
Abstract
Sigma-1 receptor (Sig-1R) is molecular chaperone regulating calcium efflux from the neuronal endoplasmic reticulum to mitochondria. Recent studies show that Sig-1R stimulation antagonizes depressive-like behaviors in animal models, but molecular mechanisms underlying this effect remain unclear. Here, we focus on the effects of Sig-1R ligands on hippocampal neurogenesis and depressive-like behaviors. Sig-1R stimulation also enhances CaMKII /CaMKIV and protein kinase B (Akt) activities in hippocampus. Therefore, we discuss the fundamental roles of Sig-1R, CaMKII /CaMKIV and protein kinase B (Akt) signaling in amelioration of depressive-like behaviors following Sig-1R stimulation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
14.1 Introduction
Sig-1R has been cloned in humans and other species [1–4], and in brain, Sig-1R protein is widely distributed in neurons and glial cells such as astrocytes , and is particularly enriched in prefrontal cortex, hippocampus and striatum [5, 6]. Sig-1R protein is primarily localized in membranes of the endoplasmic/sarcoplasmic reticulum (ER/SR), where it regulates Ca2+ signaling through the inositol 1,4,5-triphosphate receptor in close association with mitochondria [7, 8]. Sig-1R stimulation increases release of the neurotransmitters dopamine and glutamate [9, 10]. However, mechanisms underlying these activities remain unclear.
Interestingly, restricted exposure of the hippocampus to X-irradiation blocks DG (dentate gyrus) neurogenesis and compromises the ability of anti-depressants to improve depressive behaviors [11]. Consistent with this observation, in post-mortem analysis of tissues from patients with major depressive disorders, chronic treatment with tricyclic anti-depressants (TCAs) such as imipramine increases the number of neural progenitor cells in the DG [12]. Treatment with selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine or fluvoxamine also improve impaired adult hippocampal neurogenesis in the rodent DG [11, 13]. The observations that these SSRIs and imipramine bind to Sig-1R [14] and that Sig-1R null mice exhibit depressive-like behaviors [15] suggest that Sig-1R stimulation mediates neurogenesis and improvement of depression following treatment with anti-depressants. Indeed, impaired depressive-like behaviors in olfactory bulbectomized (OVX) mice improve following chronic oral administration of dehydroepiandrosterone (DHEA), an endogenous Sig-1R ligand [16, 17].
Calcium/calmodulin-dependent protein kinase IV (CaMKIV ) is a serine-threonine protein kinase activated by nuclear Ca2+ elevation that catalyzes phosphorylation of the cyclic AMP-responsive element binding protein (CREB) at residue Ser-133 [18, 19]. In rodents, this modification regulates expression of several genes, including BDNF , that function in synaptic plasticity [20], learning and memory [21–23], and emotional behaviors [24–26]. CaMKIV is widely distributed in neurons in the anterior cingulate cortex, somatosensory cortex, insular cortex, cerebellum, hippocampus, and amygdala, where it is localized primarily to nuclei [27]. As shown in Fig. 14.1, in mouse hippocampus CaMKIV is expressed in immature neurons positive for PSA-NCAM (a marker of newly generated immature granule cells) and in neurons positive for calbindin, a marker of mature granule cells. CaMKIV is also expressed in radial glia and astrocytes labeled with anti-BLBP (brain lipid binding protein) [28]. Accumulating evidence demonstrates that CaMKIV null mice display deficits in contextual and cued fear conditioning memory [29] and a decrease in anxiety-like behaviors [29, 30]. Furthermore, treatment with the typical SSRI fluoxetine fails to induce DG neurogenesis and does not have an anti-depressive effect in CaMKIV null mice [31].

CaMKIV co-localizes with the neuronal markers PSA-NCAM and calbindin but not with the glial marker brain lipid binding protein (BLBP) in the dentate gyrus. Confocal microscopy images showing double immunofluorescence staining of the adult DG for CaMKIV and BLBP, PSA-NCAM or calbindin, as indicated. Merged images show nuclear staining with 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI) (blue) (Modified from Moriguchi et al. [28])
14.2 Critical Role for Sig1-R in Depression
The depressive-like behaviors shown by Sig-1R null mice [15, 32] are associated with impaired neurogenesis in the hippocampal DG [33]. Sig-1R null male mice show depressive behaviors and reduced hippocampal neurogenesis, phenotypes not seen in female mice [34]. Enhanced estradiol (E2) levels may account for the absence of depressive-like phenotypes in female Sig-1R nulls, as E2 deprivation by ovariectomy in female mice elicits depressive-like behaviors in Sig-1R null mice [34]. E2 administration to male Sig-1R null mice rescues depressive-like behaviors, and src-dependent NMDAR phosphorylation is associated with amelioration of depressive-like behaviors in male hippocampus [34]. These findings suggest overall that NMDAR activation by Sig-1R mediates E2-induced neurogenesis and amelioration of depressive-like behaviors, either directly or indirectly.
Such phenotypes have been confirmed by pharmacological experiments. Indeed, among antidepressants, fluvoxamine and sertraline show a high affinity for Sig-1R, while fluoxetine, citalopram, and imipramine show low [35]. Specifically, the order of affinity of SSRIs for Sig-1R is: fluvoxamine (K i = 36 nM) > sertraline (K i = 57 nM) > fluoxetine (K i = 120 nM) > citalopram (K i = 292 nM) > paroxetine (K i = 1893 nM) [35]. On the other hand, inhibitory constants (Ki) for inhibition of serotonin uptake into rat brain are: paroxetine (Ki = 0.7 nM) > citalopram (Ki = 2.6 nM) > sertraline (Ki = 3.4 nM) > fluvoxamine (Ki = 6.2 nM) > fluoxetine (Ki = 14 nM) [36]. Although Sig-1R is predominantly expressed in the mitochondrion-associated ER membrane (MAM) with the IP3 receptor, once Sig-1R binds ligand, it translocates to the plasma membrane, activating NMDAR and elevating Ca2+ at postsynaptic regions .
Interestingly, Sig-1R levels are relatively decreased in hippocampus of CaMKIV null mice, and fluvoxamine or SA4503 treatment rescues those levels and improves paroxetin-resistant depressive-like behaviors in CaMKIV mutant mice (Fig. 14.2). Sig-1R is highly expressed in astrocytes in the DG subgranular zone, a region stimulated with fluvoxamine or SA4503. SA4503 completely rescues impaired neurogenesis in CaMKIV null mice (Fig. 14.3) [28]. Likewise treatment with fluvoxamine or SA4503, but not paroxetine, also rescues reduced ATP production seen in hippocampus of CaMKIV null mice . This lack of effect by paroxetine suggests that Sig-1R stimulatory action rather than inhibition of serotonin reuptake is critical for fluvaxamine’s anti-depressive activity. However, lack of amelioration by fluoxetine as reported by Sha et al. [33] cannot be explained by low affinity for Sig-1R. The Sig-1R-specific agonist SA4503 ameliorates impaired adult hippocampal neurogenesis in DG and depressive behaviors in CaMKIV null mice [28]. However, mechanisms underlying depressive behaviors in CaMKIV mice are largely unknown, although reduced CREB/BDNF activity and impaired neurogenesis seen in these mice play a role. More importantly, decreased phosphorylation of CREB, Akt and CaMKII seen in CaMKIV null mice is restored by treatment with fluvoxamine or SA4503 .

Fluvoxamine or SA4503 treatment but not paroxetine rescues decreased Sig-1R expression and ATP production in the dentate gyrus of CaMKIV null mice. (a, b) Confocal microscopy images showing double staining for Sig-1R (green), PSD95 (a) or synaptophysin (b) (red) and merged images in hippocampal slices. Far right columns show high magnification images of boxed regions in the adjacent image. (c) Representative images of immunoblots using antibodies against Sig-1R and quantitative analyses. (d) Quantitative analyses of ATP production. Vertical lines show SEM (**, p < 0.01 versus wild-type mice. ++, p < 0.01 versus CaMKIV null mice. ##, p < 0.01 versus fluvoxamine -treated CaMKIV null mice.††, < 0.01 versus SA4503-treated CaMKIV null mice. Modified from Moriguchi et al. [28]

Fluvoxamine or SA4503 but not paroxetine enhances hippocampal neurogenesis in CaMKIV null mice. (a) Confocal microscopy images showing double staining for BrdU (green), NeuN (red) and merged images in hippocampal slices from wild-type mice, CaMKIV null mice, paroxetine-treated CaMKIV null mice, fluvoxamine -treated CaMKIV null mice, SA4503-treated CaMKIV null mice, NE100 (Sig-1R antagonist) plus fluvoxamine-treated CaMKIV null mice and NE100 plus SA4503-treated CaMKIV null mice. Mice were injected with BrdU on the first day of drug treatment and then for 5 consecutive days during the 2 weeks of drug treatment. Mice were treated with paroxetine, fluvoxamine, or SA4503 treatments for 2 weeks (n = 8). (b) Quantitative analyses of the number of BrdU/NeuN double-positive cells in the DG (n = 8). Vertical lines show SEM. **, p < 0.01 versus wild-type mice. ++, p < 0.01 versus CaMKIV null mice. ##, p < 0.01 versus fluvoxamine-treated CaMKIV null mice. ††, <0.01 versus SA4503-treated CaMKIV null mice (Modified from Moriguchi et al. [28])
14.3 CaMKII Activation by Sig-1R Stimulation
It is important to understand how CaMKII is activated by Sig-1R stimulation, as CaMKII autophosphorylation is closely associated with neuronal NMDAR activity. Chronic administration of a Sig-1R agonist is required for CaMKII activation in neurons [28] and Sig-1R activation potentiates NMDAR-mediated responses in neurons [37–41]. For example, Sig-1R stimulation increases the number of NMDARs expressed at the plasma membrane. In rats, 90 minutes after intraperitoneal administration of Sig-1R agonists such as (+)-SKF10, 047, PRE-084 or (+)-pentazocine , synthesis of the NMDAR subunit proteins GluN2A and GluN2B and the postsynaptic density protein 95 (PSD-95) is enhanced hippocampus, effects totally abolished by treatment with the protein synthesis inhibitor anisomycin [41]. Although mechanisms potentially stabilizing newly synthesized NMDARs by Sig-1R remain unclear, direct interaction of Sig-1R with NMDAR has been documented: Sig-1R directly interacts with the GluN1 subunit of NMDAR through its N-terminal region [42]. When Sig-1R-FLAG is coexpressed with either GluN1 or GluN2A in embryonic kidney tsA 201 cells, only GluN1 colocalizes with Sig-1R-FLAG. In addition, the Sig-1R agonist dehydroepiandrosterone (DHEA) stimulates protein kinase C activity and promotes phosphorylation of NMDAR at GluN1 (Ser-896) in olfactory bulbectomized (OBX) mice. Increased NMDAR phosphorylation levels are closely associated with CaMKII activation in OBX mice and reportedly improve memory deficits. DHEA is an abundant, endogenous neuroactive steroid that has anti-amnesic effects through Sig-1R stimulation [43]. Dehydroepiandrosterone sulfate (DHEAS) also stimulates phosphorylation of NMDAR at GluN1 (Ser-896) through Sig-1R stimulation in spinal cord, an event that mediates NMDA-induced pain behavior in mice [44]. Taken together, Sig-1R promotes stability and intracellular trafficking of NMDAR and increases its phosphorylation through protein kinase C, thereby stimulating CaMKII activity.
Although CaMKIV has been proposed to mediate CREB (Ser-133) phosphorylation, CaMKII primarily accounts for CREB phosphorylation and BDNF expression in CaMKIV null mice, an idea confirmed by the fact that expression of BDNF mRNA containing exons I or IV is upregulated in the DG of CaMKIV null mice by Sig-1R stimulation. Likewise, Sun et al. [45] reported that unlike CaMKIV , CaMKII regulates CREB activity through phosphorylation of CREB at residue Ser-142 (in addition to Ser-133). CaMKII overexpression increases levels of BDNF transcripts containing exon IV in NG108–15 cells [46]. NMDAR stimulation [9, 47, 48] and increases in ATP production [8] by Sig-1R ligands are two of the mechanisms underlying CaMKII activation in neurons. Increased ATP production enhances Ca2+ storage in the ER by stimulating the sarcoplasmic/endoplasmic Ca2+-ATPase (SERCA) pump, which can promote Ca2+-induced Ca2+-release from the ER and in turn activate neuronal CaMKII activity. The observation of depression -like behaviors in CaMKIV null mice is important, as those behaviors are closely associated with decreased neurogenesis in the hippocampal DG, and CaMKIV is expressed highly in pyramidal neurons in both CA1 and CA3 regions and in DG granule cells [28]. Like CaMKIV null mice, CaMKII α heterozygous knockout mice show increased numbers of immature granule cells in the hippocampal DG and a decreased number of mature granule cells [49]. Moreover, analysis proliferation by BrdU incorporation shows that the number of BrdU-positive cells slightly increases in CaMKII α heterozygous knockout mice [49]. Thus, both CaMKIV and CaMKII α likely function in proliferation and/or maturation of granule cells in the mouse DG.
14.4 Sig1-R Plays a Critical Role in BDNF Expression
Enhanced adult hippocampal neurogenesis is associated with activation of both PI3K/Akt [17, 50, 51] and CREB/BDNF pathways [17, 50]. Both pathways are essential for neuronal proliferation and maturation [52], and their activation by Sig-1R agonists may antagonize depressive behaviors. For example, stimulation of Sig-1R by fluvoxamine or SA4503 markedly activates PI3K/Akt and CREB/BDNF signaling in DG of CaMKIV null mice. Akt activation by fluvoxamine and SA4503 is also associated with tyrosine kinase signaling that promotes NMDAR activation [53] or NMDAR-dependent BDNF expression though CaMKII signaling [54]. In addition to CaMKII -dependent BDNF expression, chaperone activity is crucial for BDNF maturation and release of BDNF from neurons [55, 56]. In rat neuroblastoma B104 cells, SA4503 treatment increases the secretion of BDNF (pro plus mature BDNF ) [55]. Fujimoto et al. [55] have proposed that chronic treatment with SA4503 potentiates post-translational processing of BDNF by activating Sig-1R chaperone activity at the ER membrane.
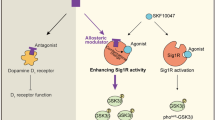
In addition, a link between Akt and CREB activities has been demonstrated in neural progenitor cells stimulated by fibroblast growth factor-2 (FGF-2), a factor is essential for proliferation of hippocampal progenitors [57]. FGF-2 and insulin-like growth factor-1 (IGF-1) also reportedly enhance proliferation of adult hippocampal neural progenitors [57]. Both mitogens stimulate Akt signaling [57]. In addition, conditional knockout of CREB in mice impairs in vivo proliferation of hippocampal neural progenitors [58]. Although the source of hippocampal FGF-2 and IGF-1 has not been defined, both mitogens are likely derived from astrocytes , based on studies of Shetty et al. [59]. In this context, our observation of immunohistochemical localization of Sig-1R in hippocampal astrocytes is particularly relevant. Cao et al. [60], using IP3 receptor type 2 transgenic mice, reported that ATP release from astrocytes is critical for anti-depressants to be effective. CaMKIV is not expressed astrocytes and co-localizes with PSA-NCAM and calbindin but not with BLBP in the DG [28]. We confirmed that CaMKIV is expressed in differentiating and mature dentate granule cells but not in neural stem cells or glial cells. Since Sig-1R is highly expressed in astrocytes of the subgranular zone and postsynaptically in CA1 and CA3 regions and its stimulation promotes hippocampal ATP production, Sig-1R stimulation of both astrocytes and postsynaptic neurons likely mediates Sig-1R stimulation-induced neurogenesis . A model of Sig-1R function in both neurogenesis and regulation of BDNF expression is shown in Fig. 14.4. Sig-1R stimulation by fluvoxamine or SA4503 promotes NMDAR function, increasing CaMKII activity. This in turn potentiates LTP through AMPAR phosphorylation and BDNF expression via CREB phosphorylation, even in the absence of CaMKIV activity. BDNF expression promotes increased Akt phosphorylation and neurogenesis. Sig-1R stimulation by fluvoxamine or SA4503 also enhances ATP production by enhancing mitochondrial Ca2+ entry. All of these activities likely antagonize depressive-like behaviors in rodent models.

Schematic representation of altered adult hippocampal neurogenesis in the DG. Sig-1R stimulation increases intracellular calcium mobilization through NMDARs in the plasma membrane or through the ER/SR via mitochondrial ATP production. Increased intracellular calcium increases CaMKII autophosphorylation and promotes CREB (Ser-133) phosphorylation and BDNF expression, which in turn increases Akt phosphorylation and promotes adult hippocampal neurogenesis (Modified from Moriguchi et al. [28])
14.5 Sig-1R Plays a Critical Role in Heart and Other Diseases
Depression is associated with substantial risk of developing heart failure and is independently associated with increased cardiovascular morbidity and mortality. Likewise, cardiovascular disease can lead severe depression . Thus, SSRI therapy has been strongly recommended to reduce cardiovascular disease-induced morbidity and mortality. We recently observed very high expression of Sig-1R in rat heart tissue [61] and determined that in rodent heart, the receptor is a direct target of SSRIs [62] and DHEA [63] in eliciting cardioprotection in both pressure overload (PO)-induced and transverse aortic constriction (TAC)-induced myocardial hypertrophy models. Our findings suggest that SSRIs such as fluvoxamine protect against PO- and TAC-induced cardiac dysfunction by upregulating Sig-1R expression and stimulating receptor-mediated Akt-eNOS signaling [63]. In addition, myocardial infarction with aortic banding elicits depressive-like behaviors in mice [64, 65]. Intracerebroventricular injection of the Sig-1R agonist PRE084 in myocardial infarction mice improved both depressive behaviors and cardiac dysfunction, with lowered sympathetic activity and recovery of Sig-1R expression in brain. Similarly, loss of Sig-1R activity mediates depressive-like behaviors in streptozotocin-induced diabetic rats [66]. The hypothalamic-pituitary-adrenal axis likely functions in perturbed central nervous system (CNS) activity mediated by Sig-1R loss in heart failure and diabetes. As yet, potential inflammatory cytokines or hormones that antagonize CNS Sig-1R signaling have not been identified. However, amelioration of depressive-like behaviors by Sig-1R agonists is particularly important for clinical therapeutics. In addition, the pathophysiological relevance of Sig-1R-mediated changes in ATP production remains unclear. To resolve the question, future studies should focus on development of the specific Sig-1R ligands useful in clinic settings.
Abbreviations
- Akt:
-
protein kinase B
- BDNF :
-
brain-derived neurotrophic factor
- BrdU:
-
bromodeoxyuridine
- CaMKII :
-
calcium/calmodulin-dependent protein kinase II
- CaMKIV :
-
calcium/calmodulin-dependent protein kinase IV
- CREB:
-
cAMP-responsive element binding protein
- DG:
-
dentate gyrus
- DHEA:
-
dehydroepiandrosterone
- ER/SR:
-
endoplasmic/sacroplasmic reticulum
- ERK:
-
extracellular signal-regulated kinase
- LTP:
-
long-term potentiation
- NMDAR:
-
N-methyl-D-aspartate receptor
- SERCA:
-
sarcoplasmic/endoplasmic Ca2+-ATPase
- Sig-1R:
-
sigma-1 receptor
- SSRIs:
-
selective serotonin reuptake inhibitors
References
Hanner M, Moebius FF, Flandorfer A et al (1996) Purification, molecular cloning, and expression of the mammalian σ1 binding site. Proc Natl Acad Sci U S A 93:8072–8077
Kekuda R, Prasad PD, Fei YJ et al (1996) Cloning and functional expression of the human type1 sigma receptor (hSigmaR1). Biochem Biophys Res Commun 229:553–558
Seth P, Fei YJ, Li HW et al (1998) Cloning and functional characterization of a σ receptor from rat brain. J Neurochem 70:922–931
Pan YX, Mey J, Xu J et al (1998) Cloning and characterization of a mouse σ1 receptor. J Neurochem 70:2279–2285
Hayashi T, Su TP (2004) Sigma-1 receptors at galactosylceramide-enriched lipid microdomains regulate oligodendrocyte differentiation. Proc Natl Acad Sci U S A 101:14949–14954
Palacios G, Muro A, Vela JM et al (2003) Immnohistochemical localization of the sigma1-receptor in oligodendrocytes in the rat central nervous system. Brain Res 961:92–99
Hayashi T, Maurice T, Su TP (2000) Ca2+ signaling via sigma1-receptors: novel regulatory mechanism affecting intracellular Ca2+ concentration. J Pharmacol Exp Ther 293:788–798
Shioda N, Ishikawa K, Tagashira H et al (2012) Expression of a truncated form of the endoplasmic reticulum chaperone protein, σ1 receptor, promotes mitochondrial energy depletion and apoptosis. J Biol Chem 287:23318–23331
Monnet FP, Debonnel G, Junien JL et al (1990) N-methyl-D- aspartate-induced neuronal activation is selectively modulated by sigma receptors. Eur J Pharmacol 179:441–445
Gonzalez-Alvear GM, Werling LL (1994) Regulation of [3H] dopamine release from rat striatal slices by sigma receptor ligands. J Pharmacol Exp Ther 271:212–219
Santarelli L, Saxe M, Gross C et al (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301(805):809
Boldrini M, Underwood MD, Hen R et al (2009) Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 34:2376–2389
Malberg JE, Duman RS (2003) Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology 28(1562):1571
Cobos EJ, Entrena JM, Nieto FR et al (2008) Pharmacology and therapeutic potential of sigma1 receptor ligands. Curr Neuropharmacol 6:344–366
Chevallier N, Keller E, Maurice T (2011) Behavioral phenotyping of knockout mice for the sigma-1 (σ1) chaperone protein revealed gender-related anxiety, depressive-like and memory alterations. J Psychopharmacol 25:960–975
Moriguchi S, Yamamoto Y, Ikuno T et al (2011) Sigma-1 receptor stimulation by dehydroepiandrosterone ameliorates cognitive impairment through activation of CaM kinase II, protein kinase C and extracellular signal-regulated kinase in olfactory bulbectomized mice. J Neurochem 117:879–891
Moriguchi S, Shinoda Y, Yamamoto Y et al (2013) Stimulation of sigma-1 receptor by DHEA enhances synaptic efficacy and neurogenesis in the hippocampal dentate gyrus of olfactory bulbectomized mice. PLoS ONE 8:e60863
Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca2+ and stimulus duration-dependent switch for hippocampal gene expression. Cell 87:1203–1214
Shaywitz AL, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68:821–861
West AE, Griffith EC, Greenberg ME (2002) Regulation of transcription factors by neuronal activity. Nat Rev Neurosci 3:921–931
Bourtchuladze R, Frenguelli B, Blendy J et al (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79:59–68
Josselyn SA, Shi C, Carlezon WAJ et al (2001) Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci 21:2404–2412
Impey S, Smith DM, Obrietan K et al (1998) Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci 1:595–601
Valverde O, Mantamadiotis T, Torrecilla M et al (2004) Modulation of anxiety-like behavior and morphine dependence in CREB-deficient mice. Neuropsychopharmacology 29:1122–1133
Maldonado R, Smadja C, Mazzucchelli M et al (1999) Altered emotional and locomotor responses in mice deficient in the transcription factor CREM. Proc Natl Acad Sci U S A 96:14094–14099
Barrot M, Olivier JD, Perrotti LI et al (2002) CREB activity in the nucleus accumbens shell controls gating of behavioral responses to stimuli. Proc Natl Acad Sci U S A 99:11435–11440
Ohmstede CA, Bland MM, Merrill BM et al (1991) Relationship of genes encoding Ca2+/calmodulin-dependent protein kinase Gr and calpermin: a gene within a page. Proc Natl Acad Sci U S A 88:5784–5788
Moriguchi S, Sakagami H, Yabuki Y et al (2015) Stimulation of sigma-1 receptor ameliorates depressive-like behaviors in CaMKIV null mice. Mol Neurobiol 52:1012–1222
Takao K, Tanda K, Nakamura K et al (2010) Comprehensive behavioral analysis of calcium/calmodulin-dependent protein kinase IV knockout mice. PLoS ONE 5:e9460
Shum FW, Ko SW, Lee YS et al (2005) Genetic alteration of anxiety and stress-like behavior in mice lacking CaMKIV. Mol Pain 1:22
Song N, Nakagawa S, Izumi T et al (2012) Involvement of CaMKIV in neurogenic effect with chronic fluoxetine treatment. Int J Neuropsychopharmacol 16:803–812
Sabino V, Cottone P, Parylak SL et al (2009) Sigma-1 receptor knockout mice display a depression-like phenotype. Behav Brain Res 198:472–476
Sha S, Qu WJ, Li L et al (2013) Sigma-1 receptor knockout impairs neurogenesis in dentate gyrus of adult hippocampus via down-regulation of NMDA receptors. CNS Neurosci Ther 19:705–713
Sha D, Hong J, Qu WJ et al (2015) Sex-related neurogenesis decrease in hippocampal dentate gyrus with depression-like behaviors in sigma-1 receptor knockout mice. Eur Neuropsychopharmacol 25:1275–1286
Narita N, Hashimoto K, Tomitaka S et al (1996) Interactions of selective serotonin reuptake inhibitors with subtypes of sigma receptors in rat brain. Eur J Pharmacol 307:117–119
Hiemke C, Hӓrtter S (2000) Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther 85:11–28
Monnet FP, Mahe V, Robel P et al (1995) Neurosteroids, via σ receptors, modulate the [3H] norepinephrine release evoked by N-methyl-D-aspartate in the rat hippocampus. Proc Natl Acad Sci U S A 92:3774–3778
Bergeron R, de Montigny C, Debonnel G (1997) Effect of short-term and long-term treatments with sigma ligands on the N-methyl-D-aspartate response in the CA3 region of the rat dorsal hippocampus. Br J Pharmacol 120:1351–1359
Debonnel G, Bergeron R, Monnet FP et al (1996) Differential effects of sigma ligands on the N-methyl-D-aspartate response in the CA1and CA3 regions of the dorsal hippocampus: effect of mossy fiber lesioning. Neuroscience 71:977–987
Martina M, Turcotte ME, Halman S et al (2007) The sigma-1 receptor modulates NMDA receptor synaptic transmission and plasticity via SK channels in rat hippocampus. J Philos 578:143–157
Pabba M, Wong AYC, Ahlskog N et al (2014) NMDA receptor are upregulated and trafficked to the plasma membrane after sigma-1 receptor activation in the rat hippocampus. J Neurosci 34:11325–11338
Balasuriya D, Stewart AP, Edwardson JM (2013) The s-1 receptor interacts directly with GluN1 but not GlunN2A in the GlunN1/Glun2A NMDA receptor. J Neurosci 33:18219–18224
Maurice T, Urani A, Phan VL et al (2001) The interaction between neuroactive steloids and the sigma1 receptor function: behavioral consequences and therapeutics opportunities. Brain Res Rev 37:116–132
Yoon SY, Roh DH, Seo HS et al (2010) An increase in spinal dehydroepiandrosterone sulfate (DHEAS) enhances NMDA-induced pain via phosphorylation of the NR1 subunit in mice: involvement of the sigma-1 receptor. Neuropharmacology 59:460–467
Sun P, Enslen H, Myung PS et al (1994) Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev 8:2527–2539
Takeuchi Y, Fukunaga K, Miyamoto E (2002) Activation of nuclear Ca2+/calmodulin-dependent protein kinase II and brain-derived neurotrophic factor gene expression by stimulation of dopamine D2 receptor in transfected NG108-15 cells. J Neurochem 82(2):316–328
Irwin RP, Lin SZ, Rogawski MA et al (1994) Steroid potentiation and inhibition of N-methyl-D-aspartate receptor-mediated intracellular Ca2+ response: structure-activity studies. J Pharmacol Exp Ther 271:677–682
Chen L, Miyamoto Y, Furuya K et al (2007) PREGS induces LTP in the hippocampal dentate gyrus of adult rats via the tyrosine phosphorylation of NR2B coupled to ERK/CREB signaling. J Neurophysiol 98:1538–1548
Yamasaki N, Maekawa M, Kobayashi K et al (2008) Alpha-CaMKII deficiency causes immature dentate gyrus, a novel candidate endophenotype of psychiatric disorders. Mol Brain 1:6
Wu H, Lu D, Jiang H et al (2008) Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma 25:130–139
Shioda N, Han F, Morioka M et al (2008) Bis(1-oxy-2-pyridinethiolato)oxovanadium(IV) enhances neurogenesis via phosphatidylinositol 3-kinase/Akt and extracellular signal regulated kinase activation in the hippocampal subgranular zone after mouse focal cerebral ischemia. Neuroscience 155:876–887
Li BS, Ma W, Zhang L et al (2001) Activation of phosphatidylinositol-3 kinase (PI-3 K) and extracellular regulated kinases (Erk1/2) is involved in muscarinic receptor-mediated DNA synthesis in neural progenitor cells. J Neurosci 21:1569–1579
Crossthwaite AJ, Valli H, Williams RJ (2004) Inhibiting Src family tyrosine kinase activity blocks glutamate signaling to ERK1/2 and Akt/PKB but not JNK in cultured striatal neurons. J Neurochem 88:1127–1139
Du J, Feng L, Yang F et al (2000) Activity- and Ca2+-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol 150:1423–1434
Fujimoto M, Hayashi T, Urfer R et al (2012) Sigma-1 receptor chaperones regulate the secretion of brain-derived neurotrophic factor. Synapse 66:630–639
Hayashi T (2015) Sigma-1 receptor: the novel intracellular target of neuropsychotherapeutic drugs. J Pharmacol Sci 127:2–5
Peltier J, O’Neill A, Schaffer DV (2007) PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol 67:1348–1361
Mantamadiotis T, Lemberger T, Bleckmann SC et al (2002) Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 31:47–54
Shetty AK, Hattiangady B, Shetty GA (2005) Stem/progenitor cell proliferation factors FGF-2, IGF-1, and VEGF exhibit early decline during the course of aging in the hippocampus: role of astrocytes. GLIA 51:173–186
Cao X, Li LP, Wang Q et al (2013) Astrocyte-derived ATP modulates depressive-like behaviors. Nat Med 19:773–777
Bhuiyan MS, Tagashira H, Shioda N et al (2010) Targeting sigma-1 receptor with fluvoxamine ameliorates pressure-overload-induced hypertrophy and dysfunctions. Expert Opin Ther Targets 14:1009–1022
Tagashira H, Bhuiyan S, Shioda N et al (2010) Sigma1-receptor stimulation with fluvoxamine ameliorates transverse aortic constriction-induced myocardial hypertrophy and dysfunction in mice. Am J Physiol Heart Circ Physiol 299:H1535–H1545
Bhuiyan MS, Tagashira H, Fukunaga K (2010) Dehydroepiandrosterone-mediated stimulation of sigma-1 receptor activates Akt-eNOS signaling in the thoracic aorta of ovariectomized rats with abdominal banding. Cardiovasc Ther 29:219–230
Ito K, Hirooka Y, Matsukawa R et al (2012) Decreased brain sigma-1 receptor contributes to the relationship between heart failure and depression. Cardiovasc Res 93:33–40
Ito K, Hirooka Y, Sunagawa K (2013) Brain sigma-1 receptor stimulation improves mental disorder and cardiac function in mice with myocardial infarction. J Cardiovasc Pharmacol 62:222–228
Lenart L, Hodreal J, Hosszu A et al (2016) The role of sigma-1 receptor and brain- derived neurotrophic factor in the development of diabetes and comorbid depression in streptozotocin-induced diabetic rats. Psychopharmacology. doi:10.1007/s00213-016-4209-x
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG (outside the USA)
About this chapter
Cite this chapter
Fukunaga, K., Moriguchi, S. (2017). Stimulation of the Sigma-1 Receptor and the Effects on Neurogenesis and Depressive Behaviors in Mice. In: Smith, S., Su, TP. (eds) Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Advances in Experimental Medicine and Biology, vol 964. Springer, Cham. https://doi.org/10.1007/978-3-319-50174-1_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-50174-1_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50172-7
Online ISBN: 978-3-319-50174-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)