. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 2012;507:187–98.
Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–60.
Cesana D, Sgualdino J, Rudilosso L, Merella S, Naldini L, Montini E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J Clin Invest. 2012;122:1667–76.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Cesana, D., Volpin, M., Serina Secanechia, Y.N., Montini, E. (2017). Safety and Efficacy of Retroviral and Lentiviral Vectors for Gene Therapy. In: Brunetti-Pierri, N. (eds) Safety and Efficacy of Gene-Based Therapeutics for Inherited Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-53457-2_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-53457-2_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53455-8
Online ISBN: 978-3-319-53457-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)