
Abstract
In recent decades, several neurodegenerative diseases have been shown to be exacerbated by systemic inflammatory processes. There is a wide range of literature that demonstrates a clear but complex relationship between the central nervous system (CNS) and the immunological system, both under naïve or pathological conditions. In diseased brains, peripheral inflammation can transform “primed” microglia into an “active” state, which can trigger stronger pathological responses. Demyelinating diseases are a group of neurodegenerative diseases characterized by inflammatory lesions associated with demyelination, which in turn induces axonal damage, neurodegeneration, and progressive loss of function. Among them, the most important are multiple sclerosis (MS) and neuromyelitis optica (NMO). In this review, we will analyze the effect of specific peripheral inflammatory stimuli in the progression of demyelinating diseases and discuss their animal models. In most cases, peripheral immune stimuli are exacerbating.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Demyelinating diseases
- Systemic inflammation
- Microglia
- Multiple sclerosis
- Neuromyelitis optica
- Experimental autoimmune encephalomyelitis
Peripheral Inflammation and Neurodegenerative Diseases
Inflammation can be viewed as one of the primary responses of the immune system to infections or body injury. Systemic inflammation is associated with several chronic diseases, including obesity, type 2 diabetes, atherosclerosis, liver disease, and cancer (reviewed in Wilson et al. 2010; Fung et al. 2012). Additionally, it may also be associated with an acute stimulus, such as infection, surgery, and acute organ injury (Ottani et al. 2009). Systemic inflammatory stimuli that circulate in the blood may induce the synthesis of cytokines in the central nervous system (CNS) (Besedovsky and del Rey 1996; Pitossi et al. 1997; Combrinck et al. 2002; Dantzer et al. 1998, 2008; Londono and Cadavid 2010). In a diseased brain, this production of proinflammatory molecules exacerbates ongoing brain damage in several neurodegenerative diseases, such as Alzheimer’s disease, multiple sclerosis (MS), Parkinson’s disease, prion disease, and stroke (Perry et al. 2002; Cunningham et al. 2005a, b; McColl et al. 2007; Palin et al. 2008; Ferrari and Tarelli 2011; Murta and Ferrari 2013). In this review, we will discuss the influence of specific systemic proinflammatory stimuli on different demyelinating diseases and animal models, and the role of several cells and molecules in this phenomenon.
Microglia as a Mediator of Systemic Inflammation and Neurodegenerative Diseases
Microglia are the resident immune cells of the CNS; their main role is monitoring the local environment and triggering an immune response after specific stimuli in the nervous tissue. As discussed in Chapters “Glial cells and Integrity of the Nervous System”, “Microglia function in the normal brain”, and “Purine Signaling and Microglial Wrapping“, microglia activation is characterized by morphological and physiological changes such as secretion of proinflammatory and anti-inflammatory cytokines. Therefore, microglia can exert either cytotoxic or repairing actions, and these are referred as the M1-like and M2-like responses (Samad et al. 2001).
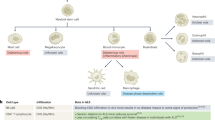
Resting microglia have a ramified morphology and represent a more quiescent basal state of this cell type. Systemic infections or mild central neurodegenerative processes can activate and prime the resting microglia. Priming of microglia precedes a further neurotoxic activation, which a secondary inflammatory stimulus can transform into an “active” state (Samad et al. 2001; Cunningham et al. 2005b; McColl et al. 2007). Microglia activation to an M1 phenotype increases neurotoxicity and, therefore, contributes to neurodegeneration through the release of free radicals such as superoxide radicals and nitric oxide (through the action of inducible nitric oxide synthase, iNOS) (Minghetti et al. 1999; Czlonkowska et al. 2002; Arimoto and Bing 2003), and immunomodulatory cytokines such as interleukin (IL) 1β, tumor necrosis factor α (TNFα), IL6, IL8, IL12, IL15, and IL10 (Kim and de Vellis 2005; Dilger and Johnson 2008; Henry et al. 2009). Therefore, ongoing inflammatory degenerative processes can be accelerated by systemic inflammation through the stimulation of “primed” microglial cells toward a more aggressive state, which in turn exacerbates damage in the nervous tissue (Fig. 1).

Schematic diagram showing the relationship between peripheral inflammation and demyelinating diseases. Demyelinating diseases are characterized by microglia activation; in which microglia change their morphology from resting (ramified) towards an activated round-shaped stage. The intermediate stage, “primed microglia,” represents the microglial stage, which precedes a further neurotoxic microglial activation as a consequence of a secondary proinflammatory stimulus. The peripheral stimuli come from the periphery either through neural or humoral pathways and influence microglia activation. Activated microglia releases proinflammatory cytokines which can, in turn, act on myelin sheath integrity, thereby inducing demyelination, axonal loss and neurodegeneration
Communication Between the Periphery and the CNS
The brain used to be considered an “immune-privileged” organ isolated from the peripheral immune system. Nowadays, it is well known that a bidirectional pathway between the brain and the peripheral immune system exists.
Circulating cytokines and other inflammatory molecules can affect the brain through several routes, mainly through the neural or humoral pathways. The neural pathway is mainly related to the transmission of peripheral inflammatory signals through the vagal afferent nerve (Perry et al. 2003; D’Mello et al. 2009; Gautron and Laye 2009; Teeling and Perry 2009; Campbell et al. 2010). The humoral pathway involves the direct action of peripheral proinflammatory cytokines (e.g., IL1β, TNFα, and IL6) and type I interferons (IFNα and IFNβ) that can initiate the synthesis of cytokines within the CNS, through blood–brain barrier (BBB) dependent or independent pathways (Perry et al. 2003; Teeling and Perry 2009).
Demyelinating Diseases
As mentioned in Chapter “Glial cells and Integrity of the Nervous System”, demyelinating diseases are a group of neurodegenerative diseases characterized by inflammatory lesions associated with demyelination, which in turn induces axonal damage, neurodegeneration, and progressive loss of function. Among them, the most important are MS, neuromyelitis optica (NMO), acute demyelinating encephalomyelitis, multifocal leukoencephalopathy, Guillain Barré syndrome, and acute disseminating encephalomyelitis. This review will mostly focus on MS and NMO, which are the most frequent in humans, and the most studied.
Multiple Sclerosis
MS is a chronic inflammatory disease characterized by multifocal and repeated inflammatory events associated with demyelination–remyelination and axonal damage, which leads to poor conduction of the nervous impulse and eventual loss of sensory and motor function.
MS follows a varied clinical course, but most patients exhibit a course of repeating exacerbation and remission from the onset (relapsing/remitting MS or RRMS) eventually leading to secondary progressive multiple sclerosis (SPMS), which worsens the patients’ quality of life (Playfair and Chain 1979; Neumann et al. 1998). A minority of patients exhibit primary progressive MS (PPMS), which is characterized by a constant decline from the onset with no recovery in neurological function (Playfair and Chain 1979; Loddick and Rothwell 2002).
Despite the fact that BBB breakdown is a major MS hallmark (McQuaid et al. 2009; Larochelle et al. 2011), some components of the inflammatory response contribute to the pathology even with an intact BBB (Buljevac et al. 2002; Lindquist et al. 2011). Although it has been proposed that in RRMS BBB breakdown allows the invasion of inflammatory cells, in the progressive forms inflammation remains enclosed behind an intact BBB (Playfair and Chain 1979).
Relapsing and Remitting MS
RRMS is the prevalent clinical type of MS and is characterized by recurrent episodes of new or worsened symptoms. Exacerbations or relapses are followed by periods of partial or complete remission, with apparent clinical stability between relapses. Relapsing episodes are unpredictable; however, peripheral inflammation may exacerbate these events (see below). Infections and other proinflammatory events have been postulated as possible triggers of the pathology and/or of relapsing episodes, and some authors have hypothesized that the autoimmune response could be a consequence of a primary central proinflammatory event (Barnett and Prineas 2004).
Progressive Multiple Sclerosis
The progressive forms of MS lead to a continuous and irreversible evolution of the disease, inducing decline of the quality of life either from the onset (PPMS) or after a course of relapsing and remitting episodes (RRMS), named SPMS. SPMS is diagnosed as a worsening after relapsing-remitting phases, with or without acute exacerbations during the progressive stage (Wagner 1996). PPMS is a distinct, non-inflammatory, or less inflammatory pathologic form of MS. The progressive forms of MS are characterized by gray matter atrophy, which could be involved in physical and cognitive disability (Rivest et al. 2000; Pocock and Kettenmann 2007; Qian et al. 2012). Cortical lesions have peculiar inflammatory and demyelinating hallmarks, characterized by lack of BBB disruption, differential inflammatory process, and reactive microglia , suggesting different immunopathogenic mechanisms (Vitkovic et al. 2000). However, anti-inflammatory or immunomodulatory therapies have no effect on neurodegeneration and cognitive impairment in the progressive forms of MS (O’Connor et al. 2005; London et al. 2013). This could be related to the fact that in progressive MS, the inflammation creates an environment that favors retention of inflammatory cells within the lesions (Konsman et al. 1999; Godbout et al. 2005).
Neuromyelitis Optica
NMO, or Devic’s disease, is a demyelinating disease characterized by inflammatory demyelinating lesions mainly in the spinal cord and optic nerve, potentially leading to paralysis and blindness. It used to be considered a subtype of MS, but the pathology and clinical features make them different diseases (Mosher et al. 2001). NMO is characterized by seropositivity for immunoglobulin G (IgG) antibodies against the astrocytic water channel aquaporin-4 (AQP4), and secondary inflammation with granulocyte and macrophage infiltration, BBB disruption, and oligodendrocyte injury. Therefore, an adaptive immune response to AQP-4 underlies the chronic demyelinating in NMO.
The etiology of the disease is still unclear, but infections and BBB permeabilizing factors could be involved in triggering the overproduction of AQP4-IgG, and its access to the CNS (Schafer et al. 1999; Galiano et al. 2001). Uzawa et al. (2010) demonstrated a significant difference in the levels of some cytokines/chemokines (e.g. IL-6 for NMO) in the cerebrospinal fluid (CSF) of patients with NMO or MS, supporting the view that different immunological and pathophysiological mechanisms exist between them.
Current NMO therapies are directed toward reducing the inflammatory response and the NMO–IgG load, such as B cell depletion and plasmapheresis. However, most MS treatments, such as IFNβ, fingolimod, and natalizumab, exacerbate NMO. Therefore, it is necessary to better comprehend the diseases’ underlying mechanisms and differentiate NMO from MS.
MS and Peripheral Inflammation
MS is a neurodegenerative disease mainly characterized by inflammatory processes. Activation of systemic immunity affects primed microglia in the CNS, reactivating lesions and increasing parenchymal inflammation. Although relapsing episodes in RRMS are unpredictable, most relapses are concomitant with peripheral inflammation (Buljevac et al. 2002). RRMS patients show increased serum levels of IL1β, IL2, IL4, IL12p70, IFNγ, and TNFα during the relapse phase (Nathan 2006; Edwards et al. 2011; Trenova et al. 2011), as well as higher numbers of IL1β, IL6, and TNFα secreting cells (Ysrraelit et al. 2008), and increased levels of T helper (Th)17 and Treg cells in the periphery (Edwards et al. 2011). Moreover, a change in CSF cytokine profile is observed during relapses; ranging from high levels of IL1β, TNFα, and transforming growth factor beta (TGFβ) to lower levels of IL-10 (Hauser et al. 1990; Edwards et al. 2011). However, the treatment of MS patients with TNFα inhibitors results in the exacerbation of central lesions (reviewed in Perry et al. 2003).
Differences in cytokine expression patterns are described when comparing progressive MS and RRMS (during relapses). SPMS patients present elevated levels of chemokine CC motif receptor 2 (CCR2) in T cells, increased serum/CSF levels of chemokine CC motif ligand 2 (CCL2) (Brinkmann et al. 2004), and decreased plasma/CSF values of TNFα and IL4 (Schmitz and Chew 2008). Peripheral blood mononuclear cells of both remitting RRMS and SPMS patients express low levels of IL10 mRNA, which return to basal levels during relapses in the RRMS form (Berkenbosch et al. 1987). Additionally, the progressive forms are characterized by a permanent peripheral type 1 immune activation, which could contribute to CNS damage during the progressive phase of the disease (Playfair and Chain 1979; Hampton et al. 1998). Thus, the peripheral blood of SPMS patients seems to reflect the inflammatory response accumulated in the CNS (Playfair and Chain 1979). On the other hand, RRMS is characterized by waves of T helper (Th)1 and Th17 cells, which are recruited into the brain causing the attacks (Neumann et al. 1998).
Inflammatory Stimuli Associated with MS
Although there is a clear association between systemic inflammation and the onset or progression of different neurodegenerative pathologies, the particular nature of these inflammatory phenomena is also relevant. Numerous studies, cited below, have investigated the role of specific systemic proinflammatory stimuli including acute or chronic stimuli, physiological imbalances, or external infections and injuries. A summary of the roles of distinct proinflammatory stimuli in MS will be addressed in the following section.
Obesity
During the last few years, a strong connection between metabolism, immunity, and inflammation was described. Obesity is considered an inflammatory disease, associated with metabolic and cardiovascular complications. Adipocyte tissue acts as an endocrine organ releasing adipocytokines, and is associated with increased levels of tissue and circulating inflammatory biomolecules (Oh et al. 1998). Excessive adipose tissue increases the number and activity of macrophages, mast cells, neutrophils, and lymphocytes (Ott et al. 1994; Kossmann et al. 1995). Moreover, leptin (an adipocyte-derived cytokine) has a role in regulating both innate and adaptive immunity (Bradl and Lassmann 2009), promoting the production of cytokines such as TNFβ, IL6, IL12, IL15, and granulocyte colony-stimulating factor in macrophages, and increasing their phagocytic activity, as well as inducing the chemotaxis of neutrophils (Bradl and Lassmann 2009; Lee et al. 2011; Golde et al. 2013; Procaccini et al. 2014). High levels of leptin have also been reported in both active inflammatory lesions and serum of MS patients (Batocchi et al. 2003).
Clinical data have demonstrated that obesity worsens the onset and progression of most autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, inflammatory bowel disease, MS, type-1 diabetes, and psoriasis. Additionally, it impairs a positive response to the treatments usually given for these diseases (Cardona et al. 2008). Data show that 18-year old obese people are twice as likely to develop MS as their normal weight age mates (Banisadr et al. 2005). However, even if it seems quite clear that obesity and diet may influence the progression of MS, few studies have linked a caloric restriction diet to reduced MS progression (Procaccini et al. 2014).
Aging
Aging processes induce a generalized proinflammatory state in the organism. This change is induced by increased immune responses in the periphery, disruption of the periphery-CNS immune communication, and an increment in “primed” microglia , which increases CNS reactivity (reviewed in Veenstra and Ransohoff 2012). Microglia in aged brains exhibit upregulated major histocompatibility complex (MHC) class II, complement receptors, toll-like receptors (TLR) 4, and cluster of differentiation (CD) 14 expression (see Chapter “Age-Dependent Changes in the Activation and Regulation of Microglia” for further reading). Therefore, peripheral innate immune stimulation induces microglial cells in aged brains to have an exaggerated inflammatory response compared with younger cohorts (Sly et al. 2001; Dilger and Johnson 2008).
PPMS and SPMS manifest around 10 years later than RRMS, therefore the timeline at which patients develop neurological deficit in PPMS and SPMS is remarkably similar, and both include aging as a major risk factor for MS progression (reviewed in Kutzelnigg et al. 2005).
Infections
Infectious pathogens have been described as important factors involved in the development of MS. Moreover, clinical studies revealed an association between infections and relapses, which worsen neurological damage even after the infection is gone (Buljevac et al. 2002; Panitch 1994). Pathogens associated with the exacerbation include bacteria (such as Mycoplasma pneumonia, Chlamydia pneumoniae, and Staphylococcus aureus-produced enterotoxins), virus (Epstein-Barr virus and human herpes virus, and human endogenous retrovirus), and the protozoan (Acanthamoeba castellanii). Viral infections that trigger MS episodes can be reduced with IFN-γ treatment (Panitch 1994; Andersen et al. 1993).
Studies of MS patients and of animal experimental models have demonstrated the influence of these infectious agents on the development and/or exacerbation of MS (Krieger et al. 1992). However, not all infections cause progression of MS, since it has been reported that infections with some parasites, such as helminthes, can protect against the exacerbation phase of the disease (Correale and Farez 2011a, b; Krieger et al. 1992). This protection is associated with the induction of CD4+, CD25+ T cells secreting IL10, and TGFβ (Correale and Farez 2011a).
Immune Regulation by the Neuroendocrine System
The neuroendocrine system exerts its action on the immune system through finely tune regulation. Glucocorticoids (GCs) induce the production of pro- and anti-inflammatory cytokines, specifically causing a shift from Th1 to Th2 immune response. GCs inhibit the production of Th1 related cytokines (IL1 and IL6, IL2, IL12, IFNγ) and increase the secretion of anti-inflammatory Th2 cytokines (IL4 and IL10) (Haak et al. 2009). However, GCs can increase both peripheral and central inflammatory responses to a systemic challenge if they are administered before the peripheral stimuli (Sorrells and Sapolsky 2010; Frank et al. 2010). Therefore, GCs can prime the immune response and, as a consequence, increase proinflammatory cytokine production and exacerbation of MS symptoms.
The Hypothalamic–Pituitary–Adrenal (HPA) Axis
Clinical and experimental studies have demonstrated that abnormalities in the HPA axis, which influences the immune response, may exacerbate MS symptoms (Hofstetter et al. 2005; Seo et al. 2013). Thus high cortisol levels are often correlated with acute relapses (Hofstetter et al. 2005; Seo et al. 2013), whereas prolactin increases the peripheral production of IFNγ and IL12 by T cells (Du and Dreyfus 2002).
The Hypothalamic–Pituitary–Gonadal (HPG) Axis
MS affects predominantly women in comparison with men, therefore, considering that gender affects the course of autoimmune diseases, the influence of sex hormones is critical (Dunn et al. 2015a, b). In particular, 17β-estradiol induces an increase of Th2 cytokines (IL10 and IL4) and a decrease of Th1 cytokines (TNFα and IFNγ) (van Riemsdijk et al. 2001; Janik et al. 1997). Estrogens, in addition to their anti-inflammatory effects, appear to be neuroprotective in CNS diseases, such as MS and Alzheimer’s, disease (Nicot 2009; Gao and Tsirka 2011). Additionally, both clinical symptoms and relapse rates of MS are decreased during pregnancy, whereas the postpartum period increases the risk for exacerbation of the disease (Ling et al. 1997).
The increased secretion of estrogen, progesterone, and cortisol during pregnancy is associated with increased production of Th2 cytokines and decreased production of Th1 cytokines (Takii et al. 1992, 1994). Additionally, progesterone also inhibits NFκB and increases IL4 production, demonstrating its anti-inflammatory effect (Piccinni et al. 1995; Nishiyori et al. 1997). Male hormones, such as testosterone, also inhibit both innate and adaptive immune systems by enhancing the production of IL5 and IL10, and decreasing IFNβ secretion, thus promoting a Th2 response (Murphy and Sturm 1923).
Environmental Factors and Peripheral Inflammation
Environmental factors have influence on most autoimmune diseases. Epidemiological risk factors for MS, including low vitamin D and elevated salt intake, are associated with peripheral inflammation. Recent studies have shown that components of the daily diet and gut microbiota can strongly affect the levels of effector T cells in the gut (Ransohoff et al. 2007).
On the other hand, high sodium chloride concentrations induced expression of serum glucocorticoid kinase 1 (SGK1) in T cells, which in turn stimulate the induction of Th17 cells from CD4+ T cells, promoting autoimmune diseases (Glabinski et al. 1997). However, direct correlation between salt intake and incidence of autoimmune disease is yet to be demonstrated (Tsai et al. 2002).
Vitamin D plays an important role in the regulation of the immune responses (Semple et al. 2010), modulating many inflammatory mechanisms including: (a) the regulation of inflammatory mediators, such as cytokines (IL1β, TNFα, IL6, TGF1β) and cyclooxygenases, (b) the interference with transcription factors, such as NFκB, and (c) the activation of signaling cascades, such as MAP kinases (Xia and Hyman 2002; Bakshi et al. 2011; Semple et al. 2010; Perry and Teeling 2013). MS exacerbation correlates with low levels of Vitamin D, whereas vitamin D supplementation has a protective effect (Aubert et al. 1995; Romeo et al. 2001; Varvel et al. 2012).
NMO and Peripheral Inflammation
There is not much evidence for peripheral inflammation affecting NMO, in contrast with MS. However, recent work shows that the peripheral immune system affects the progression of NMO. The CSF of NMO patients shows white blood cells (WBC) ≥50 cells/mm3 or ≥5 neutrophils/mm3 compared to control patients, whose counts are <5 WBC/mm3 (Campbell et al. 2008). Additionally, removing inflammatory mediators from the blood of MNO patients alleviates the symptoms (Okada et al. 2006).
NMO can occur concomitantly with systemic autoimmune disorders such as Sjogren’s syndrome and systemic lupus erythematous, which likely reflects an underlying predisposition for these patients to develop autoimmune disorders. Moreover, the presence of other systemic disease can increase the mortality rate in relapsing NMO patients (Krady et al. 2008).
Finally, the seropositivity for NMO–IgG represents a key factor for predicting future relapses; indeed, it is a prognostic marker for NMO. Additionally, humoral immune mechanisms, including the activation of B cells and the complement pathway, have been said to play a role in NMO pathogenesis (Quan et al. 2013; Kim et al. 2011).
Experimental Models of Demyelinating Diseases
Experimental models of demyelination help in understanding the pathophysiology of such demyelinating diseases as MS (Denic et al. 2011) and NMO (Linington et al. 1992). Animal models can be divided into two groups: those which attempt to replicate the disease as accurately as possible and others that provide a reductionist approach to the diseases by studying demyelination and remyelination processes (e.g., ethidium bromide, lysolecithin, and cuprizone) (reviewed in Blakemore and Franklin 2008). For MS, the most common models have been virus-induced encephalomyelitis and various forms of Experimental Autoimmune Encephalomyelitis (EAE) (reviewed in Dai et al. 2003).
A clear distinction between NMO and MS only became possible in the past decade, and nowadays the most frequently used NMO models are NMO/EAE, NMO-IgG/complement intracerebral injection, and cytokine-injection NMO (Linington et al. 1992). Some of the main features present in these experimental models are summarized in Table 1. Consistent with the human diseases, animal experimental models show the influence of peripheral inflammation on the progression of the disorders.
The importance of humoral components of the immune system is evident in EAE. For example, a specific cytokine profile appears during the different phases of acute the EAE model: decreased IL21 expression on the peak phase and high IL22 expression during the induction phase that decreases during recovery (Almolda et al. 2011). Additionally, systemic TNFα causes clinical signs to recrudesce and induces relapses in EAE (Crisi et al. 1995).
MS Animal Models and Systemic Inflammation
Obesity
Immunomodulatory effects of leptin, the adipocyte-derived hormone, are involved in the induction and progression of EAE (Matarese et al. 2001, 2008). In this context, the use of leptin antagonists improved the course of EAE (De Rosa et al. 2006). Moreover, the leptin-deficient (ob/ob) mice do not develop EAE; however, exogenous leptin treatment renders ob/ob mice susceptible to EAE development (Matarese et al. 2001). On the other hand, caloric restriction, (associated with low levels of leptin in plasma) can significantly increase the overall survival in several experimental animal models of autoimmune diseases (Oka et al. 2007).
Infections
Peripheral infection with enterotoxin A or B exacerbates clinical signs and induces relapses in EAE (Brocke et al. 1993; Crisi et al. 1995; Schiffenbauer et al. 1993). A single dose of peripheral LPS can induce increased inflammatory, demyelinating and axonal damage in EAE lesions (Serres et al. 2009; Moreno et al. 2011) as well as CD4+ cells activation (Nogai et al. 2005). Additionally, respiratory tract pathogens (Streptococcus pneumonia and Chlamydia pneumonia) aggravate EAE symptoms (Du et al. 2002; Herrmann et al. 2006; Tauber et al. 2007).
On the other hand, some data have been published demonstrating beneficial effects of peripheral LPS. In those studies, pretreatment with LPS prior to EAE induction lead to a delay in the onset of the disease by suppressing antigen presentation and altering the expression of inflammatory mediators (Buenafe and Bourdette 2007).
The presence of blood-derived peripheral polymorphonuclear neutrophils (PMN) expressing CXC chemokine receptor type 2 (CXCR2) is requisite for oligodendrocyte death, demyelination, and BBB breakdown in both EAE and cuprizone models (Liu et al. 2010; Carlson et al. 2008). Peripheral PMN are considered the first key effector leukocytes in the pathogenesis of EAE; they produce cytokines and chemokines that in turn induce lymphocyte and monocyte activation (Carlson et al. 2008).
Moreover, the importance of PMN neutrophils for the development of a demyelinating lesion in the CNS of rats has been seen in a model of chronic neuroinflammation and demyelination in response to a sustained expression of IL1β in the CNS (Ferrari et al. 2004). Furthermore, a relapsing-like lesion was achieved in the same model by inducing a peripheral sustained expression of IL1β (Murta et al. 2015). Here, the involvement of CXCR2 + PMN neutrophils from the periphery was also proven central for the development of the relapse.
Immune Regulation by the Neuroendocrine System
Estrogen inhibits clinical and histological symptoms of EAE, and pretreatment with low doses of 17beta-estradiol (E2) diminishes the symptoms of EAE by inhibiting cell migration into the CNS and promoting axon and myelin survival (Wolswijk 1998; reviewed in Murta and Ferrari 2013). Moreover, in EAE animals progesterone decreases proinflammatory cytokine secretion (IL12, IL17), increases IL10 production, and increases the CD19 + and CD8 + populations (Chang et al. 2002).
Environmental Factors
Diet also represents an important factor in experimental animal models. In EAE mice, a high salt diet increases the number of Th17 cells, worsening the disease (Tsai et al. 2002). Conversely, vitamin D (or its metabolite 1.25-dihydroxyvitamin D3) reverses the EAE symptoms by inhibiting chemokine and inducible nitric oxide synthase (iNOS) synthesis, and CD11b + monocyte trafficking into the CNS (Moynagh 2005). Moreover, this vitamin also suppresses EAE female selectivity (Byravan et al. 1994).
Another environmental factor associated with EAE progression is UV irradiation: several authors have shown that UV irradiation suppresses EAE by inducing immunosuppression through an alteration of dendritic and regulatory T cells, independently of vitamin D production (Hauser et al. 1984; Waxman 1998; Lappe-Siefke et al. 2003; Ng et al. 2013).
Moreover, the influence of the microbiome on different pathological conditions has been investigated. In some models of EAE, gut microflora-free animals are resistant to the induction of RR-EAE and have decreased Th17 and B cell responses (Tsunoda and Fujinami 2002; Tsunoda et al. 2003; Huitinga et al. 2000).
Concluding Remarks
Systemic inflammatory insults are risk factors in both the etiology and progression of demyelinating diseases. The interaction between damaged brain and systemic inflammation may be responsible for the progression of neurodegenerative diseases. However, certain systemic stimuli may be beneficial for both disease progression and repair. Primed microglial cells in the diseased CNS are viewed as one of the key components in the exacerbation of central damage due to systemic inflammatory stimuli in most CNS diseases. Additionally, the peripheral immune system contributes significantly to the pathophysiology of the demyelinating diseases discussed in the present review and their animal models, and the environment appears also to be important. Better understanding of the mechanisms of CNS and immune system communication should improve therapeutics for immune mediated diseases.
Abbreviations
- AQP4:
-
Aquaporin-4
- BBB:
-
Blood–brain barrier
- CCL2:
-
Chemokine CC motif ligand 2
- CCR2:
-
Chemokine CC motif receptor 2
- CD:
-
Cluster of differentiation
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- CXCR2:
-
CXC motif chemokine receptor type 2
- EAE:
-
Experimental autoimmune encephalomyelitis
- GC:
-
Glucocorticoids
- HPA:
-
Hypothalamic-Pituitary-Adrenal
- HPG:
-
Hypothalamic-Pituitary-Gonadal
- IFN:
-
Interferons
- IgG:
-
Immunoglobulin G
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- MHC:
-
Major histocompatibility complex
- MS:
-
Multiple sclerosis
- NMO:
-
Neuromyelitis optica
- PMN:
-
Polymorphonuclear
- PPMS:
-
Primary progressive MS
- RRMS:
-
Relapsing remitting multiple sclerosis
- SGK1:
-
Serum glucocorticoid kinase 1
- SPMS:
-
Secondary progressive multiple sclerosis
- TGF-β:
-
Transforming growth factor beta
- Th:
-
T helper
- TLR:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor α
- WBC:
-
White blood cells
References
Almolda B, Gonzalez B, Castellano B (2011) Antigen presentation in EAE: role of microglia, macrophages and dendritic cells. Front Biosci 16:1157–1171 (3781 [pii])
Andersen O, Lygner PE, Bergstrom T, Andersson M, Vahlne A (1993) Viral infections trigger multiple sclerosis relapses: a prospective seroepidemiological study. J Neurol 240(7):417–422
Arimoto T, Bing G (2003) Up-regulation of inducible nitric oxide synthase in the substantia nigra by lipopolysaccharide causes microglial activation and neurodegeneration. Neurobiol Dis 12(1):35–45
Aubert A, Vega C, Dantzer R, Goodall G (1995) Pyrogens specifically disrupt the acquisition of a task involving cognitive processing in the rat. Brain Behav Immun 9(2):129–148. doi:10.1006/brbi.1995.1013 (S0889-1591(85)71013-6 [pii])
Bakshi P, Margenthaler E, Reed J, Crawford F, Mullan M (2011) Depletion of CXCR2 inhibits gamma-secretase activity and amyloid-beta production in a murine model of Alzheimer’s disease. Cytokine 53(2):163–169 (10.1016/j.cyto.2010.10.008S1043-4666(10)00703-9 [pii])
Banisadr G, Rostene W, Kitabgi P, Parsadaniantz SM (2005) Chemokines and brain functions. Curr Drug Targets Inflamm Allergy 4(3):387–399
Barnett MH, Prineas JW (2004) Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 55(4):458–468
Batocchi AP, Rotondi M, Caggiula M, Frisullo G, Odoardi F, Nociti V, Carella C, Tonali PA, Mirabella M (2003) Leptin as a marker of multiple sclerosis activity in patients treated with interferon-beta. J Neuroimmunol 139(1–2):150–154 (S0165572803001541 [pii])
Berkenbosch F, van Oers J, del Rey A, Tilders F, Besedovsky H (1987) Corticotropin-releasing factor-producing neurons in the rat activated by interleukin-1. Science 238(4826):524–526
Besedovsky HO, del Rey A (1996) Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev 17(1):64–102
Blakemore WF (2008) Regeneration and repair in multiple sclerosis: the view of experimental pathology. J Neurol Sci 265(1–2):1–4
Blakemore WF, Franklin RJ (2008) Remyelination in experimental models of toxin-induced demyelination. Curr Top Microbiol Immunol 318:193–212
Bradl M, Lassmann H (2009) Progressive multiple sclerosis. Semin Immunopathol 31(4):455–465. doi:10.1007/s00281-009-0182-3
Bradl M, Lassmann H (2014) Experimental models of neuromyelitis optica. Brain Pathol 24(1):74–82. doi:10.1111/bpa.12098
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535 (10.1126/science.1092385303/5663/1532 [pii])
Brocke S, Gaur A, Piercy C, Gautam A, Gijbels K, Fathman CG, Steinman L (1993) Induction of relapsing paralysis in experimental autoimmune encephalomyelitis by bacterial superantigen. Nature 365(6447):642–644. doi:10.1038/365642a0
Buenafe AC, Bourdette DN (2007) Lipopolysaccharide pretreatment modulates the disease course in experimental autoimmune encephalomyelitis. J Neuroimmunol 182(1–2):32–40. doi:10.1016/j.jneuroim.2006.09.004 (S0165-5728(06)00371-7 [pii])
Buljevac D, Flach HZ, Hop WC, Hijdra D, Laman JD, Savelkoul HF, van Der Meche FG, van Doorn PA, Hintzen RQ (2002) Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain 125(Pt 5):952–960
Byravan S, Foster LM, Phan T, Verity AN, Campagnoni AT (1994) Murine oligodendroglial cells express nerve growth factor. Proc Natl Acad Sci USA 91(19):8812–8816
Campbell SJ, Anthony DC, Oakley F, Carlsen H, Elsharkawy AM, Blomhoff R, Mann DA (2008) Hepatic nuclear factor kappa B regulates neutrophil recruitment to the injured brain. J Neuropathol Exp Neurol 67(3):223–230 (10.1097/NEN.0b013e318165495700005072-200803000-00005 [pii])
Campbell SJ, Meier U, Mardiguian S, Jiang Y, Littleton ET, Bristow A, Relton J, Connor TJ, Anthony DC (2010) Sickness behaviour is induced by a peripheral CXC-chemokine also expressed in multiple sclerosis and EAE. Brain Behav Immun 24(5):738–746 (S0889-1591(10)00038-3 [pii] 10.1016/j.bbi.2010.01.011)
Cardona AE, Li M, Liu L, Savarin C, Ransohoff RM (2008) Chemokines in and out of the central nervous system: much more than chemotaxis and inflammation. J Leukoc Biol 84(3):587–594 (10.1189/jlb.1107763jlb.1107763 [pii])
Carlson T, Kroenke M, Rao P, Lane TE, Segal B (2008) The Th17-ELR + CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med 205(4):811–823 (jem.20072404 [pii] 10.1084/jem.20072404)
Combrinck MI, Perry VH, Cunningham C (2002) Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience 112(1):7–11
Correale J, Farez MF (2011a) The impact of environmental infections (parasites) on MS activity. Mult Scler 17(10):1162–1169 (17/10/1162 [pii] 10.1177/1352458511418027)
Correale J, Farez MF (2011b) The impact of parasite infections on the course of multiple sclerosis. J Neuroimmunol 233(1–2):6–11 (S0165-5728(11)00005-1 [pii] 10.1016/j.jneuroim.2011.01.002)
Crisi GM, Santambrogio L, Hochwald GM, Smith SR, Carlino JA, Thorbecke GJ (1995) Staphylococcal enterotoxin B and tumor-necrosis factor-alpha-induced relapses of experimental allergic encephalomyelitis: protection by transforming growth factor-beta and interleukin-10. Eur J Immunol 25(11):3035–3040. doi:10.1002/eji.1830251108
Cunningham C, Wilcockson DC, Boche D, Perry VH (2005a) Comparison of inflammatory and acute-phase responses in the brain and peripheral organs of the ME7 model of prion disease. J Virol 79(8):5174–5184 (79/8/5174 [pii] 10.1128/JVI.79.8.5174-5184.2005)
Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH (2005b) Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 25(40):9275–9284
Czlonkowska A, Kurkowska-Jastrzebska I, Czlonkowski A, Peter D, Stefano GB (2002) Immune processes in the pathogenesis of Parkinson’s disease—a potential role for microglia and nitric oxide. Med Sci Monit 8(8):165–177
Chang A, Tourtellotte WW, Rudick R, Trapp BD (2002) Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med 346(3):165–173 (10.1056/NEJMoa010994346/3/165 [pii])
D’Mello C, Le T, Swain MG (2009) Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 29(7):2089–2102 (29/7/2089 [pii] 10.1523/JNEUROSCI.3567-08.2009)
Dai X, Lercher LD, Clinton PM, Du Y, Livingston DL, Vieira C, Yang L, Shen MM, Dreyfus CF (2003) The trophic role of oligodendrocytes in the basal forebrain. J Neurosci 23(13):5846–5853 (23/13/5846 [pii])
Dantzer R, Bluthe RM, Laye S, Bret-Dibat JL, Parnet P, Kelley KW (1998) Cytokines and sickness behavior. Ann N Y Acad Sci 840:586–590
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9(1):46–56 (nrn2297 [pii] 10.1038/nrn2297)
De Rosa V, Procaccini C, La Cava A, Chieffi P, Nicoletti GF, Fontana S, Zappacosta S, Matarese G (2006) Leptin neutralization interferes with pathogenic T cell autoreactivity in autoimmune encephalomyelitis. J Clin Invest 116(2):447–455. doi:10.1172/JCI26523
Denic A, Johnson AJ, Bieber AJ, Warrington AE, Rodriguez M, Pirko I (2011) The relevance of animal models in multiple sclerosis research. Pathophysiology 18(1):21–29 (S0928-4680(10)00022-2 [pii] 10.1016/j.pathophys.2010.04.004)
Dilger RN, Johnson RW (2008) Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol 84(4):932–939 (jlb.0208108 [pii] 10.1189/jlb.0208108)
Du C, Yao SY, Ljunggren-Rose A, Sriram S (2002) Chlamydia pneumoniae infection of the central nervous system worsens experimental allergic encephalitis. J Exp Med 196(12):1639–1644
Du Y, Dreyfus CF (2002) Oligodendrocytes as providers of growth factors. J Neurosci Res 68(6):647–654. doi:10.1002/jnr.10245
Dunn SE, Gunde E, Lee H (2015a) Sex-based differences in multiple sclerosis (MS): part II: rising incidence of multiple sclerosis in women and the vulnerability of men to progression of this disease. Curr Top Behav Neurosci. doi:10.1007/7854_2015_370
Dunn SE, Lee H, Pavri FR, Zhang MA (2015b) Sex-based differences in multiple sclerosis (part I): biology of disease incidence. Curr Top Behav Neurosci. doi:10.1007/7854_2015_371
Edwards LJ, Sharrack B, Ismail A, Tumani H, Constantinescu CS (2011) Central inflammation versus peripheral regulation in multiple sclerosis. J Neurol 258(8):1518–1527. doi:10.1007/s00415-011-5973-5
Ferrari CC, Depino AM, Prada F, Muraro N, Campbell S, Podhajcer O, Perry VH, Anthony DC, Pitossi FJ (2004) Reversible demyelination, blood-brain barrier breakdown, and pronounced neutrophil recruitment induced by chronic il-1 expression in the brain. Am J Pathol 165(5):1827–1837
Ferrari CC, Tarelli R (2011) Parkinson’s disease and systemic inflammation. Parkinsons Dis 2011:436813. doi:10.4061/2011/436813
Frank MG, Miguel ZD, Watkins LR, Maier SF (2010) Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun 24(1):19–30 (S0889-1591(09)00386-9 [pii] 10.1016/j.bbi.2009.07.008)
Fung A, Vizcaychipi M, Lloyd D, Wan Y, Ma D (2012) Central nervous system inflammation in disease related conditions: mechanistic prospects. Brain Res 1446:144–155 (10.1016/j.brainres.2012.01.061S0006-8993(12)00164-3 [pii])
Galiano M, Liu ZQ, Kalla R, Bohatschek M, Koppius A, Gschwendtner A, Xu S, Werner A, Kloss CU, Jones LL, Bluethmann H, Raivich G (2001) Interleukin-6 (IL6) and cellular response to facial nerve injury: effects on lymphocyte recruitment, early microglial activation and axonal outgrowth in IL6-deficient mice. Eur J Neurosci 14(2):327–341 (ejn1647 [pii])
Gao Z, Tsirka SE (2011) Animal Models of MS Reveal Multiple Roles of Microglia in Disease Pathogenesis. Neurol Res Int 2011:383087. doi:10.1155/2011/383087
Gautron L, Laye S (2009) Neurobiology of inflammation-associated anorexia. Front Neurosci 3:59. doi:10.3389/neuro.23.003.2009
Glabinski AR, Tani M, Strieter RM, Tuohy VK, Ransohoff RM (1997) Synchronous synthesis of alpha- and beta-chemokines by cells of diverse lineage in the central nervous system of mice with relapses of chronic experimental autoimmune encephalomyelitis. Am J Pathol 150(2):617–630
Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW (2005) Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J 19(10):1329–1331 (05-3776fje [pii] 10.1096/fj.05-3776fje)
Golde TE, Streit WJ, Chakrabarty P (2013) Alzheimer’s disease risk alleles in TREM2 illuminate innate immunity in Alzheimer’s disease. Alzheimers Res Ther 5(3):24 (alzrt178 [pii] 10.1186/alzrt178)
Grigoriadis N, Hadjigeorgiou GM (2006) Virus-mediated autoimmunity in Multiple Sclerosis. J Autoimmune Dis 3:1
Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, Waisman A (2009) IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest 119(1):61–69 (10.1172/JCI3599735997 [pii])
Hampton MB, Kettle AJ, Winterbourn CC (1998) Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92(9):3007–3017
Hauser SL, Doolittle TH, Lincoln R, Brown RH, Dinarello CA (1990) Cytokine accumulations in CSF of multiple sclerosis patients: frequent detection of interleukin-1 and tumor necrosis factor but not interleukin-6. Neurology 40(11):1735–1739
Hauser SL, Weiner HL, Che M, Shapiro ME, Gilles F, Letvin NL (1984) Prevention of experimental allergic encephalomyelitis (EAE) in the SJL/J mouse by whole body ultraviolet irradiation. J Immunol 132(3):1276–1281
Henry CJ, Huang Y, Wynne AM, Godbout JP (2009) Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun 23(3):309–317 (S0889-1591(08)00348-6 [pii] 10.1016/j.bbi.2008.09.002)
Herrmann I, Kellert M, Schmidt H, Mildner A, Hanisch UK, Bruck W, Prinz M, Nau R (2006) Streptococcus pneumoniae Infection aggravates experimental autoimmune encephalomyelitis via Toll-like receptor 2. Infect Immun 74(8):4841–4848 (74/8/4841 [pii] 10.1128/IAI.00026-06)
Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, Gold R (2005) Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol 237(2):123–130 (S0008-8749(05)00242-X [pii] 10.1016/j.cellimm.2005.11.002)
Huitinga I, Schmidt ED, van der Cammen MJ, Binnekade R, Tilders FJ (2000) Priming with interleukin-1beta suppresses experimental allergic encephalomyelitis in the Lewis rat. J Neuroendocrinol 12(12):1186–1193 574 [pii]
Janik JE, Curti BD, Considine RV, Rager HC, Powers GC, Alvord WG, Smith JW 2nd, Gause BL, Kopp WC (1997) Interleukin 1 alpha increases serum leptin concentrations in humans. J Clin Endocrinol Metab 82(9):3084–3086
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ (2011) Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol 68(11):1412–1420 (10.1001/archneurol.2011.154archneurol.2011.154 [pii])
Kim SU, de Vellis J (2005) Microglia in health and disease. J Neurosci Res 81(3):302–313
Konsman JP, Kelley K, Dantzer R (1999) Temporal and spatial relationships between lipopolysaccharide-induced expression of Fos, interleukin-1beta and inducible nitric oxide synthase in rat brain. Neuroscience 89(2):535–548 (S0306-4522(98)00368-6 [pii])
Kossmann T, Hans VH, Imhof HG, Stocker R, Grob P, Trentz O, Morganti-Kossmann C (1995) Intrathecal and serum interleukin-6 and the acute-phase response in patients with severe traumatic brain injuries. Shock 4(5):311–317
Krady JK, Lin HW, Liberto CM, Basu A, Kremlev SG, Levison SW (2008) Ciliary neurotrophic factor and interleukin-6 differentially activate microglia. J Neurosci Res 86(7):1538–1547. doi:10.1002/jnr.21620
Krieger M, Brunner T, Bischoff SC, von Tscharner V, Walz A, Moser B, Baggiolini M, Dahinden CA (1992) Activation of human basophils through the IL-8 receptor. J Immunol 149(8):2662–2667
Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H (2005) Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128(Pt 11):2705–2712 (awh641 [pii] 10.1093/brain/awh641)
Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33(3):366–374 (10.1038/ng1095ng1095 [pii])
Larochelle C, Alvarez JI, Prat A (2011) How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett 585(23):3770–3780 (S0014-5793(11)00336-X [pii] 10.1016/j.febslet.2011.04.066)
Lee M, Schwab C, McGeer PL (2011) Astrocytes are GABAergic cells that modulate microglial activity. Glia 59(1):152–165. doi:10.1002/glia.21087
Lindquist S, Hassinger S, Lindquist JA, Sailer M (2011) The balance of pro-inflammatory and trophic factors in multiple sclerosis patients: effects of acute relapse and immunomodulatory treatment. Mult Scler 17(7):851–866 (1352458511399797 [pii] 10.1177/1352458511399797)
Ling PR, Schwartz JH, Bistrian BR (1997) Mechanisms of host wasting induced by administration of cytokines in rats. Am J Physiol 272(3 Pt 1):E333–E339
Linington C, Engelhardt B, Kapocs G, Lassman H (1992) Induction of persistently demyelinated lesions in the rat following the repeated adoptive transfer of encephalitogenic T cells and demyelinating antibody. J Neuroimmunol 40(2–3):219–224
Liu L, Belkadi A, Darnall L, Hu T, Drescher C, Cotleur AC, Padovani-Claudio D, He T, Choi K, Lane TE, Miller RH, Ransohoff RM (2010) CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci 13(3):319–326 (nn.2491 [pii] 10.1038/nn.2491)
Loddick S, Rothwell N (2002) Cytokines and neurodegeneration. In: Loddick S, Rothwell N (eds) Immune an inflammatory responses in the nervous system. Oxford University Press, Oxford, pp 90–105
London A, Cohen M, Schwartz M (2013) Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci 7:34. doi:10.3389/fncel.2013.00034
Londono D, Cadavid D (2010) Bacterial lipoproteins can disseminate from the periphery to inflame the brain. Am J Pathol 176(6):2848–2857 (ajpath.2010.091235 [pii] 10.2353/ajpath.2010.091235)
Matarese G, Di Giacomo A, Sanna V, Lord GM, Howard JK, Di Tuoro A, Bloom SR, Lechler RI, Zappacosta S, Fontana S (2001) Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J Immunol 166(10):5909–5916
Matarese G, Procaccini C, De Rosa V (2008) The intricate interface between immune and metabolic regulation: a role for leptin in the pathogenesis of multiple sclerosis? J Leukoc Biol 84(4):893–899 (jlb.0108022 [pii] 10.1189/jlb.0108022)
McColl BW, Rothwell NJ, Allan SM (2007) Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci 27(16):4403–4412
McQuaid S, Cunnea P, McMahon J, Fitzgerald U (2009) The effects of blood-brain barrier disruption on glial cell function in multiple sclerosis. Biochem Soc Trans 37(Pt 1):329–331
Minghetti L, Polazzi E, Nicolini A, Greco A, Levi G (1999) Possible role of microglial prostanoids and free radicals in neuroprotection and neurodegeneration. Adv Exp Med Biol 468:109–119
Moreno B, Jukes JP, Vergara-Irigaray N, Errea O, Villoslada P, Perry VH, Newman TA (2011) Systemic inflammation induces axon injury during brain inflammation. Ann Neurol 70(6):932–942. doi:10.1002/ana.22550
Mosher B, Dean R, Harkema J, Remick D, Palma J, Crockett E (2001) Inhibition of Kupffer cells reduced CXC chemokine production and liver injury. J Surg Res 99(2):201–210 (10.1006/jsre.2001.6217S0022-4804(01)96217-1 [pii])
Moynagh PN (2005) The interleukin-1 signalling pathway in astrocytes: a key contributor to inflammation in the brain. J Anat 207(3):265–269 (JOA445 [pii] 10.1111/j.1469-7580.2005.00445.x)
Murphy JB, Sturm E (1923) Conditions Determining the Transplantability of Tissues in the Brain. J Exp Med 38(2):183–197
Murta V, Farias MI, Pitossi FJ, Ferrari CC (2015) Chronic systemic IL-1beta exacerbates central neuroinflammation independently of the blood-brain barrier integrity. J Neuroimmunol 278:30–43 (10.1016/j.jneuroim.2014.11.023S0165-5728(14)00984-9 [pii])
Murta V, Ferrari CC (2013) Influence of Peripheral inflammation on the progression of multiple sclerosis: evidence from the clinic and experimental animal models. Mol Cell Neurosci 53:6–13 (10.1016/j.mcn.2012.06.004S1044-7431(12)00108-X [pii])
Nathan C (2006) Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 6(3):173–182 nri1785 [pii] 10.1038/nri1785
Neumann H, Misgeld T, Matsumuro K, Wekerle H (1998) Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc Natl Acad Sci USA 95(10):5779–5784
Ng RL, Scott NM, Strickland DH, Gorman S, Grimbaldeston MA, Norval M, Waithman J, Hart PH (2013) Altered immunity and dendritic cell activity in the periphery of mice after long-term engraftment with bone marrow from ultraviolet-irradiated mice. J Immunol 190(11):5471–5484 (10.4049/jimmunol.1202786jimmunol.1202786 [pii])
Nicot A (2009) Gender and sex hormones in multiple sclerosis pathology and therapy. Front Biosci (Landmark Ed) 14:4477–4515 (3543 [pii])
Nishiyori A, Minami M, Takami S, Satoh M (1997) Type 2 interleukin-1 receptor mRNA is induced by kainic acid in the rat brain. Brain Res Mol Brain Res 50(1–2):237–245
Nogai A, Siffrin V, Bonhagen K, Pfueller CF, Hohnstein T, Volkmer-Engert R, Bruck W, Stadelmann C, Kamradt T (2005) Lipopolysaccharide injection induces relapses of experimental autoimmune encephalomyelitis in nontransgenic mice via bystander activation of autoreactive CD4 + cells. J Immunol 175(2):959–966 (175/2/959 [pii])
O’Connor JC, Satpathy A, Hartman ME, Horvath EM, Kelley KW, Dantzer R, Johnson RW, Freund GG (2005) IL-1beta-mediated innate immunity is amplified in the db/db mouse model of type 2 diabetes. J Immunol 174(8):4991–4997 (174/8/4991 [pii])
Oh JW, Van Wagoner NJ, Rose-John S, Benveniste EN (1998) Role of IL-6 and the soluble IL-6 receptor in inhibition of VCAM-1 gene expression. J Immunol 161(9):4992–4999
Oka Y, Ibuki T, Matsumura K, Namba M, Yamazaki Y, Poole S, Tanaka Y, Kobayashi S (2007) Interleukin-6 is a candidate molecule that transmits inflammatory information to the CNS. Neuroscience 145(2):530–538 (S0306-4522(06)01470-9 [pii] 10.1016/j.neuroscience.2006.10.055)
Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H (2006) Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med 12(7):829–834 (nm1425 [pii] 10.1038/nm1425)
Ott L, McClain CJ, Gillespie M, Young B (1994) Cytokines and metabolic dysfunction after severe head injury. J Neurotrauma 11(5):447–472
Ottani A, Giuliani D, Mioni C, Galantucci M, Minutoli L, Bitto A, Altavilla D, Zaffe D, Botticelli AR, Squadrito F, Guarini S (2009) Vagus nerve mediates the protective effects of melanocortins against cerebral and systemic damage after ischemic stroke. J Cereb Blood Flow Metab 29(3):512–523 (10.1038/jcbfm.2008.140jcbfm2008140 [pii])
Palin K, Cunningham C, Forse P, Perry VH, Platt N (2008) Systemic inflammation switches the inflammatory cytokine profile in CNS Wallerian degeneration. Neurobiol Dis 30(1):19–29 (S0969-9961(07)00267-7 [pii] 10.1016/j.nbd.2007.11.012)
Panitch HS (1994) Influence of infection on exacerbations of multiple sclerosis. Ann Neurol 36(Suppl):S25–S28
Perry V, Cunningham C, Boche D (2002) Atypical inflammation in the central nervous system in prion disease. Curr Opin Neurol 15:349–354
Perry VH, Newman TA, Cunningham C (2003) The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci 4(2):103–112
Perry VH, Teeling J (2013) Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. doi:10.1007/s00281-013-0382-8
Piccinni MP, Giudizi MG, Biagiotti R, Beloni L, Giannarini L, Sampognaro S, Parronchi P, Manetti R, Annunziato F, Livi C et al (1995) Progesterone favors the development of human T helper cells producing Th2-type cytokines and promotes both IL-4 production and membrane CD30 expression in established Th1 cell clones. J Immunol 155(1):128–133
Pitossi F, del Rey A, Kabiersch A, Besedovsky H (1997) Induction of cytokine transcripts in the central nervous system and pituitary following peripheral administration of endotoxin to mice. J Neurosci Res 48(4):287–298
Playfair JHL, Chain BM (1979) Immunology at a glance. Blackwell Science Publishing, London
Pocock JM, Kettenmann H (2007) Neurotransmitter receptors on microglia. Trends Neurosci 30(10):527–535 (S0166-2236(07)00211-1 [pii] 10.1016/j.tins.2007.07.007)
Procaccini C, Pucino V, De Rosa V, Marone G, Matarese G (2014) Neuro-endocrine networks controlling immune system in health and disease. Front Immunol 5:143 (10.3389/fimmu.2014.00143)
Qian J, Zhu L, Li Q, Belevych N, Chen Q, Zhao F, Herness S, Quan N (2012) Interleukin-1R3 mediates interleukin-1-induced potassium current increase through fast activation of Akt kinase. Proc Natl Acad Sci USA 109(30):12189–12194 (10.1073/pnas.12052071091205207109 [pii])
Quan C, Yu H, Qiao J, Xiao B, Zhao G, Wu Z, Li Z, Lu C (2013) Impaired regulatory function and enhanced intrathecal activation of B cells in neuromyelitis optica: distinct from multiple sclerosis. Mult Scler 19(3):289–298 (10.1177/13524585124547711352458512454771 [pii])
Ransohoff RM, Liu L, Cardona AE (2007) Chemokines and chemokine receptors: multipurpose players in neuroinflammation. Int Rev Neurobiol 82:187–204 (S0074-7742(07)82010-1 [pii] 10.1016/S0074-7742(07)82010-1)
Rivest S, Lacroix S, Vallieres L, Nadeau S, Zhang J, Laflamme N (2000) How the blood talks to the brain parenchyma and the paraventricular nucleus of the hypothalamus during systemic inflammatory and infectious stimuli. Proc Soc Exp Biol Med 223(1):22–38 (pse22304 [pii])
Romeo HE, Tio DL, Rahman SU, Chiappelli F, Taylor AN (2001) The glossopharyngeal nerve as a novel pathway in immune-to-brain communication: relevance to neuroimmune surveillance of the oral cavity. J Neuroimmunol 115(1–2):91–100 (S0165572801002703 [pii])
Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ (2001) Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410(6827):471–475 (10.1038/3506856635068566 [pii])
Schafer KH, Mestres P, Marz P, Rose-John S (1999) The IL-6/sIL-6R fusion protein hyper-IL-6 promotes neurite outgrowth and neuron survival in cultured enteric neurons. J Interferon Cytokine Res 19(5):527–532. doi:10.1089/107999099313974
Schiffenbauer J, Johnson HM, Butfiloski EJ, Wegrzyn L, Soos JM (1993) Staphylococcal enterotoxins can reactivate experimental allergic encephalomyelitis. Proc Natl Acad Sci USA 90(18):8543–8546
Schmitz T, Chew LJ (2008) Cytokines and myelination in the central nervous system. Sci World J 8:1119–1147. doi:10.1100/tsw.2008.140
Semple BD, Bye N, Ziebell JM, Morganti-Kossmann MC (2010) Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiol Dis 40(2):394–403 (10.1016/j.nbd.2010.06.015S0969-9961(10)00211-1 [pii])
Seo JH, Miyamoto N, Hayakawa K, Pham LD, Maki T, Ayata C, Kim KW, Lo EH, Arai K (2013) Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J Clin Invest 123(2):782–786 (10.1172/JCI6586365863 [pii])
Serres S, Anthony DC, Jiang Y, Broom KA, Campbell SJ, Tyler DJ, van Kasteren SI, Davis BG, Sibson NR (2009) Systemic inflammatory response reactivates immune-mediated lesions in rat brain. J Neurosci 29(15):4820–4828 (29/15/4820 [pii] 10.1523/JNEUROSCI.0406-09.2009)
Sly LM, Krzesicki RF, Brashler JR, Buhl AE, McKinley DD, Carter DB, Chin JE (2001) Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res Bull 56(6):581–588 (S0361923001007304 [pii])
Sorrells SF, Sapolsky RM (2010) Glucocorticoids can arm macrophages for innate immune battle. Brain Behav Immun 24(1):17–18 (S0889-1591(09)00468-1 [pii] 10.1016/j.bbi.2009.10.004)
Takii T, Akahoshi T, Kato K, Hayashi H, Marunouchi T, Onozaki K (1992) Interleukin-1 up-regulates transcription of its own receptor in a human fibroblast cell line TIG-1: role of endogenous PGE2 and cAMP. Eur J Immunol 22(5):1221–1227. doi:10.1002/eji.1830220517
Takii T, Hayashi H, Marunouchi T, Onozaki K (1994) Interleukin-1 down-regulates type I interleukin 1 receptor mRNA expression in a human fibroblast cell line TIG-1 in the absence of prostaglandin E2 synthesis. Lymphokine Cytokine Res 13(3):213–219
Tauber SC, Nau R, Gerber J (2007) Systemic infections in multiple sclerosis and experimental autoimmune encephalomyelitis. Arch Physiol Biochem 113(3):124–130 (782870844 [pii] 10.1080/13813450701531227)
Teeling JL, Perry VH (2009) Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: underlying mechanisms. Neuroscience 158(3):1062–1073 (S0306-4522(08)01045-2 [pii] 10.1016/j.neuroscience.2008.07.031)
Trenova AG, Manova MG, Kostadinova II, Murdjeva MA, Hristova DR, Vasileva TV, Zahariev ZI (2011) Clinical and laboratory study of pro-inflammatory and antiinflammatory cytokines in women with multiple sclerosis. Folia Med (Plovdiv) 53(2):29–35
Tsai HH, Frost E, To V, Robinson S, Ffrench-Constant C, Geertman R, Ransohoff RM, Miller RH (2002) The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell 110(3):373–383 (S0092867402008383 [pii])
Tsunoda I, Fujinami RS (2002) Inside-Out versus Outside-In models for virus induced demyelination: axonal damage triggering demyelination. Springer Semin Immunopathol 24(2):105–125. doi:10.1007/s00281-002-0105-z
Tsunoda I, Kuang LQ, Libbey JE, Fujinami RS (2003) Axonal injury heralds virus-induced demyelination. Am J Pathol 162(4):1259–1269 (S0002-9440(10)63922-3 [pii] 10.1016/S0002-9440(10)63922-3)
Uzawa A, Mori M, Sato Y, Hayakawa S, Masuda S, Taniguchi J, Kuwabara S (2010) Cytokine and chemokines profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler 16(12):1443–52. doi:10.1177/1352458510379247
van Riemsdijk IC, Baan CC, Loonen EH, Knoop CJ, Navarro Betonico G, Niesters HG, Zietse R, Weimar W (2001) T cells activate the tumor necrosis factor-alpha system during hemodialysis, resulting in tachyphylaxis. Kidney Int 59(3):883–892 (kid571 [pii] 10.1046/j.1523-1755.2001.059003883.x)
Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, Brawek B, Thal DR, Charo IF, Heppner FL, Aguzzi A, Garaschuk O, Ransohoff RM, Jucker M (2012) Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci USA 109(44):18150–18155 (10.1073/pnas.12101501091210150109 [pii])
Veenstra M, Ransohoff RM (2012) Chemokine receptor CXCR2: physiology regulator and neuroinflammation controller? J Neuroimmunol 246(1–2):1–9 (10.1016/j.jneuroim.2012.02.016S0165-5728(12)00064-1 [pii])
Vitkovic L, Konsman JP, Bockaert J, Dantzer R, Homburger V, Jacque C (2000) Cytokine signals propagate through the brain. Mol Psychiatry 5(6):604–615
Wagner JA (1996) Is IL-6 both a cytokine and a neurotrophic factor? J Exp Med 183(6):2417–2419
Waxman SG (1998) Demyelinating diseases–new pathological insights, new therapeutic targets. N Engl J Med 338(5):323–325. doi:10.1056/NEJM199801293380610
Wilkins A, Chandran S, Compston A (2001) A role for oligodendrocyte-derived IGF-1 in trophic support of cortical neurons. Glia 36(1):48–57 (10.1002/glia.1094 [pii])
Wilson EH, Weninger W, Hunter CA (2010) Trafficking of immune cells in the central nervous system. J Clin Invest 120(5):1368–1379 (10.1172/JCI4191141911 [pii])
Wolswijk G (1998) Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci 18(2):601–609
Xia M, Hyman BT (2002) GROalpha/KC, a chemokine receptor CXCR2 ligand, can be a potent trigger for neuronal ERK1/2 and PI-3 kinase pathways and for tau hyperphosphorylation-a role in Alzheimer’s disease? J Neuroimmunol 122(1–2):55–64 (S0165572801004635 [pii])
Ysrraelit MC, Gaitan MI, Lopez AS, Correale J (2008) Impaired hypothalamic-pituitary-adrenal axis activity in patients with multiple sclerosis. Neurology 71(24):1948–1954 (10.1212/01.wnl.0000336918.32695.6b 71/24/1948 [pii])
Acknowledgments
Carina C. Ferrari and Verónica Murta are members of the Research Career of the National Council of Scientific and Technological Research (CONICET), Argentina. CF is supported by CONICET (PIP 2012-2014, 11220110100560) and National Agency of Science and Technology of Argentina (ANPCyT) (PICT 2012-2014).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Murta, V., Ferrari, C. (2016). Peripheral Inflammation and Demyelinating Diseases. In: von Bernhardi, R. (eds) Glial Cells in Health and Disease of the CNS. Advances in Experimental Medicine and Biology, vol 949. Springer, Cham. https://doi.org/10.1007/978-3-319-40764-7_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-40764-7_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-40762-3
Online ISBN: 978-3-319-40764-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)