
Abstract
There has been a continued interest in translational research focused on both natural products and manipulation of functional groups on these compounds to create novel derivatives with higher desired activities. Oleanolic acid, a component of traditional Chinese medicine used in hepatitis therapy, was modified by chemical processes to form 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO). This modification increased anti-inflammatory activity significantly and additional functional groups on the CDDO backbone have shown promise in treating conditions ranging from kidney disease to obesity to diabetes. CDDO’s therapeutic effect is due to its upregulation of the master antioxidant transcription factor Nuclear factor erythroid 2-related factor 2 (Nrf2) through conformational change of Nrf2-repressing, Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1 (Keap1) and multiple animal and human studies have verified subsequent activation of Nrf2-controlled antioxidant genes via upstream Antioxidant Response Element (ARE) regions. At the present time, positive results have been obtained in the laboratory and clinical trials with CDDO derivatives treating conditions such as lung injury, inflammation and chronic kidney disease. However, clinical trials for cancer and cardiovascular disease have not shown equally positive results and further exploration of CDDO and its derivatives is needed to put these shortcomings into context for the purpose of future therapeutic modalities.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Triterpenoids are natural saponin compounds (such as cholesterol and phytosterols) with a multi-carbon skeleton that can be manipulated synthetically, adding various chemical groups for functionality. CDDO, or 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, is a synthetic triterpenoid derived from oleanolic acid that was purposely constructed for anti-inflammatory purposes in macrophages and has, over time, been modified with methyl, amine, and imidazolide groups to further affect various signaling pathways such as FLIP/TRAIL, caspase, SMAD, and mTOR. However, the primary target of CDDO and its related compounds is the Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1 (Keap1) that regulates nuclear factor erythroid 2-related factor 2 (Nrf2) that, in turn, acts as a master transcription factor for upregulation of antioxidant response genes such as heme oxygenase-1 (HO-1) and NAD(P)H:quinone oxidoreductase (NQO1). Modulation of Nrf2 by CDDO and related compounds has been repeatedly shown in the literature to positively affect a multitude of disease states in animal models, including amyotrophic lateral sclerosis (ALS), various cancers (breast, prostate, etc.), inflammatory shock, and cardiovascular damage. Phases I and II trials with CDDO in chronic kidney disease have shown much promise. However, well-controlled Phase I cancer trials in humans have not shown dramatic improvements in the disease outcomes. The reasons for these variable results are not well understood, and much work remains to be done in determining the minute details of CDDO-affected signaling pathways in humans. This review will explore a wide range of the literature to provide a framework of understanding about CDDO’s chemical properties, its signaling targets, and therapeutic efficacy in animal models. Although CDDO compounds may not yet be the “silver bullet” for some diseases, the clear effect of CDDO treatment on crucial cellular pathways in multiple disease models makes it useful in the laboratory and more work may yet find a derivative compound that proves efficacious in treating human disease.
2 Chemical Properties and Pharmacokinetics
2.1 Chemical Synthesis and Characteristics
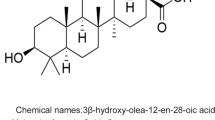
Oleanolic acid has been used in China as a therapy for hepatitis and serves as the backbone of CDDO [1]. CDDO was first reported in a series of papers by Honda et al. that stepwise-converted oleanolic and ursolic acids into a compound capable of both inhibiting inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) production in mouse macrophages and growth in an NRP.152 prostate cell line [2–4]. CDDO was originally synthesized from modification of the A and C rings of the saponin compounds with 1-en-3-one functionality, but the C-2 position was found to be critical for activity of the compound [3]. Further studies revealed that α,β-unsaturated carbonyl moieties boost the effect of the compound by several fold [5]. Briefly, oleanolic acid was formylated in the presence of sodium methoxide, with tetrahydrofuran (THF) added. After an isoxazole intermediate was formed via methoxide cleavage and alkali hydrolysis, the nitrile intermediate. A nitrile intermediate was reacted in dimethylfuran (DMF) with lithium iodide in a halogenolysis reaction to form CDDO [3]. A later report from Fu and Gribble [6] introduced a scalable and much more efficient synthesis protocol for CDDO-Me that could prove useful in clinical studies. The polar structure of CDDO lends well to solubility in DMSO for the laboratory while human trials used microcrystalline preparations in gelatin capsules [7]. Functional groups can be added to CDDO, such as a C28 methyl ester, imidazolide, and various amides (ethyl, diethyl, and trifluoroethyl amides) [8, 9]. Each of these compounds has specific kinetics and, taken as a whole group, offer an arsenal of compounds to test in the laboratory.
2.2 Pharmacokinetics of CDDO
CDDO, as synthesized by Honda and Suh [3], has an IC50 of just 0.0004 µM in mouse macrophages, over a thousand times stronger than its parent compound oleanolic acid. Derivatives such as CDDO-Me and CDDO-Im have induced strong NQO1 activity at single doses of as little as 10 µmol/kg in mice while other reports have seen 5–130 nM inhibitory effects in tumor cell lines with CDDO amides [8, 9].
A study undertaken by Noker et al. [10] in rats and dogs found that CDDO is eliminated from plasma in 2 stages, with a mean half-life of 0.06 h for α-phase and 1.95 h for β-phase in rats with total clearance being roughly 9 L/m2/h. Dogs showed a much faster clearance with an α-phase of 0.02 h and a β-phase of 0.65 h with a total clearance of 44.6 L/m2/h [10] Total results indicated linear pharmacokinetics with side effects (diarrhea, piloerection) but no toxicity even at doses of 50 mg/m2/h with total maximum tolerated dosages of 2160 mg/m2 in rats and 6000 mg/m2 in dogs [10]. This low toxicity with lack of catastrophic side effects makes CDDO an ideal compound for in vivo animal studies. Further studies on methodology for bioanalytical methods for in vivo studies with CDDO found that its electrophilic nature caused it to react with glutathione (GSH) and nucleophilic groups in proteins as well as N-acetylcysteine, forming covalently adducted metabolites that can be measured with protein precipitation, Edman degradation, and ammonium hydroxide [11]. Clearly, CDDO’s main mode of chemical action is for the functional electrophilic groups to attack sulfhydryl and cysteine moieties on target proteins. A clinical trial in humans by Hong et al. [7] explored the more complex pharmacokinetics in chronic kidney disease patients treated with CDDO-Me, finding a maximum tolerated dosage to be 900 mg/day and a maximum peak plasma time of 4 h with a mean half-life of about 39 ± 20 h. Hong et al. concluded that 900 mg/day was the appropriate dosage for any further Phase II trials. Clinical testing has been done in human volunteers with an amorphous spray-dried dispersion (SDD) microcrystalline version of CDDO-Me to increase bioavailability and this was found to be superior in bioavailability to the microcrystalline version used in the Hong trial [12] (Table 1).
3 Signaling Pathways Affected by CDDO
3.1 Antioxidant Response and Nrf2 Regulation
Although CDDO compounds may directly interact with proteins in multiple signaling pathways, the primary mode of CDDO action is the upregulation of Nrf2. Nrf2 is in the Cap “n” Collar (CNC) family of basic leucine zipper (bZip) transcription factors constitutively expressed in the cell and resident in the cytoplasm [13, 14]. The forked Keap1, a substrate adaptor of the E3 ubiquitin ligase Cullin3 complex, contains two large spheres (β-propeller regions) that repress Nrf2 constitutively and represents the most popular target of treatments to modulate Nrf2 activity [15–17]. Upon ubiquitination, Nrf2 is rapidly degraded by the proteasome, but CDDO (and some other small electrophilic molecules) can bind to a key cysteine residue in the Broad complex, Tramtrack, and Bric-a-Brac (BTB) domain of Keap1 to inhibit ubiquitination and proteasomal degradation of Nrf2 [15, 18–20]. Other signaling pathways of Keap1 regulation have been reported, specifically that p21Cip1/WAF1 can compete with Nrf2 for Keap1 binding, increasing levels of free Nrf2 to localize to the nucleus and that p62 directly binds to Keap1 on three specific arginine residues to inhibit Keap-1-mediated Nrf2 ubiquitination (linking Nrf2 and autophagy) [21, 22]. Whether or not these alternate pathways may be affected by CDDO is still unclear. If Keap1 is active, the half-life of Nrf2 is very short, on the order of about 20 min [23]. Although this may make probing for nominal levels of Nrf2 difficult, the rapid turnover allows for rapid response. Once translocated to the nucleus, Nrf2 binds with the adaptor protein Maf and binds to antioxidant response elements (ARE) which attracts CREB and p300 to form a complex that can attract RNA polymerases to transcribe antioxidant genes such as superoxide dismutase (SOD), glutathione peroxidase (GPx), gamma-glutamylcysteine synthetase (γ-GCS), HO-1, and NQO1 [13, 24–27]. The Keap1-Nrf2 axis in antioxidant response has been extensively reviewed and, as CDDO has a potent ability to effect a conformational change in Keap1 to reduces its ability to catalyze Nrf2 for K48 ubiquitination, this axis serves as the basis for CDDO’s use in disease therapy models [13, 16, 24–26, 28]. Therefore, it is safe to say that any use of CDDO will involve Nrf2 as an upstream modulator of genes of interest, made possible by Nrf2’s master ability to affect downstream pathways by ARE activity. Note that natural compounds such as α-lipoic acid and polyphenols like quercetin have also been extensively shown to increase Nrf2 activity by upstream pathways such as PI3 K/Akt, especially in liver and cardiovascular studies [29–33] (Fig. 1).

a The Keap1-Nrf2 regulatory pathway features Cul3-mediated K48-linked polyubiquitination of Nrf2, causing subsequent degradation in the proteasome. b Damage by ROS or interaction with CDDO on Cysteine 151 of Keap1 releases Nrf2 to the nucleus, where it upregulates antioxidant protective factors that can counter inflammation and related disease states. c Nrf2 regulates many apoptotic factors in cancer cells that it does not in healthy cells. This may be due to defects in cellular metabolism that render cancer cells uniquely vulnerable to Nrf2-mediated apoptosis
4 Reactive Oxygen Species and Cell Death: The Primary Target of CDDO
4.1 Apoptosis and Necrosis: CDDO Affects Cell Death via Nrf2
The two known types of cell death are apoptosis and necrosis. Cellular damage from reactive oxygen species (ROS) or nitrogen species (NOS) can affect mitochondria, membranes, and cell nuclei, triggering checkpoint genes such as p53 to induce apoptosis or system wide damage, resulting in necrosis [34]. In apoptosis, death occurs in an “implosive” style with programmed and sequential events shutting down the cell to avoid damage to surrounding cells. Necrosis, on the other hand, can be considered “explosive,” where cellular debris (especially highly reactive mitochondrial cytochrome c) and cytokine release cause inflammation and damage to cascade into surrounding tissue [35]. Nrf2 is directly involved in reducing necrotic cell death by upregulation of antioxidant factors such as HO-1, super oxide dismutase (SOD), and NQO1, but can actually cause apoptosis in cancer cells by affecting the upregulation of apoptotic factors such as Snail, slug, TCF-/ZEB1, and Bax [27, 36]. If Nrf2 can control so many factors critical in apoptosis and necrosis, it stands to reason that upregulation of Nrf2 by CDDO compounds may provide protection and/or ablation of cytotoxic ROS-induced death in normal cells while causing cancerous or abnormal cells to die.
In prostate cancer cells, CDDO-Me was reported to activate caspases 3, 8, and 9 while disrupting NF-κB signaling through direct inhibition of iκB kinase, killing the cancer cells [37, 38]. It was also documented that CDDO-Me induced prostate cancer cell death via suppression of Akt [39]. Similar results were found in ovarian cancer cells and rat kidney reperfusion injury [40–42]. In acute myeloid leukemia (AML) cells, CDDO-Me has been reported to suppress phosphorylation of ERK1/2 through the activation of p38/MAPK (43). Intriguingly, another report showed that, in AML, CDDO-Me could sensitize cells to pro-apoptotic TRAIL while downregulating anti-apoptotic FLIP levels and that CDDO strongly upregulates caspase 8 [44, 45]. In iMycEµ mice that are prone to B and plasma cell neoplasms (viz. lymphoma), CDDO-Im treatment caused upregulation of Fmo4 and P450 oxygenases with downregulation of c-Myc and apoptosis [46]. Yates et al. demonstrated that CDDO-Im mitigates the aflatoxin-induced oxidative stress in the liver with an increase in GSTA2, GSTA5, AFAR, and EPHX1 antioxidant genes [47, 48]. Bey et al. [49] reported that NQO1, which is a primary transcriptional target of Nrf2 and is upregulated upon CDDO treatment, is required for PARP1-programmed necrosis in breast cancers, which can be apoptosis resistant. On the flip side of ROS-induced injury and cellular protection, Li et al. [50] have reported that doxorubicin-induced cardiac necrosis can be ameliorated by Nrf2 increases. Treatment with CDDO could provide the increased Nrf2 necessary to ablate doxorubicin-induced cardiac injury. In the normal liver, caspases intimately associated with apoptosis and necrosis have been shown to be downregulated by lipoic acid which activates Nrf2, especially caspase-3 [31]. In normal hearts and in early-stage heart disease, Nrf2 is also very protective [16, 27, 51]. It is therefore important to note that CDDO compounds in published studies show protective effects to normal or injured cells but kill cancerous cells. Interestingly, a report that compared Keap1 knockout mice to CDDO-Im-treated mice show that both genetic and pharmacological activation of Nrf2 regulates many metabolism genes, including lipid and carbohydrate metabolism (particularly the pentose phosphate pathway to sustain Nrf2 activity) [52, 53]. This indicates that cancerous cells, which often show defects in metabolic pathways, may be adversely affected by Nrf2 upregulation forcing upregulation of apoptotic factors in these and not normal cells or cells with minor injury. In fact, a recent report by Qin et al. [54] has found a solid link between the action of Nrf2 and the functional status of autophagy: Nrf2 activation is cardioprotective when myocardial autophagy is intact whereas Nrf2 acts as a mediator of cardiac maladaptive remodeling and dysfunction when myocardial autophagy is impaired. Bernstein et al. showed a similar effect in B-cell lymphoma cells via CDDO treatment/Nrf2 activation and inhibition of the Lon protease system, which clears mitochondrial proteins [55–57]. This proves that a critical link exists between metabolism (specifically autophagy) and the effect of Nrf2 on a cell. Upon Nrf2 activation, normal cells (and even the retina) with normal autophagic function receive protection in the form of upregulated antioxidant defense while abnormal cells with autophagy impairment may see deleterious effects [58, 59]. Other oleanolic acid derivatives have also been shown to cause this induction of autophagy in normal cells that, in cells with dysfunctional autophagic machinery, can cause early death [60, 61].
5 Other Targets of CDDO: Heat Shock Protein, Telomerase, and MTOR
Although the strongest transcriptional effects from CDDO treatment come from Nrf2 activation, there have been several other direct targets reported in the literature. Suh et al. [62] reported that CDDO-Im and CDDO-ethyl amide were able to induce chondrogenesis in newborn mice by upregulating SOX9 and collagen. However, as there was no in-depth exploration as to the effect of Nrf2 on these genes, it is yet unclear as to whether or not CDDO had direct effects on upstream elements in the chondrogenic pathway. Telomerase (hTERT) activity is a critical part of cancer cell proliferation and there is a report that CDDO-Me targets hTERT in prostate cancer cells, with knockdown of hTERT increasing apoptosis upon CDDO treatment [63]. Heat shock protein 90 (hsp90) was found by Qin et al. [64] to directly target hsp90 in an ovarian cancer cell model, inhibiting it and reducing cell proliferation. This was shown by thermal shift assay and can be blocked by dithiothreitol [64]. Although several putative targets have been reported, only the hsp90 interaction has been shown to be a direct effect of CDDO and not as a Nrf2-mediated effect. Although these several reports have shown some potential of CDDO to target alternate pathways directly, the majority of evidence points to a primarily Nrf2-mediated mechanism of action. However, the specificity of CDDO for Nrf2 makes it useful in isolating Nrf2-related pathways and mechanisms with little interference from other upstream regulators.
6 Diseases
The goal of CDDO as a therapy is to exploit its ability to upregulate Nrf2 and Nrf2’s ability to protect healthy cells from necrosis while destroying abnormal ones by apoptosis. Because CDDO and its derivatives are fairly nontoxic, they lend themselves well to testing as therapies for various diseases.
6.1 Cardiac Disease/Vascular Dysfunction
Much work has been done in animal models with CDDO in the prevention of cardiopulmonary disease and injury. Sussan et al. [16] reported that, in mice, cigarette smoke-induced cardiac dysfunction and emphysema modeling chronic obstructive pulmonary disease (COPD) could be reduced by CDDO-Im treatment. CDDO-Im has also been shown to ameliorate obesity in mice fed a high-fat diet, showing potential for reducing a major cause of cardiovascular disease [65]. CDDO could also play a key role in attenuating lesion formation and loss of tone in vascular disease, especially as FLIP/TRAIL and Myc, lesion forming factors regulated by Nrf2, play a role in VSMC-mediated neointimal formation [46, 66–68]. Wang reviews several preclinical trials that indicate that CDDO-Me can reduce blood vessel inflammatory responses by regulating the endothelin pathway and that this may be due to involvement of NF-κB in the endothelin pathway which Nrf2 can counter [69]. It is well established in the literature that iNOS and cytokines like IFNγ produced by macrophages activated by periodontal diseases or LPS can cause inflammatory damage in blood vessels, recruiting more macrophages in an M1 response and amplifying vascular damage [3, 70, 71]. CDDO-dhTFEA (dh404) and CDDO-Me have been shown to suppress the inflammatory responses in macrophages, thereby providing protection to the vascular system [71, 72]. Regarding future cardiac studies, vital cardiac adaptation has been shown to rely on a Nrf2/autophagy axis, while clearance of toxic proteins relies on Nrf2, making CDDO treatment a distinct possibility to upregulate those protective features [51, 73].
Studies in humans with CDDO compounds have been completed up to Phase II. A review by Wang lists several Phase I studies that evaluated several pharmacokinetic parameters of CDDO-Me administration in healthy volunteers [69]. Subsequent Phase II trials have either been terminated or withdrawn. A CDDO-Me evaluation in patients with pulmonary hypertension is currently recruiting patients (clinicaltrials.gov NCT02036970). Clearly, CDDO usage in humans carries some kind of cardiovascular risk and the knowledge of autophagic sufficiency for CDDO efficacy in the cardiovascular system may provide a critical insight on targeting upstream regulators of both Nrf2 and autophagy.
6.2 Kidney Disease
Another realm of intense research into CDDO and related compounds is in chronic kidney disease (CKD). Impacting almost 13 % of the US population, CKD is a reduction in estimated glomerular filtration rate (eGFR) along with albuminuria (protein in the urine) that eventually results in a need for renal replacement and sequelae such as anemia and metabolic bone disease [74]. In fact, cardiovascular complications from CKD anemia such as left ventricular hypertrophy due to maladaptation can affect the kidneys further, creating a vicious loop of escalating damage termed “cardiorenal anemia syndrome” that drastically reduces patient long-term survivability by 30 % [74]. To ameliorate the multiple effects of CKD, CDDO in its multiple forms has been extensively tested in animals and clinical trials have been held in humans. In rats, CKD studies have shown improvement in the areas of ROS, inflammation, and fibrosis, with CDDO-TFEA and CDDO-Me [41, 75]. Liu et al. [76] have found CDDO-Im to protect kidneys from ischemic reperfusion injury, which models the damage of CKD, and this protection is entirely dependent on Nrf2 as Nrf2-knockout mice treated with CDDO-Im showed no improvement. Wu et al. [77] showed that CDDO was able to ameliorate lupus-induced nephritis by reduction in ERK, STAT3, and Nf-κB, resulting in decreased CD4 T cell activation. Shelton et al. [78] conducted an extensive proteomic and transcriptomic analysis of wild type and Nrf2 knockout mice treated with CDDO-Me and found that CDDO-Me via Nrf2 upregulation positively regulated proteins related to redox homeostasis and NADPH regulation. The authors also concluded that CDDO would be useful in countering xenobiotics (such as cisplatin or cyclosporin) that generate large amounts of nephrotoxic molecules as well as chronic kidney insults from heavy metals [78, 79]. This clearly points to CDDO’s potential to protect the kidneys during chemotherapy which Aleksunes et al. [80] explored in mice, finding that CDDO-Im could protect from cisplatin-induced nephrotoxicity. On the structural level, Aminzadeh et al. [81] found that CDDO-TFEA could ameliorate damage due to the cardiorenal axis with restoration of endothelial function in CKD rat aortic rings as measured by acetylcholine-mediated relaxation response. Additionally, CKD-induced aortic upregulation of MCP-1 angiotensin II and NADPH oxidases was all ameliorated by CDDO-TFEA [81].
These animal studies serve to illustrate that CDDO compounds are of great value in renal protection against chronic diseases. Indeed, in a Phase I trial conducted by Hong et al. in 2012, 47 patients diagnosed with solid tumors and lymphomas were given CDDO-Me in microcrystalline form for 21 consecutive days out of a 28-day cycle, with multiple cycles and saw an increase in eGFR estimated at 26 % in all patients with a 33.9 % increase in the highest dosage [7]. Clearly, CDDO-Me improved kidney health that may have been ravaged by antineoplastics and other chemotherapeutics. A Reata Pharmaceuticals-funded Phase II trial from 2008 to 2009 (clincaltrials.gov NCT00811889) found significant improvement in eGFR in patients treated with CDDO-Me at 24 weeks, with up to 10.5 additional ml (per minute per 1.73 m2 body surface area) in the 75 mg dosage range [82]. Another Phase II study in 2010 by Reata (clinicaltrials.gov NCT01053936) evaluating CDDO-Me in eGFR in type 2 diabetes as well as another study (clinicaltrials.gov NCT00664027) was completed with no published data. Unfortunately, after initial Phase II success, a Phase III trial in 2014 (clinicaltrials.gov NCT013516750) was terminated due to an increase in cardiovascular adverse events, even though eGFR, renal function, and body weight improved significantly [83] (Table 2).
6.3 AML
Leukemia, especially acute myeloid leukemia, is defined as an increase in myeloid cells within the bone marrow and a subsequent insufficiency of the hematopoietic cells due to their failure to mature [84]. Ito et al. [45] reported that human AML cells underwent caspase 8-mediated apoptosis when treated with CDDO. Subsequent reports found that CDDO-Me could induce apoptosis in acute myelogenous leukemia, suppress MAPK in these cells, increase TRAIL sensitization, downregulate FLIP, promote hematopoietic progenitor expansion, and upregulate p42 CCAAT enhancer-binding protein alpha in granulocytes [43, 44, 85–87]. Although not yet used in human clinical trials, CDDO compounds show a clear effect in promoting aberrant leukocytes to undergo apoptosis while pushing differentiation/maturation of immature cells forward (Table 3).
6.4 Retinal Blindness
Damage to the eyes of diabetic patients is a well-known diabetic complication. Age-related macular degeneration due to oxidative damage may also be a concern in a rapidly aging population. A cytoprotective effect of CDDO-TFEA, CDDO-Im, and CDDO-Me was seen in retinal cell lines to protect against oxidation-induced retinal degeneration and the lipid phosphatase PTEN was inhibited in mice treated with CDDO-TFEA [59]. Wei et al. [88] found that Nrf2 protects both neurons and capillaries from retinal ischemia-reperfusion injury so the action of CDDO is protective to the retina via upregulation of Nrf2. Xu et al. [89] verified this in a separate report, showing that Nrf2 knockout mice experienced a greater loss of retinal neuron function. Other reports verified that Nrf2 can modulate cigarette smoke-induced complement activation in the retina and that Nrf2 is a critical modulator of oxidation-induced death in the retinal ganglion [90, 91]. Experiments in astrocytes, microglia, and neurons showed that all CDDO compounds are protective against oxidative damage and upregulate antioxidant genes via Nrf2 [92]. Taken together, these results show that CDDO may hold promise for protecting the brain from age-related oxidative stress as well as treating such chronic eye diseases as macular degeneration and tobacco smoke-induced injury.
6.5 Cancer
CDDO compounds have been rigorously tested in multiple rodent cancer models, as its ability to force abnormal cells into apoptosis could form the basis of a chemotherapeutic approach that, unlike current xenobiotics and chemotherapeutics, also simultaneously protects the liver and kidneys. CDDO-Me has been used in colitis-associated colon cancer models in mice to interrupt inflammation-driven downregulation of prostaglandin dehydrogenase as well as the entire suite of inflammatory cytokines such as IL-6, iNOS, IL-1β, and TNFα [93]. A study by Alabran et al. that used CDDO compounds against multiple human neuroblastoma cell lines found that these cells underwent rapid arrest in S-phase, Bax was activated (apoptosis induction), and that CDDO compounds were effective against these cells in low concentrations (IC50 5–170 nM) [9]. CDDO-Me has also been found to downregulate telomerase activity (hTERT) and induce cell death in pancreatic cancer cell lines [94]. However, it is unclear as to whether this hTERT inhibition is a direct interaction or an Nrf2-associated effect. A review by Shanmugam et al. details oleanolic acid derivatives and the genes they primarily affect, such as mTOR, AKT, STAT3, ICAM1, and PADP polymerase—all of which are critical for regulating cellular homeostasis and which may be compromised in transformed cells [95, 96]. As cancer cells use vascular endothelial growth factor (VEGF)-driven angiogenesis to form new capillary feeder networks, it is interesting to note that CDDO-Me has been reported in mice to suppress Matrigel plug angiogenesis in picomolar concentrations [18]. Coupled with induced arrest and apoptosis, a one-two punch of cutting off the blood supply and then inducing tumor cell death is an attractive prospect for a chemotherapeutic drug. It is important to note that CDDO compounds may harm cancerous cells but it must be remembered that normal cells are protected against insult [97]. Again, this may be due to a need for autophagic competency to accompany increased Nrf2 levels in order to be protected or in the mitochondrial Lon protease system, which is inhibited by CDDO [57]. In a rat liver cancer model involving aflatoxin, CDDO-Im proved a powerful and complete protection, with damage almost completely ablated by treatment [98]. Liby et al. [99] found that CDDO-Me and CDDO-ethyl amide could protect A/J mice against vinyl carbamate-induced lung cancer. This proves that CDDO compounds may be a powerful prophylactic against specific types of environmental carcinogens that transform cells by ROS damage.
Not only can CDDO protect against somatic cancers but increasing evidence shows that it is effective against gender-specific cancers, as well. Interestingly, a report by Gao et al. revealed that CDDO-Me controls apoptosis in ovarian cancer by inhibiting AKT, NF-κB, and mTOR signaling without affecting PDK1 kinase or PP2A activity [39, 100]. Mammary carcinogenesis in polyoma middle T mice was slowed by CDDO-Me, extending lifespan by roughly 5 weeks, and BRCA1-mediated cancers in mice are delayed by CDDO-Me, as well [101, 102]. In prostate cancer, CDDO-Me regulates Bcl and other survival signals in TRAMP mice and hTERT can also be targeted by CDDO-Me in prostate cancer [63, 103]. There is also evidence that Hsp90 might be targeted by CDDO-Me in cancer cells [64].
There is a recent theory of cancer stem cells, which are cells from a tumor that possess stem cell abilities to differentiate into different cancer types. Current reports show that CDDO-Me can suppress stemness in esophageal cancer lines and triple negative breast cancer cells [36, 104].
Other oleanolic acid derivatives, such as SZC017, CDDO-2P-Im, CDDO-3P-Im, and HIMOXOL, are being evaluated for anticancer effects such as apoptotic induction ability and cancer cell arrest [60, 61, 105]. Also, vehicles for efficient delivery of CDDO by nanoparticles have been explored [106] (Table 4).
There have been several clinical trials for cancer treatment using CDDO-Me. Hong et al. accomplished a Phase I trial which showed promise in hepatoprotection but did not show significant improvement in tumor size as seen in animal models [7]. However, two Phase II studies in patients with advanced solid tumors (clinicaltrials.gov NCT00508807, NCT00529438) were completed but results were not disclosed. Although in vitro and animal models show great promise in using CDDO compounds as cancer treatments, a lack of published data in the two completed clinical trials makes it difficult to evaluate CDDO as a promising chemotherapeutic.
6.6 Liver
The liver is the most critical organ in the body for metabolic processes and detoxification by the cytochrome enzymatic pathways. This makes it susceptible to not just ROS damage but other hepatotoxic molecules that may be generated by medicines, chemical compounds, or alcohols. Shah et al. [107] found that CDDO-Im could protect HepG2 cells against acrolein-induced toxicity by GSH upregulation as well as reducing levels of death markers such as protein carbonyls. A report by Liu as far back as 1993 established that oleanolic acid could rescue large-dosage acetaminophen liver damage in mice and upregulate GSH levels [1]. This led to the logical conclusion that oleanolic acid derivates with even higher Nrf2 stimulating activity could protect the liver even better. As such, CDDO-dhTFEA was reported to induce hepatoprotective genes including thioredoxin reductase (Txnrd), glutamate cysteine ligase catalytic and modifier subunits (Gclc and Gclm), gamma-glutamyl transpeptidase 1 (Ggt1), heme oxygenase-1 (Ho-1), and NAD(P)H quinone oxidoreductase 1 (Nqo1), while also increasing bile flow in rats, as bile is related to GSH levels [108]. A review by Klaassen collates over 15 separate studies in rats and mice where hepatotoxic compounds such as acetaminophen, concavalin A, and high-fat diets saw their damage ameliorated by CDDO-Im or oleanolic acid [26]. This clearly indicates that Nrf2 is strongly hepatoprotective, especially against hepatic damage induced by medications for chronic diseases (e.g., large doses of acetaminophen for arthritis) or metabolic syndrome. Also of note in the Klaassen review is a summary of 6 Nrf2 knockout rodent models for use in studying liver disease and CDDO treatment, such as carbon tetrachloride and arsenic [26]. A comprehensive proteomic report by Walsh et al. [109] has found that CDDO-Me in mice induces cytochrome P4502A5, glutathione-S-transferase, UDP-glucose-6-dehydrogenase, and epoxide hydrolase, prompting the liver to begin detoxification with P450 enzymes while simultaneously protecting from subsequent free radical activity by inducing antioxidant response enzymes. Most importantly, they found that 97 % of the proteins induced by CDDO-Me were specific for Nrf2 signaling, reassuring researchers that non-Nrf2-targeting effects during treatment would be kept to a minimum [78]. As seen in other organs, CDDO compounds provide strong hepatoprotective abilities that may be useful in combatting damage from medications, environmental chronic exposure, and metabolic syndrome.
6.7 Sepsis/Sickle Cell/Lupus
The presence of lipopolysaccharides (LPS) in the bloodstream may produce an aggressive immune response including inflammation. LPS, along with active bacteria, may also induce a massive cytokine release, dilating the capillary network, and causing a severe and often fatal drop in blood pressure. This septic shock is often fatal. Noel et al. [110] explored ex vivo Nrf2 activation by CDDO-Me administration to monocytes obtained from human patients with septic shock. Their results showed a differential activation between purified and peripheral monocytes, with purified monocytes decreasing in IL-6 production and peripheral monocytes increasing IL-6 production [110]. Another study using neutrophils and peripheral monocytes showed strong activation of antioxidant response and attenuation of inflammatory cytokine response (TNFα,MLP, etc.) upon treatment with CDDO-Im and CDDO-Me, revealing that Nrf2 activation may be protective against challenge with LPS [111]. An interesting report by Keleku-Lukwete et al. [112] showed that Nrf2 could modulate the clearance of plasma heme in a mouse sickle cell model with administration of CDDO-Im relieving organ inflammation and failure in the model mice. These studies indicate that CDDO can be useful in sepsis as well as in chronic inflammatory diseases such as sickle cell anemia.
7 Conclusion
Oleanolic acid has been used in China as traditional medicine for liver problems and can be found in many plants and foods [113]. Having shown promise in multiple organ systems, CDDO compounds (by virtue of the ability to strongly upregulate Nrf2) seemed set to serve as a panacea for chronic diseases. In the liver, kidneys, retina, blood, and bone marrow, CDDO works to upregulate Nrf2, which is highly protective against oxidative damage and also to modulate a host of signaling pathways, including Akt/PI3 K, mTOR, FLIP/TRAIL, and Myc. CDDO-mediated upregulation of Nrf2 in these organs and in cancer cell lines and animal models induces apoptosis of aberrant cells and protects normal cells against insult from both oxidative stress and environmental insult.
Unfortunately, CDDO has not shown promise in the amelioration of cardiovascular disease or human cancers. Although in vitro and in vivo animal studies have shown excellent results, systemic effects of CDDO have not been fully elucidated and, in human clinical trials, CDDO has actually shown deleterious effects in the heart. Although multiple studies have shown that, at least early on in heart disease, Nrf2 upregulation is cardioprotective, a seminal finding in mice has shown very strong evidence that a lack of Nrf2 response in later stage heart disease is actually protective [51, 114, 115]. Recent literature has dubbed this effect as “reductive stress” (RS) as opposed to “oxidative stress” (OS) and that too many antioxidants can actually cause damage to the tissue as they reduce (donate electrons to reduce charge as opposed to steal them and add charge) [116]. DNA, with a negatively charged backbone, could be easily damaged by reductive attack, damaging it repair. Clinical reports are filtering in that finding reductive stress can occur in post-surgical complications such as restenosis of balloon angioplasty and stenting (BAS) and post-exercise [117, 118]. Additionally, in cancer, high antioxidant responses may be protective to the cancer cells against ROS-inducing chemotherapy as well as ROS generated by their much higher basal metabolic rate. Nrf2 can also cause upregulation of genes that metabolize chemotherapeutics as well as upregulate the pentose phosphate pathway, increasing tumor cell proliferation and survival [28, 52, 53, 78].
Intriguingly, recent reports have found that the functional integrity of autophagy is needed to reap positive benefits from Nrf2 regulation, with upregulation of Nrf2 and autophagic dysfunction associated with negative outcomes [119]. It may be that Nrf2 upregulation can increase scavenging of free radicals but can also damage cells by reductive stress from which they cannot recover without autophagy to recycle reduced membrane or other cell components. In fact, since some antioxidants can be more damaging than free radicals (e.g., ECGC as reported by Lu et al.), it is entirely possible that a lack of autophagy is a necrotic death sentence for a reductively damaged cell [120]. In this case, it would be critical to regulate both Nrf2 and autophagy simultaneously by regulating a common upstream element. The deubiquitinase CYLD is a master key in the NF-κB pathway regulating inflammation and immune development, but has been shown to affect many other pathways such as TLR-mediated signaling, Wnt/Catenin, and Snail [121]. CYLD has been recently shown in a seminal report to regulate Nrf2 transcriptionally, repressing it via downregulation of the p38 MAPK/ERK pathway [122]. If CYLD could also be shown to negatively regulate autophagy, then it would be possible to downregulate CYLD locally in the heart and preserve autophagic function along with Nrf2 activation which would be the best of both worlds. Since there are critical autophagic components, such as p62 and HDAC6, that are K63 polyubiquitinated, it may be possible that CYLD can control autophagy by enzymatic action on p62 or some other component of the pathway [123, 124]. In conclusion, CDDO holds some promise for certain types of chronic diseases, but cardiovascular and cancer therapies might benefit if master upstream elements like CYLD could be exploited to control both Nrf2 and autophagic mechanisms to prevent damage from reductive stress.
Abbreviations
- CDDO:
-
2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid
- CDDO-Im:
-
CDDO imidazolide
- CDDO-Me:
-
CDDO-methyl ester
- CDDO-dhTFEA:
-
CDDO-dihydrotrifluoroamide
References
Liu J, Liu Y, Madhu C, Klaassen CD (1993) Protective effects of oleanolic acid on acetaminophen-induced hepatotoxicity in mice. J Pharmacol Exp Ther 266(3):1607–1613
Honda T, Finlay HJ, Gribble GW, Suh N (1997) MBS. New enone derivatives of oleanolic acid and ursolic acid as inhibitors of nitric oxide production in mouse macrophages. Bioorg Med Chem Lett 7(13):1623–1628
Honda T, Suh N (1998) Bioorganic Med Chem Lett 8:2711–2714
Suh N, Wang Y, Honda T, Gribble GW, Dmitrovsky E, Hickey WF et al (1999) A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res 59(2):336–341
Couch RD, Browning RG, Honda T, Gribble GW, Wright DL, Sporn MB et al (2005) Studies on the reactivity of CDDO, a promising new chemopreventive and chemotherapeutic agent: implications for a molecular mechanism of action. Bioorganic Med Chem Lett. 15(9):2215–2219
Fu L, Gribble GW (2013) Efficient and scalable synthesis of bardoxolone methyl (cddo-methyl ester). Org Lett 15(7):1622–1625
Hong DS, Kurzrock R, Supko JG, He X, Naing A, Wheler J et al (2012) A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin Cancer Res 18(12):3396–3406
Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T et al (2007) Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther 6(1):154–162
Alabran JL, Cheuk A, Liby K, Sporn M, Khan J, Letterio J et al (2008) Human neuroblastoma cells rapidly enter cell cycle arrest and apoptosis following exposure to C-28 derivatives of the synthetic triterpenoid CDDO. Cancer Biol Ther 7(5):709–717
Noker Patricia E, Gorman Gregory S, Schweikart Karen M, Tomaszewski Joseph E, Sporn MB, Page JG (2004) Pharmacokinetics and toxicity of CDDO, a synthetic triterpenoid, in rats and dogs. Cancer Res 45:471
Perez HL, Junnotula V, Knecht D, Nie H, Sanchez Y, Boehm JC et al (2014) Analytical approaches for quantification of a Nrf2 pathway activator: overcoming bioanalytical challenges to support a toxicity study. Analyst. 139(8):1902–1912
Thomas M (2012) A preliminary evaluation of bardoxolone methyl for the treatment of diabetic nephropathy. Expert Opin Drug Metab Toxicol. 8(8):1015–1022
Jaramillo MC, Zhang DD (2013) The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev 27(20):2179–2191
Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426
Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA et al (2014) Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS ONE 9(6):e98896
Sussan TE, Rangasamy T, Blake DJ, Malhotra D, El-Haddad H, Bedja D et al (2009) Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci USA 106(1):250–255
Ogura T, Tong KI, Mio K, Maruyama Y, Kurokawa H, Sato C et al (2010) Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc Natl Acad Sci USA 107(7):2842–2847
Vannini N, Lorusso G, Cammarota R, Barberis M, Noonan DM, Sporn MB et al (2007) The synthetic oleanane triterpenoid, CDDO-methyl ester, is a potent antiangiogenic agent. Mol Cancer Ther 6(12 Pt 1):3139–3146
Winkel AF, Engel CK, Margerie D, Kannt A, Szillat H, Glombik H et al (2015) Characterization of RA839, a non-covalent small-molecule binder to Keap1 and selective activator of Nrf2 signalling. J Biol Chem 17:jbc.M115.678136
Kobayashi A, Kang M-I, Okawa H, Ohtsuji M, Zenke Y, Chiba T et al (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24(16):7130–7139
Chen W, Sun Z, Wang X-J, Jiang T, Huang Z, Fang D et al (2009) Direct Interaction between Nrf2 and p 21Cip1/WAF1 upregulates the Nrf2-mediated antioxidant response. Mol Cell 34(6):663–673
Lau A, Wang X-J, Zhao F, Villeneuve NF, Wu T, Jiang T et al (2010) A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 30(13):3275–3285
Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M (2003) Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8(4):379–391
Sporn MB, Liby KT (2012) NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer 12(8):564–571
Kansanen E, Kuosmanen SM, Leinonen H, Levonen A-L (2013) The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 1(1):45–49
Klaassen CD, Reisman S (2010) Nrf2 the rescue: effects of the antioxidative/electrophilic response on the liver. Toxicol Appl Pharmacol 244(1):57–65
Xing Y, Niu T, Wang W, Li J, Li S, Janicki JS et al (2012) Triterpenoid dihydro-CDDO-trifluoroethyl amide protects against maladaptive cardiac remodeling and dysfunction in mice: a critical role of Nrf2. PLoS ONE 7(9):e44899
Leinonen HM, Kansanen E, Pölönen P, Heinäniemi M, Levonen A-L (2014) Role of the Keap1-Nrf2 pathway in cancer. Adv Cancer Res 122:281–320
Tanigawa S, Fujii M, Hou D-X (2007) Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic Biol Med 42(11):1690–1703
Ahmed MAE, El-Awdan SA (2015) Lipoic acid and pentoxifylline mitigate nandrolone decanoate-induced neurobehavioral perturbations in rats via re-balance of brain neurotransmitters, up-regulation of Nrf2/HO-1 pathway, and down-regulation of TNFR1 expression. Horm Behav 73:186–199
Valdecantos MP, Prieto-Hontoria PL, Pardo V, Módol T, Santamaría B, Weber M et al (2015) Essential role of Nrf2 in the protective effect of lipoic acid against lipoapoptosis in hepatocytes. Free Radic Biol Med 84:263–278
Deng C, Sun Z, Tong G, Yi W, Ma L, Zhao B et al (2013) α-Lipoic acid reduces infarct size and preserves cardiac function in rat myocardial ischemia/reperfusion injury through activation of PI3 K/Akt/Nrf2 pathway. PLoS ONE 8(3):e58371
Na H-K, Surh Y-J (2008) Modulation of Nrf2-mediated antioxidant and detoxifying enzyme induction by the green tea polyphenol EGCG. Food Chem Toxicol 46(4):1271–1278
Moallem SA, Hales BF (1998) The role of p53 and cell death by apoptosis and necrosis in 4-hydroperoxycyclophosphamide-induced limb malformations. Development 125(16):3225–3234
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B et al (2012) Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 45(15):487–498
Wang Y-Y, Yang Y-X, Zhao R, Pan S-T, Zhe H, He Z-X et al (2015) Bardoxolone methyl induces apoptosis and autophagy and inhibits epithelial-to-mesenchymal transition and stemness in esophageal squamous cancer cells. Drug Des Devel Ther 9:993–1026
Wang Y-Y, Zhe H, Zhao R (2014) Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol Cancer 13:30
Yore MM, Liby KT, Honda T, Gribble GW, Sporn MB (2006) The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor- B activation through direct inhibition of I B kinase. Mol Cancer Ther 5(12):3232–3239
Liu Y, Gao X, Deeb D, Gautam SC (2012) Oleanane triterpenoid CDDO-Me inhibits Akt activity without affecting PDK1 kinase or PP2A phosphatase activity in cancer cells. Biochem Biophys Res Commun. 417(1):570–575
Gao X, Liu Y, Deeb D, Arbab AS, Guo AM, Dulchavsky SA et al (2011) Synthetic oleanane triterpenoid, CDDO-Me, induces apoptosis in ovarian cancer cells by inhibiting prosurvival AKT/NF-κB/mTOR signaling. Anticancer Res 31(11):3673–3681
Kocak C, Kocak EF, Akcilar R, Bayat Z, Aras B, Metineren MH et al (2015) Effects of captopril, telmisartan, and bardoxolone methyl (CDDO-Me) in ischemia reperfusion-induced acute kidney injury in rats: an experimental comparative study. Clin Exp Pharmacol Physiol
Deeb D, Gao X, Dulchavsky SA, Gautam SC (2007) CDDO-me induces apoptosis and inhibits Akt, mTOR and NF-kappaB signaling proteins in prostate cancer cells. Anticancer Res 27(5A):3035–3044
Konopleva M, Contractor R, Kurinna SM, Chen W, Andreeff M, Ruvolo PP (2005) The novel triterpenoid CDDO-Me suppresses MAPK pathways and promotes p 38 activation in acute myeloid leukemia cells. Leukemia 19(8):1350–1354
Suh W-S, Kim YS, Schimmer AD, Kitada S, Minden M, Andreeff M et al (2003) Synthetic triterpenoids activate a pathway for apoptosis in AML cells involving downregulation of FLIP and sensitization to TRAIL. Leuk Off J Leuk Soc Am Leuk Res Fund UK 17(11):2122–2129
Ito Y, Pandey P, Place A, Sporn MB, Gribble GW, Honda T et al (2000) The novel triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid induces apoptosis of human myeloid leukemia cells by a caspase-8-dependent mechanism. Cell Growth Differ 11(5):261–267
Han S-S, Peng L, Chung S-T, DuBois W, Maeng S-H, Shaffer AL et al (2006) CDDO-Imidazolide inhibits growth and survival of c-Myc-induced mouse B cell and plasma cell neoplasms. Mol Cancer. 5:22
Shen H, Liu J, Wang Y, Lian H, Wang J, Xing L et al (2013) Aflatoxin G1-induced oxidative stress causes DNA damage and triggers apoptosis through MAPK signaling pathway in A549 cells. Food Chem Toxicol 62:661–669
Yates MS (2006) Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res 66(4):2488–2494
Bey EA, Reinicke KE, Srougi MC, Varnes M, Anderson VE, Pink JJ et al (2013) Catalase abrogates β-lapachone-induced PARP1 hyperactivation-directed programmed necrosis in NQO1-positive breast cancers. Mol Cancer Ther 12(10):2110–2120
Li S, Wang W, Niu T, Wang H, Li B, Shao L et al (2014) Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid Med Cell Longev
Wang W, Li S, Wang H, Li B, Shao L, Lai Y et al (2014) Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J Mol Cell Cardiol 72:305–315
Yates MS, Tran QT, Dolan PM, Osburn WO, Shin S, McCulloch CC et al (2009) Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 30(6):1024–1031
Heiss EH, Schachner D, Zimmermann K, Dirsch VM (2013) Glucose availability is a decisive factor for Nrf2-mediated gene expression. Redox Biol 1(1):359–365
Qin Q, Qu C, Niu T, Zang H, Qi L, Lyu L et al (2016) Nrf2-mediated cardiac maladaptive remodeling and dysfunction in a setting of autophagy insufficiency. Hypertension 67(1):107–117
Bernstein SH, Venkatesh S, Li M, Lee J, Lu B, Hilchey SP et al (2012) The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 119(14):3321–3329
Ngo JK, Pomatto LCD, Davies KJA (2013) Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 1:258–264
Gibellini L, Pinti M, Bartolomeo R, De Biasi S, Cormio A, Musicco C et al (2015) Inhibition of Lon protease by triterpenoids alters mitochondria and is associated to cell death in human cancer cells. Oncotarget. 6(28):25466–25483
Hsieh M-J, Yang S-F, Hsieh Y-S, Chen T-Y, Chiou H-L (2012) Autophagy inhibition enhances apoptosis induced by dioscin in huh7 cells. Evid Based Complement Alternat Med 2012:134512
Pitha-Rowe I, Liby K, Royce D, Sporn M (2009) Synthetic triterpenoids attenuate cytotoxic retinal injury: cross-talk between Nrf2 and PI3K/AKT signaling through inhibition of the lipid phosphatase PTEN. Invest Ophthalmol Vis Sci 50(11):5339–5347
Gao L, Wang Y, Xu Z, Li X, Wu J, Liu S et al (2015) SZC017, a novel oleanolic acid derivative, induces apoptosis and autophagy in human breast cancer cells. Apoptosis 20(12):1636–1650
Lisiak N, Paszel-Jaworska A, Bednarczyk-Cwynar B, Zaprutko L, Kaczmarek M, Rybczyńska M (2014) Methyl 3-hydroxyimino-11-oxoolean-12-en-28-oate (HIMOXOL), a synthetic oleanolic acid derivative, induces both apoptosis and autophagy in MDA-MB-231 breast cancer cells. Chem Biol Interact 5(208):47–57
Suh N, Paul S, Lee HJ, Yoon T, Shah N, Son AI et al (2012) Synthetic triterpenoids, CDDO-Imidazolide and CDDO-Ethyl amide, induce chondrogenesis. Osteoarthritis Cartilage 20(5):446–450
Liu Y, Gao X, Deeb D, Arbab AS, Gautam SC (2012) Telomerase reverse transcriptase (TERT) is a therapeutic target of oleanane triterpenoid CDDO-Me in prostate cancer. Molecules 17(12):14795–14809
Qin D-J, Tang C-X, Yang L, Lei H, Wei W, Wang Y-Y et al (2015) Hsp90 is a novel target molecule of CDDO-Me in inhibiting proliferation of ovarian cancer cells. PLoS ONE 10(7):e0132337
Shin S, Wakabayashi J, Yates MS, Wakabayashi N, Dolan PM, Aja S et al (2009) Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-Imidazolide. Eur J Pharmacol 620(1–3):138–144
Clempus RE, Griendling KK (2006) Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res 71:216–225
Chan J, Prado-Lourenco L, Khachigian LM, Bennett MR, Di Bartolo BA, Kavurma MM (2010) TRAIL promotes VSMC proliferation and neointima formation in a FGF-2-, Sp1 phosphorylation-, and NFkappaB-dependent manner. Circ Res 106(6):1061–1071
Mannion JD, Ormont ML, Magno MG, O’Brien JE, Shi Y, Zalewski A (1998) Sustained reduction of neointima with c-myc antisense oligonucleotides in saphenous vein grafts. Ann Thorac Surg 66(6):1948–1952
Wang Y-Y, Yang Y-X, Zhe H, He Z-X, Zhou S-F (2014) Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties. Drug Des Devel Ther 8:2075–2088
Miyajima S, Naruse K, Kobayashi Y, Nakamura N, Nishikawa T, Adachi K et al (2014) Periodontitis-activated monocytes/macrophages cause aortic inflammation. Sci Rep. 4:5171
Li B, Abdalrahman A, Lai Y, Janicki JS, Ward KW, Meyer CJ et al (2014) Dihydro-CDDO-trifluoroethyl amide suppresses inflammatory responses in macrophages via activation of Nrf2. Biochem Biophys Res Commun 444(4):555–561
Onyango EO, Fu L, Cao M, Liby KT, Sporn MB, Gribble GW (2014) Synthesis and biological evaluation of amino acid methyl ester conjugates of 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid against the production of nitric oxide (NO). Bioorg Med Chem Lett 24(2):532–534
Li S, Wang W, Niu T, Wang H, Li B, Shao L et al (2014) Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid Med Cell Longev 2014:748524
Thomas R, Kanso A, Sedor JR (2008) Chronic kidney disease and its complications. Prim Care Clin Off Pract 35(2):329–344
Aminzadeh MA, Reisman SA, Vaziri ND, Khazaeli M, Yuan J, Meyer CJ (2014) The synthetic triterpenoid RTA dh404 (CDDO-dhTFEA) restores Nrf2 activity and attenuates oxidative stress, inflammation, and fibrosis in rats with chronic kidney disease. Xenobiotica 44(6):570–578
Liu M, Reddy NM, Higbee EM, Potteti HR, Noel S, Racusen L et al (2014) The Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys from ischemia-reperfusion injury in mice. Kidney Int 85(1):134–141
Wu T, Ye Y, Min S-Y, Zhu J, Khobahy E, Zhou J et al (2014) Prevention of murine lupus nephritis by targeting multiple signaling axes and oxidative stress using a synthetic triterpenoid. Arthritis Rheumatol (Hoboken, NJ) 66(11):3129–3139
Shelton LM, Lister A, Walsh J, Jenkins RE, Wong MHL, Rowe C et al (2015) Integrated transcriptomic and proteomic analyses uncover regulatory roles of Nrf2 in the kidney. Kidney Int 1261–1273
Shelton LM, Park BK, Copple IM (2013) Role of Nrf2 in protection against acute kidney injury. Kidney Int 84(6):1090–1095
Aleksunes LM, Goedken MJ, Rockwell CE, Thomale J, Manautou JE, Klaassen CD (2010) Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J Pharmacol Exp Ther 335(1):2–12
Aminzadeh MA, Reisman SA, Vaziri ND, Shelkovnikov S, Farzaneh SH, Khazaeli M et al (2013) The synthetic triterpenoid RTA dh404 (CDDO-dhTFEA) restores endothelial function impaired by reduced Nrf2 activity in chronic kidney disease. Redox Biol 1:527–531
Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB et al (2011) Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365(4):327–336
de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H et al (2013) Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369(26):2492–2503
Lowenberg B, Downing JR, Burnett A (1999) Acute myeloid leukemia. N Engl J Med 341(14):1051–1062
Konopleva M (2002) Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood 99(1):326–335
Koschmieder S, D’Alò F, Radomska H, Schöneich C, Ji SC, Konopleva M et al (2007) CDDO induces granulocytic differentiation of myeloid leukemic blasts through translational up-regulation of p42 CCAAT enhancer-binding protein alpha. Blood 110(10):3695–3705
Ames E, Harouna S, Meyer C, Welniak LA, Murphy WJ (2012) The triterpenoid CDDO-Me promotes hematopoietic progenitor expansion and myelopoiesis in mice. Biol Blood Marrow Transplant 18(3):396–405
Wei Y, Gong J, Yoshida T, Eberhart CG, Xu Z, Kombairaju P et al (2011) Nrf2 has a protective role against neuronal and capillary degeneration in retinal ischemia-reperfusion injury. Free Radic Biol Med 51(1):216–224
Xu Z, Cho H, Hartsock MJ, Mitchell KL, Gong J, Wu L et al (2015) Neuroprotective role of Nrf2 for retinal ganglion cells in ischemia-reperfusion. J Neurochem 133(2):233–241
Wang L, Kondo N, Cano M, Ebrahimi K, Yoshida T, Barnett BP et al (2014) Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free Radic Biol Med 70:155–166
Himori N, Yamamoto K, Maruyama K, Ryu M, Taguchi K, Yamamoto M et al (2013) Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J Neurochem 127(5):669–680
Graber DJ, Park PJ, Hickey WF, Harris BT (2011) Synthetic triterpenoid CDDO derivatives modulate cytoprotective or immunological properties in astrocytes, neurons, and microglia. J Neuroimmune Pharmacol. 6(1):107–120
Choi SH, Kim B-G, Robinson J, Fink S, Yan M, Sporn MB et al (2014) Synthetic triterpenoid induces 15-PGDH expression and suppresses inflammation-driven colon carcinogenesis. J Clin Invest 124(6):2472–2482
Deeb D, Gao X, Liu Y, Kim S-H, Pindolia KR, Arbab AS et al (2012) Inhibition of cell proliferation and induction of apoptosis by oleanane triterpenoid (CDDO-Me) in pancreatic cancer cells is associated with the suppression of hTERT gene expression and its telomerase activity. Biochem Biophys Res Commun 422(4):561–567
Shanmugam MK, Dai X, Kumar AP, Tan BKH, Sethi G, Bishayee A (2014) Oleanolic acid and its synthetic derivatives for the prevention and therapy of cancer: preclinical and clinical evidence. Cancer Lett 346(2):206–216
Taguchi K, Motohashi H, Yamamoto M (2011) Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 16(2):123–140
El-Ashmawy M, Delgado O, Cardentey A, Wright WE, Shay JW (2014) CDDO-Me protects normal lung and breast epithelial cells but not cancer cells from radiation. PLoS ONE 9(12):e115600
Johnson NM, Egner PA, Baxter VK, Sporn MB, Wible RS, Sutter TR et al (2014) Complete protection against aflatoxin B(1)-induced liver cancer with a triterpenoid: DNA adduct dosimetry, molecular signature, and genotoxicity threshold. Cancer Prev Res (Phila) 7(7):658–665
Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, Honda T et al (2007) The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res 67(6):2414–2419
Gao X, Liu Y, Deeb D, Arbab AS, Guo AM, Dulchavsky SA et al (2011) Synthetic oleanane triterpenoid, CDDO-Me, induces apoptosis in ovarian cancer cells by inhibiting prosurvival AKT/NF-kappaB/mTOR signaling. Anticancer Res 31(11):3673–3681
Tran K, Risingsong R, Royce D, Williams CR, Sporn MB, Liby K (2012) The synthetic triterpenoid CDDO-methyl ester delays estrogen receptor-negative mammary carcinogenesis in polyoma middle T mice. Cancer Prev Res (Phila) 5(5):726–734
Kim E-H, Deng C, Sporn MB, Royce DB, Risingsong R, Williams CR et al (2012) CDDO-methyl ester delays breast cancer development in BRCA1-mutated mice. Cancer Prev Res (Phila) 5(1):89–97
Deeb D, Gao X, Liu Y, Jiang D, Divine GW, Arbab AS et al (2011) Synthetic triterpenoid CDDO prevents the progression and metastasis of prostate cancer in TRAMP mice by inhibiting survival signaling. Carcinogenesis 32(5):757–764
So JY, Lin JJ, Wahler J, Liby KT, Sporn MB, Suh N (2014) A synthetic triterpenoid CDDO-Im inhibits tumorsphere formation by regulating stem cell signaling pathways in triple-negative breast cancer. PLoS ONE 9(9):e107616
Cao M, Onyango EO, Williams CR, Royce DB, Gribble GW, Sporn MB, et al. Novel synthetic pyridyl analogues of CDDO-Imidazolide are useful new tools in cancer prevention. Pharmacol Res. Elsevier Ltd; 2015;100:135–47
Zhao Y, Huo M, Xu Z, Wang Y, Huang L (2015) Nanoparticle delivery of CDDO-Me remodels the tumor microenvironment and enhances vaccine therapy for melanoma. Biomaterials 68:54–66
Shah H, Speen AM, Saunders C, Brooke EA, Nallasamy P, Zhu H et al (2015) Protection of HepG2 cells against acrolein toxicity by 2-cyano-3,12-dioxooleana-1,9-dien-28-imidazolide via glutathione-mediated mechanism. Exp Biol Med (Maywood) 240(10):1340–1351
Reisman SA, Ward KW, Klaassen CD, Meyer CJ (2013) CDDO-9,11-dihydro-trifluoroethyl amide (CDDO-dhTFEA) induces hepatic cytoprotective genes and increases bile flow in rats. Xenobiotica 43(7):571–578
Walsh J, Jenkins RE, Wong M, Olayanju A, Powell H, Copple I et al (2014) Identification and quantification of the basal and inducible Nrf2-dependent proteomes in mouse liver: biochemical, pharmacological and toxicological implications. J Proteomics. 28(108):171–187
Noel S, Zheng L, Navas-Acien A, Fuchs RJ (2014) The effect of ex vivo CDDO-Me activation on nuclear factor erythroid 2-related factor 2 pathway in white blood cells from patients with septic shock. Shock. 42(5):392–399
Thimmulappa RK, Fuchs RJ, Malhotra D, Scollick C, Traore K, Bream JH et al (2007) Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxid Redox Signal 9(11):1963–1970
Keleku-Lukwete N, Suzuki M, Otsuki A, Tsuchida K, Katayama S, Hayashi M et al (2015) Amelioration of inflammation and tissue damage in sickle cell model mice by Nrf2 activation. Proc Natl Acad Sci 112(39):12169–12174
Hastings J, de Matos P, Dekker A, Ennis M, Harsha B, Kale N et al (2013) The ChEBI reference database and ontology for biologically relevant chemistry: enhancements for 2013. Nucleic Acids Res 41(Database issue):D456–D463
Zhou S, Sun W, Zhang Z, Zheng Y (2014) The role of Nrf2-mediated pathway in cardiac remodeling and heart failure. Oxid Med Cell Longev 2014:260429
Kannan S, Muthusamy VR, Whitehead KJ, Wang L, Gomes AV, Litwin SE et al (2013) Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc Res 100(1):63–73
Seifirad S, Ghaffari A, Amoli MM (2014) The antioxidants dilemma: are they potentially immunosuppressants and carcinogens? Front Physiol 5:245
de Haan JB (2014) Limiting reductive stress for treating in-stent stenosis: the heart of the matter? J Clin Invest 124(12):5092–5094
Margaritelis NV, Kyparos A, Paschalis V, Theodorou AA, Panayiotou G, Zafeiridis A et al (2014) Reductive stress after exercise: the issue of redox individuality. Redox Biol 2:520–528
Qin Q, Qu C, Niu T, Zang H, Qi L, Lyu L et al (2016) Nrf2-mediated cardiac maladaptive remodeling and dysfunction in a setting of autophagy insufficiency. Hypertension 67(1):107–117
Lu LY, Ou N, Lu Q-B (2013) Antioxidant induces DNA damage, cell death and mutagenicity in human lung and skin normal cells. Sci Rep. 3:3169
Mathis BJ, Lai Y, Qu C, Janicki JS, Cui T (2015) CYLD-mediated signaling and diseases. Curr Drug Targets 16(4):284–294
Wang H, Lai Y, Mathis BJ, Wang W, Li S, Qu C et al (2015) Deubiquitinating enzyme CYLD mediates pressure overload-induced cardiac maladaptive remodeling and dysfunction via downregulating Nrf2. J Mol Cell Cardiol 84:143–153
Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT (2013) K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 51(3):283–296
Yao T-P (2010) The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer. 1(7):779–786
Wang Y-Y, Zhang C-Y, Ma Y-Q, He Z-X, Zhe H, Zhou S-F (2015) Therapeutic effects of C-28 methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO-Me; bardoxolone methyl) on radiation-induced lung inflammation and fibrosis in mice. Drug Des Devel Ther 9:3163–3178
Ai Y, Kang F, Huang Z, Xue X, Lai Y, Peng S et al (2015) Synthesis of CDDO–amino acid-nitric oxide donor trihybrids as potential antitumor agents against both drug-sensitive and drug-resistant colon cancer. J Med Chem 58(5):2452–2464
To C, Ringelberg CS, Royce DB, Williams CR, Risingsong R, Sporn MB et al (2015) Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis 36(7):769–781
Liby KT (2014) Synthetic triterpenoids can protect against toxicity without reducing the efficacy of treatment with carboplatin and paclitaxel in experimental lung cancer. Dose Response 12(1):136–151
Gao X, Deeb D, Liu Y, Liu P, Zhang Y, Shaw J et al (2015) CDDO-Me inhibits tumor growth and prevents recurrence of pancreatic ductal adenocarcinoma. Int J Oncol 47(6):2100–2106
Kitsukawa M, Tsuchiyama H, Maeda A, Oshida K, Miyamoto Y (2014) Immunosuppressive potential of bardoxolone methyl using a modified murine local lymph node assay (LLNA). J Toxicol Sci 39(4):545–550
Perez HL, Junnotula V, Knecht D, Nie H, Sanchez Y, Boehm JC et al (2014) Analytical approaches for quantification of a Nrf2 pathway activator: overcoming bioanalytical challenges to support a toxicity study. Analyst. 139:1902–1912
Fitzpatrick LR, Stonesifer E, Small JS, Liby KT (2014) The synthetic triterpenoid (CDDO-Im) inhibits STAT3, as well as IL-17, and improves DSS-induced colitis in mice. Inflammopharmacology 22(6):341–349
Reisman SA, Buckley DB, Tanaka Y, Klaassen CD (2009) CDDO-Im protects from acetaminophen hepatotoxicity through induction of Nrf2-dependent genes. Toxicol Appl Pharmacol 236(1):109–114
Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB et al (2008) Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol Sci 104(1):218–227
Kensler KH, Slocum SL, Chartoumpekis DV, Dolan PM, Johnson NM, Ilic Z et al (2014) Genetic or pharmacologic activation of Nrf2 signaling fails to protect against aflatoxin genotoxicity in hypersensitive GSTA3 knockout mice. Toxicol Sci 139(2):293–300
Zhang F, Wang S, Zhang M, Weng Z, Li P, Gan Y et al (2012) Pharmacological induction of heme oxygenase-1 by a triterpenoid protects neurons against ischemic injury. Stroke 43(5):1390–1397
Furusawa Y, Uruno A, Yagishita Y, Higashi C, Yamamoto M (2014) Nrf2 induces fibroblast growth factor 21 in diabetic mice. Genes Cells 19(12):864–878
Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M et al (2011) Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic Biol Med 51(1):88–96
Getachew Y, Cusimano FA, Gopal P, Reisman SA, Shay JW (2015) The synthetic triterpenoid RTA 405 (CDDO-EA) halts progression of liver fibrosis and reduces hepatocellular carcinoma size resulting in increased survival in an experimental model of chronic liver injury. Toxicol Sci 405:kfv213
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Mathis, B.J., Cui, T. (2016). CDDO and Its Role in Chronic Diseases. In: Gupta, S., Prasad, S., Aggarwal, B. (eds) Drug Discovery from Mother Nature. Advances in Experimental Medicine and Biology, vol 929. Springer, Cham. https://doi.org/10.1007/978-3-319-41342-6_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-41342-6_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41341-9
Online ISBN: 978-3-319-41342-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)