
Abstract
Like in most other areas of cellular metabolism, the functions of the ubiquitin-like modifier SUMO in the maintenance of genome stability are manifold and varied. Perturbations of global sumoylation causes a wide spectrum of phenotypes associated with defects in DNA maintenance, such as hypersensitivity to DNA-damaging agents, gross chromosomal rearrangements and loss of entire chromosomes. Consistent with these observations, many key factors involved in various DNA repair pathways have been identified as SUMO substrates. However, establishing a functional connection between a given SUMO target, the cognate SUMO ligase and a relevant phenotype has remained a challenge, mainly because of the difficulties involved in identifying important modification sites and downstream effectors that specifically recognize the target in its sumoylated state. This review will give an overview over the major pathways of DNA repair and genome maintenance influenced by the SUMO system and discuss selected examples of SUMO’s actions in these pathways where the biological consequences of the modification have been elucidated.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- SUMO
- DNA repair
- Genome stability
- Homologous recombination
- Base excision repair
- DNA replication
- Telomeres
- Chromosome segregation
- Poly-SUMO chains
- SUMO-targeted ubiquitin ligases
1 Introduction
Our cells face the constant challenge of protecting their DNA from spontaneous and exogenous insults that include single- and double-strand breaks (DSB s), various base adducts (Lindahl 1993), replication blocks and topological stress . Dealing with these problems is essential for the maintenance of genome stability because mutations arising from unrepaired DNA can lead to loss or incorrect transmission of genetic information, which in turn can predispose to cancer (Hanahan and Weinberg 2011). Therefore, cells have evolved many mechanistically diverse DNA repair and genome maintenance pathways that are able to respond to damage rapidly, ensuring that mutations do not become fixed in the genome (Hoeijmakers 2001). One way of achieving this responsiveness is the activation or modulation of the properties of key DNA repair factors through post-translational modifications, which usually result in changes in their activities, localization or interactions with other cellular proteins (Huang and D’Andrea 2006).
Like other post-translational modifiers such as phosphate groups and ubiquitin , the ubiquitin-like protein SUMO is important for the maintenance of genome stability (Huang and D’Andrea 2006). On one hand, budding ( Saccharomyces cerevisiae ) and fission yeast (Schizosaccharomyces cerevisiae) mutants of the SUMO E1 , E2 and some E3 enzymes are hypersensitive to various DNA damaging agents, accumulate gross chromosomal rearrangements, lose mini-chromosome s frequently and fail to maintain telomere s and segregate chromatids properly (Tanaka et al. 1999; Ho and Watts 2003; Maeda et al. 2004; Xhemalce et al. 2004, 2007; Andrews et al. 2005; Zhao and Blobel 2005; Motegi et al. 2006; Takahashi et al. 2006; Watts et al. 2007). Altering the normal regulation of sumoylation is also detrimental to some pathways of DNA repair in human cells (Li et al. 2000; Potts and Yu 2005). On the other hand, proteins involved in many of the main DNA repair pathways have been shown to be sumoylated. These include components of nucleotide excision repair [e.g. XPC and XRCC4 (Wang et al. 2005; Yurchenko et al. 2006)], base excision repair [e.g. thymine DNA glycosylase, (Hardeland et al. 2002)], homologous recombination [HR , e.g. Rad52 , PCNA and the RECQ family of DNA helicases (Kawabe et al. 2000; Hoege et al. 2002; Eladad et al. 2005; Sacher et al. 2006)] and non-homologous end joining [NHEJ , e.g. Ku70 (Zhao and Blobel 2005)]. Sumoylation also modulates the functions of proteins that are not directly involved in DNA repair but that nonetheless play a role in preserving genome stability, such as DNA replication factors (Wei and Zhao 2016), topoisomerase II (Bachant et al. 2002) and many proteins essential to protect telomeres (Potts and Yu 2007).
Recent advances in mass spectrometry have revealed so many new sumoylation substrates that our insight into SUMO’s mechanism of action is lagging far behind the number of its known targets (Hendriks et al. 2014; Lamoliatte et al. 2014; Tammsalu et al. 2014). For some of the proteins mentioned above, however, we understand how SUMO alters their functions. Beginning with a brief overview over the components of the SUMO system relevant to genome maintenance, this review will highlight such cases, focusing on DNA replication, homologous recombination, base-excision repair, telomere maintenance, and chromosome segregation pathways. From these examples, it will become clear that whatever the downstream effect of sumoylation may be, it usually involves a change in the affinity of the modified proteins for either other proteins or DNA.
2 Components of the SUMO Pathway
Rather than giving a full account of the SUMO system here, the intention of this section is to provide a brief mechanistic overview over those features relevant for understanding SUMO metabolism and highlight those components that play prominent roles in genome maintenance.
2.1 SUMO Proteins
SUMO belongs to the family of ubiquitin -like modifiers, which share a common three-dimensional structure and a C-terminal di-glycine motif needed for attachment to a lysine residue via an isopeptide bond (van der Veen and Ploegh 2012). In contrast to other ubiquitin -like modifiers, SUMO possesses a long flexible N-terminal tail. While only one SUMO paralogue is present in budding or fission yeast (Smt 3 or Pmt3, respectively), human cells have four different SUMO isoforms, SUMO1–4. All are translated as longer precursors that need to be cleaved to obtain the corresponding mature forms. SUMO1 shares about 48% sequence identity with SUMO2, while SUMO2 and SUMO3 are highly similar with 95% sequence identity (Saitoh and Hinchey 2000). Therefore, these two isoforms , which are most closely related to the fungal proteins, cannot be distinguished via immune-staining and are usually referred to as SUMO2/3. SUMO4 seems to be processed to its mature form only under rare conditions and has so far only been described to be conjugated to other proteins in serum-starved cells (Wei et al. 2008).
Like ubiquitin , SUMO2/3, Smt 3 and Pmt3 can form polymeric chains on their substrates (Tatham et al. 2001; Bylebyl et al. 2003; Matic et al. 2008; Windecker and Ulrich 2008). These are predominantly linked via lysine residues in the N-terminal tail. SUMO chains play important roles in genome maintenance, as demonstrated by the fact that budding yeast mutants accumulating them show pleiotropic phenotypes, including hypersensitivity to genotoxins (Bylebyl et al. 2003). While mono-sumoylation can act antagonistically to ubiquitylation (Desterro et al. 1998), poly-SUMO chains can induce ubiquitylation and subsequent proteasome -mediated degradation, with important implications for genome stability (see Sect. 4.2.4).
2.2 SUMO Ligases
SUMO ligases boost the efficiency and determine the substrate specificity of sumoylation events mediated by the sole SUMO-specific E2 , UBC9 . The largest, most conserved category of SUMO E3s is the PIAS /SIZ family of proteins. In mammals it includes PIAS1, PIAS2 (PIASx), PIAS3 and PIAS4 (PIASy), which were initially described as protein inhibitors of the activated JAK-STAT signaling pathway. Fungal PIAS /SIZ proteins are Siz1 and Siz2 in budding yeast, and Pli1 in fission yeast. Both in vitro and in vivo, these enzymes show a significant amount of redundancy (Reindle et al. 2006). Mms21 (also known as Nse2 in fission yeast and NSMCE2 in humans) also contains an SP-RING domain , but does not strictly belong to the PIAS /SIZ family of proteins. This E3 will be discussed in detail in Sect. 4.5.1.
Structurally, PIAS /SIZ proteins share a modular architecture that consists of four domains. The N-terminal SAP domain interacts with DNA but is dispensable for catalytic activity (Okubo et al. 2004; Takahashi et al. 2005; Parker et al. 2008; Suzuki et al. 2009). The PINIT motif directly contacts certain sumoylation substrates and helps determine the selectivity for both the target protein and the target site (Takahashi et al. 2005; Yunus and Lima 2009). The SIZ/PIAS RING (SP-RING) finger harbors the catalytic activity (Kotaja et al. 2002; Takahashi et al. 2005; Yunus and Lima 2009). It resembles the RING finger of ubiquitin E3s but, unlike such folds, which sport two zinc-coordinating loops, the SP-RING domain contains only one. The second loop is instead held together by hydrogen bonds and Van der Waals forces (Duan et al. 2009; Yunus and Lima 2009). Like RING-type ubiquitin ligases, PIAS /SIZ proteins enhance sumoylation likely by facilitating the interaction between the SUMO-loaded E2 and its substrates. At their C-termini, PIAS /SIZ E3s contain a SUMO-interacting motif (SIM ). This motif is not essential for catalytic activity but promotes SUMO conjugation, probably by contacting the SUMO appendage of the charged UBC9 (Takahashi et al. 2005; Yunus and Lima 2009).
PIAS1 and PIAS4 play critical roles in the response to DNA DSB s. In human cells, these lesions trigger a cascade of events controlled by different types of post-translational modifications, such as phosphorylation and ubiquitylation, which leads to the formation of microscopically visible repair foci and culminates in the recruitment of the repair factor BRCA1 (Jackson and Bartek 2009). SUMO1, SUMO2/3, UBC9 , PIAS1, PIAS4 and MMS21 are all recruited to such DNA repair foci, and depleting PIAS1 or PIAS4, but not MMS21, obstructs their formation. However, PIAS1 and PIAS4 do not act redundantly. While PIAS4 is required for the recruitment of SUMO2/3, PIAS1 is necessary to recruit SUMO1, and each E3 appears to mediate the accumulation of a different set of additional signaling factors (Galanty et al. 2009; Morris et al. 2009). Consistent with a role in controlling DNA repair , depletion of PIAS1 or PIAS4 renders cells sensitive to various genotoxins and reduces their ability to mend DSBs by HR and NHEJ . The relevant substrates remain unclear, but one of them could be BRCA1 itself. Although depletion of PIAS1/4 also affects factors upstream of BRCA1 in the pathway, BRCA1 is modified by SUMO1 and SUMO2/3 in a PIAS1/4-dependent manner following exposure to genotoxic stress (Galanty et al. 2009; Morris et al. 2009). Sumoylation likely enhances BRCA1’s ubiquitin ligase activity, as mutating a sumoylation consensus motif within BRCA1 reduces the formation of K6-linked ubiquitin chains in vivo, a chain type characteristic for BRCA1 activity. Consistent with these results, in vitro sumoylation of BRCA1 enhances its activity by an order of magnitude (Morris et al. 2009).
2.3 SUMO Proteases
SUMO proteases catalyze both the maturation of SUMO and its deconjugation from target proteins. The largest category of SUMO proteases is the family of sentrin -specific proteases (SENPs ). It comprises two members in budding yeast, Ulp1 and Ulp2, and six members in mammalian cells, SENP1, −2, −3, −5, −6 and −7 (Hickey et al. 2012; Nayak and Muller 2014). These proteases have varying preferences for the different SUMO paralogues and chain lengths and exhibit distinctive localizations within the cell, which largely determines their substrate specificity. Ulp1 is targeted to the nuclear pore and processes a broad range of substrates. It is also responsible for the maturation of most of the SUMO translational fusions (Li and Hochstrasser 2003). Ulp2, on the other hand, is nucleoplasmic and has a strong preference for poly-sumoylated target proteins (Li and Hochstrasser 2000; Bylebyl et al. 2003). The mammalian homologue of Ulp2, SENP6, and its closest relative, SENP7, preferentially deconjugate SUMO chains in biochemical assays and are distributed throughout the nucleoplasm (Drag et al. 2008; Lima and Reverter 2008). SENP1 and SENP2 localize to the nuclear pore, and SENP3 and SENP5 show preferential retention at the nucleolus (Hang and Dasso 2002; Gong and Yeh 2006) .
2.4 SUMO-Targeted Ubiquitin Ligases
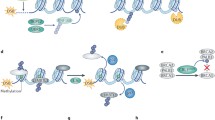
SUMO-targeted ubiquitin ligases (STUbLs) are ubiquitin E3s that recognize SUMO moieties via internal SIMs and thereby specifically ubiquitylate sumoylated proteins (Fig. 4.1). Three STUbLs have been described in yeasts. S. cerevisiae Uls1 is a large RING finger protein that binds to SUMO via four internal SIMs (Fig. 4.1a). Although uls1 mutant cells accumulate SUMO conjugates, efficient ubiquitylation of sumoylated proteins by Uls1 has so far not been validated biochemically (Hannich et al. 2005; Uzunova et al. 2007). Budding yeast Rad18 exhibits a highly specific STUbL-like function towards its substrate, PCNA , and will be discussed in detail in Sect. 4.5.4 (Parker and Ulrich 2012). The third and major STUbL, which is present in both budding and fission yeasts, is the RING E3 Slx 8 (Mullen et al. 2001; Ii et al. 2007a; Uzunova et al. 2007; Xie et al. 2007). Its RING domain forms an obligatory complex with a SUMO-binding subunit, Slx5 in budding yeast, and Rfp1 (or the redundant Rfp2) in fission yeast (Prudden et al. 2007; Sun et al. 2007; Uzunova et al. 2007; Xie et al. 2007; Mullen and Brill 2008). Slx 5’s preference for poly-SUMO chains (Uzunova et al. 2007) is likely shared by Rfp1 and Rfp2 because they, like Slx5, possess multiple SIMs (Prudden et al. 2007; Sun et al. 2007). Accordingly, Slx5/8 and Rfp1/2-Slx8 efficiently ubiquitylate a model substrate in vitro only if it is sumoylated (Sun et al. 2007; Mullen and Brill 2008). In vivo, the presence of proteins that are simultaneously sumoylated and ubiquitylated strictly depends on Slx 5/8 and the ability of SUMO to form chains (Uzunova et al. 2007).

Ubiquitylation of poly-sumoylated proteins via SUMO-targeted ubiquitin ligases . (a) Domain architecture of the SUMO-targeted ubiquitin ligases (STUbLs ) from yeast and humans. (b) In response to a signal, a poly-SUMO (S) chain may form on a certain sub-population of protein X. By means of its SIMs , the Slx 5/8 complex recognizes such poly-sumoylated substrate and ubiquitylates it (U). Following ubiquitylation, the modified substrate is degraded by the proteasome . Whether the SUMO moieties are degraded together with the substrate or deconjugation occurs before proteolysis is unknown. Adapted from Ulrich (2008). (c) RPA binds to ssDNA during replication. Under undisturbed replication conditions, RPA is kept in a hyposumoylated state through associating with the SUMO protease SENP6. After DNA double-strand formation during replication, SENP6 dissociates from the chromatin , allowing poly-sumoylation of RPA. On the one hand, this induces the recruitment of RAD51 to DNA DSB s. On the other hand, it promotes binding of RNF4 to the poly-SUMO chains via its internal SIMs and poly-ubiquitylation of RPA. The poly-ubiquitin chains serve as a signal for proteasomal degradation of RPA, allowing its replacement by Rad51 needed for repair of the DSBs via homologous recombination
In budding and fission yeasts, inactivating the Slx 8 complex results in increased levels of sumoylated species (Burgess et al. 2007; Ii et al. 2007b; Uzunova et al. 2007; Wang et al. 2006; Xie et al. 2007; Mullen and Brill 2008). A similar phenotype results from defects in ubiquitin conjugation or proteasome activity (Uzunova et al. 2007), indicating that Slx8 generally mediates the ubiquitin-dependent degradation of poly-sumoylated proteins (Uzunova et al. 2007, Fig. 4.1b). The ability of Slx 8-like complexes to modify sumoylated proteins and potentially target them for degradation is important for genome stability. In budding and fission yeasts, Slx 8-complex mutants are hypersensitive to various genotoxins such as hydroxyurea (HU) and methyl-methanesulfonate (MMS) (Mullen et al. 2001; Zhang et al. 2006; Kosoy et al. 2007; Ii et al. 2007b; Prudden et al. 2007; Sun et al. 2007; Xie et al. 2007; Mullen and Brill 2008). They also show a high incidence of events that may arise from the repair of spontaneous DSB s by HR , such as Rad52 foci during S phase, gross chromosomal rearrangements, gene conversion events and small point mutations (Zhang et al. 2006; Burgess et al. 2007; Prudden et al. 2007; Nagai et al. 2008).
It should be noted that the Slx 8 complex also comprises SUMO-independent functions as demonstrated for the transcriptional repressor Matɑ2, needed to control mating and differentiation. For example, Slx 5/8 was shown to ubiquitylate unmodified Matɑ2 and was able to trigger proteasomal turnover of Matɑ2 also in the absence of a functional sumoylation system (Xie et al. 2010).
Despite a striking difference in structure and size, the small mammalian RING finger protein RNF 4 can rescue the genome stability defects of Slx 5/8-deficient yeast cells, clearly demonstrating that the function of STUbLs is evolutionary conserved among eukaryotes (Prudden et al. 2007; Sun et al. 2007). In addition to its RING domain, RNF4 contains four SIMs (SIM1–4), which explains its preference for poly-SUMO chains (Tatham et al. 2008, Fig. 4.1a). SIM2 and SIM3 have been shown to be necessary and sufficient for the binding to chains of at least two SUMO moieties, while SIM4 only contributes to interactions with longer chains. SIM1, on the other hand, is likely irrelevant for SUMO binding, resulting in three functional SIMs in RNF4 (Keusekotten et al. 2014). Binding to SUMO chains induces homodimerization and thereby activation of RNF4, promoting the transfer of ubiquitin to the distal SUMO of the chain (Rojas-Fernandez et al. 2014; Xu et al. 2014).
RNF4 has been extensively characterized in the context of arsenic -induced degradation of PML and its oncogenic variant PML-RARɑ, a fusion protein expressed in acute promyelocytic leukemia (Lallemand-Breitenbach et al. 2008; Tatham et al. 2008; Weisshaar et al. 2008). In RNF4-depleted cells, sumoylated PML and mixed poly-ubiquitin and poly-SUMO chains highly accumulate in the nucleus, clearly indicating that RNF4 targets these substrates for degradation (Tatham et al. 2008). Other proteins that co-localize with PML in so-called PML bodies carry poly-SUMO chains, suggesting a more global function for RNF4 in PML body turnover. While these findings suggest that RNF4 mainly catalyzes the attachment of K48-linked ubiquitin chains for proteasomal degradation of target proteins, depletion of RNF4 has also been described to result in a decrease in K63 ubiquitin chains (Yin et al. 2012). In line with this finding, RNF4 can cooperate with the K63-specific ubiquitin E2 UBC13-UEV1 in vitro to poly-ubiquitylate an N-terminally mono-ubiquitylated SUMO2 (Tatham et al. 2013). Rather than leading to degradation, modification with these chains is generally assumed to facilitate complex assembly and signal transduction.
Apart from a striking increase in SUMO chains, RNF4-deficient cells as well as RNF4 knockout mice show increased sensitivity to a variety of DNA damaging agents (Luo et al. 2012; Yin et al. 2012; Vyas et al. 2013). Additionally, RNF4 −/− mice show impaired spermatogenesis , as described after depletion of other regulators of DSB repair. These studies implicate a key role for RNF4 in the assembly and disassembly of DNA repair complexes at the sites of DNA DSBs . Without RNF4, important DNA damage response factors such as MDC1, RNF8, 53BP1, and BRCA1 are still recruited to DSBs but their removal from repair foci is delayed (Luo et al. 2012; Yin et al. 2012).
A second mammalian STUbL, RNF111/Arkadia , has been identified by computational means (Sun and Hunter 2012). Similar to RNF4, Arkadia contains a RING domain and three clustered SIMs (Erker et al. 2013). While the SIMs do not seem to be required for Arkadia’s function in the TGF -β pathway, they are essential for its interaction with sumoylated PML after arsenic treatment. Indeed, sumoylated PML highly accumulates after depletion of Arkadia, suggesting a destabilizing effect of Arkadia on PML bodies similar to RNF4 (Erker et al. 2013). However, Arkadia and RNF4 do not form heterodimers, but seem to act independently on PML. Apart from its proteolytic activity towards PML, Arkadia catalyzes the formation of K63-linked ubiquitin chains on sumoylated XPC, a key regulator of nucleotide excision repair, after UV treatment. Ubiquitylation by Arkadia strongly induces XPC recruitment to UV lesions, revealing an essential non-proteolytic function for this STUbL in DNA repair (Poulsen et al. 2013).
2.5 SUMO-Targeted Ubiquitin Proteases
Given the importance of STUbLs , it is not surprising to find a class of proteases that catalyze the reverse reaction, i.e. the removal of ubiquitin from SUMO or sumoylated proteins. One such enzyme is Wss1, a metalloprotease from budding yeast. It was initially linked to SUMO by the observation that its deletion suppresses the phenotypes of a SUMO mutant, smt3–331 (Biggins et al. 2001). Indeed, Wss1 directly binds to SUMO and efficiently deubiquitylates ubiquitin-SUMO hybrid chains and also ubiquitin-SUMO fusion proteins in vitro. Although Wss1 also interacts with proteasomal subunits, it exhibits direct proteolytic activity towards poly-SUMO chains. In contrast, its activity on ubiquitylated substrates and poly-ubiquitin chains is not as pronounced. Hence, it is not entirely clear whether Wss1 acts as a SUMO protease or a SUMO-targeted ubiquitin protease (Mullen et al. 2010). Wss1 helps the formation of SUMO chains at sites of DNA damage through forming a complex with the ubiquitin-dependent protein segregase Cdc48/p97 and its adaptor Doa1. It then further promotes auto-cleavage and proteolytic degradation of associated proteins, resulting in extraction of sumoylated proteins from the chromatin (Balakirev et al. 2015).
In mammalian cells two potential SUMO-targeted ubiquitin proteases have been described, USP 11 and USP7. USP11 is a functional interactor of RNF4 and specifically deubiquitylates ubiquitin-SUMO hybrid chains. It also stabilizes PML bodies through deubiquitylation of sumoylated PML, thereby directly antagonizing RNF4 function (Wu et al. 2014; Hendriks et al. 2015b). Downregulation of USP11 expression confers PML destabilization and several malignant characteristics to a glioma tumor cell line, such as increased proliferation, invasiveness and tumor growth, implying that USP 11 might serve as an interesting novel cancer drug target (Wu et al. 2014).
USP7 deubiquitylates SUMO2 in vitro and in vivo and appears to specifically act on SUMO substrates at replication forks (Lecona et al. 2016). Inhibiting USP7 in cells increases the amount of sumoylated and ubiquitylated proteins on newly replicated chromatin, and additional inhibition of the Cdc48/p97 segregase further enhances this effect. This indicates that USP7 limits ubiquitylation of SUMO targets on chromatin, thus preventing their subsequent extraction by Cdc48/p97. In this way, USP7 appears to regulate the balance of post-translational modifications at and around replication forks that leads to a high concentration of SUMO at active forks, while ubiquitin conjugates dominate on mature chromatin (Lopez-Contreras et al. 2013). Although USP 7 is clearly important for efficient DNA replication , its exact substrates in this context remain unknown .
3 SUMO Proteomics
The number of proteomic screens for SUMO targets has greatly expanded within the last decade. Due to substantial methodological improvements in mass spectrometry , hundreds of SUMO target proteins and thousands of SUMO sites are known to date, many of which imply far-reaching consequences for genome maintenance pathways. One recurring problem in the identification of SUMO targets is the observation that the fraction of sumoylated protein for a given target is usually minute and can strongly vary between different cell cycle stages or cellular responses. Therefore, the sumoylated fraction often has to be enriched via appropriate treatment of the cells and subsequent affinity purification. In addition, SUMO proteases are highly potent and therefore have to be inactivated by working with denaturing buffers or specific protease inhibitors.
To be able to efficiently purify endogenous SUMO targets from a wide range of samples, such as patient material or rare tissues, monoclonal antibodies raised against human SUMO1 and SUMO2 have been used for immunoprecipitation (Barysch et al. 2014). So far, almost 600 human SUMO targets have been identified in this manner (Becker et al. 2013). An issue with this method is that it requires relatively large amounts of starting material. For a much more efficient purification of SUMO targets, several proteomic approaches made use of N-terminally tagged SUMO alleles exogenously expressed in cells. Altogether, more than 3000 human proteins have been reported to be sumoylated in such studies (Vertegaal et al. 2006; Schimmel et al. 2008; Golebiowski et al. 2009; Matic et al. 2010; Tatham et al. 2011; Hendriks et al. 2014; Impens et al. 2014; Lamoliatte et al. 2014; Schimmel et al. 2014; Schou et al. 2014; Bursomanno et al. 2015; Hendriks et al. 2015a; Sohn et al. 2015 ; Tammsalu et al. 2014, 2015; Xiao et al. 2015). However, this approach is usually restricted to cultured cells, and overexpression of SUMO might lead to false positive results. Therefore, potential SUMO targets identified in this manner should first be validated via purification of the endogenous proteins.
The identification of sumoylation sites is even more challenging, since it relies on the detection of a proteolytic remnant of SUMO’s C-terminus on the modified lysine residue, after digestion with a protease specific for basic amino acids. For ubiquitin targets, marked by a di-glycine remnant, this approach has been highly successful; however, the corresponding remnant of SUMO is too large to be efficiently identified via mass spectrometry . To overcome this difficulty, several approaches have made use of an additional proteolytic cleavage site introduced via point mutations close to SUMO’s C-terminus (Matic et al. 2010; Hendriks et al. 2014; Impens et al. 2014; Lamoliatte et al. 2014; Schimmel et al. 2014; Tammsalu et al. 2014, 2015; Bursomanno et al. 2015; Hendriks et al. 2015a; Sohn et al. 2015; Xiao et al. 2015). These mutant alleles can be exogenously expressed as tagged versions in the sample of interest. In this manner, more than 7000 SUMO sites have been reported under various conditions in human cells to date. Proteome-wide identification of SUMO sites under completely endogenous conditions, however, still remains unsolved.
Thanks to these developments, many new general characteristics of sumoylation have been revealed by analyzing the dynamics of the SUMO proteome during different cell cycle stages and in response to specific external stimuli (Golebiowski et al. 2009; Psakhye and Jentsch 2012; Hendriks et al. 2014; Schimmel et al. 2014; Cubenas-Potts et al. 2015; Hendriks et al. 2015c; Xiao et al. 2015). These studies strongly support the concept of SUMO group modification, i.e. the collective modification of an ensemble of functionally related proteins at their site of action (Johnson 2004; Matunis et al. 2006; Jentsch and Psakhye 2013). This concept, according to which SUMO acts as a “molecular glue” that promotes local protein-protein interactions in a relatively redundant manner, was first systematically substantiated for the HR -mediated repair of DSB s in yeast (Psakhye and Jentsch 2012), but appears to apply to other pathways relevant to genome maintenance, such as nucleotide excision repair and DNA replication , and may well turn out to be a common theme in protein sumoylation. In human cells, treatment with MMS not only triggers sumoylation of HR proteins, but also affects many chromatin remodelers and transcriptional regulators, suggesting an important role of sumoylation in changing chromatin dynamics and the transcriptional program in response to MMS (Hendriks et al. 2015c). Similarly, two proteomic screens that analyzed sumoylation after replication stress showed a dynamic sumoylation response on several components of the DNA replication machinery and on factors involved in DNA repair (Bursomanno et al. 2015; Xiao et al. 2015).
In conclusion, the recent advances in SUMO proteomics demonstrate that in contrast to those post-translational modifiers that mostly target specific proteins, SUMO can act on large protein complexes and functional networks to elicit a global cellular response to external stimuli.
4 Effects of SUMO on DNA Replication and Replication Stress
Accurate and complete DNA replication is essential for genome maintenance even in the absence of exogenous damage, as both over- and under-replication of the genome will inevitably lead to problems with subsequent chromosome segregation. Moreover, most types of DNA damage strongly interfere with the progression of replication forks. Hence, the response to replication stress appears to be a finely tuned reaction ranging from subtle effects that can be viewed as part of the normal replication process up to a full-blown damage response that follows from replication fork collapse and the emergence of replication-associated DSB s. The SUMO system has been shown to contribute to this process at several levels (reviewed by Garcia-Rodriguez et al. 2016).
4.1 SUMO in Replication Initiation
DNA replication initiates at characteristic sequences named origins of replication, which are marked as such by the association of the origin recognition complex (ORC). In preparation for replication, origins are primed for activation by the assembly of the pre-replicative (pre-RC) complex, which includes the hexameric MCM2–7 complex as a precursor of the replicative helicase . Conversion to the active helicase at the entry into S phase requires phosphorylation of the complex and association of additional subunits, Cdc45 and the GINS complex, which then allows DNA unwinding, recruitment of DNA polymerases and other accessory factors, and finally initiation of DNA synthesis (Fragkos et al. 2015). SUMO has recently been reported to exert a subtle, but measurable, negative influence on this process. In budding yeast, all subunits of the MCM complex are subject to sumoylation (Wei and Zhao 2016). Interestingly, different E3s appear to act on the various subunits, and the modification patterns vary somewhat over the cell cycle (de Albuquerque et al. 2016). Overall, sumoylation was shown to exhibit a pattern complementary to MCM phosphorylation, i.e. it was found highest at the pre-RC stage upon loading of the inactive complex onto DNA, and diminished in the course of S phase. Inhibition of one of the relevant kinases, DDK, or interference with origin firing by other means prevented desumoylation of the MCM complex. In contrast, local enhancement of sumoylation by means of tethering a strong SUMO-binding domain to Mcm6 compromised helicase activation and thus inhibited origin firing, likely via the SUMO-dependent recruitment of a phosphatase, Glc7. This enzyme appears to preferentially interact with the sumoylated form of Mcm6, thereby preventing essential phosphorylation events required for helicase activation (Wei and Zhao 2016). The significance of such a complex sumoylation pattern of the different MCM subunits for replication initiation is not yet understood, and apparently the inhibitory effect of SUMO cannot be ascribed to the modification of an individual subunit.
In vertebrate systems, a similar effect may apply, although it appears to be regulated in a different fashion. In Xenopus egg extracts, SUMO exerts a negative influence on replication initiation, as inhibition of sumoylation caused an increase in origin firing (Bonne-Andrea et al. 2013). Here, the sumoylation target responsible for the effect was cyclin E, which was modified upon its recruitment to pre-RCs. The mechanistic details of this phenomenon have not been elucidated, but its recurrence in different organisms suggests that SUMO may contribute to limiting excessive origin firing. In this context, it is interesting that many pre-RC components, including ORC subunits, have been found to be sumoylated (Golebiowski et al. 2009), thus possibly indicating a case of group sumoylation at replication origins.
4.2 SUMO at Replication Forks and in Replication Stress
In human cells, SUMO has been shown to be strongly enriched at replication forks (Lopez-Contreras et al. 2013), and in budding yeast, numerous components of the replication machinery are sumoylated, such as subunits of DNA polymerases , the replicative clamp loader and the Rad27 flap endonuclease (Cremona et al. 2012). The relevance of this enrichment is not entirely clear yet, but the maintenance of appropriate sumoylation levels appears to be important for efficient replication, given the actions of the human SUMO-targeted ubiquitin protease USP 7 at replication forks (Lecona et al. 2016). As described above (see Sect. 4.2.5), USP7 appears to counteract RNF4 , which would otherwise target sumoylated replication factors for ubiquitylation and extraction from chromatin by the ubiquitin -dependent chaperone Cdc48/p97. In budding yeast, the STUbL complex Slx 5/8 also appears to influence events at replication forks; however, here it seems more relevant as a response to fork damage. This is particularly important for refractive sequences such as CAG triplet repeats, which are prone to fragility and instability. During replication, these regions tend to localize to the nuclear periphery, where they have been suggested to undergo processing and fork restart by HR in a Slx 5/8-dependent manner (Su et al. 2015). This principle of damage re-localization not only applies to damaged replication forks, but also to DSB s (see Sect. 4.5).
A cross-talk between SUMO and ubiquitin is also observed in the Fanconi anemia (FA) pathway, a system for the resolution of replication fork problems as well as DNA interstrand crosslinks (reviewed by Walden and Deans 2014; Coleman and Huang 2016). The FA pathway coordinates the cooperation between components of different repair systems, involving nucleotide excision repair, HR , and translesion synthesis. FA pathway mutations are associated with a rare hereditary disease, Fanconi anemia, which is associated with bone marrow and congenital abnormalities as well as cancer predisposition (reviewed by Kee and D’Andrea 2012). Two components of this pathway, FANCI and FANCD2, form a heterodimer, the ID complex, which is loaded onto chromatin after stalling of replication forks. This is accompanied by several post-translational modification events, including phosphorylation and ubiquitylation, which facilitates the recruitment of downstream factors. After being loaded onto chromatin , both FANCI and FANCD2 are also sumoylated in a PIAS1/4-dependent manner. This promotes poly-ubiquitylation of the complex by RNF4 and subsequent extraction from the chromatin by Cdc48/p97 (Gibbs-Seymour et al. 2015). SENP6 antagonizes PIAS1/4-dependent sumoylation of FANCI and FANCD2, thus stabilizing the ID complex at stalled replication forks by abolishing RNF4-mediated ubiquitylation (Gibbs-Seymour et al. 2015).
Another sumoylation target within the FA pathway is FANCA, a subunit of the FA core complex, which acts as an ubiquitin ligase on the ID complex at stalled replication forks. A patient-derived point mutation in FANCA abolishes the interaction of this protein with another core complex subunit, FAAP20, and increases FANCA sumoylation (Xie et al. 2015). This in turn induces ubiquitylation of FANCA by RNF4 and subsequent proteasomal degradation, which prevents efficient execution of downstream events. Interestingly, not only the patient-derived mutant, but also wild-type (WT) FANCA, is sumoylated and targeted by RNF4, even though to a lesser extent, possibly suggesting that a regulated release of FANCA from the FA core complex is physiologically relevant. In conclusion, the extensive crosstalk between ubiquitylation and sumoylation fine-tunes the FA pathway at multiple levels.
5 Effects of SUMO on Homologous Recombination
HR involves the exchange or replacement of genetic information between homologous DNA regions, which is vital to repair DSB s and damaged replication forks, but also for the correct pairing and segregation of chromosomes during meiosis . When a DSB occurs, its ends are initially clipped by the MRX/MRN complex (Mre11-Rad50-Xrs2/Nbs1) and Sae1/CtIP and subsequently resected further by Exo1 and Sgs1 to produce 3′ single-stranded DNA (ssDNA) overhangs. This DNA is coated by the ssDNA-binding trimeric replication protein A (RPA, Rfa1–3), which is exchanged for Rad51 by means of Rad52 (or BRCA2 in vertebrates). The resulting Rad51-ssDNA filaments search DNA molecules for regions of homology. These are subsequently invaded by displacing the homologous strand. Following strand extension and capture of the second end, four-way DNA structures called Holliday junctions are generated, which migrate along the DNA to create extended heteroduplex regions. The junctions are eventually resolved by specific nucleases to yield two intact DNA molecules (reviewed by San Filippo et al. 2008). In addition to the proteins involved in the core pathway described above, additional factors can control when and where HR takes place. These factors include anti-recombinogenic helicases such as Sgs1 , Srs2 and Rrm3 in budding yeast and WRN, BLM and RECQ5 in mammals (Branzei and Foiani 2007; Bachrati and Hickson 2008).
Sumoylation plays important roles in controlling HR at several stages. It affects overall damage-induced recombination rates in mammalian and yeast cells (Li et al. 2000; Maeda et al. 2004), but it also controls the initial resection/clipping of DSB s (Cremona et al. 2012). SUMO targets many proteins with well-established roles in this repair pathway in both budding yeast and human cells, such as the MRN /MRX complex, Sae2, Rad52 , Rad59 and many more (Golebiowski et al. 2009; Cremona et al. 2012; Psakhye and Jentsch 2012). In response to DNA damage, several, although not all, of these proteins are synchronously sumoylated (Cremona et al. 2012; Psakhye and Jentsch 2012). This modification “wave” probably occurs due to the coordinated recruitment of multiple HR factors and a suitable SUMO E3 to DNA. On one hand, the process strictly depends on the resection of a DSB to ssDNA, which is necessary for HR proteins to accrue on damaged DNA. In fact, deleting mre11, exo1 or sgs1 significantly reduces the sumoylation of recombination factors, while mutations that accumulate unusually high amounts of ssDNA, such as cdc13 ts, enhance the modification. On the other hand, the coordinated sumoylation of recombination factors requires the SUMO E3 Siz2 and its recruitment to DNA. The latter is likely mediated by a combination of two features: a direct binding of Siz2 to DNA via its SAP domain and a SIM -mediated interaction of Siz2’s C-terminus with sumoylated Mre11. Although it remains to be determined whether the interaction between Siz2 and Mre11 actually depends on the sumoylation of Mre11 itself, this model would explain why deletion of MRE11, but not an allele encoding a catalytically inactive mutant, abolishes collective sumoylation of HR proteins (Cremona et al. 2012; Psakhye and Jentsch 2012).
Sumoylation apparently also influences where in the nucleus HR takes place. As described above for damaged replication forks, DSB s also re-localize to the nuclear envelope in budding yeast and cannot be efficiently processed in mutants where the integrity of the nuclear pore is compromised (Nagai et al. 2008). Recent findings demonstrate that the relocation of DSBs to the nuclear pore is dependent on poly-sumoylation mediated by the E3s Siz2 and Mms21 in G1 phase, which leads to the recruitment of Slx 5/8 to DSBs. This STUbL then promotes the relocation of lesions to the nuclear envelope (Horigome et al. 2016). Accordingly, Slx 5 colocalizes with Rad52 and Rad9 at repair foci in a SIM - and Slx 8-dependent manner (Cook et al. 2009). Interestingly, when Slx 5 is artificially targeted to undamaged DNA, it is sufficient to induce relocalization of these loci to nuclear pores, independently of previous sumoylation. While this essential function of Slx 5/8 seems to be specific for repair processes in G1 phase, DSBs arising in S phase appear to trigger Mms21 -dependent mono-sumoylation and subsequent re-localization to the nuclear periphery, but not the nuclear pore. In this case, association is mediated by the membrane protein Mps3 and is promoted by, but not dependent on, the presence of Slx 5 (Horigome et al. 2016). This finding might also explain why another study found that deletion of SLX8 does not affect the survival of cells where replication forks are transiently stalled or collapsed (Zhang et al. 2006).
Some aspects of SUMO with particular relevance to HR have been characterized in detail and will be discussed below: (1) the SUMO ligase activity of Mms21 , (2) the sumoylation of the ssDNA-binding RPA complex, (3) of the recombinase Rad52 , (4) of the eukaryotic DNA polymerase processivity factor PCNA , and (5) of the helicase Sgs1/BLM .
5.1 MMS21 -Dependent Sumoylation
Mms21 (also called Nse2 or NSMCE2) is part of an essential complex defined by two structural maintenance of chromosome (SMC) proteins, Smc5 and Smc6, and several non-SMC elements, called Nse1–6 in yeast (Stephan et al. 2011). In addition to Smc5/6, eukaryotes possess two additional SMC complexes: cohesin (Smc1/Smc3) and condensin (Smc2/Smc4). SMC proteins share a common structure that consists of a central coiled coil, which brings their globular N- and C-termini together to form an ATPase domain, and a hinge region that mediates heterodimerization. It is generally accepted that SMC heterodimers encircle DNA providing structural support to chromosomes and possibly targeting non-SMC partners to relevant loci (Lehmann et al. 1995; Fousteri and Lehmann 2000; Lehmann 2005; Zhao and Blobel 2005; Taylor et al. 2008; Uhlmann 2016).
Mms21 is essential in almost all species tested so far, except for Arabidopsis thaliana and chicken DT40 cells, where SMC5 itself is also dispensable (Giaever et al. 2002; McDonald et al. 2003; Huang et al. 2009; Kliszczak et al. 2012; Jacome et al. 2015). Mutating or removing Mms21’s catalytic domain is compatible with viability, but slows growth, sensitizes cells to various genotoxins and leads to increased levels of chromosome mis-segregation in both mitosis and meiosis , thus pointing to a specific role in genome maintenance (McDonald et al. 2003; Pebernard et al. 2004; Andrews et al. 2005; Potts and Yu 2005; Zhao and Blobel 2005; Behlke-Steinert et al. 2009; Rai et al. 2011; Xaver et al. 2013; Liu et al. 2014; Yuan et al. 2014).
In budding yeast, these phenotypes most probably derive from the formation of toxic sister chromatid junctions at damaged replication forks that likely represent HR intermediates (Branzei et al. 2006). Presently, the Mms21 targets responsible for these phenotypes have not been identified. In fission yeast, the processing of damaged replication forks also seems to involve Mms21-dependent sumoylation (Pebernard et al. 2008). In a mutant where replication forks are induced to irreversibly collapse, the Smc5/6 complex re-localizes to sub-telomeric regions, which are sequences particularly prone to fork stalling. A similar re-localization is observed in MMS-treated WT cells. Additionally, a functional Smc5/6 complex is required for efficient HR and the repair of collapsed replication forks and DSB s (Ampatzidou et al. 2006; Potts et al. 2006). These phenotypes closely resemble those seen for other Smc5/6 complex mutants, which indicates that Mms21, as an integral component of this complex, is required to prevent DNA damage or that its absence creates toxic DNA structures. Accordingly, a budding yeast mms21 mutant lacking the SP-RING domain not only shows a mild, but constitutive, activation of the DNA damage checkpoint, but it also requires a functional checkpoint to grow properly (Rai et al. 2011). Mms21 itself is phosphorylated upon activation of the S phase checkpoint during DNA replication . Inhibiting this modification causes a mild increase in the rate of chromosome loss after DNA damage and reduces sumoylation of Mms21 targets, suggesting that phosphorylation is required for full activation of this E3 (Carlborg et al. 2015).
Structural studies on budding yeast Mms21 show that it interacts with Smc5’s coiled-coil domain via its N-terminus (Duan et al. 2009; Duan et al. 2011), while its C-terminus contains the catalytic SP-RING finger . Disrupting the Mms21-Smc5 interaction recapitulates many of the defects observed in mms21 or Smc5/6 mutants, such as gross defects in chromosome segregation and reduced sumoylation of Mms21 targets (Bermudez-Lopez et al. 2015). A Smc5 mutant that proficiently binds to Mms21 and chromatin , but is defective in ATP binding, also impairs Mms21 ligase function. Considering that ATP binding seems to change the structure of the Smc5-Mms21 complex in vitro (Bermudez-Lopez et al. 2015), this suggests that an ATP-driven conformational change within the Smc5/6 complex could contribute to activating the E3 function of Mms21 (Bermudez-Lopez et al. 2015). The observation that SMC5 and NSE2 are epistatic in chicken DT40 cells with respect to DNA damage sensitivity supports this idea (Kliszczak et al. 2012).
Mms21 contributes to sumoylation of several proteins with known roles in DNA damage and repair, such as fission yeast Smc6, Nse3 and Nse4 (Andrews et al. 2005; Pebernard et al. 2008) and budding yeast Ku70, Smc5 and Bir1 (Zhao and Blobel 2005; Montpetit et al. 2006; Yong-Gonzales et al. 2012). In human cells, MMS21 also modifies SMC6, and several components of the telomeric shelterin complex (Potts and Yu 2005; Potts et al. 2006; 2007; see Sect. 4.7). Other prominent substrates of Mms21 include yeast and human cohesin subunits (Almedawar et al. 2012; McAleenan et al. 2012; Wu et al. 2012), as described in more detail below (see Sect. 4.7.1).
Although the consequences of Mms21-dependent sumoylation are often poorly understood, it appears that in many instances the sumoylated targets are subject to subsequent STUbL -mediated ubiquitylation and possibly proteasomal degradation. Accordingly, budding yeast Slx 5/8 was shown to act on many Mms21-dependent SUMO conjugates (Albuquerque et al. 2013). One of these is Bir1, a component of the chromosome passenger complex, which regulates several key mitotic events, including activation of the spindle assembly checkpoint (SAC; Carmena et al. 2012). Upon mild replicative stress induced by a dysfunctional allele of the replication factor Mcm10, deletion of SLX5 caused a SAC-mediated mitotic block and accumulation of sumoylated Bir1 (Thu et al. 2016). Moreover, inhibition of the proteasome led to a similar accumulation of Bir1 SUMO conjugates, consistent with a model where the joint action of Mms21 and the Slx 5/8 complex suppresses SAC activation via degradation of Bir1, thus allowing progression through mitosis in the presence of tolerable replicative stress (Thu et al. 2016).
5.2 Sumoylation of RPA
RPA serves as a platform for various ssDNA-associated protein complexes during a multitude of DNA transactions, including HR . The largest subunit of the human complex, RPA1 (RPA70), is sumoylated, but is kept in a hyposumoylated state during unperturbed S phase by means of a tight interaction with the SUMO protease SENP6 (Dou et al. 2010). In response to DSB s, SENP6 dissociates from RPA1, which thus becomes sumoylated at K449 and K577. This in turn leads to an increase in the number of HR events (Dou et al. 2010). On one hand, sumoylation of RPA1 boosts the interaction with RAD51 in vitro, suggesting that the modification could promote the assembly of the recombinogenic filament by means of enhancing the RPA1-RAD51 interaction (Dou et al. 2010). On the other hand, some of the consequences of RPA sumoylation may be mediated by RNF4 . This STUbL is essential for the removal of RPA1 from resected DNA to allow the subsequent loading of RAD51. Hence, formation of RAD51 repair foci is abolished and RPA1 association is prolonged in cells depleted of RNF4. Similarly, an unsumoylatable mutant of RPA1 remains associated with chromatin after DSB formation (Galanty et al. 2012). RPA1 interacts with RNF4 in a SIM -dependent manner, and association with the proteasomal subunit PSMD4 is observed after DNA damage in the presence of RNF4. Although biochemical evidence for a preferential action of RNF4 on sumoylated RPA1 is still needed, these results strongly suggest that the extraction of RPA1 from the chromatin is mediated by sumoylation-induced, RNF4-dependent, ubiquitylation and subsequent proteasomal degradation (Galanty et al. 2012). Taken together, both mechanisms, i.e. RPA’s induced binding to RAD51 and its extraction from damaged DNA after sumoylation, likely promote the formation of RAD51 filaments and might jointly facilitate DSB repair by HR (Fig. 4.1c).
5.3 Sumoylation of RAD52
Sumoylation of Rad52 is a widely conserved phenomenon observable in both budding and fission yeasts, Xenopus laevis egg extracts and human cells (Ho and Watts 2003; Leach and Michael 2005; Sacher et al. 2006; Ohuchi et al. 2008). However, the process is best understood in S. cerevisiae . Budding yeast Rad52 is sumoylated at K10, K11 and K220 both in vivo (via Siz2) and in vitro (in the absence of any E3). Whereas in vitro K220 is the predominant target and K10 and K11 appear to be modified as a consequence of K220 sumoylation, in vivo all three lysine residues are required for efficient sumoylation. This phenomenon may reflect the actions of an E3 in cells (Sacher et al. 2006). In vitro, sumoylation of Rad52 requires its C-terminal DNA-binding domain and is stimulated by naked or RPA -covered ssDNA, but not by Rad51 filaments (Altmannova et al. 2010). In vivo, the modification is boosted by DSBs induced during meiotic recombination or by DNA-damaging agents (Sacher et al. 2006). It also requires the presence of Mre11, but not its nuclease activity, and is enhanced by deleting RAD51, but not other factors involved in later steps of HR (Ohuchi et al. 2008). This suggests that Rad52 sumoylation may occur just before or at the time of Rad51 recruitment to a DSB . Artificially tethering Rad52 to DNA, via a sequence-specific DNA-binding domain, also promotes its sumoylation, even in the absence of exogenous DNA damage.
Functionally, sumoylation appears to mildly modulate the known properties of Rad52. In vitro, SUMO does not affect Rad52’s oligomerization state or its interaction with Rad51 or RPA , but it reduces its affinity for both ssDNA and double-stranded DNA (dsDNA), and it slightly impairs its ssDNA annealing activity. In vivo, cells that carry an unsumoylatable rad52 mutant (rad52 K10,11,220R) are not sensitive to DNA-damaging agents (Sacher et al. 2006; Silva et al. 2016) and are proficient in forming Rad52 foci as a mark of ongoing HR , albeit with a slightly reduced half-life and an altered distribution (Altmannova et al. 2010; Yong-Gonzales et al. 2012). Overall, Rad52 sumoylation appears to influence not so much the efficiency of HR , but rather the type of recombination pathway that is used for repair, i.e. the balance between single-stranded annealing, gene conversion and break-induced replication events. However, not all studies agree on the direction or magnitude of these phenotypes (Sacher et al. 2006; Ohuchi et al. 2008; Altmannova et al. 2010).
Sumoylation also appears to affect the stability of Rad52, but different studies report contrasting results. Sacher et al. (2006) report that SUMO protects Rad52 from accelerated proteasomal degradation. In contrast, Su et al. (2015) show that the STUbL Slx 5/8, which preferentially targets sumoylated Rad52 in vitro (Xie et al. 2007), promotes the degradation of a Rad52-SUMO fusion upon DNA damage in vivo. Moreover, slx8Δ is epistatic with the non-sumoylatable rad52 K10,11,220R mutant with respect to recombination rates at sequences that interfere with DNA replication (Su et al. 2015).
Although the majority of the phenotypes caused by preventing Rad52 sumoylation are minor, some are more obvious, and these relate to how Rad52 interacts with the anti-recombinogenic helicases Rrm3 and Srs2. While Srs2 acts globally, Rrm3 specifically prevents recombination and facilitates replication fork restart within the rDNA (Veaute et al. 2003; Torres et al. 2004a; Torres et al. 2004b). An rrm3∆ srs2∆ double mutant is inviable, but can be rescued by deleting RAD52, indicating that unrestrained recombination at the rDNA locus causes the lethality (Torres et al. 2004a; Sacher et al. 2006). Inhibiting Rad52 sumoylation also suppresses the inviability of rrm3∆ srs2∆ cells, suggesting that the modification may selectively affect the role of Rad52 in rDNA recombination (Sacher et al. 2006). Although the rad52 K10,11,220R mutant is proficient in rDNA recombination, it causes Rad52 foci to form within the nucleolus, while in WT cells Rad52 foci assemble outside of this compartment , possibly due to a transient re-localization of the break. These observations therefore suggest that Rad52 sumoylation could be critical to exclude the core HR machinery from the nucleolus. Hence, the rescue of the rrm3∆ srs2∆ mutant lethality by rad52 K10,11,220R may be due to a facilitated access of HR factors to the nucleolus, which might allow replication fork restart in the rDNA even in the absence of Rrm3 and Srs2 (Torres-Rosell et al. 2007). Presently, it is unknown how sumoylation affects Rad52’s accessibility to the nucleolus, but it may involve a SUMO-dependent change in interactions between Rad52 and its partners. Surprisingly, the consequences of altering Rad52 sumoylation for the single srs2Δ mutant are strikingly different from those observed for rrm3∆ srs2∆ cells: preventing Rad52 sumoylation slightly aggravates the damage sensitivity of the srs2Δ mutant, while a Rad52-SUMO fusion fully rescues it (Esta et al. 2013). Further evidence for a direct role of Rad52 sumoylation in controlling Srs2 functions is that overexpressing SIZ2, encoding Rad52’s cognate E3, also rescues the srs2Δ mutant phenotypes as long as Rad52 can be sumoylated (Esta et al. 2013). Given the well-established role of Srs2 in disassembling Rad51-ssDNA complexes, it is therefore likely that Rad52 sumoylation prevents the formation of excessive or defective nucleoprotein filaments by modulating the interactions of Rad52 with Rad51. The observations that Rad51 contains a SIM within its C-terminus that enhances its interaction with Rad52, and a Rad52-SUMO fusion protein binds to Rad51 somewhat better than unmodified Rad52 support this hypothesis (Bergink et al. 2013).
Rad52 sumoylation may not just control the properties of Rad51 filaments directly, but it could recruit other proteins to do so, such as Cdc48/p97 with its cofactors Ufd1-Npl4. This segregase is well-known for its roles in extracting proteins from complexes. It interacts preferentially with sumoylated Rad52 via SIMs in both Ufd1 and Cdc48, therefore suggesting that it could compete with Rad51 for binding to Rad52. Epistasis between a hypomorphic allele of cdc48 and a rad51 SIM mutant with respect to DNA damage sensitivity favors this model. Also, Cdc48/p97 can displace Rad51/Rad52 from DNA, and it does so more effectively when Rad52 is fused to SUMO (Bergink et al. 2013).
Identification of a robust function for Rad52 sumoylation has probably been hampered by the fact that it represents only one of many sumoylation events that coordinately target and therefore likely jointly regulate the HR pathway (see Sect. 4.3). Mechanistically, this phenomenon could involve SUMO acting as a “molecular glue” to control the interactions amongst the relevant proteins (Matunis et al. 2006). In fact, Rad52 preferentially interacts with the sumoylated forms of Rfa1, as part of RPA , and Rad59 (Psakhye and Jentsch 2012; Silva et al. 2016). Likewise, Rfa1 preferentially interacts with the sumoylated forms of Rad52 (Psakhye and Jentsch 2012). Fusing SUMO to either Rad52 or Rad59, to mimic constitutively modified versions of these proteins, also enhances their respective interactions, but, surprisingly, occluding their sumoylation does not appreciably inhibit it (Psakhye and Jentsch 2012; Silva et al. 2016). Phenotypically, mutations of the known sumoylation sites of RPA (in Rfa1, Rfa2 and Rfa3), Rad52, and Rad59 impair growth upon chronic exposure to MMS and, unlike the rad52 K10,11,220R single mutant, significantly reduce the rates of both spontaneous and damage-induced interchromosomal recombination (Psakhye and Jentsch 2012). As expected from the function of Siz2 in mediating bulk sumoylation of HR factors, siz2Δ is epistatic with the “SUMO-less” RPA/Rad52/Rad59 mutant (Psakhye and Jentsch 2012). Overall, these results suggest that the DNA damage-induced and -coordinated sumoylation of recombination factors, including Rad52, stabilizes the interactions amongst such proteins, promoting repair.
Like in yeast, human RAD52 is also sumoylated in vivo and in vitro, at K411 and K412, which are close to the C-terminus of this protein . In vitro, this modification does not affect Rad52’s binding to ssDNA or dsDNA, its ssDNA annealing activity or its interaction with Rad51. In vivo, mutating K411 and K412 to arginine restricts RAD52, which is normally a nuclear protein, to the cytoplasm . It remains to be determined whether this phenotype actually results from loss of sumoylation, or from a disruption of RAD52’s nuclear localization signal , which overlaps with K411 and K412 (Saito et al. 2010).
5.4 Sumoylation of PCNA
In budding yeast, the homotrimeric DNA polymerase processivity factor PCNA is sumoylated mainly at K164 by Siz1 , and to a lesser extent at K127 (Hoege et al. 2002; Stelter and Ulrich 2003). PCNA sumoylation normally takes place during S phase (Hoege et al. 2002), which is consistent with the observation that the protein is efficiently modified only when loaded onto DNA (Parker et al. 2008). In fact, the relative abundance of loaded and sumoylated PCNA suggests that a large proportion, if not all, of the loaded trimer could be modified during unperturbed replication (Parker et al. 2008). Yet, abolishing PCNA sumoylation does not compromise the replication process per se. PCNA is not only targeted by SUMO: in response to DNA damage it is also modified by mono- and poly-ubiquitin by a set of ubiquitin E2 s and E3s known as the RAD6 pathway (Hoege et al. 2002). PCNA mono-ubiquitylation at K164 by the E3 Rad18 promotes the bypass of DNA lesions by recruiting damage-tolerant polymerases to stalled replication forks, while poly-ubiquitylation activates a poorly understood error-free damage avoidance pathway that likely involves template switching (Ulrich 2005). Although they target the same site on PCNA, sumoylation does not seem to compete or act antagonistically with ubiquitylation. Instead, SUMO appears to channel damage processing away from HR into the RAD6 pathway via two cooperating mechanisms (Fig. 4.2).

SUMO modification of PCNA in budding yeast. During S phase, PCNA is loaded onto DNA, thus becoming a favorable substrate for sumoylation by Ubc9 and Siz1 . The resulting PCNA -SUMO conjugate recruits the Srs2 helicase to replication forks, where this helicase counteracts the accumulation of recombinogenic Rad51 filaments. Sumoylation of PCNA also contributes to the recruitment of Elg1, which facilitates the unloading of PCNA from the chromatin . After replication fork stalling due to DNA damage, the E2 -E3 complex Rad6-Rad18 associates preferentially with sumoylated PCNA through a SIM within Rad18. Rad6-Rad18-dependent ubiquitylation of PCNA then allows DNA damage bypass via translesion synthesis or template switching. (S) = SUMO, (U) = ubiquitin
The first clue to the roles of PCNA sumoylation counteracting HR came from the observation that an unsumoylatable PCNA mutant (pol30 K127,164R) or a deletion of SIZ1 strongly suppresses the DNA damage sensitivity of rad18Δ cells, where PCNA cannot be ubiquitylated (Papouli et al. 2005; Pfander et al. 2005). Interestingly, deleting SRS2 suppresses the rad18∆ phenotype to a similar extent (Lawrence and Christensen 1979), suggesting that Srs2 may act in the same pathway as PCNA sumoylation (Papouli et al. 2005; Pfander et al. 2005). In fact, deleting SRS2 in rad18∆ siz1 ∆ or rad18∆ pol30 K127,164R cells does not suppress their damage sensitivity any further, and the effects of both SRS2 and SIZ1 were found to depend on the presence of an intact HR pathway. This genetic relationship between PCNASUMO and SRS2 was elaborated mechanistically by showing that sumoylation enhances the affinity of Srs2 for PCNA both in vivo and in vitro, due to a tandem receptor motif consisting of a PCNA-interacting protein box (PIP-box) and a SUMO interaction motif (SIM ) at the C-terminus of Srs2 (Papouli et al. 2005; Pfander et al. 2005; Armstrong et al. 2012). These findings have given rise to a model whereby sumoylated PCNA recruits Srs2 to replication forks, where the helicase counteracts the assembly of Rad51 filaments onto DNA. Conversely, when PCNA cannot be sumoylated, Srs2 fails to associate with replication forks efficiently, resulting in an increased rate of sister chromatid recombination due to the elevated levels of Rad51 on DNA (Robert et al. 2006). In addition, Srs2 recruitment by PCNASUMO has been shown to induce the dissociation of Polδ and Polη from a recombination intermediate (Burkovics et al. 2013).
At the same time, the attachment of SUMO to PCNA greatly enhances the activity of the ubiquitin ligase Rad18 towards the DNA-bound clamp (Parker and Ulrich 2012, Fig. 4.2). This effect is attributable to a SIM in Rad18 and suggests that PCNASUMO is Rad18’s physiological substrate. Altogether these observations indicate that sumoylation of PCNA, by means of recruiting the anti-recombinogenic Srs2 and the ubiquitin E3 Rad18, may allow stalled replication forks to use PCNA ubiquitylation for damage bypass rather than the possibly deleterious recombination pathway (Papouli et al. 2005; Pfander et al. 2005; Parker and Ulrich 2012).
There is also good evidence for a more direct contribution of SUMO to the regulation of Srs2. Sumoylation of Srs2, mediated through the C-terminal SIM of this protein, has been described to inhibit the interaction with PCNASUMO, thus likely ensuring an appropriate balance between PCNA-bound and free Srs2 (Kolesar et al. 2012). Furthermore, the SUMO-like domain protein Esc2 can counteract the Srs2-mediated inhibition of HR by promoting Srs2 turnover on chromatin and thereby facilitating Rad51 recruitment (Urulangodi et al. 2015).
Sumoylation of PCNA is not limited to budding yeast, but has also been observed in Xenopus egg extracts, chicken DT40 B lymphocytes and more recently human cells (Leach and Michael 2005; Arakawa et al. 2006; Gali et al. 2012; Moldovan et al. 2012). Although SUMO-dependent Rad18 recruitment and stimulation of PCNA ubiquitylation do not appear to be conserved in vertebrates (Parker and Ulrich 2012), a potential homologue of Srs2 has been identified: similar to Srs2 in yeast, the protein PARI harbors a UvrD-like helicase domain, interacts with Rad51 and preferentially binds to a PCNA-SUMO1 fusion construct via a C-terminal PIP-box and a SIM . Depletion of PARI in U2OS or DT40 cells significantly stimulates HR rates, suggesting that vertebrate PARI acts like Srs2 in the suppression of inappropriate HR events (Moldovan et al. 2012). In accordance with these findings, overexpression of a PCNA-SUMO1 fusion inhibits recombination at stalled replication forks, and overexpression of sumoylation-deficient PCNA mutants induces DNA DSB s (Gali et al. 2012). The relevance of this mechanism in a physiological setting needs to be determined, however, as sumoylation of human PCNA has been detected only after overexpression of tagged SUMO1 in 293T or HeLa cells (Gali et al. 2012; Moldovan et al. 2012).
In addition to enhancing mono-ubiquitylation via Rad18 and controlling HR via Srs2, SUMO may affect other aspects of PCNA biology. The yeast protein Elg1 bears homology to the largest subunit of the replication factor C (RFC) complex, and it has been proposed to act as an unloader of PCNA in complex with the Rfc1–4 subunits (Kubota et al. 2013, Fig. 4.2). As a consequence, both sumoylated PCNA and Srs2 strongly accumulate on the chromatin of elg1 mutants. Similar to Srs2, Elg1 preferentially binds to sumoylated PCNA via two SIMs (Parnas et al. 2010); however, the Elg1 complex likewise acts on unmodified PCNA.
Finally, SUMO modification of PCNA can also interfere with the binding of interaction partners, such as Rfc1 and Eco1 (Moldovan et al. 2006). It has been noted that K127 is located within a region of PCNA that serves as its major partner-interaction site. Sumoylation of such residue might therefore compromise the association of PCNA with its partners, essentially acting as an “off-switch” to clear the clamp (Moldovan et al. 2006).
5.5 Sumoylation of SGS1/BLM
The budding yeast RecQ-like helicase Sgs1 is an important player in the repair of DSB s via HR . A sgs1 knockout mutant accumulates Rad51 foci at damaged replication forks, a phenotype that has similarly been described for a ubc9 mutant (Liberi et al. 2005; Branzei et al. 2006). This indicates that both sumoylation and Sgs1 functions are needed for the regulation of HR at damaged replication forks. Sgs1 has been identified as a SUMO target; however, this sumoylation event per se does not affect HR efficiency (Lu et al. 2010). Recently, the STUbL Slx 5/8 has been reported to interact with Sgs1 and to negatively affect the formation of Sgs1 foci after replication fork stalling, indicating that Sgs1 might be removed from damaged replication forks in a STUbL -dependent manner (Bohm et al. 2015). However, overall protein levels of Sgs1 after damage induction remain unaltered, suggesting that removal of Sgs1 does not involve proteasomal degradation. Interestingly, expression of the human STUbL RNF4 in a slx8Δ mutant background also reduces the formation of Sgs1 foci. A triple slx5Δ slx8Δ sgs1Δ mutant is synthetically lethal, but overexpression of the protease Wss1 in this background rescues this phenotype (Tong et al. 2001; Pan et al. 2006; Mullen et al. 2010). A wss1Δ sgs1Δ double mutant also exhibits synthetic lethality, suggesting that Slx 8 and Wss1 act synergistically in this pathway.
Accumulation of the human homologue of Sgs1, BLM, in repair foci is also diminished after depletion of RNF4 in human cells, clearly indicating that the phenomenon of STUbL -mediated modulation of RecQ-like helicases is conserved in evolution (Bohm et al. 2015). Accordingly, BLM is indeed sumoylated, and cells producing an unsumoylatable BLM mutant accumulate higher levels of DNA damage after replication fork stalling than those expressing the WT protein, indicating that the modification is important to resolve replication problems (Eladad et al. 2005). Abolishing sumoylation of BLM prevents the recruitment of RAD51 and subsequent HR at stalled replication forks. Indeed, RAD51 preferentially binds to sumoylated BLM in vitro, providing a first mechanistic explanation for how sumoylation of BLM may stimulate HR at stalled replication forks (Ouyang et al. 2009).
5.6 Sumoylation of Thymidine DNA Glycosylase in Base Excision Repair
Base excision repair processes a variety of chemical lesions inflicted on the nitrogenous bases of the DNA. It relies on several highly specialized glycosylases that recognize and cleave a narrow spectrum of damaged or modified bases. The resulting abasic sites, regardless of the enzyme that generated them, feed into a common core pathway that involves the initial displacement of the glycosylase from DNA and the nicking of the damaged duplex by the APE1 endonuclease. DNA polymerase β then removes the baseless sugar residue and polymerizes across the gap. Finally, the XRCC1-ligase III complex seals the nick in the DNA (reviewed by Memisoglu and Samson 2000; Barnes and Lindahl 2004).
Thymidine DNA glycosylase (TDG) is best known for its ability to protect DNA against C → T transitions by recognizing thymine or uracil within G•T and G•U mismatches arising from spontaneous deamination of 5-methyl-cytosine or cytosine, respectively (Barnes and Lindahl 2004). More recently, TDG has also been implicated in regulating DNA methylation. TDG actively demethylates DNA by excising 5-carboxylcytosine and 5-formylcytosine, the products of iterative oxidation of 5-methyl-cytosine by TET dioxygenases (Cortazar et al. 2011; Cortellino et al. 2011; He et al. 2011; Maiti and Drohat 2011; Kohli and Zhang 2013). This process is critical for the development of higher eukaryotes and could explain why, unlike other DNA glycosylases, TDG is essential for viability in mice (Hu et al. 2010).
Human TDG is sumoylated at K330 in vivo and in vitro. In vitro, TDG is preferentially modified by SUMO1, and to a lesser extent by SUMO2, under equivalent reaction conditions. This could be partly explained by the observation that TDG also non-covalently interacts with SUMO1 through a SIM at the C-terminus of the central catalytic core, and possibly a second one within the N-terminal regulatory domain (Hardeland et al. 2002; Baba et al. 2005; Steinacher and Schar 2005; Takahashi et al. 2005; Mohan et al. 2007; Smet-Nocca et al. 2011; Coey et al. 2014). Both covalent and non-covalent interactions influence the functions of TDG .
Initially, sumoylation was proposed to reduce TDG ’s affinity for DNA, thereby relieving the strong product inhibition exhibited by this enzyme and promoting catalytic turnover. The crystal structure of a central region of TDG conjugated to SUMO1 appears to support this model because it shows that the covalent and non-covalent interactions between SUMO and TDG may result in the protrusion of a helix from the surface of the glycosylase. When DNA is modeled into this structure, the protruding helix sterically clashes with the duplex, suggesting that a SUMO-induced conformational change may force the enzyme to dissociate from DNA (Baba et al. 2006, 2005). This conformational change does not strictly depend on the covalent modification of TDG, but can apparently also be triggered by the non-covalent binding of SUMO to the glycosylase (Smet-Nocca et al. 2011). An interaction between SUMO and the N-terminus of TDG , which is required for tight binding to G•T, may also be involved in this process because deleting this domain enhances TDG turnover in a way that is “epistatic” with SUMO modification. Consistently, early studies show that the N-terminus of TDG undergoes a conformational change in response to sumoylation of the enzyme (Steinacher and Schar 2005). More recently, however, NMR analysis reported no change in the structure of TDG’s N-terminal regulatory domain upon sumoylation. It rather seems that the interaction between SUMO and the C-terminal SIM of TDG competes with its regulatory domain for binding to the catalytic domain. Therefore, SUMO could dislodge the regulatory domain from the catalytic interface of TDG , leading to an extended conformation that is poised for catalysis (Smet-Nocca et al. 2011). Observations showing that sumoylation reduces the affinity of TDG for DNA and thereby stimulates its catalytic turnover also corroborated the above-described model (Hardeland et al. 2002). However, subsequent studies show that sumoylated TDG can still bind to DNA fairly tightly, albeit less so than the unmodified enzyme (Coey et al. 2014). In addition, in contrast to what the model described above would predict, DNA-bound TDG is not sumoylated more efficiently than the free enzyme, at least in vitro and in the absence of an E3 (Coey et al. 2014). Free SUMO can also boost the catalytic turnover of TDG in vitro in a SIM -independent manner, which possibly suggests a more indirect influence of SUMO on TDG activity (Smet-Nocca et al. 2011). In vivo, sumoylation does not appear to be important for TDG activity either, as neither preventing sumoylation nor disrupting non-covalent interactions with SUMO compromise TDG ’s ability to excise 5-carboxylcytosine. Likewise, overexpressing SUMO or altering the cellular sumoylation/desumoylation balance does not affect TDG’s in vivo activity (McLaughlin et al. 2016).
Given that APE1 can also relieve product inhibition of TDG (Waters et al. 1999; Fitzgerald and Drohat 2008; McLaughlin et al. 2016), it is conceivable that TDG sumoylation may actually regulate some other process, e.g. binding to other proteins. In fact, in exponentially growing cells TDG is found exclusively in the nucleus and is enriched within PML nuclear bodies. This localization relies on the interaction of TDG’s two SIMs with sumoylated PML (Takahashi et al. 2005; Mohan et al. 2007; McLaughlin et al. 2016). Consistently, TDG preferentially binds to sumoylated PML in vitro (Takahashi et al. 2005). This association appears to be incompatible with DNA binding (Mohan et al. 2007). As a consequence, deleting the DNA-binding N-terminus of TDG enhances co-localization with PML, probably by exposing TDG ’s SIMs (Mohan et al. 2007). At the same time, TDG sumoylation prevents the non-covalent, intermolecular interaction with a SUMO moiety on PML (Mohan et al. 2007). Taken together, these observations suggest that when TDG is released from DNA, it exposes its SIMs that would mediate its translocation to PML bodies unless the intermolecular interaction with sumoylated PML is prevented by sumoylation of TDG itself (Mohan et al. 2007). Why unsumoylated TDG localizes to PML bodies is unclear, but it may involve CBP/p300 , an acetyl-transferase responsible for the transcriptional activation of several genes in mammalian cells (Goodman and Smolik 2000). CBP/p300 can interact with and acetylate TDG (Tini et al. 2002), but only when the glycosylase is unmodified, suggesting that TDG localization to PML bodies may promote its acetylation (Mohan et al. 2007).
The STUbL RNF4 may also affect the functions of sumoylated TDG , as determined by work on DNA methylation. Overexpressing RNF4 reduces the methylation levels of a methylation-sensitive reporter promoter, leading to its activation. This effect requires both the SUMO-binding and ubiquitin ligase activities of RNF4, as well as TDG and APE1. Vice versa, RNF4−/− mouse embryonic fibroblasts (MEFs) show increased levels of global and locus-specific DNA methylation compared to WT cells. Via its SIM -containing N-terminal region, RNF4 physically interacts with TDG and APE1, synergizing with them in the activation of DNA demethylation (Hu et al. 2010). Therefore, RNF4 controls DNA demethylation via TDG/APE1 as a STUbL . It remains to be determined whether these functions actually depend on the sumoylation and subsequent ubiquitylation of TDG or on its SUMO-binding activity: although the interaction between RNF4 and TDG can be recapitulated in vitro, it does not apparently require, or is enhanced by, prior sumoylation of the glycosylase (Moriyama et al. 2014).
6 SUMO in the Maintenance of Telomere Function
Telomeres are structural elements at the ends of chromosomes that protect these from being recognized as DSB s and provide a solution to the end replication problem, which would otherwise cause a shortening of linear DNA molecules after every round of replication (Watson 1972; Verdun and Karlseder 2007; Arnoult and Karlseder 2015). Telomeric sequences consist of tandemly repeated dsDNA that terminates in a G-rich single-stranded 3′-overhang (Blackburn et al. 2015). They are covered by a group of proteins collectively called shelterin complex that, together with a range of accessory factors, controls telomere length and function (Fig. 4.3). Telomere length is maintained by an RNA -dependent DNA polymerase named telomerase, an enzyme that uses an RNA cofactor as a template to elongate telomeres (Autexier and Lue 2006). In the absence of telomerase, chromosome ends progressively shorten, leading to senescence and/or cell death. Hence, all immortal cell lines appear to have acquired some mechanism to maintain telomeres. Frequently, this involves re-expression of telomerase (Granger et al. 2002), but it is also possible by means of a mechanism called alternative lengthening of telomeres (ALT), as observed in a few cancer s. Although the exact molecular aspects of ALT remain unclear, increasing evidence suggests that this process involves some type of HR -mediated DNA replication that uses telomeric DNA, in cis or in trans, as a template (Pickett and Reddel 2015).

Telomere composition and contributions of the SUMO system in yeast and human cells. (a) In fission yeast, the dsDNA-binding protein Taz1 coats double-stranded telomeric repeats (Spink et al. 2000) and interacts with Rap1 and Poz1. Via Tpz1, Poz1 interacts with Pot1, which directly recognizes the 3′ telomeric overhang (Baumann and Cech 2001). (b) In budding yeast, dsDNA telomeric repeats are bound by Rap1, which interacts with Rif1/2, while the CST complex recognizes the 3′-overhangs via Cdc13. (c) Human telomere s harbor a set of proteins similar to those found in fission yeast, which are collectively called shelterin complex: TRF 1 and TRF2 (the orthologues of Taz1) bind to RAP1 and TIN2. In turn, TIN2 recognizes TPP1, which interacts with POT1 (de Lange 2005). In addition to shelterin, another complex is important for telomere regulation. It is called CST, after its constituent human proteins CTC1 (Cdc13 in budding yeast), STN1 and TEN1, and it is reminiscent of RPA. (S) = SUMO
SUMO plays an important role in telomere biology. Not only do telomeres become longer than usual in budding and fission yeast sumoylation mutants (Tanaka et al. 1999; Xhemalce et al. 2004; Zhao and Blobel 2005; Hang et al. 2011), but in S. cerevisiae compromising sumoylation causes cells to senesce more quickly than normal (Chavez et al. 2010). Conversely, senescent cells also show increased levels of total sumoylation (Chavez et al. 2010).
In both budding and fission yeast, sumoylation controls telomerase activity. Early studies hinted at this possibility by showing that the unusually long telomeres observed in S. pombe SUMO mutants probably originate from extension of the telomeric 3′-overhangs, a hallmark of uncontrolled telomerase activity (Xhemalce et al. 2007). The main player in this process appears to be the shelterin factor Tpz1. Tpz1 sumoylation peaks with telomere replication in late S phase and is catalyzed by the SUMO E3 Pli1 (Garg et al. 2014; Miyagawa et al. 2014). An unsumoylatable allele, tpz1 K242R does not affect shelterin stability but leads to longer-than-usual telomeres. This phenotype is suppressed by loss of telomerase activity but is not further enhanced by loss of pli1 or pmt3, indicating that Tpz1 likely is the main sumoylation target in telomere homeostasis in fission yeast, and that this process is mediated via telomerase. In fact, tpz1 K242R cells show both a loss of the telomerase-inhibitory complex Stn1/Ten1 from telomeres and an increased association of telomerase. Stn1 binds non-covalently and independently to both SUMO and Tpz1, resulting in a synergistic enhancement of binding to a covalent SUMO-Tpz1 fusion. These observations suggest a model where Tpz1 sumoylation prevents telomere elongation by recruiting Stn1/Ten1 to telomeres and thereby restraining telomerase activity (Garg et al. 2014; Miyagawa et al. 2014). In support of this model, fusing Stn1 to SUMO or even directly to Tpz1, to mimic a constitute interaction between these two proteins, suppresses the telomeric phenotypes of pmt3Δ cells (Miyagawa et al. 2014, Fig. 4.3a).
A surprisingly similar contribution of SUMO to telomere homeostasis is observed in budding yeast (Hang et al. 2011), although both the molecular mechanism and the key modification target are probably different. In S. cerevisiae , several telomeric proteins are sumoylated: yKu70, Rap1, the helicase Pif1 and the 3′ overhang binding protein Cdc13 (Zhao and Blobel 2005; Hang et al. 2011; Hang et al. 2014; Chymkowitch et al. 2015). Similar to sumoylation of Tpz1 in S. pombe , budding yeast Cdc13 sumoylation peaks in late S phase. The modification, catalyzed by Siz1 and Siz2, occurs at K909 within Cdc13’s Stn1-binding domain. Cells bearing an unsumoylatable cdc13 K909R allele have longer-than-usual telomeres but show normal levels of telomeric 3′-overhangs, indicating that chromosome end protection is unaffected. The cdc13 K909R allele is epistatic with siz1 /siz2 and stn1, strongly suggesting that Cdc13 is the main target of Siz1 /Siz2-dependent sumoylation in telomere control in budding yeast, and that the modification acts via Stn1. In fact, inhibiting Cdc13 sumoylation reduces the binding of Cdc13 to Stn1 in the yeast two-hybrid system, while a permanent fusion of SUMO to Cdc13 increases the interaction in vitro and leads to shorter telomeres in vivo. Given that the telomeric phenotype of cdc13 K909R cells depends on active telomerase and is suppressed by overexpressing STN1, Cdc13 sumoylation has been proposed to facilitate Stn1/Ten1 recruitment to telomeres (Hang et al. 2011). However, unlike fission yeast Stn1, budding yeast Stn1 does not appear to bind appreciably to SUMO; therefore, it is unlikely that it directly recognizes sumoylated Cdc13 (Hang et al. 2011). Rather, sumoylation may change other properties of Cdc13, such as its structure or DNA binding, which could ultimately facilitate association of Stn1 with telomeres (Fig. 4.3b).
In budding yeast, Siz2-dependent sumoylation appears to regulate telomerase activity by a second mechanism, involving telomere clustering (Ferreira et al. 2011; Ferreira et al. 2013). As in a number of other organisms (Ferreira et al. 2013), chromosome ends cluster at the nuclear envelope in S. cerevisiae . This effect is mediated by two pathways: one dependent on the partially redundant Sir2/3/4 proteins and the other on Yku70/80 (yeast Ku70/80; Hediger et al. 2002). Both Sir4 and Yku80, and to some extent Yku70, are sumoylated in a Siz2-dependent manner (Ferreira et al. 2011), and deletion of SIZ2 leads to a loss of telomere clustering. Both pathways appear to be affected, because siz2Δ cells are unable to tether a reporter locus to the nuclear envelope via either Yku70/80 or Sir4. Clustering can be rescued by permanently fusing SUMO to Yku70 or Yku80 to mimic a constitutively sumoylated Yku70/80 complex, indicating that sumoylated Yku70/80 directly controls telomere clustering. Sumoylation of Sir4 does not seem to be as critical. Telomere lengthening upon loss of siz2 is epistatic with deletion of PIF1, encoding a helicase with an inhibitory effect on telomerase. It has therefore been proposed that SUMO-dependent clustering of telomeres at the nuclear envelope may antagonize telomerase activity (Ferreira et al. 2011). Although interesting, definitive proof for this model will require a demonstration that the increase in telomere length observed in siz2Δ cells actually depends on, for example, Yku70 sumoylation, rather than another functionally analogous sumoylation event, such as the modification of Cdc13.
The roles of SUMO in telomere regulation go beyond controlling telomerase activity. For instance, sumoylation of the anti-recombinogenic helicase Sgs1 by Siz1 /Siz2 at K621 stimulates recombination between telomeres (Lu et al. 2010). In addition, sumoylation of Yku70 by Mms21 , which appears to be functionally distinct from that catalyzed by Siz2, may also play a role in this process. This modification event, which occurs at a cluster of lysines within the DNA-binding domain of Yku70, does not in fact act via telomerase (Hang et al. 2014). In addition, a yku70 mutant that shows reduced levels of Yku70 sumoylation has shortened, rather than elongated, telomeres and an increased length of telomeric 3′-overhangs. These results are consistent with the Yku70/80 complex’s well-established ability to inhibit end resection by DNA binding, thereby preventing HR and promoting NHEJ . Inhibiting sumoylation of Yku70 reduces its binding to DNA ends at both telomeres and DSB s and, consequently, has been reported to speed up resection of the latter. It follows that Yku70 sumoylation could facilitate the association of the Yku70/80 complex with DNA ends in general and thereby contribute to telomere maintenance (Hang et al. 2014). The exact mechanism of this process is unclear but could involve HR , because inhibiting Mms21 E3 activity boosts the levels of recombination intermediates at telomeres in senescing telomerase-deficient mutant yeast (Chavez et al. 2010).
SUMO’s roles in regulating NHEJ at budding yeast telomeres also involves STUbL s. Cells devoid of Uls1 show increased levels of chromosomal end-to-end fusions (Lescasse et al. 2013). This phenotype requires a functional NHEJ pathway and the ability to form poly-SUMO chains and is recapitulated by inhibiting Uls1’s ubiquitin E3 activity (Lescasse et al. 2013). This implies that Uls1’s poly-sumoylation-mediated ubiquitylation activity protects telomeres by inhibiting NHEJ at these loci. Consistently, Uls1 binds to telomeres. The relevant SUMO target in this pathway might be Rap1, as it was not only shown to be sumoylated, but its modification increases upon loss of uls1. Also, a rap1 K240,246R allele, which largely but not completely abolishes sumoylation, can partially suppress the end -to-end fusions observed in uls1 cells (Lescasse et al. 2013). Although it seems clear that Uls1-dependent degradation of Rap1 is important for inhibiting NHEJ at telomere fusions, the molecular details of this mechanism remain to be determined.
Another STUbL contributing to telomere function is the Slx 5/8 complex. Like human cells, budding yeast cells devoid of telomerase accumulate critically short telomeres and eventually undergo senescence . Under these conditions, eroded telomeres relocalize from their typical position around the nuclear envelope to the nuclear pore complex (Khadaroo et al. 2009). This accrual of dysfunctional telomeres is thought to facilitate HR amongst them, thereby allowing some cells to maintain telomeres without telomerase by a mechanism similar to ALT in human cells. These cells can escape senescence and are called “survivors”. Mutants devoid of STUbL activity (slx5Δ) poorly concentrate telomeres to the nuclear pore complex and also show reduced levels of survivors (Churikov et al. 2016). A similar, although not as penetrant, phenotype is observed in the siz1 siz2 mutant (Churikov et al. 2016), suggesting that Slx 5/8 controls recombination at dysfunctional telomeres by mechanisms that are dependent on its ability to ubiquitylate poly-sumoylated proteins. Accordingly, Slx 5/8 has been suggested to recruit telomeres to the nuclear pore complex, since it also associates with the latter (Nagai et al. 2008). This model appears, however, unlikely because artificially tethering a telomere to the nuclear pore complex cannot restore recombination at this locus in the slx5Δ mutant (Churikov et al. 2016). On the other hand, both SUMO and Slx 8 are recruited to eroding telomeres with similar kinetics (Churikov et al. 2016). The relevant sumoylated proteins on which Slx 5/8 acts in this process are unclear. RPA has been suggested as a candidate, because Rfa1 is sumoylated in response to telomere attrition, and deleting SLX5 also boosts this modification (Churikov et al. 2016). However, this scenario seems improbable because RPA is sumoylated not exclusively upon telomere erosion, but also in response to other kinds of DNA damage (see Sects. 4.5.2 and 4.5.3), all of which produce large amounts of ssDNA as repair intermediates. Therefore, ssDNA is the likely trigger for RPA sumoylation, rather than eroded telomeres per se.
Sumoylation plays an important role at human telomeres as well (Fig. 4.3c): several components of human shelterin are sumoylated. The main SUMO E3 involved in this process is the SMC5/6-associated ligase MMS21 , which appears to be of particular importance for the ALT pathway (Potts and Yu 2007). In ALT cells only, telomeres co-localize with specialized PML bodies containing many HR factors, suggesting that they could act as sites of telomere elongation (Yeager et al. 1999). This co-localization depends on both an intact SMC5/6 complex and MMS21 -dependent sumoylation. Consistently, MMS21 can sumoylate four (TRF 1, TRF2, TIN2 and RAP1) out of the six components of the shelterin complex. In ALT cells, over-producing unsumoylatable TRF1 or TRF 2 mutants reduces the recruitment of telomeres to PML bodies. Blocking shelterin sumoylation by silencing MMS21 also inhibits recombination between sister telomeres in these cells, possibly explaining why they develop progressively shorter telomeres and eventually senesce (Potts and Yu 2007). The functions of shelterin sumoylation could be mediated, at least partially, via SLX4, an interaction partner of TRF 2. SLX4 acts as a scaffold protein by interacting with several complexes involved in different DNA repair pathways, possibly orchestrating their activities. SLX4 contains three SIMs that allow it to interact preferentially with SUMO2/3 chains and have been shown to contribute to the SLX4-TRF2 interaction in ALT cells (Ouyang et al. 2015). Overall, these results suggest that sumoylation of telomeric factors, including but not limited to TRF 2, helps telomeres to associate with specialized PML bodies and SLX4 in ALT-positive cells.
TRF 2 (and TRF1) is also modified by PIAS1 with SUMO1 and SUMO2/3, but the relevant target lysines are different from those sumoylated by MMS21 (Her et al. 2015). The molecular consequences are also distinct. Poly-sumoylation of TRF2, together with its ability to physically interact with the STUbL RNF4, leads to its poly -ubiquitylation and subsequent proteasome -mediated degradation (Her et al. 2015). The exact downstream functions of this process are unknown, but they could be related to TRF 2’s roles in telomere protection because depleting RNF4 leads to telomere end-to-end fusions (Groocock et al. 2014).
7 Sumoylation in Chromosome Topology
Not only DNA repair pathways, but also the mechanisms that control chromosome segregation, contribute to genome maintenance, as errors in the distribution of the genetic material during cell division result in aneuploidy and thus genomic instability. Not surprisingly, SUMO plays a prominent role in this area as well. Accordingly, SUMO has been detected in association with essentially the entire chromosome segregation apparatus, i.e. centromeres and kinetochores , the chromosomal passenger complex and the mitotic spindle , and numerous components within these structures have been identified as SUMO targets by mass spectrometry in yeast and mammalian cells. The functions of SUMO in controlling chromosome segregation, especially in the context of centromere and kinetochore function, have been the subject of several excellent reviews (Dasso 2008; Wan et al. 2012) and are also covered in more detail elsewhere in this volume [see Chap. 9 ]. Here we will only discuss those aspects that directly impinge on the genetic material itself, namely DNA damage-induced cohesion and the processing of topological stress by means of topoisomerase II.
7.1 Sumoylation of Cohesin
In S. cerevisiae , both core components of the coh esin complex (Smc1 and Smc3) and its ancillary factors (Mdc1/Scc1, Scc3 and Pds5) are sumoylated (Almedawar et al. 2012). Although all three mitotic SUMO E3s (Siz1 , Siz2 and Mms21 ) contribute to modification of Scc1, Mms21 appears to be responsible for the bulk of it. During unchallenged growth, modification peaks at the beginning of S phase (Almedawar et al. 2012). As determined by genetically arresting cohesin at different stages of its cohesion cycle, modification occurs at a point between loading of cohesin onto chromatin and DNA entrapment (Almedawar et al. 2012). Functionally, SUMO regulates the core activities of cohesin. Overproducing Scc1 fused to the catalytic domain of Ulp1 (Scc1-Ulp1CD), as a means to reduce the sumoylation of adjoining proteins, is toxic to cells, but only when such an appendage is catalytically active. Although it is not possible to unambiguously ascribe this effect to the sumoylation of cohesion itself, as Ulp1CD could also promote the desumoylation of other proteins, it strongly suggests that SUMO is required to establish cohesion during unchallenged growth. In fact, cells carrying an unsumoylatable mutant of Scc1 experience defects in sister chromatid cohesion (McAleenan et al. 2012). Cohesin sumoylation is also induced by exposure to DNA damaging agents, such as MMS (McAleenan et al. 2012). Since nucleotide depletion by HU treatment does not induce this modification, it may be triggered by genuine DNA damage. In fact, a single DSB is sufficient to boost the reaction (McAleenan et al. 2012). Hence, SUMO appears to affect cohesin’s ability to facilitate DSB repair by promoting cohesion around them. Inhibiting the SUMO E3 activity of Mms21 , compromising the integrity of the Smc5/6 complex (by using the smc6–9 allele), or rendering Scc1 refractory to sumoylation reduces the recruitment of cohesin to a DSB and impairs the establishment of damage-induced cohesion at such lesions (McAleenan et al. 2012).
Human Scc1 is also sumoylated at several lysines by MMS21 but, unlike in yeast, this modification is not induced by DSBs (Wu et al. 2012). Although an unsumoylatable SCC1 mutant (SCC1K15R) can fully rescue the chromatid separation phenotype exhibited by SCC1-depleted cells, it cannot suppress the increased rate of sister chromatid exchanges that such cells experience, and it also sensitizes them to ionizing radiation (Wu et al. 2012). Also, in contrast to the yeast system, sumoylation is dispensable for recruiting human cohesin to DNA damage because SCC1 proficiently localizes to DNA lesions upon depletion of MMS21 or SMC5 (Wu et al. 2012). Instead, sumoylation of human SCC1 appears to exert its functions through the negative regulator of cohesion, WAPL. Knocking down this protein suppresses both the increased sensitivity to ionizing radiation and the defects in sister chromatid recombination caused by depleting MMS21 or rendering SCC1 unsumoylatable (Wu et al. 2012).
7.2 Sumoylation of Topoisomerase II
During DNA replication , the topological stress associated with opening the template DNA generates catenanes between homologous chromosomes (Lucas et al. 2001). Catenanes are inter-locked structures of two topologically constrained DNA molecules. They pose a serious problem to cells because they have to be resolved prior to chromosome segregation in order to avoid damage to or partial loss of the genome. Topoisomerase II (Topo II) resolves catenanes by covalently binding to both strands of a DNA helix and creating a DSB that is used as a gate to allow passage of a second duplex. It then reseals the initial break to avoid lasting damage (reviewed by Champoux 2001). Inhibiting Topo II results in metaphase arrest and extensive chromosome bridging, which likely arises from a defect in chromosome disjunction due to impaired decatenation (DiNardo et al. 1984; Downes et al. 1991; Shamu and Murray 1992).
Analysis of Topo II function, especially in higher eukaryotes, has relied on the use of pharmacologically relevant inhibitors such as ICRF-193, which traps the enzyme on its target DNA and prevents strand cleavage (Roca et al. 1994), and poisons such as etoposide, which stabilizes the covalent DNA-Topo II complex (Froelich-Ammon and Osheroff 1995). Topo II sumoylation is conserved from budding yeast to metazoans. Although the way in which this modification is regulated is analogous in the two systems, its functions appear to be distinct. A pivotal role for the centromere may in fact be the only similarity. In Xenopus egg, mouse and human cell extracts Topo II is preferentially sumoylated by SUMO2/3 in metaphase (Azuma et al. 2003; Agostinho et al. 2008; Dawlaty et al. 2008). PIAS4 facilitates this reaction in Xenopus egg extracts and possibly in some human cell lines (Azuma et al. 2005; Diaz-Martinez et al. 2006). In Xenopus egg extracts, depletion of PIAS4 eliminates nearly the entire pool of SUMO conjugates on mitotic chromosomes, demonstrating a general role for this SUMO ligase during mitotic progression (Azuma et al. 2005). Depletion of PIAS4 in HeLa cells blocks the localization of Topo II at centromeres and thereby faithful chromosomal segregation (Diaz-Martinez et al. 2006). Conversely, in MEFs , RanBP2 , but not PIAS4, promotes Topo II sumoylation (Dawlaty et al. 2008). Agostinho et al. (2008) also showed, by using chemicals that arrest Topo II at different catalytic stages, that the modification occurs at the covalent DNA-Topo II complex state, implying that sumoylation may depend on Topo II activity.
Regardless of the mechanism of regulation, PIAS4-depleted HeLa cells or Xenopus egg extracts and RanBP2 -deficient MEFs show a similar phenotype: chromosomes correctly condense, attach to the spindle and align onto the metaphase plate but then fail to disjoin properly (Azuma et al. 2005; Diaz-Martinez et al. 2006; Dawlaty et al. 2008). When anaphase naturally occurs or is induced, chromosomes show extensive bridging, and lagging chromosomes and mis-segregation frequently cause aneuploidy (Diaz-Martinez et al. 2006; Dawlaty et al. 2008). This phenotype is independent of cohesin because in cells co-depleted of PIAS 4 and the cohesin guardian hSGO1 chromosomes are still unable to separate (Diaz-Martinez et al. 2006). Such studies demonstrate that in the absence of PIAS4/RanBP2 -mediated sumoylation, chromosome segregation is impaired most likely because of an unusually high level of catenation. This phenotype in turn suggests that Topo II function is compromised in the absence of sumoylation. In support of this view, Topo II cannot localize to the axial core of chromosomes and centromer es in both PIAS4-depleted Xenopus egg extracts and HeLa cells and in RanBP2 -deficient MEFs (Azuma et al. 2005; Diaz-Martinez et al. 2006; Agostinho et al. 2008; Dawlaty et al. 2008). In the latter system, an over-produced SUMO-Topo II fusion protein localizes to centromeres and prevents chromosome bridging, suggesting that Topo II is indeed the relevant substrate whose sumoylation is required for efficient chromosome decatenation (Dawlaty et al. 2008). Interestingly, sumoylation of Topo II strongly inhibits decatenation activity in vitro (Ryu et al. 2010). Using a mass spectrometric approach, X. laevis Topo II has been found to be sumoylated within its DNA-binding domain at K660. Modification is enhanced by DNA binding. Mutation of K660 abolishes the inhibitory effect on decatenation, suggesting that SUMO specifically modifies active Topo II on centromeric DNA and thereby regulates decatenation activity (Ryu et al. 2010). Additional SUMO sites at the C-terminus of Topo II have no influence on the decatenation activity, but facilitate its interaction with other proteins, such as Claspin , an essential regulator of checkpoint arrest (Ryu et al. 2015). Sumoylation of Topo II and SIMs within Claspin are needed for the efficient recruitment of Claspin to centromeric DNA. Thus, sumoylation has different effects on the functionality and interactome of Topo II.
In budding yeast, the roles of Topo II sumoylation are somewhat different. Siz1 and Siz2 mediate the modification of the enzyme at K1220, K1246 and K1277 during metaphase (Bachant et al. 2002; Takahashi et al. 2006). A yeast strain carrying an unsumoylatable Topo II mutant (top2 K1220,1246,1277R) grows normally and shows only a mild chromosomal phenotype. This includes low levels of bridged chromosomes (Bachant et al. 2002), which possibly explains why top2 K1220,1246,1277R cells lose artificial mini-chromosomes at a slightly higher rate than the WT (Takahashi et al. 2006). The most notable phenotype of the top2 K1220,1246,1277R mutant is an increased stretching of centromeres during precocious separation of chromatids (Warsi et al. 2008). In budding yeast, as homologues pair during metaphase, they separate and recoil over a ~10 kbp region surrounding the centromere (Goshima and Yanagida 2000). It has been proposed that cells may use this mechanism to sense and control for correct tension at kinetochores and bi-orientation of homologous chromosomes prior to anaphase (Yeh et al. 2008). The top2 K1220,1246,1277R allele is also able to stabilize spindle attachment in a genetic background where kinetochores are weakened. Both of these phenotypes occur in the absence of catenation (Warsi et al. 2008). Thus, these observations indicate that in budding yeast sumoylation impinges on a new role of Topo II in the maintenance of centromere compaction and separation that is independent of its function in decatenation.
8 Conclusion
In this chapter we have reviewed the contributions of the SUMO system to several aspects of genome stability, including homologous recombination , base excision repair , telomere maintenance and chromosome segregation. On one hand, these examples illustrate the diversity of the effects that the modifier can exert on cellular metabolism. Here, the pervasive influence of a few key factors, such as the SUMO-targeted ubiquitin ligases , throughout the various pathways of genome maintenance is particularly noteworthy. On the other hand, the cases discussed here exemplify two distinct mechanistic principles in the action of sumoylation: by becoming attached to a substrate, the modifier can either create a binding surface for a particular downstream effector that recognizes SUMO through a dedicated interaction motif (SIM ), as observed in the recruitment of Srs2 through sumoylated PCNA or the intricate modulation of inter- and intramolecular interactions involving human TDG . Alternatively, there are many examples of SUMO targets for which the consequences of the modification are much less well understood because of the difficulties in identifying the modification sites and/or relevant SUMO-interacting proteins. Here, a clear-cut one-to-one relationship between a SUMO target and its downstream effector may not even apply. This appears to be particularly relevant for those SUMO substrates that are part of multi-protein complexes or macromolecular assemblies where more than one subunit is modified, such as the nuclear PML bodies or the chromatin -associated HR machinery. In these cases, SUMO is believed to either maintain the structural integrity of a subcellular compartment via multiple covalent and non-covalent interactions among the different components or mediate effective recruitment and complex formation of an ensemble of factors involved in a common process, thus acting as a “molecular glue” in a relatively substrate-independent fashion. Therefore, in the future it will be necessary to differentiate between the target-specific and the bulk interaction effects of sumoylation on a case-by-case basis. Last, but not least, a better understanding of how SUMO protects DNA will require assigning the phenotypes exhibited by global sumoylation defects to the relevant target proteins.
Abbreviations
- ALT:
-
alternative lengthening of telomeres
- DSB:
-
double-strand break
- dsDNA:
-
double-stranded DNA
- E1 :
-
SUMO (or ubiquitin) activating enzyme
- E2 :
-
SUMO (or ubiquitin) conjugating enzyme
- E3:
-
SUMO (or ubiquitin) protein ligase
- FA:
-
Fanconi anemia
- HR :
-
homologous recombination
- HU:
-
hydroxyurea
- MEF :
-
mouse embryonic fibroblast
- MMS:
-
methyl methanesulfonate
- NHEJ:
-
non-homologous end joining
- ORC:
-
origin recognition complex
- PIP:
-
PCNA-interacting protein
- pre-RC:
-
pre-replication complex
- rDNA:
-
ribosomal DNA
- RFC:
-
replication factor C
- RPA:
-
replication protein A
- SAC:
-
spindle assembly checkpoint
- SENP :
-
sentrin -specific protease
- SIM:
-
SUMO-interacting motif
- SMC:
-
structural maintenance of chromosomes
- SP-RING:
-
Siz/PIAS really interesting new gene
- ssDNA:
-
single-stranded DNA
- STUbL:
-
SUMO-targeted ubiquitin ligase
- WT:
-
wild-type
References
Agostinho M, Santos V, Ferreira F, Costa R, Cardoso J, Pinheiro I, Rino J, Jaffray E, Hay RT, Ferreira J (2008) Conjugation of human topoisomerase 2 alpha with small ubiquitin-like modifiers 2/3 in response to topoisomerase inhibitors: cell cycle stage and chromosome domain specificity. Cancer Res 68:2409–2418
Albuquerque CP, Wang G, Lee NS, Kolodner RD, Putnam CD, Zhou H (2013) Distinct SUMO ligases cooperate with Esc2 and Slx5 to suppress duplication-mediated genome rearrangements. PLoS Genet 9:e1003670
Almedawar S, Colomina N, Bermudez-Lopez M, Pocino-Merino I, Torres-Rosell J (2012) A SUMO-dependent step during establishment of sister chromatid cohesion. Curr Biol 22:1576–1581
Altmannova V, Eckert-Boulet N, Arneric M, Kolesar P, Chaloupkova R, Damborsky J, Sung P, Zhao X, Lisby M, Krejci L (2010) Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res 38:4708–4721
Ampatzidou E, Irmisch A, O’Connell MJ, Murray JM (2006) Smc5/6 is required for repair at collapsed replication forks. Mol Cell Biol 26:9387–9401
Andrews EA, Palecek J, Sergeant J, Taylor E, Lehmann AR, Watts FZ (2005) Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol Cell Biol 25:185–196
Arakawa H, Moldovan GL, Saribasak H, Saribasak NN, Jentsch S, Buerstedde JM (2006) A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol 4:e366
Armstrong AA, Mohideen F, Lima CD (2012) Recognition of SUMO-modified PCNA requires tandem receptor motifs in Srs2. Nature 483:59–63
Arnoult N, Karlseder J (2015) Complex interactions between the DNA-damage response and mammalian telomeres. Nat Struct Mol Biol 22:859–866
Autexier C, Lue NF (2006) The structure and function of telomerase reverse transcriptase. Annu Rev Biochem 75:493–517
Azuma Y, Arnaoutov A, Dasso M (2003) SUMO-2/3 regulates topoisomerase II in mitosis. J Cell Biol 163:477–487
Azuma Y, Arnaoutov A, Anan T, Dasso M (2005) PIASy mediates SUMO-2 conjugation of Topoisomerase-II on mitotic chromosomes. EMBO J 24:2172–2182
Baba D, Maita N, Jee JG, Uchimura Y, Saitoh H, Sugasawa K, Hanaoka F, Tochio H, Hiroaki H, Shirakawa M (2005) Crystal structure of thymine DNA glycosylase conjugated to SUMO-1. Nature 435:979–982
Baba D, Maita N, Jee JG, Uchimura Y, Saitoh H, Sugasawa K, Hanaoka F, Tochio H, Hiroaki H, Shirakawa M (2006) Crystal structure of SUMO-3-modified thymine-DNA glycosylase. J Mol Biol 359:137–147
Bachant J, Alcasabas A, Blat Y, Kleckner N, Elledge SJ (2002) The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol Cell 9:1169–1182
Bachrati CZ, Hickson ID (2008) RecQ helicases: guardian angels of the DNA replication fork. Chromosoma 117:219–233
Balakirev MY, Mullally JE, Favier A, Assard N, Sulpice E, Lindsey DF, Rulina AV, Gidrol X, Wilkinson KD (2015) Wss1 metalloprotease partners with Cdc48/Doa1 in processing genotoxic. SUMO Conjugates Elife 4:e06763
Barnes DE, Lindahl T (2004) Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet 38:445–476
Barysch SV, Dittner C, Flotho A, Becker J, Melchior F (2014) Identification and analysis of endogenous SUMO1 and SUMO2/3 targets in mammalian cells and tissues using monoclonal antibodies. Nat Protoc 9:896–909
Baumann P, Cech TR (2001) Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 292:1171–1175
Becker J, Barysch SV, Karaca S, Dittner C, Hsiao HH, Berriel Diaz M, Herzig S, Urlaub H, Melchior F (2013) Detecting endogenous SUMO targets in mammalian cells and tissues. Nat Struct Mol Biol 20:525–531
Behlke-Steinert S, Touat-Todeschini L, Skoufias DA, Margolis RL (2009) SMC5 and MMS21 are required for chromosome cohesion and mitotic progression. Cell Cycle 8:2211–2218
Bergink S, Ammon T, Kern M, Schermelleh L, Leonhardt H, Jentsch S (2013) Role of Cdc48/p97 as a SUMO-targeted segregase curbing Rad51-Rad52 interaction. Nat Cell Biol 15:526–532
Bermudez-Lopez M, Pocino-Merino I, Sanchez H, Bueno A, Guasch C, Almedawar S, Bru-Virgili S, Gari E, Wyman C, Reverter D, Colomina N, Torres-Rosell J (2015) ATPase-dependent control of the Mms21 SUMO ligase during DNA repair. PLoS Biol 13:e1002089
Biggins S, Bhalla N, Chang A, Smith DL, Murray AW (2001) Genes involved in sister chromatid separation and segregation in the budding yeast Saccharomyces cerevisiae. Genetics 159:453–470
Blackburn EH, Epel ES, Lin J (2015) Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science 350:1193–1198
Bohm S, Mihalevic MJ, Casal MA, Bernstein KA (2015) Disruption of SUMO-targeted ubiquitin ligases Slx5-Slx8/RNF4 alters RecQ-like helicase Sgs1/BLM localization in yeast and human cells. DNA Repair 26:1–14
Bonne-Andrea C, Kahli M, Mechali F, Lemaitre JM, Bossis G, Coux O (2013) SUMO2/3 modification of cyclin E contributes to the control of replication origin firing. Nat Commun 4:1850
Branzei D, Foiani M (2007) RecQ helicases queuing with Srs2 to disrupt Rad51 filaments and suppress recombination. Genes Dev 21:3019–3026
Branzei D, Sollier J, Liberi G, Zhao X, Maeda D, Seki M, Enomoto T, Ohta K, Foiani M (2006) Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 127:509–522
Burgess RC, Rahman S, Lisby M, Rothstein R, Zhao X (2007) The Slx5-Slx8 complex affects sumoylation of DNA repair proteins and negatively regulates recombination. Mol Cell Biol 27:6153–6162
Burkovics P, Sebesta M, Sisakova A, Plault N, Szukacsov V, Robert T, Pinter L, Marini V, Kolesar P, Haracska L, Gangloff S, Krejci L (2013) Srs2 mediates PCNA-SUMO-dependent inhibition of DNA repair synthesis. EMBO J 32:742–755
Bursomanno S, Beli P, Khan AM, Minocherhomji S, Wagner SA, Bekker-Jensen S, Mailand N, Choudhary C, Hickson ID, Liu Y (2015) Proteome-wide analysis of SUMO2 targets in response to pathological DNA replication stress in human cells. DNA Repair 25:84–96
Bylebyl GR, Belichenko I, Johnson ES (2003) The SUMO isopeptidase Ulp2 prevents accumulation of SUMO chains in yeast. J Biol Chem 278:44113–44120
Carlborg KK, Kanno T, Carter SD, Sjogren C (2015) Mec1-dependent phosphorylation of Mms21 modulates its SUMO ligase activity. DNA Repair 28:83–92
Carmena M, Wheelock M, Funabiki H, Earnshaw WC (2012) The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 13:789–803
Champoux JJ (2001) DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 70:369–413
Chavez A, George V, Agrawal V, Johnson FB (2010) Sumoylation and the structural maintenance of chromosomes (Smc) 5/6 complex slow senescence through recombination intermediate resolution. J Biol Chem 285:11922–11930
Churikov D, Charifi F, Eckert-Boulet N, Silva S, Simon MN, Lisby M, Geli V (2016) SUMO-dependent relocalization of eroded telomeres to nuclear porecComplexes controls telomere recombination. Cell Rep 15:1242–1253
Chymkowitch P, Nguea AP, Aanes H, Koehler CJ, Thiede B, Lorenz S, Meza-Zepeda LA, Klungland A, Enserink JM (2015) Sumoylation of Rap1 mediates the recruitment of TFIID to promote transcription of ribosomal protein genes. Genome Res 25:897–906
Coey CT, Fitzgerald ME, Maiti A, Reiter KH, Guzzo CM, Matunis MJ, Drohat AC (2014) E2-mediated small ubiquitin-like modifier (SUMO) modification of thymine DNA glycosylase is efficient but not selective for the enzyme-product complex. J Biol Chem 289:15810–15819
Coleman KE, Huang TT (2016) How SUMOylation fine-tunes the Fanconi anemia DNA repair pathway. Front Genet 7:61
Cook CE, Hochstrasser M, Kerscher O (2009) The SUMO-targeted ubiquitin ligase subunit Slx5 resides in nuclear foci and at sites of DNA breaks. Cell Cycle 8:1080–1089
Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, Wirz A, Schuermann D, Jacobs AL, Siegrist F, Steinacher R, Jiricny J, Bird A, Schar P (2011) Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470:419–423
Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A (2011) Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146:67–79
Cremona CA, Sarangi P, Yang Y, Hang LE, Rahman S, Zhao X (2012) Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol Cell 45:422–432
Cubenas-Potts C, Srikumar T, Lee C, Osula O, Subramonian D, Zhang XD, Cotter RJ, Raught B, Matunis MJ (2015) Identification of SUMO-2/3-modified proteins associated with mitotic chromosomes. Proteomics 15:763–772
Dasso M (2008) Emerging roles of the SUMO pathway in mitosis. Cell Div 3:5
Dawlaty MM, Malureanu L, Jeganathan KB, Kao E, Sustmann C, Tahk S, Shuai K, Grosschedl R, van Deursen JM (2008) Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell 133:103–115
de Albuquerque CP, Liang J, Gaut NJ, Zhou H (2016) Molecular circuitry of the SUMO (Small Ubiquitin-like Modifier) pathway in controlling sumoylation homeostasis and suppressing genome rearrangements. J Biol Chem 291:8825–8835
de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19:2100–2110
Desterro JM, Rodriguez MS, Hay RT (1998) SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell 2:233–239
Diaz-Martinez LA, Gimenez-Abian JF, Azuma Y, Guacci V, Gimenez-Martin G, Lanier LM, Clarke DJ (2006) PIASgamma is required for faithful chromosome segregation in human cells. PLoS One 1:e53
DiNardo S, Voelkel K, Sternglanz R (1984) DNA topoisomerase II mutant of Saccharomyces cerevisiae: topoisomerase II is required for segregation of daughter molecules at the termination of DNA replication. Proc Natl Acad Sci U S A 81:2616–2620
Dou H, Huang C, Singh M, Carpenter PB, Yeh ET (2010) Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol Cell 39:333–345
Downes CS, Mullinger AM, Johnson RT (1991) Inhibitors of DNA topoisomerase II prevent chromatid separation in mammalian cells but do not prevent exit from mitosis. Proc Natl Acad Sci U S A 88:8895–8899
Drag M, Mikolajczyk J, Krishnakumar IM, Huang Z, Salvesen GS (2008) Activity profiling of human deSUMOylating enzymes (SENPs) with synthetic substrates suggests an unexpected specificity of two newly characterized members of the family. Biochem J 409:461–469
Duan X, Sarangi P, Liu X, Rangi GK, Zhao X, Ye H (2009) Structural and functional insights into the roles of the Mms21 subunit of the Smc5/6 complex. Mol Cell 35:657–668
Duan X, Holmes WB, Ye H (2011) Interaction mapping between Saccharomyces cerevisiae Smc5 and SUMO E3 ligase Mms21. Biochemistry 50:10182–10188
Eladad S, Ye TZ, Hu P, Leversha M, Beresten S, Matunis MJ, Ellis NA (2005) Intra-nuclear trafficking of the BLM helicase to DNA damage-induced foci is regulated by SUMO modification. Hum Mol Genet 14:1351–1365
Erker Y, Neyret-Kahn H, Seeler JS, Dejean A, Atfi A, Levy L (2013) Arkadia, a novel SUMO-targeted ubiquitin ligase involved in PML degradation. Mol Cell Biol 33:2163–2177
Esta A, Ma E, Dupaigne P, Maloisel L, Guerois R, Le Cam E, Veaute X, Coic E (2013) Rad52 sumoylation prevents the toxicity of unproductive Rad51 filaments independently of the anti-recombinase Srs2. PLoS Genet 9:e1003833
Ferreira HC, Luke B, Schober H, Kalck V, Lingner J, Gasser SM (2011) The PIAS homologue Siz2 regulates perinuclear telomere position and telomerase activity in budding yeast. Nat Cell Biol 13:867–874
Ferreira HC, Towbin BD, Jegou T, Gasser SM (2013) The shelterin protein POT-1 anchors Caenorhabditis elegans telomeres through SUN-1 at the nuclear periphery. J Cell Biol 203:727–735
Fitzgerald ME, Drohat AC (2008) Coordinating the initial steps of base excision repair. Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J Biol Chem 283:32680–32690
Fousteri MI, Lehmann AR (2000) A novel SMC protein complex in Schizosaccharomyces pombe contains the Rad18 DNA repair protein. EMBO J 19:1691–1702
Fragkos M, Ganier O, Coulombe P, Mechali M (2015) DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16:360–374
Froelich-Ammon SJ, Osheroff N (1995) Topoisomerase poisons: harnessing the dark side of enzyme mechanism. J Biol Chem 270:21429–21432
Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP (2009) Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 462:935–939
Galanty Y, Belotserkovskaya R, Coates J, Jackson SP (2012) RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev 26:1179–1195
Gali H, Juhasz S, Morocz M, Hajdu I, Fatyol K, Szukacsov V, Burkovics P, Haracska L (2012) Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res 40:6049–6059
Garcia-Rodriguez N, Wong RP, Ulrich HD (2016) Functions of ubiquitin and SUMO in DNA replication and replication stress. Front Genet 7:87
Garg M, Gurung RL, Mansoubi S, Ahmed JO, Dave A, Watts FZ, Bianchi A (2014) Tpz1TPP1 SUMOylation reveals evolutionary conservation of SUMO-dependent Stn1 telomere association. EMBO Rep 15:871–877
Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391
Gibbs-Seymour I, Oka Y, Rajendra E, Weinert BT, Passmore LA, Patel KJ, Olsen JV, Choudhary C, Bekker-Jensen S, Mailand N (2015) Ubiquitin-SUMO circuitry controls activated Fanconi anemia ID complex dosage in response to DNA damage. Mol Cell 57:150–164
Golebiowski F, Matic I, Tatham MH, Cole C, Yin Y, Nakamura A, Cox J, Barton GJ, Mann M, Hay RT (2009) System-wide changes to SUMO modifications in response to heat shock. Sci Signal 2:ra24
Gong L, Yeh ET (2006) Characterization of a family of nucleolar SUMO-specific proteases with preference for SUMO-2 or SUMO-3. J Biol Chem 281:15869–15877
Goodman RH, Smolik S (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev 14:1553–1577
Goshima G, Yanagida M (2000) Establishing biorientation occurs with precocious separation of the sister kinetochores, but not the arms, in the early spindle of budding yeast. Cell 100:619–633
Granger MP, Wright WE, Shay JW (2002) Telomerase in cancer and aging. Crit Rev Oncol Hematol 41:29–40
Groocock LM, Nie M, Prudden J, Moiani D, Wang T, Cheltsov A, Rambo RP, Arvai AS, Hitomi C, Tainer JA, Luger K, Perry JJ, Lazzerini-Denchi E, Boddy MN (2014) RNF4 interacts with both SUMO and nucleosomes to promote the DNA damage response. EMBO Rep 15:601–608
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hang J, Dasso M (2002) Association of the human SUMO-1 protease SENP2 with the nuclear pore. J Biol Chem 277:19961–19966
Hang LE, Liu X, Cheung I, Yang Y, Zhao X (2011) SUMOylation regulates telomere length homeostasis by targeting Cdc13. Nat Struct Mol Biol 18:920–926
Hang LE, Lopez CR, Liu X, Williams JM, Chung I, Wei L, Bertuch AA, Zhao X (2014) Regulation of Ku-DNA association by Yku70 C-terminal tail and SUMO modification. J Biol Chem 289:10308–10317
Hannich JT, Lewis A, Kroetz MB, Li SJ, Heide H, Emili A, Hochstrasser M (2005) Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiae. J Biol Chem 280:4102–4110
Hardeland U, Steinacher R, Jiricny J, Schar P (2002) Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J 21:1456–1464
He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333:1303–1307
Hediger F, Neumann FR, Van Houwe G, Dubrana K, Gasser SM (2002) Live imaging of telomeres: yKu and Sir proteins define redundant telomere-anchoring pathways in yeast. Curr Biol 12:2076–2089
Hendriks IA, D’Souza RC, Yang B, Verlaan-de Vries M, Mann M, Vertegaal AC (2014) Uncovering global SUMOylation signaling networks in a site-specific manner. Nat Struct Mol Biol 21:927–936
Hendriks IA, Schimmel J, Eifler K, Olsen JV, Vertegaal AC (2015a) Ubiquitin-specific Protease 11 (USP11) Deubiquitinates hybrid small ubiquitin-like modifier (SUMO)-ubiquitin chains to counteract RING finger protein 4 (RNF4). J Biol Chem 290:15526–15537
Hendriks IA, Treffers LW, Verlaan-de Vries M, Olsen JV, Vertegaal AC (2015b) SUMO-2 orchestrates chromatin modifiers in response to DNA damage. Cell Rep 10:1778–1791
Hendriks IA, D’Souza RC, Chang JG, Mann M, Vertegaal AC (2015c) System-wide identification of wild-type SUMO-2 conjugation sites. Nat Commun 6:7289
Her J, Jeong YY, Chung IK (2015) PIAS1-mediated sumoylation promotes STUbL-dependent proteasomal degradation of the human telomeric protein TRF2. FEBS Lett 589:3277–3286
Hickey CM, Wilson NR, Hochstrasser M (2012) Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol 13:755–766
Ho JC, Watts FZ (2003) Characterization of SUMO-conjugating enzyme mutants in Schizosaccharomyces pombe identifies a dominant-negative allele that severely reduces SUMO conjugation. Biochem J 372:97–104
Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419:135–141
Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411:366–374
Horigome C, Bustard DE, Marcomini I, Delgoshaie N, Tsai-Pflugfelder M, Cobb JA, Gasser SM (2016) PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev 30:931–945
Hu XV, Rodrigues TM, Tao H, Baker RK, Miraglia L, Orth AP, Lyons GE, Schultz PG, Wu X (2010) Identification of RING finger protein 4 (RNF4) as a modulator of DNA demethylation through a functional genomics screen. Proc Natl Acad Sci U S A 107:15087–15092
Huang TT, D’Andrea AD (2006) Regulation of DNA repair by ubiquitylation. Nat Rev Mol Cell Biol 7:323–334
Huang L, Yang S, Zhang S, Liu M, Lai J, Qi Y, Shi S, Wang J, Wang Y, Xie Q, Yang C (2009) The Arabidopsis SUMO E3 ligase AtMMS21, a homologue of NSE2/MMS21, regulates cell proliferation in the root. Plant J 60:666–678
Ii T, Fung J, Mullen JR, Brill SJ (2007a) The yeast Slx5-Slx8 DNA integrity complex displays ubiquitin ligase activity. Cell Cycle 6:2800–2809
Ii T, Mullen JR, Slagle CE, Brill SJ (2007b) Stimulation of in vitro sumoylation by Slx5-Slx8: evidence for a functional interaction with the SUMO pathway. DNA Repair 6:1679–1691
Impens F, Radoshevich L, Cossart P, Ribet D (2014) Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proc Natl Acad Sci U S A 111:12432–12437
Isik S, Sano K, Tsutsui K, Seki M, Enomoto T, Saitoh H, Tsutsui K (2003) The SUMO pathway is required for selective degradation of DNA topoisomerase IIbeta induced by a catalytic inhibitor ICRF-193(1). FEBS Lett 546:374–378
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461:1071–1078
Jacome A, Gutierrez-Martinez P, Schiavoni F, Tenaglia E, Martinez P, Rodriguez-Acebes S, Lecona E, Murga M, Mendez J, Blasco MA, Fernandez-Capetillo O (2015) NSMCE2 suppresses cancer and aging in mice independently of its SUMO ligase activity. EMBO J 34:2604–2619
Jentsch S, Psakhye I (2013) Control of nuclear activities by substrate-selective and protein-group SUMOylation. Annu Rev Genet 47:167–186
Johnson ES (2004) Protein modification by SUMO. Annu Rev Biochem 73:355–382
Kawabe Y, Seki M, Seki T, Wang WS, Imamura O, Furuichi Y, Saitoh H, Enomoto T (2000) Covalent modification of the Werner’s syndrome gene product with the ubiquitin-related protein, SUMO-1. J Biol Chem 275:20963–20966
Kee Y, D’Andrea AD (2012) Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest 122:3799–3806
Keusekotten K, Bade VN, Meyer-Teschendorf K, Sriramachandran AM, Fischer-Schrader K, Krause A, Horst C, Schwarz G, Hofmann K, Dohmen RJ, Praefcke GJ (2014) Multivalent interactions of the SUMO-interaction motifs in RING finger protein 4 determine the specificity for chains of the SUMO. Biochem J 457:207–214
Khadaroo B, Teixeira MT, Luciano P, Eckert-Boulet N, Germann SM, Simon MN, Gallina I, Abdallah P, Gilson E, Geli V, Lisby M (2009) The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat Cell Biol 11:980–987
Kliszczak M, Stephan AK, Flanagan AM, Morrison CG (2012) SUMO ligase activity of vertebrate Mms21/Nse2 is required for efficient DNA repair but not for Smc5/6 complex stability. DNA Repair 11:799–810
Kohli RM, Zhang Y (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502:472–479
Kolesar P, Sarangi P, Altmannova V, Zhao X, Krejci L (2012) Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res 40:7831–7843
Kosoy A, Calonge TM, Outwin EA, O’Connell MJ (2007) Fission yeast Rnf4 homologs are required for DNA repair. J Biol Chem 282:20388–20394
Kotaja N, Karvonen U, Janne OA, Palvimo JJ (2002) PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol Cell Biol 22:5222–5234
Kubota T, Nishimura K, Kanemaki MT, Donaldson AD (2013) The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol Cell 50:273–280
Lallemand-Breitenbach V, Jeanne M, Benhenda S, Nasr R, Lei M, Peres L, Zhou J, Zhu J, Raught B, de The H (2008) Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat Cell Biol 10:547–555
Lamoliatte F, Caron D, Durette C, Mahrouche L, Maroui MA, Caron-Lizotte O, Bonneil E, Chelbi-Alix MK, Thibault P (2014) Large-scale analysis of lysine SUMOylation by SUMO remnant immunoaffinity profiling. Nat Commun 5:5409
Lawrence CW, Christensen RB (1979) Metabolic suppressors of trimethoprim and ultraviolet light sensitivities of Saccharomyces cerevisiae rad6 mutants. J Bacteriol 139:866–876
Leach CA, Michael WM (2005) Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J Cell Biol 171:947–954
Lecona E, Rodriguez-Acebes S, Specks J, Lopez-Contreras AJ, Ruppen I, Murga M, Munoz J, Mendez J, Fernandez-Capetillo O (2016) USP7 is a SUMO deubiquitinase essential for DNA replication. Nat Struct Mol Biol 23:270–277
Lehmann AR (2005) The role of SMC proteins in the responses to DNA damage. DNA Repair 4:309–314
Lehmann AR, Walicka M, Griffiths DJ, Murray JM, Watts FZ, McCready S, Carr AM (1995) The rad18 gene of Schizosaccharomyces pombe defines a new subgroup of the SMC superfamily involved in DNA repair. Mol Cell Biol 15:7067–7080
Lescasse R, Pobiega S, Callebaut I, Marcand S (2013) End-joining inhibition at telomeres requires the translocase and polySUMO-dependent ubiquitin ligase Uls1. EMBO J 32:805–815
Li SJ, Hochstrasser M (2000) The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol Cell Biol 20:2367–2377
Li SJ, Hochstrasser M (2003) The Ulp1 SUMO isopeptidase: distinct domains required for viability, nuclear envelope localization, and substrate specificity. J Cell Biol 160:1069–1081
Li W, Hesabi B, Babbo A, Pacione C, Liu J, Chen DJ, Nickoloff JA, Shen Z (2000) Regulation of double-strand break-induced mammalian homologous recombination by UBL1, a RAD51-interacting protein. Nucleic Acids Res 28:1145–1153
Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M (2005) Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19:339–350
Lima CD, Reverter D (2008) Structure of the human SENP7 catalytic domain and poly-SUMO deconjugation activities for SENP6 and SENP7. J Biol Chem 283:32045–32055
Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362:709–715
Liu M, Shi S, Zhang S, Xu P, Lai J, Liu Y, Yuan D, Wang Y, Du J, Yang C (2014) SUMO E3 ligase AtMMS21 is required for normal meiosis and gametophyte development in Arabidopsis. BMC Plant Biol 14:153
Lopez-Contreras AJ, Ruppen I, Nieto-Soler M, Murga M, Rodriguez-Acebes S, Remeseiro S, Rodrigo-Perez S, Rojas AM, Mendez J, Munoz J, Fernandez-Capetillo O (2013) A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep 3:1105–1116
Lu CY, Tsai CH, Brill SJ, Teng SC (2010) Sumoylation of the BLM ortholog, Sgs1, promotes telomere-telomere recombination in budding yeast. Nucleic Acids Res 38:488–498
Lucas I, Germe T, Chevrier-Miller M, Hyrien O (2001) Topoisomerase II can unlink replicating DNA by precatenane removal. EMBO J 20:6509–6519
Luo K, Zhang H, Wang L, Yuan J, Lou Z (2012) Sumoylation of MDC1 is important for proper DNA damage response. EMBO J 31:3008–3019
Maeda D, Seki M, Onoda F, Branzei D, Kawabe Y, Enomoto T (2004) Ubc9 is required for damage-tolerance and damage-induced interchromosomal homologous recombination in S. cerevisiae. DNA Repair 3:335–341
Maiti A, Drohat AC (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem 286:35334–35338
Matic I, van Hagen M, Schimmel J, Macek B, Ogg SC, Tatham MH, Hay RT, Lamond AI, Mann M, Vertegaal AC (2008) In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Mol Cell Proteomics 7:132–144
Matic I, Schimmel J, Hendriks IA, van Santen MA, van de Rijke F, van Dam H, Gnad F, Mann M, Vertegaal AC (2010) Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Mol Cell 39:641–652
Matunis MJ, Zhang XD, Ellis NA (2006) SUMO: the glue that binds. Dev Cell 11:596–597
McAleenan A, Cordon-Preciado V, Clemente-Blanco A, Liu IC, Sen N, Leonard J, Jarmuz A, Aragon L (2012) SUMOylation of the alpha-kleisin subunit of cohesin is required for DNA damage-induced cohesion. Curr Biol 22:1564–1575
McDonald WH, Pavlova Y, Yates JR 3rd, Boddy MN (2003) Novel essential DNA repair proteins Nse1 and Nse2 are subunits of the fission yeast Smc5-Smc6 complex. J Biol Chem 278:45460–45467
McLaughlin D, Coey CT, Yang WC, Drohat AC, Matunis MJ (2016) Characterizing requirements for small ubiquitin-like modifier (SUMO) modification and binding on base excision repair activity of thymine-DNA glycosylase in vivo. J Biol Chem 291:9014–9024
Memisoglu A, Samson L (2000) Base excision repair in yeast and mammals. Mutat Res 451:39–51
Miyagawa K, Low RS, Santosa V, Tsuji H, Moser BA, Fujisawa S, Harland JL, Raguimova ON, Go A, Ueno M, Matsuyama A, Yoshida M, Nakamura TM, Tanaka K (2014) SUMOylation regulates telomere length by targeting the shelterin subunit Tpz1(Tpp1) to modulate shelterin-Stn1 interaction in fission yeast. Proc Natl Acad Sci U S A 111:5950–5955
Mohan RD, Rao A, Gagliardi J, Tini M (2007) SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment. Mol Cell Biol 27:229–243
Moldovan GL, Pfander B, Jentsch S (2006) PCNA controls establishment of sister chromatid cohesion during S phase. Mol Cell 23:723–732
Moldovan GL, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ, D’Andrea AD (2012) Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell 45:75–86
Montpetit B, Hazbun TR, Fields S, Hieter P (2006) Sumoylation of the budding yeast kinetochore protein Ndc10 is required for Ndc10 spindle localization and regulation of anaphase spindle elongation. J Cell Biol 174:653–663
Moriyama T, Fujimitsu Y, Yoshikai Y, Sasano T, Yamada K, Murakami M, Urano T, Sugasawa K, Saitoh H (2014) SUMO-modification and elimination of the active DNA demethylation enzyme TDG in cultured human cells. Biochem Biophys Res Commun 447:419–424
Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, Ng T, Solomon E (2009) The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 462:886–890
Motegi A, Kuntz K, Majeed A, Smith S, Myung K (2006) Regulation of gross chromosomal rearrangements by ubiquitin and SUMO ligases in Saccharomyces cerevisiae. Mol Cell Biol 26:1424–1433
Mullen JR, Brill SJ (2008) Activation of the Slx5-Slx8 ubiquitin ligase by poly-small ubiquitin-like modifier conjugates. J Biol Chem 283:19912–19921
Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ (2001) Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157:103–118
Mullen JR, Chen CF, Brill SJ (2010) Wss1 is a SUMO-dependent isopeptidase that interacts genetically with the Slx5-Slx8 SUMO-targeted ubiquitin ligase. Mol Cell Biol 30:3737–3748
Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ (2008) Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 322:597–602
Nayak A, Muller S (2014) SUMO-specific proteases/isopeptidases: SENPs and beyond. Genome Biol 15:422
Ohuchi T, Seki M, Branzei D, Maeda D, Ui A, Ogiwara H, Tada S, Enomoto T (2008) Rad52 sumoylation and its involvement in the efficient induction of homologous recombination. DNA Repair 7:879–889
Okubo S, Hara F, Tsuchida Y, Shimotakahara S, Suzuki S, Hatanaka H, Yokoyama S, Tanaka H, Yasuda H, Shindo H (2004) NMR structure of the N-terminal domain of SUMO ligase PIAS1 and its interaction with tumor suppressor p53 and A/T-rich DNA oligomers. J Biol Chem 279:31455–31461
Ouyang KJ, Woo LL, Zhu J, Huo D, Matunis MJ, Ellis NA (2009) SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol 7:e1000252
Ouyang J, Garner E, Hallet A, Nguyen HD, Rickman KA, Gill G, Smogorzewska A, Zou L (2015) Noncovalent interactions with SUMO and ubiquitin orchestrate distinct functions of the SLX4 complex in genome maintenance. Mol Cell 57:108–122
Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD (2006) A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell 124:1069–1081
Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD (2005) Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 19:123–133
Parker JL, Ulrich HD (2012) A SUMO-interacting motif activates budding yeast ubiquitin ligase Rad18 towards SUMO-modified PCNA. Nucleic Acids Res 40:11380–11388
Parker JL, Bucceri A, Davies AA, Heidrich K, Windecker H, Ulrich HD (2008) SUMO modification of PCNA is controlled by DNA. EMBO J 27:2422–2431
Parnas O, Zipin-Roitman A, Pfander B, Liefshitz B, Mazor Y, Ben-Aroya S, Jentsch S, Kupiec M (2010) Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J 29:2611–2622
Pebernard S, McDonald WH, Pavlova Y, Yates JR 3rd, Boddy MN (2004) Nse1, Nse2, and a novel subunit of the Smc5-Smc6 complex, Nse3, play a crucial role in meiosis. Mol Biol Cell 15:4866–4876
Pebernard S, Schaffer L, Campbell D, Head SR, Boddy MN (2008) Localization of Smc5/6 to centromeres and telomeres requires heterochromatin and SUMO, respectively. EMBO J 27:3011–3023
Pfander B, Moldovan GL, Sacher M, Hoege C, Jentsch S (2005) SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436:428–433
Pickett HA, Reddel RR (2015) Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat Struct Mol Biol 22:875–880
Potts PR, Yu H (2005) Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol Cell Biol 25:7021–7032
Potts PR, Yu H (2007) The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol 14:581–590
Potts PR, Porteus MH, Yu H (2006) Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J 25:3377–3388
Poulsen SL, Hansen RK, Wagner SA, van Cuijk L, van Belle GJ, Streicher W, Wikstrom M, Choudhary C, Houtsmuller AB, Marteijn JA, Bekker-Jensen S, Mailand N (2013) RNF111/Arkadia is a SUMO-targeted ubiquitin ligase that facilitates the DNA damage response. J Cell Biol 201:797–807
Prudden J, Pebernard S, Raffa G, Slavin DA, Perry JJ, Tainer JA, McGowan CH, Boddy MN (2007) SUMO-targeted ubiquitin ligases in genome stability. EMBO J 26:4089–4101
Psakhye I, Jentsch S (2012) Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151:807–820
Rai R, Varma SP, Shinde N, Ghosh S, Kumaran SP, Skariah G, Laloraya S (2011) Small ubiquitin-related modifier ligase activity of Mms21 is required for maintenance of chromosome integrity during the unperturbed mitotic cell division cycle in Saccharomyces cerevisiae. J Biol Chem 286:14516–14530
Reindle A, Belichenko I, Bylebyl GR, Chen XL, Gandhi N, Johnson ES (2006) Multiple domains in Siz SUMO ligases contribute to substrate selectivity. J Cell Sci 119:4749–4757
Robert T, Dervins D, Fabre F, Gangloff S (2006) Mrc1 and Srs2 are major actors in the regulation of spontaneous crossover. EMBO J 25:2837–2846
Roca J, Ishida R, Berger JM, Andoh T, Wang JC (1994) Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc Natl Acad Sci U S A 91:1781–1785
Rojas-Fernandez A, Plechanovova A, Hattersley N, Jaffray E, Tatham MH, Hay RT (2014) SUMO chain-induced dimerization activates RNF4. Mol Cell 53:880–892
Ryu H, Furuta M, Kirkpatrick D, Gygi SP, Azuma Y (2010) PIASy-dependent SUMOylation regulates DNA topoisomerase IIalpha activity. J Cell Biol 191:783–794
Ryu H, Yoshida MM, Sridharan V, Kumagai A, Dunphy WG, Dasso M, Azuma Y (2015) SUMOylation of the C-terminal domain of DNA topoisomerase IIalpha regulates the centromeric localization of Claspin. Cell Cycle 14:2777–2784
Sacher M, Pfander B, Hoege C, Jentsch S (2006) Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat Cell Biol 8:1284–1290
Saito K, Kagawa W, Suzuki T, Suzuki H, Yokoyama S, Saitoh H, Tashiro S, Dohmae N, Kurumizaka H (2010) The putative nuclear localization signal of the human RAD52 protein is a potential sumoylation site. J Biochem 147:833–842
Saitoh H, Hinchey J (2000) Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem 275:6252–6258
San FJ, Sung P, Klein H (2008) Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77:229–257
Schimmel J, Larsen KM, Matic I, van Hagen M, Cox J, Mann M, Andersen JS, Vertegaal AC (2008) The ubiquitin-proteasome system is a key component of the SUMO-2/3 cycle. Mol Cell Proteomics 7:2107–2122
Schimmel J, Eifler K, Sigurethsson JO, Cuijpers SA, Hendriks IA, Verlaan-de Vries M, Kelstrup CD, Francavilla C, Medema RH, Olsen JV, Vertegaal AC (2014) Uncovering SUMOylation dynamics during cell-cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol Cell 53:1053–1066
Schou J, Kelstrup CD, Hayward DG, Olsen JV, Nilsson J (2014) Comprehensive identification of SUMO2/3 targets and their dynamics during mitosis. PLoS One 9:e100692
Shamu CE, Murray AW (1992) Sister chromatid separation in frog egg extracts requires DNA topoisomerase II activity during anaphase. J Cell Biol 117:921–934
Silva S, Altmannova V, Eckert-Boulet N, Kolesar P, Gallina I, Hang L, Chung I, Arneric M, Zhao X, Buron LD, Mortensen UH, Krejci L, Lisby M (2016) SUMOylation of Rad52-Rad59 synergistically change the outcome of mitotic recombination. DNA Repair 42:11–25
Smet-Nocca C, Wieruszeski JM, Leger H, Eilebrecht S, Benecke A (2011) SUMO-1 regulates the conformational dynamics of thymine-DNA Glycosylase regulatory domain and competes with its DNA binding activity. BMC Biochem 12:4
Sohn SY, Bridges RG, Hearing P (2015) Proteomic analysis of ubiquitin-like posttranslational modifications induced by the adenovirus E4-ORF3 protein. J Virol 89:1744–1755
Spink KG, Evans RJ, Chambers A (2000) Sequence-specific binding of Taz1p dimers to fission yeast telomeric DNA. Nucleic Acids Res 28:527–533
Steinacher R, Schar P (2005) Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr Biol 15:616–623
Stelter P, Ulrich HD (2003) Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425:188–191
Stephan AK, Kliszczak M, Morrison CG (2011) The Nse2/Mms21 SUMO ligase of the Smc5/6 complex in the maintenance of genome stability. FEBS Lett 585:2907–2913
Su XA, Dion V, Gasser SM, Freudenreich CH (2015) Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev 29:1006–1017
Sun H, Hunter T (2012) Poly-small ubiquitin-like modifier (PolySUMO)-binding proteins identified through a string search. J Biol Chem 287:42071–42083
Sun H, Leverson JD, Hunter T (2007) Conserved function of RNF4 family proteins in eukaryotes: targeting a ubiquitin ligase to SUMOylated proteins. EMBO J 26:4102–4112
Suzuki R, Shindo H, Tase A, Kikuchi Y, Shimizu M, Yamazaki T (2009) Solution structures and DNA binding properties of the N-terminal SAP domains of SUMO E3 ligases from Saccharomyces cerevisiae and Oryza sativa. Proteins 75:336–347
Takahashi H, Hatakeyama S, Saitoh H, Nakayama KI (2005) Noncovalent SUMO-1 binding activity of thymine DNA glycosylase (TDG) is required for its SUMO-1 modification and colocalization with the promyelocytic leukemia protein. J Biol Chem 280:5611–5621
Takahashi Y, Yong-Gonzalez V, Kikuchi Y, Strunnikov A (2006) SIZ1/SIZ2 control of chromosome transmission fidelity is mediated by the sumoylation of topoisomerase II. Genetics 172:783–794
Tammsalu T, Matic I, Jaffray EG, Ibrahim AF, Tatham MH, Hay RT (2014) Proteome-wide identification of SUMO2 modification sites. Sci Signal 7:rs2
Tammsalu T, Matic I, Jaffray EG, Ibrahim AF, Tatham MH, Hay RT (2015) Proteome-wide identification of SUMO modification sites by mass spectrometry. Nat Protoc 10:1374–1388
Tanaka K, Nishide J, Okazaki K, Kato H, Niwa O, Nakagawa T, Matsuda H, Kawamukai M, Murakami Y (1999) Characterization of a fission yeast SUMO-1 homologue, pmt3p, required for multiple nuclear events, including the control of telomere length and chromosome segregation. Mol Cell Biol 19:8660–8672
Tatham MH, Jaffray E, Vaughan OA, Desterro JM, Botting CH, Naismith JH, Hay RT (2001) Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem 276:35368–35374
Tatham MH, Geoffroy MC, Shen L, Plechanovova A, Hattersley N, Jaffray EG, Palvimo JJ, Hay RT (2008) RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat Cell Biol 10:538–546
Tatham MH, Matic I, Mann M, Hay RT (2011) Comparative proteomic analysis identifies a role for SUMO in protein quality control. Sci Signal 4:rs4
Tatham MH, Plechanovova A, Jaffray EG, Salmen H, Hay RT (2013) Ube2W conjugates ubiquitin to alpha-amino groups of protein N-termini. Biochem J 453:137–145
Taylor EM, Copsey AC, Hudson JJ, Vidot S, Lehmann AR (2008) Identification of the proteins, including MAGEG1, that make up the human SMC5-6 protein complex. Mol Cell Biol 28:1197–1206
Thu YM, Van Riper SK, Higgins L, Zhang T, Becker JR, Markowski TW, Nguyen HD, Griffin TJ, Bielinsky AK (2016) Slx5/Slx8 promotes replication stress tolerance by facilitating mitotic progression. Cell Rep 15:1254–1265
Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P (2002) Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell 9:265–277
Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294:2364–2368
Torres JZ, Bessler JB, Zakian VA (2004a) Local chromatin structure at the ribosomal DNA causes replication fork pausing and genome instability in the absence of the S. cerevisiae DNA helicase Rrm3p. Genes Dev 18:498–503
Torres JZ, Schnakenberg SL, Zakian VA (2004b) Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: viability of rrm3 cells requires the intra-S-phase checkpoint and fork restart activities. Mol Cell Biol 24:3198–3212
Torres-Rosell J, Sunjevaric I, De Piccoli G, Sacher M, Eckert-Boulet N, Reid R, Jentsch S, Rothstein R, Aragon L, Lisby M (2007) The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9:923–931
Uhlmann F (2016) SMC complexes: from DNA to chromosomes. Nat Rev Mol Cell Biol. doi:10.1038/nrm.2016.30
Ulrich HD (2005) The RAD6 pathway: control of DNA damage bypass and mutagenesis by ubiquitin and SUMO. Chembiochem 6:1735–1743
Ulrich HD (2008) The fast-growing business of SUMO chains. Mol Cell 32:301–305
Urulangodi M, Sebesta M, Menolfi D, Szakal B, Sollier J, Sisakova A, Krejci L, Branzei D (2015) Local regulation of the Srs2 helicase by the SUMO-like domain protein Esc2 promotes recombination at sites of stalled replication. Genes Dev 29:2067–2080
Uzunova K, Gottsche K, Miteva M, Weisshaar SR, Glanemann C, Schnellhardt M, Niessen M, Scheel H, Hofmann K, Johnson ES, Praefcke GJ, Dohmen RJ (2007) Ubiquitin-dependent proteolytic control of SUMO conjugates. J Biol Chem 282:34167–34175
van der Veen AG, Ploegh HL (2012) Ubiquitin-like proteins. Annu Rev Biochem 81:323–357
Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F (2003) The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423:309–312
Verdun RE, Karlseder J (2007) Replication and protection of telomeres. Nature 447:924–931
Vertegaal AC, Andersen JS, Ogg SC, Hay RT, Mann M, Lamond AI (2006) Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol Cell Proteomics 5:2298–2310
Vyas R, Kumar R, Clermont F, Helfricht A, Kalev P, Sotiropoulou P, Hendriks IA, Radaelli E, Hochepied T, Blanpain C, Sablina A, van Attikum H, Olsen JV, Jochemsen AG, Vertegaal AC, Marine JC (2013) RNF4 is required for DNA double-strand break repair in vivo. Cell Death Differ 20:490–502
Walden H, Deans AJ (2014) The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys 43:257–278
Wan J, Subramonian D, Zhang XD (2012) SUMOylation in control of accurate chromosome segregation during mitosis. Curr Protein Pept Sci 13:467–481
Wang QE, Zhu Q, Wani G, El-Mahdy MA, Li J, Wani AA (2005) DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res 33:4023–4034
Wang Z, Jones GM, Prelich G (2006) Genetic analysis connects SLX5 and SLX8 to the SUMO pathway in Saccharomyces cerevisiae. Genetics 172:1499–1509
Warsi TH, Navarro MS, Bachant J (2008) DNA topoisomerase II is a determinant of the tensile properties of yeast centromeric chromatin and the tension checkpoint. Mol Biol Cell 19:4421–4433
Waters TR, Gallinari P, Jiricny J, Swann PF (1999) Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J Biol Chem 274:67–74
Watson JD (1972) Origin of concatemeric T7 DNA. Nat New Biol 239:197–201
Watts FZ, Skilton A, Ho JC, Boyd LK, Trickey MA, Gardner L, Ogi FX, Outwin EA (2007) The role of Schizosaccharomyces pombe SUMO ligases in genome stability. Biochem Soc Trans 35:1379–1384
Wei L, Zhao X (2016) A new MCM modification cycle regulates DNA replication initiation. Nat Struct Mol Biol 23:209–216
Wei W, Yang P, Pang J, Zhang S, Wang Y, Wang MH, Dong Z, She JX, Wang CY (2008) A stress-dependent SUMO4 sumoylation of its substrate proteins. Biochem Biophys Res Commun 375:454–459
Weisshaar SR, Keusekotten K, Krause A, Horst C, Springer HM, Gottsche K, Dohmen RJ, Praefcke GJ (2008) Arsenic trioxide stimulates SUMO-2/3 modification leading to RNF4-dependent proteolytic targeting of PML. FEBS Lett 582:3174–3178
Windecker H, Ulrich HD (2008) Architecture and assembly of poly-SUMO chains on PCNA in Saccharomyces cerevisiae. J Mol Biol 376:221–231
Wu N, Kong X, Ji Z, Zeng W, Potts PR, Yokomori K, Yu H (2012) Scc1 sumoylation by Mms21 promotes sister chromatid recombination through counteracting Wapl. Genes Dev 26:1473–1485
Wu HC, Lin YC, Liu CH, Chung HC, Wang YT, Lin YW, Ma HI, Tu PH, Lawler SE, Chen RH (2014) USP11 regulates PML stability to control Notch-induced malignancy in brain tumours. Nat Commun 5:3214
Xaver M, Huang L, Chen D, Klein F (2013) Smc5/6-Mms21 prevents and eliminates inappropriate recombination intermediates in meiosis. PLoS Genet 9:e1004067
Xhemalce B, Seeler JS, Thon G, Dejean A, Arcangioli B (2004) Role of the fission yeast SUMO E3 ligase Pli1p in centromere and telomere maintenance. EMBO J 23:3844–3853
Xhemalce B, Riising EM, Baumann P, Dejean A, Arcangioli B, Seeler JS (2007) Role of SUMO in the dynamics of telomere maintenance in fission yeast. Proc Natl Acad Sci U S A 104:893–898
Xiao Z, Chang JG, Hendriks IA, Sigurethsson JO, Olsen JV, Vertegaal AC (2015) System-wide analysis of SUMOylation dynamics in response to replication stress reveals novel small ubiquitin-like modified target proteins and acceptor lysines relevant for genome stability. Mol Cell Proteomics 14:1419–1434
Xie Y, Kerscher O, Kroetz MB, McConchie HF, Sung P, Hochstrasser M (2007) The yeast Hex3.Slx8 heterodimer is a ubiquitin ligase stimulated by substrate sumoylation. J Biol Chem 282:34176–34184
Xie Y, Rubenstein EM, Matt T, Hochstrasser M (2010) SUMO-independent in vivo activity of a SUMO-targeted ubiquitin ligase toward a short-lived transcription factor. Genes Dev 24:893–903
Xie J, Kim H, Moreau LA, Puhalla S, Garber J, Al Abo M, Takeda S, D’Andrea AD (2015) RNF4-mediated polyubiquitination regulates the Fanconi anemia/BRCA pathway. J Clin Invest 125:1523–1532
Xu Y, Plechanovova A, Simpson P, Marchant J, Leidecker O, Kraatz S, Hay RT, Matthews SJ (2014) Structural insight into SUMO chain recognition and manipulation by the ubiquitin ligase RNF4. Nat Commun 5:4217
Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR (1999) Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res 59:4175–4179
Yeh E, Haase J, Paliulis LV, Joglekar A, Bond L, Bouck D, Salmon ED, Bloom KS (2008) Pericentric chromatin is organized into an intramolecular loop in mitosis. Curr Biol 18:81–90
Yin Y, Seifert A, Chua JS, Maure JF, Golebiowski F, Hay RT (2012) SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev 26:1196–1208
Yong-Gonzales V, Hang LE, Castellucci F, Branzei D, Zhao X (2012) The Smc5-Smc6 complex regulates recombination at centromeric regions and affects kinetochore protein sumoylation during normal growth. PLoS One 7:e51540
Yuan D, Lai J, Xu P, Zhang S, Zhang J, Li C, Wang Y, Du J, Liu Y, Yang C (2014) AtMMS21 regulates DNA damage response and homologous recombination repair in Arabidopsis. DNA Repair 21:140–147
Yunus AA, Lima CD (2009) Structure of the Siz/PIAS SUMO E3 ligase Siz1 and determinants required for SUMO modification of PCNA. Mol Cell 35:669–682
Yurchenko V, Xue Z, Sadofsky MJ (2006) SUMO modification of human XRCC4 regulates its localization and function in DNA double-strand break repair. Mol Cell Biol 26:1786–1794
Zhang C, Roberts TM, Yang J, Desai R, Brown GW (2006) Suppression of genomic instability by SLX5 and SLX8 in Saccharomyces cerevisiae. DNA Repair 5:336–346
Zhao X, Blobel G (2005) A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci U S A 102:4777–4782
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Zilio, N., Eifler-Olivi, K., Ulrich, H.D. (2017). Functions of SUMO in the Maintenance of Genome Stability. In: Wilson, V. (eds) SUMO Regulation of Cellular Processes. Advances in Experimental Medicine and Biology, vol 963. Springer, Cham. https://doi.org/10.1007/978-3-319-50044-7_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-50044-7_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50043-0
Online ISBN: 978-3-319-50044-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)