
Abstract
Glutamate is the most important excitatory neurotransmitter in the brain. The N-methyl-D-aspartate (NMDA) subtype of glutamate receptor is found both in neurons and glial cells such as oligodendrocytes, which have been shown to be dysfunctional in schizophrenia. For this reasons, the oligodendrocyte MO3.13 cell line has been used to study glutamatergic dysfunction as a model of schizophrenia using the NMDA receptor antagonists such as MK-801 to block receptor function. Here, we describe a comprehensive protocol for culturing and carrying out proteomic analyses of MK-801-treated MO3.13 cells as a means of identifying potential new biomarkers and targets for drug discovery in schizophrenia research.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The brain is the main organ of the central nervous system (CNS) and is composed of a heterogeneous group of cells, comprised of neurons and glia. Glial cells consist of astrocytes, oligodendrocytes, and microglia. For over a century, neurons have been perceived as being the most important cell type for brain function, and glia were seen simply as tissue glue. However, recent studies have clearly indicated the important role of glia in brain function. Glial cells are responsible for many functions, such as power support, neuronal control, maintenance, and plasticity of synaptic contacts [1]. Many studies have shown that neurons and glial cells communicate bidirectionally at both structural and functional levels. Thus, glial cells may act as partners of neurons in the formation of information processing mediated by glutamate, the most abundant excitatory neurotransmitter in the brain [1,2,3].
Glutamatergic transmission occurs throughout the CNS and is responsible for numerous essential brain functions such as cognition, learning, and memory. The inhibition of glutamatergic transmission can lead to disabling of the entire nervous system. Each glutamatergic synaptic event is initiated by an action potential that leads to Ca2+ influx in the presynaptic terminal, resulting in the release of the neurotransmitter into the synaptic cleft. After being released, glutamate rapidly crosses the synaptic cleft and activates specific receptors in the postsynaptic membrane [3]. The interaction of glutamate with glial cells is basically mediated by four types of receptors: G protein-coupled metabotropic receptors and three families of ionotropic glutamate receptors, comprised of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainate, and N-methyl-D-aspartate (NMDA) receptors [1, 4, 5]. Following the emergence of phencyclidine (PCP) around 1950, NMDA receptors have been implicated in the pathophysiology of neuropsychiatric disorders [6].
The NMDA receptor is a glutamate-activated cation channel, encoded by seven genes. It has four sites of pharmacological relevance: the glutamate recognition site, the glycine modulating site, the binding site for PCP and its analogues, and a cation-binding site within the channel, where magnesium acts [3, 7]. In Alzheimer’s disease, impaired memory and learning has been linked to perturbations in NMDA receptor function [8]. Studies using PCP have also led to the glutamatergic hypothesis of schizophrenia due to similarities regarding symptomatic manifestations of PCP-induced psychosis and symptoms of this psychiatric illness [2, 7]. However, it has also been shown that the use of NMDA antagonists in patients with chronic pain reduced the need of opioids and these compounds also exhibit anticonvulsant properties in epilepsy patients [9].
In addition to PCP, other pharmacological agents have been used to study the pathophysiology of schizophrenia through the modulation of neurotransmitter systems. One of these is dizocilpine (also known as MK-801), which is the most powerful antagonist of the NMDA receptor [10]. Studies have revealed that neurons, astrocytes, and oligodendrocytes are affected by MK-801 treatment, although in a differential manner. For example, glycolysis appears to be more affected in MK-801-treated oligodendrocytes compared to the other cell types [11]. Furthermore, recent proteomic studies found that many proteins associated with energy metabolism were upregulated in the MK-801-treated oligodendrocyte cell line, MO3.13 [12].
The interest in understanding the role of oligodendrocytes in human disease led to development of oligodendroglial models for testing hypotheses under a controlled environment. The MO3.13 cell line was developed via fusion of a 6-thioguanine-resistant mutant of the human rhabdomyosarcoma RD with adult human oligodendrocytes cultured from a surgical specimen [13, 14]. Thus, proteomic profiling of these cells could help in understanding the mechanisms of action of pharmacological agents in studies of psychiatric disorders such as schizophrenia. Here, we present a protocol for culturing the MO3.13 oligodendrocyte cell line, treatment with MK-801, and mass spectrometry-based proteomic analysis.
2 Materials (See Note 1)
2.1 MO3.13 Cell Culture
-
1.
Human oligodendroglia cell line MO3.13 (Cedarlane; Burlington, NC, USA)
-
2.
Culture medium: DMEM, high glucose supplemented with 10% fetal bovine serum (FBS), and 1% 5000 U/mL penicillin-streptomycin (ThermoFisher Scientific; Waltham, MA, USA)
-
3.
Flask Nunclon Delta-treated Vent/Close 75 (Sigma-Aldrich; São Paulo, SP, Brazil)
-
4.
MAXYMum Recovery™ PCR Tubes (Axygen Scientific; Radnor, PA, USA)
-
5.
Pipette tips (Axygen Scientific)
-
6.
Serological pipette 25 mL (Sigma-Aldrich)
-
7.
15 mL conical centrifuge tubes (ThermoFisher Scientific)
2.2 Cell Collection
-
1.
Phosphate-buffered saline (PBS)
-
2.
Sarstedt cell scraper (Sigma-Aldrich)
-
3.
Freezing medium: 60% DMEM, 30% FBS, 10% DMSO
2.3 Cell Treatment
-
1.
MK-801 hydrogen maleate (Sigma-Aldrich)
2.4 Lysis, Reduction, Alkylation, and Digestion
-
1.
Ultrasonic homogenizer (Cole Parmer Instrument Co.)
-
2.
Lysis and reduction buffer: 6 M urea, 2 M thiourea, 10 mM dithiothreitol (DTT), 0.1 mM sodium pervanadate
-
3.
Hydrogen peroxide solution
-
4.
Protease and phosphatase inhibitors (cOmplete ULTRA) (Sigma-Aldrich)
-
5.
Alkylation buffer: 20 mM iodoacetamide in 200 mM triethylammonium bicarbonate buffer (TEAB)
-
6.
Digestion buffer: 2% sequencing grade modified trypsin (Promega; Madison, WI, USA)
-
7.
Stop digestion buffer: 100% formic acid (FA)
2.5 Desalting and Concentration of Peptides
-
1.
Oasis® HLB Short Cartridge (Waters Corporation; Milford, MA, USA)
-
2.
Qubit® assay tubes (Thermo Fisher)
-
3.
Qubit® protein assay kit (Thermo Fisher)
-
4.
Concentrator plus (Eppendorf; Westbury, NY, USA)
-
5.
Activation solution 1: 100% methanol (HPLC grade)
-
6.
Activation solution 2: 100% LC-MS grade acetonitrile (ACN)
-
7.
Reverse phase (RP) loading solution: 0.1% trifluoroacetic acid (TFA)
-
8.
RP elution solution: 70% ACN, 01% TFA
2.6 NanoLC-MS/MS Analyses
-
1.
Solvent A: 0.1% FA in water
-
2.
Solvent B: 0.1% FA in ACN
-
3.
Lock Spray solution: 100 fmol/μL [Glu1]-fibrinopeptide B standard (Waters Corporation) in methanol/water/FA (50:50:0.1%)
-
4.
Ammonium hydroxide
-
5.
ACQUITY UPLC M-Class system with 2D Technology: binary solvent manager; auxiliary solvent manager; sample manager (Waters Corporation)
-
6.
First-dimension analytical column: M-Class peptide 130 Å, 5 μm, 300 μm × 50 mm BEH C18 trap column
-
7.
Second dimension analytical columns: M-Class Symmetry 100 Å, 5 μm, 180 μm × 20 mm C18 Trap Column and 1.8 μm, 75 μm × 150 mm V/M and M-Class HSS T3 Column (Waters Corporation)
-
8.
Autosampler vials: total recovery glass vials (Waters Corporation)
-
9.
Instrument control software for UPLC: MassLynx (version 4.1; Waters Corporation)
-
10.
SYNAPT G2-Si high-definition mass spectrometer (Waters Corporation)
-
11.
NanoLock Spray dual electrospray ion source (Waters Corporation)
-
12.
Pre-Cut Picotip emitter (Waters Corporation)
-
13.
MS instrument control software: MassLynx (version 4.1; Waters Corporation)
2.7 Data Processing
-
1.
Software for raw data processing, database searching, and label-free quantification: Progenesis QI for Proteomics version 3.0 (Nonlinear Dynamics; Waters Corporation)
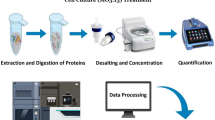
3 Methods
3.1 Cell Culture (See Note 2)
-
1.
Take off the cryogenic vial from liquid nitrogen and unfreeze quickly in a 37 °C water bath (see Note 3).
-
2.
Promptly transfer the content to a centrifuge tube containing 10 mL of DMEM and centrifuge for 5 min at 1200 × g (see Note 4).
-
3.
Discard the supernatant and distribute the cell pellet in a T75 cell culture flask with 25 mL of culture medium (see Note 5).
-
4.
Grow MO3.13 cells at 37 °C in 5% CO2 atmosphere.
-
5.
Change the culture medium every 2–3 days, depending on rate of growth (see Note 6).
-
6.
Remove cells from the flask using approximately 5 mL 0.25% trypsin-EDTA solution and leave for 3 min at 37 °C.
-
7.
Transfer the cells to a centrifuge tube containing 10 mL DMEM.
-
8.
Centrifuge for 5 min at 1,200 × g and discard the supernatant.
-
9.
Divide the cell pellet into four to five new T75 flasks to continue cell culture growth or freeze the cells.
3.2 Freezing (See Note 7)
-
1.
Prepare the number of cryogenic vials according to the amount of cells to be frozen.
-
2.
Take the flask containing the cells and discard the medium.
-
3.
Remove the cells from the flask by leaving in 5 mL 0.25% trypsin-EDTA for 3 min at 37 °C and then transfer the cells into a centrifuge tube containing 10 mL DMEM.
-
4.
Centrifuge for 5 min at 1,200 × g and discard the supernatant.
-
5.
Resuspend the cells with the target concentration of 105/mL of freezing medium.
-
6.
Freeze the vials first by placing in a −20 °C freezer for 2–3 h and then into a −80 °C freezer.
-
7.
Transfer the vials the next day into a liquid nitrogen tank (see Note 8).
3.3 Acute 8 h Treatment with MK-801 (See Note 9)
-
1.
Dilute MK-801 in HCL solution.
-
2.
Dilute the MK-801 solution with 15 mL DMEM to a final concentration of 50 μM.
-
3.
Discard the medium and transfer 5 mL new medium containing 50 μM MK-801 in each flask.
-
4.
After 8 h, collect the cells.
3.4 Collecting the Cells
-
1.
Discard the culture medium.
-
2.
Add 600 μL PBS to the flask and harvest cells by scraping them off the flask.
-
3.
Wash the cells with 600 μL PBS 1x containing protease and phosphatase inhibitors (optional).
-
4.
Collect cells into 15 mL falcon tube and pellet them by 5 min centrifugation (1,200 x g).
-
5.
Remove supernatant and snap-froze the cell pellet containing lysis buffer in liquid nitrogen until further sample preparation.
3.5 Lysis, Reduction, Alkylation, and Digestion (See Note 10)
-
1.
Lyse the cells by adding 100 μL of lysis buffer containing protease and phosphatase inhibitors to the MO3.13 pellet (see Note 11).
-
2.
Stir well and incubate for 2 h at 37 °C (see Note 12).
-
3.
After incubation, dilute the sample 10x with 20 mM TEAB pH 7.5 and sonicate on ice (see Note 13).
-
4.
Add 100 μL 200 mM iodoacetamide in 20 mM triethylammonium bicarbonate to achieve final concentration of 20 mM iodoacetamide, and incubate the sample for 20 min in the dark at room temperature.
-
5.
After incubation, digest the sample using 5.5 μL of 2% trypsin at an enzyme/substrate ratio of 1:50 overnight (12–16 h) at 37 °C.
-
6.
To stop the reaction, add 100% FA to a final concentration of 5% and leave for 5 min at room temperature.
-
7.
Centrifuge for 45 min at 14,000 × g at 4 °C to remove pellet lipids and other vestiges.
-
8.
Transfer the supernatant to another tube (see Note 14).
3.6 Desalting and Concentration of Peptides
-
1.
Use 0.1% TFA to dilute the peptide sample achieving a final volume of 1 mL and adjust the pH to 2.0.
-
2.
Wash the cartridge with 1 mL activation solution 1 followed by 1 mL of activation solution 2 (see Note 15).
-
3.
Balance the cartridge twice using 2 mL of 0.1% TFA.
-
4.
Load the sample onto the cartridge slowly and collect the flow through (FT).
-
5.
Apply the FT again slowly to the same cartridge.
-
6.
Wash the cartridge twice with 1 mL 0.1% TFA.
-
7.
Elute the peptides into a new microtube with 1 mL RP elution solution.
-
8.
Dry the sample in a concentrator or lyophilizer.
-
9.
Reconstitute the sample in 20 mM ammonium formate (pH 10).
3.7 NanoLC-MS/MS Analysis (See Note 16)
-
1.
Create the LC-MS/MS method.
-
2.
Load samples containing 500 ng protein into a M-Class HSS T3 column.
-
3.
Set peptide elutions using ACN gradient from 7% to 40% for 90 min at a flow rate of 0.4 μL/min directly into a Synapt G2-Si HDMS.
-
4.
Use 100 fmol/μL [Glu1]-fibrinopeptide B as a lock mass compound, and use the auxiliary pump to deliver it to the reference sprayer of the NanoLock Spray source at 0.5 μL/min.
-
5.
Turn MS acquisition on in the LC software and align the gradient with the beginning of MS acquisition.
-
6.
Perform MS analysis in DIA mode using ion mobility separation and CID fragmentation.
-
7.
Ramp the transfer cell collision energy from 25 to 55 eV in the elevated energy scan.
-
8.
Perform triplicate LC-MS analysis of each sample (see Note 17).
3.8 Data Processing
-
1.
Perform initial signal processing of continuum LC-IMS-MSE data using Progenesis QI for Proteomics, and create a new project for your experiment.
-
2.
Add the acquired raw data files to the project.
-
3.
Provide lock mass m/z as 785.8426 to perform the calibration.
-
4.
Set up processing parameters: MSe experiment, 150 counts for the low energy threshold, 50.0 counts for the elevated energy threshold, and 750 counts for the intensity threshold.
-
5.
Import the data.
-
6.
Start automatic processing, selecting for automatic alignment of the runs (see Note 18).
-
7.
Proceed with automatic peak picking using 8 as the maximum ion charge, and adjust the sensitivity method of the automatic peak picking algorithm to 4.
-
8.
Define your experiment design (optional).
-
9.
Set the parameters for automatic peptide identification: choose your target-decoy database for peptide and protein identification; trypsin should be selected as the digestion enzyme; and one missed cleavage can be allowed.
-
10.
Set carbamidomethyl of cysteine and oxidation of methionine as fixed and variable modifications, respectively.
-
11.
Choose relative quantitation using Hi-N and three peptides to measure per protein.
-
12.
Use protein grouping.
4 Notes
-
1.
Make sure that all materials are ready for use. For example, the medium should already be at the right temperature (37 °C).
-
2.
All the solutions and buffers should be prepared with Milli-Q water (UHQ), analytical grade reagents, and highest purity chemicals. Organic solutions should be prepared fresh.
-
3.
During unfreezing, it is indicated to move the cryovial circularly inside the bath.
-
4.
For centrifuge cells, it is better to use mobile centrifuge rotor angle.
-
5.
Depending on the number of cells, it is possible to divide the pellet in more than one T75 cell culture flask.
-
6.
Cultures should be split at ~90% confluency.
-
7.
It is recommended to freeze a few aliquots of the cells promptly after the initial growth/split to help maintain a stock of the cell line.
-
8.
It is recommended to test the cells for regrowth after freezing to be sure that the freezing procedure was performed correctly.
-
9.
It is recommended to grow MO3.13 cells until a confluency of 90% is reached in T25 cell culture flasks to initiate the treatment. It is also suggested to treat the cells in triplicate and ensure that everything is clean.
-
10.
Topics 3.5 and 3.6 are optimized versions of Melo-Braga et al. [15].
-
11.
The lysis buffer volume used depends on the pellet size. Here we used 107 cells.
-
12.
Mix by vortexing and pipetting the cell pellet up and down. The cell lysate will form a viscous solution due to the presence of DNA.
-
13.
Probes/tips must be submerged properly into the solution for efficient sonication. If the tip is not submerged enough, the sample will foam or bubble. If the tip is too deep, it will not circulate the sample effectively. Foaming can also be caused when the amplitude setting on the sonication device is too high. In addition, the tip must not touch the sides of the tube to avoid releasing plastic into the sample.
-
14.
It is better use microtubes with low protein retention (i.e., LoBind from Eppendorf or Maxymum Recovery from Axygen).
-
15.
The option of cartridges depends on the quantity of material. For peptide samples with quantity higher or equal to 500 μg, the Oasis HLB cartridges are usually a good choice.
-
16.
Before starting a gradient for peptide separation, make sure that your LC system is set up properly and that you use freshly prepared and degassed solvents. Set and keep the temperature of the sample manager at a constant temperature of 6 °C, while samples are stored therein.
-
17.
This is carried out to increase accuracy and reproducibility.
-
18.
Use this to assess all runs in the experiment for suitability.
References
Verkhratsky A, Kirchhoff F (2007) Glutamate-mediated neuronal-glial transmission. J Anat 210:651–660
Hashimoto K, Malchow B, Falkai P, Schmitt A (2013) Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur Arch Psychiatry Clin Neurosci 263:367–377
Verkhratsky A, Kirchhoff F (2016) NMDA receptors in glia. Neuroscientist 13:28–37
Mayer ML, Armstrong N (2004) Structure and function of glutamate receptor Ion channels. Annu Rev Physiol 66:161–181. doi:10.1146/annurev.physiol.66.050802.084104
Meador-Woodruff JH, Healy DJ (2000) Glutamate receptor expression in schizophrenic brain. Brain Res Brain Res Rev 31:288–294
Krystal JH, D’Souza DC, Petrakis IL, Belger A, Berman RM, Charney DS et al (1999) NMDA agonists and antagonists as probes of glutamatergic dysfunction and pharmacotherapies in neuropsychiatric disorders. Harv Rev Psychiatry 7:125–143
Coyle JT (1996) The glutamatergic dysfunction hypothesis for schizophrenia. Harv Rev Psychiatry 3:241–253
Greenamyre JT, Young AB (1989) Excitatory amino acids and Alzheimer’s disease. Neurobiol Aging 10:593–602
Rogawski MA (1992) The NMDA receptor, NMDA antagonists and epilepsy therapy. A status report. Drugs 44:279–292
Lodge D, Mercier MS (2015) Ketamine and phencyclidine: the good, the bad and the unexpected. Br J Pharmacol 172:4254–4276
Guest PC, Iwata K, Kato TA, Steiner J, Schmitt A, Turck CW et al (2015) MK-801 treatment affects glycolysis in oligodendrocytes more than in astrocytes and neuronal cells: insights for schizophrenia. Front Cell Neurosci 9:1–10
Cassoli JS, Iwata K, Steiner J, Guest PC, Turck CW, Nascimento JM et al (2016) Effect of MK-801 and clozapine on the proteome of cultured human oligodendrocytes. Front Cell Neurosci 10:52. doi:10.3389/fncel.2016.00052
Mclaurin J, Trudel GC, Shaw IT, Antel JP, Cashman NR (1993) A human glial hybrid cell line differentially expressing genes subserving oligodendrocyte and astrocyte phenotype. J Neurobiol 26:283–293
Buntinx M, Vanderlocht J, Hellings N, Vandenabeele F, Lambrichts I, Raus J et al (2003) Characterization of three human oligodendroglial cell lines as a model to study oligodendrocyte injury: morphology and oligodendrocyte-specific gene expression. J Neurocytol 32:25–38
Melo-Braga MN, Ibáñez-Vea M, Larsen MR, Kulej K (2015) Comprehensive protocol to simultaneously study protein phosphorylation, acetylation, and N-linked sialylated glycosylation. Methods Mol Biol 1295:275–292
Acknowledgments
CBT, JSC, and DMS are funded by FAPESP (São Paulo Research Foundation, grants 2015/23049-0, 2014/14881-1, 2013/08711-3, and 2014/10068-4).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Brandão-Teles, C., Martins-de-Souza, D., Guest, P.C., Cassoli, J.S. (2017). MK-801-Treated Oligodendrocytes as a Cellular Model to Study Schizophrenia. In: Guest, P. (eds) Proteomic Methods in Neuropsychiatric Research. Advances in Experimental Medicine and Biology(), vol 974. Springer, Cham. https://doi.org/10.1007/978-3-319-52479-5_25
Download citation
DOI: https://doi.org/10.1007/978-3-319-52479-5_25
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52478-8
Online ISBN: 978-3-319-52479-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)