
Abstract
Mitochondria arose from bacterial endosymbionts. One of the consequences of the endosymbiosis event was that the ancestral bacterial genome underwent deep transformations, including massive gene transfer to the host nucleus and gene losses and rearrangements. Upon eukaryotic origin, the gene content, size and shape of mitochondrial genomes evolved differently throughout eukaryote’s radiation into many lineages. As a consequence of this phenomenon, the mechanisms for gene expression in mitochondria have also co-evolved and diverged from the ancestral bacterial systems. Mitochondria possess the complete machinery to translate reduced sets of messenger RNAs transcribed in these organelles. Although this machinery retained many features from bacteria, it has also undergone modifications across eukaryote lineages, rendering this process highly specialized and regulated. In this chapter, we summarize and discuss the general characteristics of the mitochondrial translation machinery. We also discuss the current knowledge on mitochondrial translation across different eukaryotic phyla and compare it with its bacterial counterpart to throw light on the evolution of translation in mitochondria.
Aldo E. García-Guerrero and Angélica Zamudio-Ochoa have contributed equally to this work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Mitochondrial Genome
- Land Plant
- Small Ribosomal Subunit
- Mitochondrial Translation
- Mammalian Mitochondrion
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Around 1.5 billion years ago a bacterial cell related to modern α-proteobacteria established a symbiosis with a eukaryote that originated mitochondria [1]. It is well-established that mitochondrial origin is monophyletic (i.e., it happened only once in evolution) and that the organelle arose from an α-proteobacterium with identity yet to be established [2, 3]. The symbiotic event was followed by extensive reduction of the organelle’s genetic material, either by gene loss or gene transfer to the nuclear genome. In addition, mitochondrial DNA (mtDNA) from different lineages diverged extensively in shape, size, content, mutation rate, and gene expression mechanisms. What mitochondria from different lineages have in common is that more than 1000 proteins are present in the organelle [4–7]. However, only a very limited number of proteins are encoded in mtDNA. For example, mtDNAs from the Phylum Apicomplexa have only three protein-coding genes [8], animal mitochondria code (in general) for 13 proteins [9], land plants code for more than 30–40 proteins [10], and members of the jakobid protists, which are considered to be relics of the endosymbiont bacterial ancestor, code around 65 proteins [11]. Thus, the majority of proteins necessary for function are imported into mitochondria from the cytosol (for a review, see [12]). The organization of mtDNA among lineages has also diverged. Some organisms have extended non-coding regions, including type I and type II introns, as is the case for land plants, while others, like metazoan, have very compact mitochondrial genomes with only a few hundreds non-coding regions [9]. While fungal and apicomplexan mtDNAs are lineal molecules, animals and some protists have circular mtDNA, trypanosome mtDNA is composed of minicircles and maxicircles, which are topologically intertwined [13], and some Amoebidium have several hundred linear DNA molecules with different gene contents in each molecule [14]. In land plants, mtDNA is arranged in circular molecules of DNA whose composition varies constantly as a high frequency of recombination events occurs in this clade [10].
Independent of the shape, coding capacity and size of mtDNA, these organelles contain a complete gene expression system that comprises DNA replication and maintenance, transcription, post-transcriptional processing, translation and post-translation functions, such as protein assembly and prosthetic group additions. Much of the mitochondrial expression machinery is nucleus-encoded, while only a limited set of mtDNA genes is coded in the organelle. The Phylum Apicomplexa has only two ribosomal RNAs coded by the mtDNA. Metazoans have around 22 tRNAs and the small and large subunits rRNAs, while land plant mtDNA in addition to tRNAs and rRNAs codes for a varied number of ribosomal proteins. Protists from the jakobid lineage code in addition a translation factor (TufA), 5S rRNA, RNA polymerase and a sigma factor, and three chaperones for protein processing [11]. In general, mitochondrial genomes code for subunits of respiratory complexes and ATP synthase. Apicomplexan mtDNAs code for only subunits 1 and 3 of cytochrome c oxidase and cytochrome b from complex bc1 [8]. In contrast, the jakobid Phylum codes for 12 subunits from complex I (NADH dehydrogenase), 3 for complex II (succinate dehydrogenase), 1 for bc1 complex, 3 for cytochrome c oxidase and 6 for ATP synthase [11].
Since mitochondria evolved from an α-proteobacterial ancestor, one might expect that the mtDNA expression mechanisms have conserved bacterial features. Even when this is the case, many novel mechanisms to control mtDNA expression have emerged and diverged among the eukaryotic groups. Some are conserved among certain lineages, but others appeared later during eukaryote divergence. In the present chapter, we describe the most prominent features of the mitochondrial translation machinery across different eukaryotic lineages. This knowledge allows us to better understand the evolution of the translation process in mitochondria.
2 The Mitochondrial Genetic Code
Translation in jakobid and land plant mitochondria uses the universal genetic code in mitochondria [11, 15]. However, at least 27 genetic code alterations (i.e., codon reassignments) are detected in mitochondrial systems of diverse eukaryote lineages (reviewed in [16, 17]). One of the most common changes in mitochondrial genetic code is the reassignment of termination codons to sense codons, such as the use of the canonical UGA stop codon to decode tryptophan in numerous biological groups [18] (Table 1). Other cases include the use of the typical UAG triplet as a leucine codon in chlorophycean algae [19] and the codon UAA, which decodes tyrosine in the nematode Radopholus similis [20]. Non-standard stop codons are used in mitochondria from some lineages. For example, the chlorophycean algae Scenedesmus obliquus uses the TCA codon as a translation stop signal [21]. In bryophytes and vascular plants, the codons CAA, CGA and GGU are reassigned stop codons, while AAA and AAU are recognized as stop codons in Oryza sativa [22]. In vertebrate mitochondria, the AGA and AGG codons, which are universally assigned to arginine, were thought to become stop codons [23]. However, recent studies indicate that these codons are unassigned [24]. Other prevalent reassignment is the use of AUA in Saccharomyces cerevisiae, vertebrates and some invertebrates to decode methionine instead of the canonical isoleucine [25, 26]. The standard arginine codons AGA/AGG were reassigned to serine in certain invertebrate groups (Nematoda, Arthropoda) and decode glycine in Ascidians [27]. In S. cerevisiae, the typical arginine codons CGA/CGC are unassigned, and the triplets CUU/CUC/CUA/CUG are used for threonine instead of leucine [28]. In some invertebrates (flat and round worms), AAA was reassigned, from lysine to asparagine (for an example, see [29]). Atypical start codons are also present in mitochondrial systems. For example, humans use AUA and AUU as start alternatives [24]; other cases of alternative start codons occur in trypanosomatids, which use UUA, UUG, CUG and the ciliate Tetrahymena with AUU, AUA or AUG [30] and certain nematodes that use the UUG triplet to initiate protein translation [31].
Why did mitochondria acquire modified codon assignments during evolution? One explanation is that codon reassignments might be a consequence of the organelle genome reduction, which encodes for a small set of proteins, and in most cases for a small number of tRNAs [8–10]. The diversity of mitochondrial genetic codes across eukaryotic groups might also reflect differential mutational rates in mtDNAs, a general increase in AT content and a diversification of genome expression mechanisms [17]. Interestingly, in silico studies suggest that genome size is not correlated to incident mutations that could lead to codon reassignments (i.e., the size of mitochondrial genomes does not correlate with mutation rates) [16]. The tRNAs’ structure, the mitochondrial-targeted aminoacyl tRNA synthetases and in general the translation machinery are adjusted to the mitochondrial genetic code of each eukaryotic group. For example, reassignment of UGA for tryptophan (instead of the stop codon) is mediated by a tRNA where the wobble position carries a modified uridine. Modifications include 5-taurinomethyluridine (τm5U), 5-carboxymethylaminomethyl-2-thio-uridine (cmnm5s2U) or 5-carboxymethylaminomethyluridine (cmnm5U). These modifications expand the decoding capacity to R-ending codons, enabling the decoding of UGG and UGA as tryptophan [32]. Decoding of mammalian AUG and AUA as methionine is possible because the met-tRNAMet(CAU) has a 5-formylcytidine (f5C) in the wobble position [33, 34]. Some theories try to explain how reassignments in the mitochondrial genetic code might have occurred during evolution. Two of the most established theories are the Codon Capture and Ambiguous Intermediate models.
The Codon Capture, also termed the Codon Disappearance theory, proposed by Osawa and Jukes in 1989, postulates that genetic code alterations are the result of neutral changes associated with the GC/AT content balance [35–38]. The theory posits that the disappearances of both the codon and the decoding tRNA are fundamental steps for further codon reassignment. Later, the “lost codon” can be reintroduced into the system by new mutations, but now is decoded at a relatively low efficiency by a different, noncognate tRNA, but with a similar anticodon sequence that allows the “capture” of the recently reestablished codon. Some reassignments are consistent with this Codon Capture model, such as the case of the frequent reuse of the UGA triplet to decode tryptophan [16]. A prediction derived from this model is that in mitochondrial genomes, which are high in AT content, GC-rich codons disappear at higher frequencies than AT-rich codons [36, 38, 39]. However, some codon reassignments in mtDNA do not follow the predictions of the GC/AT content balance. Thus, the Codon Capture theory does not explain satisfactorily the use of GC-rich codons in genomes with high AT content or the fact that some codons seem to be unassigned in some mtDNA genetic systems.
The Ambiguous Intermediate theory, proposed by Schultz and Yarus [40, 41], suggests that codon reassignment is the result of selective mechanisms that favor ambiguity in codon recognition during protein translation. The model postulates that codon recognition ambiguity, associated with structural changes in the tRNA molecules, is fundamental for the codon reassignment. The idea is that the codon in the spotlight is suddenly decoded by two different tRNAs, namely the “original” and the new “mutant,” which is now able to form a cognate pair with the codon. Later the “mutant” tRNA takes over the codon in a selection-driven process. Thus, the triplet is reassigned to a new amino acid. During mitochondrial evolution, many repeated tRNAs for each amino acid were lost, and in general mitochondria contain only one tRNA for each amino acid [42]. In contrast to the Codon Capture theory, in this model the initial loss of the codon before the reassignment is not necessary [37]. Some examples consistent with the Ambiguous Intermediate theory are the reassignments of leucine to threonine in yeast mitochondria [17] and from serine to lysine in Arthropoda [43].
Overall, both models are not mutually exclusive, as reassignments might have arisen from combinatory events during evolution [17, 44]. Some changes in the mitochondrial genetic code are explained by the Codon Capture theory, while others by the Ambiguous Intermediate theory.
3 Mitochondrial tRNAs
Translation of mitochondrial mRNAs requires around 20 tRNAs, but the exact number varies depending on the wobble rules and the genetic code in each species. Mitochondrial tRNAs have nuclear and mitochondrial origins. Depending on the organism, the proportion of nuclear and mitochondrial tRNAs varies. While human and the jakobid Andalucia godoyi [9, 11] encode a complete set of mitochondrial encoded tRNAs for reading all codons in mtDNA, protist-like Trypanosoma brucei and Plasmodium falciparum have no mtDNA-coded tRNAs [8, 45] and therefore have to import all tRNAs necessary for translation. Interestingly, the number of mtDNA-derived tRNAs among closely related organisms is variable. For example, in chlorophycean algae, Chlamydomonas reinhardtii codes for only 3 tRNAs in mitochondria, while Nephroselmis olivacea codes for a full set of 26 tRNAs [46]. It is expected that mitochondria would import only the necessary number of tRNAs to complete the ~22 tRNAs necessary for translation. However, in some cases, import of redundant tRNAs can take place. For example, in the yeast S. cerevisiae a tRNALys(CUU) is imported from cytosol even when mtDNA codes for the full set of tRNAs necessary to decode all codons [28, 47]. This tRNA is particularly important to decode codons under stress conditions [48]. Mammalian mitochondria can also import redundant cytosolic tRNAs [49]. The unicellular algae C. reinhardtii imports 31 tRNAs instead of the expected 22 tRNAs necessary to decode all codons [50]. The mechanisms to import cytosolic tRNAs are particular to each eukaryotic group, indicating that import of tRNAs into mitochondria is a process that emerged independently several times during evolution (for a review, see [51]). Delivery of tRNAs to mitochondria is mediated by proteins, usually with a previously described function. S. cerevisiae Eno2 (involved in glycolysis) delivers the charged tRNALys(CUU) to the mitochondrial surface, where the mitochondrial lysyl-tRNA synthetase binds it and co-transports it via the general import machinery. In land plants, aminoacyl-tRNA synthetases might be involved in the delivery of tRNAs to mitochondria, and the Voltage Dependent Anion Channel (VDAC), together with the outer membrane receptors Tom20 and Tom40, functions in tRNA import. In Trypanosoma, the cytosolic EF1a, together with the import component Tim17 and Hsp70, Hsp60 and Hsp20 might participate in the delivery and import of cytosolic tRNAs.
The structure, sequence and post-transcriptional modifications of mitochondrial tRNAs have conserved features with cytosolic RNAs. However, many of these features have amazingly diverged in different eukaryotic groups and among specific tRNAs from the same organism. According to the structural characteristics, mitochondrial tRNAs are classified into five groups, named 1–5 [51]. Group 1 shares the most conserved features with cytosolic tRNAs. They carry canonical T and D arms, anticodon and acceptor arms, and L1/l2 connectors (involved in joining the acceptor and anticodon helices) [52]. This class of tRNAs is present in mitochondria from amoebozoans, alveolates, plants and fungi. Group 2 carries conserved anticodon and acceptor arms. However, T/D arms may be smaller in size and may have less conservation on bases involved in D/T-loop interactions (mainly bases G18, G19, U55 and C56). These tRNAs are present in amoebozoans, alveolates, plants, fungi and some metazoans (including mammals). Group 3 consists of tRNAs where the acceptor arm may be 1–3 nucleotides shorter; they are T-armless and carry a shorter D-loop. The L2 connector is also shorter (6–7 nt instead of 21–30 nt). This class of tRNAs is present in some nematodes, bryozoan and arachnid species. Group 4 is represented by some insect and bryozoan species and by mammals. They are D-armless and carry shorter T arms. The L1 connector is also shorter (5–12 nt instead of 19–20 nt). Group 5 carries both shorter anticodon and acceptor arms; they are T- and D-armless and have shorter L1 and L2 connectors. These minimalist tRNAs are found in acaria and some nematodes. In this group, the only conserved features with cytosolic tRNAs are the presence of an acceptor arm with the 3′-single-stranded CCA terminus and an anticodon arm with the canonical anticodon loop of seven nucleotides. The shortest mitochondrial tRNA so far is 54-nt long (tRNASer(UCU)) from the nematode Ascaris suum [53, 54].
As a universal feature, mitochondrial tRNAs are also post-transcriptionally modified to define the structure and decoding capabilities. The best understood model is Bos taurus, where all mitochondrial tRNAs were isolated and analyzed. There are 15 types of modifications at 118 positions (representing 7.5 % abundance in mitochondrial tRNA bases) [55]. However, the occurrence of modified nucleotides can be as low as one residue in mitochondrial tRNASer of the rodent Mesocricetus auratus (representing 1.7 % abundance) [56]. To date, 15 out of 18 conserved modifications (present throughout kingdoms of life) are observed in mitochondrial tRNAs, with the exception of ac4C, m3U and m66A, which are not yet detected (reviewed in [51]). Comparative analyses of tRNA sequences indicate that mitochondria have the highest number of modified positions that are not universally conserved. The acceptor stem is particularly rich in Ψ residues, and the number of modifications located in positions 46–50, 5′ to the T arm, is also relatively low in mitochondrial tRNAs [55, 56]. There are mitochondria-specific base modifications, like τm5U and τm5s2U, discovered in ascidian mitochondria [32], f5C, f5Cm, present at the wobble position 34 in bovine and the nematode A. suum [33, 54], and k2C in potato [57].
4 Mitochondrial mRNAs
Mitochondrial mRNAs have conserved some prokaryotic features, but some other characteristics have diverged. Mitochondrial mRNAs from some lineages, such as jakobid protists, have a putative Shine-Dalgarno-like sequence to locate the ribosome at the correct AUG start codon [11]. Other lineages lack a Shine-Dalgarno-like sequence and therefore must have different, unknown mechanisms to initiate translation. This is the case for flowering plants [58] and mammal mitochondria [59]. Similar to what is observed in prokaryotes, mitochondrial mRNAs do not have a 7-methylguanylate cap (5′-cap), as is found in cytosol mRNAs. Moreover, mitochondrial mRNAs undergo post-transcriptional modifications before they are ready for translation. The major post-transcriptional RNA processing events in mitochondria include 3′-end polyadenylation, intron/exon splicing and editing. Polyadenylation of RNA is present in all kingdoms of life and is a near-universal feature of RNA metabolism, although it can trigger different signals among cells and organelles. Today, the function of polyadenylation in mitochondrial gene expression is not fully understood. An additional interesting feature of mitochondrial mRNAs from some lineages is the requirement of RNA edition before translation. RNA editing might be important to correct transcript sequences that otherwise would affect the translation product’s function [59, 60]. An example is editing in land plants, where the amino acid encoded by an edited mRNA is frequently more conserved than the one predicted from the gene sequence.
4.1 Polyadenylation of Mitochondrial mRNAs
Polyadenylation is the non-template addition of adenosine residues to the 3′ end of RNAs. In the eukaryotic cytoplasm, the majority of nuclear-encoded mRNAs require a poly(A) tail for stability, nuclear export and translatability (for reviews, see [61, 62]). In contrast, in prokaryotes, RNA polyadenylation functions to tag the mRNA for exonucleolytic degradation [63, 64]. Although mitochondria have a monophyletic origin, many features of polyadenylation have extensively diverged within eukaryotes.
In mammalian mitochondria, 12 out of 13 mRNAs have stable poly(A) tails of 45 nt on average. However, there are slight variations between cell types and between transcripts within the same cell type [65]. For example, only the ND6 transcript lacks a poly(A) tail [66]. The precise function of polyadenylation is not entirely understood. However, one function of polyadenylation is to complete the UAA codon, since several mammalian RNAs contain incomplete translational stop codons. The same feature is observed in general in metazoans, where some coding regions lack a complete UAA stop codon, suggesting that polyadenylation also plays an important role in translation [67, 68]. Although polyadenylation produces stable transcripts [66, 69], truncated, adenylated transcripts may coexist, suggesting that human mitochondria use transient poly(A) tails to degrade RNA [70]. The mechanism of a possible differential polyadenylation on stabilizing and destabilizing RNAs remains to be elucidated. In plants, similarly to the bacterial system, addition of a poly(A) tail targets exonucleolytic degradation of RNA [71]. In trypanosomatid mitochondria, most protein-coding transcripts suffer a massive edition (insertion or deletion of uridines) necessary to render translatable mRNAs [59]. The addition of a poly(A) tail in these organisms seems to render both stable and unstable transcripts. Polyadenylation in these organisms has an intricate relation to edition and translation. Poly(A) tails are 20–200 nt long, and the length of the tail seems to correlate with the state of edition. Short tails (~20 nt) stabilize edited or non-edited mRNAs. Long (100–200 nt) poly(A/U) tails are added to fully edited RNAs, and this extension might render the transcript translationally competent [72, 73].
Yeast mitochondria are so far the only organelles that do not polyadenylate their mRNAs. This was found to be the case in S. cerevisiae, Schizosaccharomyces pombe and Candida albicans [74–77], suggesting that it might be a general phenomenon of fungal mitochondria. Instead, the 3′ ends of some, but not all fungal mitochondrial mRNAs possess a conserved dodecamer sequence that is encoded in the mitochondrial genome and seems to be vital for mRNA stability and translatability [75, 76, 78, 79].
4.2 Edition of Mitochondrial mRNAs
Some organisms require mitochondrial (and plastid) transcript edition before they can be translated. RNA editing consists of nucleotide substitutions, post-transcriptional or co-transcriptional insertion/deletions. These three processes occur in very different taxonomic groups, suggesting that they arose as several independent acquisitions [80]. This process is present in dinoflagellates (variable one-nucleotide substitutions), excavates (U insertions/deletions), unikonts (co-transcriptional insertion of 1 or 2 nucleotides), metazoa (U to C substitution) and archaeplastida (U to C and C to U substitutions) [80]. Editions throughout a transcript can be limited in number, as is the case for land plants [60, 81]. In other cases, extensive edition of a transcript is required to transform an unrecognizable sequence into a conserved protein sequence, as is the case of trypanosomatids [59, 82] and calcaronean sponges [83].
In land plants, C to U (and less frequently U to C) editing often results in changes of the amino acid sequence from what the genomic sequence predicts. This process evolved in land plants [84] and was most likely subsequently lost in some marchantiid liverworts [85]. The number of edited nucleotides among plant lineages is: Physcomitrella patens edits 11 sites [86], Arabidopsis thaliana edits 600 cytidines [87], while the lycophytes Isoetes engelmanii and Selaginella moellendorfii edit more than 1,700 and 2,100 nucleotides, respectively [88, 89]. The composition of the RNA editosome is not yet fully understood, although cis- and trans-factors are essential for the editing process. The cis elements that specify the editing of the C target are present in close proximity to the edition site. Trans-factors include members of the pentatricopeptide repeat (PPR) motif-containing family, which are site-specific recognition factors. While the cytidine deaminase catalyzing C-to-U conversion has not been identified, considerable evidence points to the C-terminal DYW domain found on some PPR proteins, which exhibits sequence similarity to known cytidine deaminase motifs (for reviews, see [60, 80, 81]).
In trypanosomatids, edition is a post-transcriptional process, where uridines are inserted or deleted from mRNA precursors [90]. Edition introduces start and stop codons, restores frame shifts and often completes the coding sequence of mRNAs. Mitochondrial editing can occur at different extensions: transcripts that are never edited, transcripts where edition is restricted to a small region, with minimal edition, and transcripts that are extensively edited or pan-edited, where a single mRNA is altered by 553 insertions and 89 deletions [80]. The process in trypanosomatids includes mRNA cleavage, U deletion or insertion, and mRNA ligation [91]. The maxicircle molecules of mtDNA code for guide RNAs (gRNAs), which are derived from scattered intergenic regions. A partial hybrid is formed between the 5′ portion of the gRNA and the complementary sequence on the pre-edited mRNA. Cleavage of the mRNA at the 3′ end of the first base that is not paired with the gRNA leaves a free 3′OH. The uridine addition or deletion is followed by immediate relegation of the two molecules. Many proteins have been implicated in the edition process (reviewed in [59, 82]). However, these proteins are not related to the proteins involved on plant edition.
5 The Mitoribosome
Mitochondrial ribosomes (mitoribosomes) are located in the matrix, and are closely associated with the inner membrane [92, 93]. This location facilitates the insertion of newly synthesized products, which are mainly hydrophobic proteins. All mitochondrial genomes currently sequenced encode ribosomal RNAs (rRNAs). In contrast, almost all mitochondrial ribosomal proteins (MRPs) are nuclear encoded. Thus, assembly of functional ribosomes requires a coordinated expression of both genomes and a proper import of the necessary components into the organelle [94, 95]. The mechanism of this process is almost unknown, but evidence supports that several MRPs assemble with rRNAs in a co-transcriptional fashion [96, 97].
In contrast to the cytosolic ribosomes, mitoribosome composition is highly variable between different eukaryotic lineages. Their sedimentation coefficient ranges from 80S in ciliates, to 70–74S in fungi, to 77–78S in vascular plants and 55S in animals. These variable sedimentation values are the result of the difference in the protein:RNA ratio, while bacterial ribosomes contain a protein:RNA proportion of 1:2, in mitoribosomes this proportion varies from 1:1 in yeast to 2:1 in bovine [98].
The α-proteobacterial ribosome is composed of 54 proteins [99], which were also likely to be present in the ancestor of mitochondria. It is proposed that, in the earliest stage of eukaryotic evolution, several novel proteins were recruited for ribosomal function, and only one, Rps20, was lost, resulting in an ancestral mitoribosome of 72 proteins (Fig. 1) [100]. An interesting feature of several mitoribosomal proteins of bacterial origin is that they increased in length sequence. Accordingly, this stage in mitoribosome evolution is known as the “constructive phase”, as the total size of the ribosome was increased considerably [101].

Reconstruction of the evolutionary history of the mitochondrial ribosome proteome. Incoming and outgoing arrows indicate the gains and losses of the ribosomal proteins that are showed in the box. This figure is based on the data given by [102] and [100]. The models considered for the construction of this figure were: for fungi Neurospora crassa, Aspergillus fumigatus, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Cryptococcus neoformans, Ustilago maydis and Encephalitozoon cuniculi; for metazoa Mus musculus, Homo sapiens, Danio rerio, Drosophila melanogaster, Caenorhabditis elegans and Monosiga brevicolis; for amoebozoa Dictyostelium discoideum and Entamoeba histolytica; for Archaeplastida Arabidopsis thaliana, Oryza sativa, Chlamydomonas reinhardtii, Ostreococcus tauri and Cyanidioschyzon merolae; for Strameopila Thalassiosira pseudonana and Phytophthora ramorum; for Alveolata Tetrahymena thermophila, Paramecium tetraurelia, Theileria annulata, Plasmodium falciparum, Plasmodium yoelii and Cryptosporidium parvum and for Excavata Leishmania brasilensis, Leishmania infantum, Leishmania major, Trypanosoma cruzi, Trypanosoma brucei, Naegleria gruberi, Trichomonas vaginalis, Giardia lamblia and Reclinomonas americana
The cause of the constructive phase of the mitoribosome is proposed to be the accumulation of slightly deleterious mutations on the mitochondrial genome, as this genome, with the exception of land plants, presents a higher mutation rate than the nuclear one [103, 104]. Slightly deleterious mutations could trigger the recruitment of new proteins because a mutation in an original component of the complex is compensated by the interaction with a new component [105]. This process is called Constructive Neutral Evolution (CNE), a universal evolutionary ratchet that leads to complexity [106]. Accordingly, genes coding for MRPs show higher levels of amino acid replacements than cytoplasmic ribosomal proteins, which suggests a compensatory modification [107, 108]. Gain of complexity throughout the evolution of mitochondria is not exclusive to the mitoribosome. The respiratory chain complexes have also acquired new proteins that are usually important for regulation, assembly and stability [101]. These eukaryotic subunits are in general localized in the peripheral regions of the enzymes. This feature is also observed for the mitoribosomes [109, 110]. The extensive gain of protein mass observed for mitoribosomes does not reflect the fate of all endosymbiotic organelles, as the plastid ribosomes only gained approximately 170 kDa [111, 112]. Several evolutionary mechanisms have led to the increase of protein mass in the mitoribosome. One of them was the recruitment of existent proteins, such as the case of Mrpl45, a homolog of Tim44 (a subunit of the mitochondrial protein translocase machinery), which is present in several bacteria but is not part of the prokaryote ribosome [102]. Mrpl39, a metazoan protein, was recruited later in evolution and is homologous to threonyl-tRNA synthetases [113]. It is proposed that addition of Mrpl39 to the mitoribosome compensated for the loss of bacterial proteins involved in tRNA binding [102]. Numerous new ribosomal proteins emerged through gene duplication. For instance, Mrps10 gave rise to Mrpl48 through this process in metazoans. Interestingly, the duplicated gene product became part of the other ribosomal subunit [102]. Another case is Mrps18, which in Caenorhabditis elegans has three variants originated by gene duplication. It is believed that each ribosome contains only one copy of the protein, suggesting that mitoribosomes exist in heterogeneous populations [102].
The increment in protein mass in mitoribosomes is not only due to the addition of new subunits, but also to the gain of new domains in the prokaryotic proteins. MRPs are sometimes almost twice the size of their bacterial counterparts [102]. In P. falciparum, Mrpl4 has an AAA domain, which is not present in the bacterial counterpart. This domain is known to participate in chaperone-like functions [114]. Another case is the presence of an RRM (RNA recognition motif) domain in Mrps19 of A. thaliana, which could be involved in the association of the protein with rRNAs [115]. In the yeast S. cerevisiae, the carboxyl-terminal end of Mrp20, which is mitochondria-specific, plays a role in ribosome assembly [116].
The evolution of mitochondria involved numerous independent losses of ribosomal proteins in different lineages (Fig. 1). Bacterial-exclusive S20 protein seems to have been lost early during mitoribosome evolution. This protein is not essential for bacterial growth. However, its absence causes a decrease in the association of the ribosomal subunits [117, 118]. In contrast, S1 protein, which was lost early in the evolution of unikonts, is an essential protein in bacteria [119]. Moreover, there is no apparent pattern favoring protein loss from either bacterial or eukaryotic origin, suggesting that there is no tendency in protein dispensability [100].
Whereas protein gain in mitoribosomes is a general phenomenon in all lineages, the rRNA content varies greatly. While bacteria have an rRNA content of 1.4 MDa, in mitochondria this number varies from 0.5 MDa in C. elegans to 1.6 MDa in Neurospora crassa. Since animals show an important reduction of rRNA, it was previously thought that the proteins acquired during mitoribosome evolution replaced the lost helices of rRNA [120]. However, now it is clear that the high content of proteins in mitoribosomes is not a consequence of the lower concentration of rRNA, as the increase in MRPs occurred previously to the reductive phase of rRNA [101]. This is consistent with the structural data in which the extra proteins of the ribosome do not substitute the lost portions of rRNA [121–123]. Furthermore, it is proposed that rRNA reduction might be driven by the reduction of the mitochondrial genome size and not necessarily by adaptive changes of the translational machinery [124].
Reduction of rRNA had triggered an important mitoribosome remodeling. For example, the bacterial ribosomal protein L24 contacts the helices H7 and H19 of the 23S rRNA, stabilizing its binding to the 39S subunit. The mammalian mitochondrial counterpart, Mrpl24, lacks both helices. However, mitochondria-specific protein elements maintain Mrpl24 in the same place and orientation as the prokaryotic counterpart [125].
An almost general phenomenon in mitochondria is the loss of 5S rRNA, which is present only in plants and some algae. An extension of the 23S rRNA replaces the resulting gap in N. crassa. On the contrary, in the mammalian mitoribosome this space is occupied by protein [121–123].
An interesting aspect of the evolution of mitoribosomes is their assembly mechanisms. This process has probably evolved differently in each lineage, as the components of the ribosome are partially different among eukaryote groups. In some lineages mitochondrial-encoded rRNAs are fragmented, need edition or lack 5S rRNA [8, 9, 126]. As stated above, the composition of proteins also diverged among eukaryote lineages. The understanding of ribosome assembly, the order of rRNA processing and protein addition, the chaperones involved in such events and the role of mitochondrial RNA granules in ribosome biogenesis are just starting to emerge, especially in mammals and yeast models [97, 127].
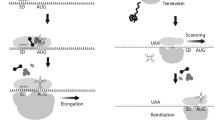
6 Mitochondrial Translation Initiation
Translation initiation in bacteria is carried out by three conserved factors: IF1, IF2 and IF3 [128]. There are important differences between prokaryote and mitochondrial initiation factors. While mitochondrial IF2mt is universally present, IF3mt is semi-universal and IF1mt was completely lost from the mitochondrial machinery [129]. In addition, there are important structural variations in the mitochondrial initiation factors. In agreement with the prokaryotic origin, mitochondria seem to initiate translation with formylated methionine, at least for the studied cases. Initially, by in vitro experiments, it was demonstrated that the initiation machinery in mammals does not need a formylated Met-tRNA. However, recent experiments demonstrate that a failure in formylation is a cause of disease in humans [130, 131]. In the yeast S. cerevisiae it was previously shown that a mutant Δfmt1 (coding for a methionyl-tRNA formyltransferase) does not affect translation initiation [132]. However, an accessory factor, Aep3, was compensating the lack of Δfmt1. Double mutant Δfmt1 and Δaep3 affect respiratory growth [133]. The mechanisms of translation initiation regulation have extensively diverged from the bacterial counterpart. Despite the differences between bacterial and mitochondrial translation initiation factors, the general steps for initiation are conserved.
6.1 Structural and Functional Conservation of IF2mt
In bacteria, IF2 interacts with initiator fMet-tRNA and promotes binding with the small ribosomal subunit and with mRNA. It also contains a GTPase activity to release all initiation factors from the completely assembled ribosome into the mRNA. IF2 triggers the binding of tRNA to the incomplete P site on the 30S subunit. After binding of the 50S subunit to the initiation complex, IF2 GTP hydrolysis assists the release of all initiation factors from the completely assembled ribosome [128]. Bacterial IF2 contains six domains (I–VI). To date, the function of domain I is not completely understood. Domain II stabilizes the interaction of IF2 with the ribosomal 30S subunit; this region is not conserved among bacterial species. Domain III is a linker between domains II and IV. Domain IV contains the GTPase activity. Domain V interacts with the ribosomal 30S subunit, and domain VI recognizes the fMet-tRNA [134]. In mitochondria, IF2mt consists only of domains III–VI (Fig. 2). This short version of IF2mt is not particular for mitochondrial systems, as shorter versions of IF2 factors are also present in some bacterial groups, like extremophiles [135]. Instead, in mammals IF2mt interaction with the 28S is performed by domain III, and this interaction is even stronger when GTP is bound to domain V [136]. Domain III of IF2mt is not conserved in all eukaryote lineages, but its function might be compensated by the differences in the small ribosomal subunit protein and RNA content among eukaryotes [137]. Domain IV is the most conserved region of IF2mt in structure and sequence similarity, sharing 99 % (metazoans) to 50 % (fungi) identity with bacterial counterparts [138–140]. Domain V in IF2mt is modestly conserved with the bacterial IF2, sharing identity of 35–50 %. However, the function of this region is not completely understood [140]. It might be important for interaction with the small ribosomal subunit because structural modeling of IF2mt suggests that this region is similar to domain II of EF-Tu and EF-G. This region is important for contact with small ribosomal subunits [141]. Interestingly, domain V in metazoa IF2mt contains an insertion of variable length and sequence (Fig. 2). This region might perform the same function as IF1 [142] (discussed below). Domain VI in bacteria and mitochondria is divided into subdomains C1 and C2. Subdomain C2 is important for binding of IF2 to the fMet-tRNA [143]. Mutagenic analysis in Bacillus stearothermophilus shows that there are two critical cysteines at the 668 and 714 positions necessary for this interaction [144, 145]. These amino acids are usually present in IF2mt, suggesting that IF2mt subdomain C2 conserved the same function as in bacteria. IF2mt contains the C1 subdomain. However, as in bacteria, the function of this domain is still unknown. By NMR studies it was suggested that this subdomain from Bacillus stearothermophilus has a similar structure as domain III from eukaryotic eIF5B. This region is implicated in transmitting and amplifying structural changes to the G-domain after GTP binding [144, 146].

Alignment of the insertion sequence on mIF2 among species from different phyla. Variations in domain composition between E. coli IF2 and Homo sapiens mIF2 are presented in the upper panel. Numbers indicate amino acid positions. The insertion sequence (IS) is amplified, and an alignment among different species is shown. Alignments of the insertion sequences were made with the MAFFT software, with the Blosum70 matrix as in [129]
6.2 The Mystery of the Lost IF1 in Mitochondria
In bacteria, IF1 plays an important role in the recognition of the correct AUG initiation codon. IF1 interacts with the A site of the 30S ribosomal subunit and prevents binding of the initiator aminoacyl-tRNA to the A site. In addition, bacterial IF1 increases the affinity of IF2 for 30S, has a role in small subunit dissociation and assists the release of IF2 form the 70S complex [128, 147]. So far, biochemical and bioinformatic approaches have failed to identify mitochondrial IF1. This suggests that IF1 was lost at the earliest stage of eukaryotic evolution [129, 148]. Mitochondria may be able to bypass the need for IF1: Experimental evidence indicates that, in the presence of mammalian IF2mt and IF3mt, the bacterial ribosome does not need IF1 for the formation of the 70S particle or translation in general [149]. As discussed above, metazoan IF2mt contains an insertion between domains V and VI [129] (Fig. 2). Even though there is no conservation of the insertion sequence among eukaryotic IF2mts, it is possible that this insertion substitutes the function of IF1, at least in metazoa. Through cryo-electron microscopy and nuclease digestion experiments, it was observed that bacterial IF2 associates with the interphase of the 30S subunit [134, 148]. Modeling of mammalian IF2mt suggests that the extension is close to the small subunit A site, similar to bacterial IF1 [140]. How the need for IF1 is bypassed in other eukaryotic lineages remains an open question.
6.3 Mitochondrial IF3mt
Bacterial IF3 plays a critical role in translation initiation. It binds the 30S ribosomal subunit in order to prevent association with the 50S subunit. This interaction is necessary for the initiation complex to recognize the Shine-Dalgarno sequence in the mRNA and enhances the interaction and activity of IF2 [140, 150]. Simple BLAST-P analysis failed to detect orthologs of IF3 in mitochondria. However, the existence of IF3mt was hypothesized because orthologs of the bacterial ribosomal proteins S7, S11 and S18, which are in proximity to IF3, are present in mitochondria [129]. More sensitive searching algorithms, like PSI-BLAST, identified IF3mt in fungi, animal, plant and excavates mitochondria [129, 140]. Structural data show that the IF3 C-terminal end is necessary for interaction with the 30S subunit through residues in two helical segments, designated H3 and H4. In most IF3mt orthologs the C-terminal domain is the least conserved region of the protein. However, some residues from the H3 segments are conserved in IF3mt [140, 151, 152]. Biochemical and structural approaches will clarify the mechanism of action of IF3mt.
6.4 How Is the AUG Start Codon Recognized in Mitochondria?
As discussed above, most mitochondrial mRNAs seem to lack Shine-Dalgarno-like sequences to direct the ribosome to the AUG start codon. This is the case for metazoans, flowering plants and fungi [57, 58, 153]. In the case of metazoans it is even more puzzling because the start codon locates at or very near the 5′ end of the mRNA. This implicates that cells developed different mechanisms to localize the ribosome to the correct start codon.
Mammalian mitochondria have developed an initiation codon selection that relies on leaderless mRNAs. Addition of three nucleotides prior to the COX2 5′ AUG decreased translation by 40 %, and addition of 12 nucleotides reduced translation by 80 % [154]. It is proposed that the movement of the ribosome is paused after the first 17 nucleotides of the mRNA enter the ribosome. The small subunit then inspects the mRNA 5′ end. If there is a start codon at the P site, then a stable initiation complex is formed [58, 154].
Study of the mechanisms for initiation codon selection in the yeast S. cerevisiae has made important progress. A group of proteins, named translational activators, plays a role in the localization of the mitoribosome in the correct AUG start codon. Each one of these proteins interacts with specific mitochondrial mRNAs and with the ribosome to pose it on the start codon [155–157]. In addition, translational activators interact with each other and with the mitochondrial inner membrane, probably to tether translation initiation to the site where nascent peptides will be inserted [158–160] (Table 2). Many of these proteins are members of the pentatricopeptide repeat (PPR) family or RNA recognition motif (RRM). Other translational activators have no detectable RNA-binding motifs whatsoever. Many efforts have been made to find orthologs of these proteins in other organisms. Some translational activators may be present in other fungi [161–163] and probably also in humans [164]: However, in mammalian mitochondria, the mechanisms of action of the putative activators remains to be elucidated, as human mRNAs have either very short or no 5′-UTRs. Translational activation is also observed in plastids [165–167], suggesting that this mechanism arose several times during eukaryotic evolution.
7 Translation Elongation
Translation elongation in mitochondria is highly conserved with bacteria. During this process three elongation factors (EF) assist the mitoribosome for addition of new residues to the nascent polypeptide chain. EF-Tumt forms a ternary complex with the aminoacylated tRNA and GTP and enters the mitoribosome A site. Cognate codon-anticodon pairing triggers GTP hydrolysis by EF-Tumt and release of EF-Tumt-GDP. The mitoribosome catalyzes the peptide bond formation at the PTC. Thus, deacetylated tRNA is left in the P site and the elongated peptidyl-tRNA in the A site of the ribosome. This process is assisted by EF-G1mt, which catalyzes the translocation of peptidyl-tRNA from the A to the P site, and removing the deacetylated tRNA from the ribosome. EF-Tsmt exchanges GDP to GTP from EF-Tumt to allow a new round of elongation [58, 140]. Mitochondrial elongation, at least for mammalian and yeast models, seems to be a more conserved process than initiation and ribosome recycling. However, many components of the translation machinery have extensively diverged in different phyla, leading to adaptations of the elongation machinery. For example, as previously discussed, the structure of tRNAs and of mitoribosomes has diverged from the bacterial ancestor. In the next section, we discuss the main changes observed in the elongation factors.
7.1 Mitochondrial EF-Tumt
EF-Tumt must be able to bind tRNA with canonical conformations (e.g., fungi, plants and some protist lineages) and shorter tRNAs versions (metazoans) [51]. In some cases the tRNAs reduction is so extensive that EF-Tumt should use alternative binding modes. The divergence of EF-Tu is evident inside the nematode group: nematodes have 2 EF-Tumt homologs [53]. While EF-Tu1mt is unable to bind cloverleaf type tRNAs, it is the only factor that binds T-armless tRNAs [170, 171]. C. elegans EF-Tu1mt has a C-terminal extension of around 60 amino acids that likely interacts with the D arm of T-armless tRNAs [172]. In the Trichinella lineage EF-Tu1mt binds T-armless tRNAs, D-armless tRNA and cloverleaf type tRNAs [170, 171]. Nematode EF-Tu2mt has a short C-terminal extension of 7–15 amino acids that is necessary for interaction with the D-armless tRNASer [170]. C. elegans mt EF-Tu2mt is unique because it interacts with phosphates on the T arm on the opposite side from where canonical EF-Tu binds [173].
In trypanosomatids, EF-Tumt has a highly charged insertion of approximately 30 amino acids near the C terminus. This trypanosomatid-specific motif is dispensable for the union of EF-Tsmt, but critical for EF-Tumt function [174]. This extension might be necessary for interaction with tRNAs or with the mitoribosome, which has less RNA content than mammalian ribosomes [175]. Since trypanosomatid mitochondrial tRNAs are imported, EF-Tumt has evolved to interact with eukaryotic-type tRNAs, suggesting that the appearance of this motif is an adaptation of the mitochondrial machinery to recognize imported tRNAs [174]. Interestingly, complete loss of tRNA genes from mtDNA is also observed in apicomplexans [176, 177], and therefore their EF-Tumts have to bind imported tRNAs. However, in this case EF-Tumt is closer to the bacterial factor, indicating that each group has their own mechanisms for imported tRNA-EF-Tumt binding [174]. Another distinctive feature of EF-Tumt is found in hemi-ascomycete yeasts, where EF-Tsmt seems to be lost [178]. S. cerevisiae EF-Tumt displays greater affinity for GTP, like the self-recycling GTPases EF-G or IF2 [179]. It is functionally equivalent to the S. pombe EF-Tumt/EF-Tsmt [180].
7.2 Mitochondrial EF-G1mt and EF-G2mt
Bacterial EF-G participates in translation elongation and ribosome recycling. However, in some bacterial groups these functions are separated in two specialized paralogs. This is the case of the majority of Spirochaetes, Planctomycetes, Lentisphaera and some species of δ-proteobacteria. Mitochondria of most organisms have specialized EF-Gmts paralogs as well, and these are phylogenetically related to the specialized bacterial EF-Gs. EF-Gmts probably were acquired before the eukaryotic last common ancestor, since at least one EF-Gmt paralog is present in all mitochondriate eukaryotes [181].
So far, aerobic eukaryotes that possess a unique EF-Gmt (which is an EF-G1mt paralog) are the plastid/apicoplast-carrying eukaryotes: Archaeplastida, stramenopile algae and Apicomplexa [181]. Interestingly, instead of a second EF-Gmt, these species have a plastid/apicoplast-targeted EF-G (termed EF-Gcp or EF-Gapi). Outside of these groups, some Cryptococcus species have only one EF-G1mt. Curiously, they do not have plastids. Until now, it is estimated that all mitochondriate eukaryotes have either two specialized EF-Gs or one EF-G1mt and an EF-Gcp/EF-Gapi [181, 182]. Until now, it is estimated that all mitochondriate eukaryotes have either two specialized EF-Gmts or one EF-Gmt and an EF-Gcp/EF-Gapi [181, 182].
There are limited studies about the function of each mitochondrial paralog. Mammalian EF-G1mt is specialized in translation elongation, while EF-G2mt participates in ribosome recycling [183]. In contrast, A. thaliana EF-G1mt carries both functions, translocation and ribosome recycling [182]. P. falciparum EF-G1mt participates in ribosome recycling, although its translocation activity was not investigated [184, 185].
8 Termination and Ribosomal Recycling
Translation ends when the ribosome reaches one of three stop codons, UGA, UAA, or UAG. These codons are recognized by releases factors (RF) that enter the ribosomal A site and induce release of the nascent peptide (class-I RFs). Bacterial RF1 recognizes UAA and UAG, while RF2 recognizes UAA and UGA [186]. These factors assist the hydrolysis of ester bonds on the peptidyl-tRNA, which is located in the ribosomal P site, releasing the newly synthesized protein. Bacterial class-II RFs are GTPases that trigger dissociation of the class-I RF from the ribosome after peptide release. RFs have a conserved GGQ motif that is involved in ester bond hydrolysis (peptidyl-hydrolase domain, PTH), whereas RF1 has a PAT or PVT motif and RF2 SPF motif important for stop codon recognition (codon-recognition domain, CR) (reviewed in [187]). As mentioned in a previous section, mitochondria recognize non-conventional codons as stop codons (Table 1). Thus, understanding the mechanisms of termination and stop codon recognition is a fertile ground for research. Despite recent advances (mostly in mammalian mitochondria), it is still unclear how translation terminates in mitochondria. The most challenging subject is to understand how non-standard stop codons are decodified in mitochondria.
Mitochondrial release factors divide in five distinct subfamilies: mtRF1a, mtRF2a and ICT1, derived from bacterial ancestors, C12orf65 and mtRF1, so far found only in vertebrates. While mtRF1a, mtRF1 and mtRF2a conserved both the PTH and CR domains, ICT1 and C12orf65 have lost the CR domain [188]. Because the release factor family seems particularly prone to genetic expansion and functional divergence [188], there are high probabilities that the mechanism of translation termination varies among different phyla.
-
mtRF1a is present in every eukaryotic organism and evolved from an α-protobacterial ancestor [188]. This protein recognizes UAA/UAG stop codons, both in vitro and in vivo [189, 190].
-
mtRF1 is a vertebrate-specific mitochondrial protein [190], and it may originate from duplication of the mtRF1a gene at the root of this clade [191]. The function of mtRF1 is controversial. It may recognize the non-standard stop codons AGA and AGG [191]. However, posterior structural predictions and experimental data could not find evidence that mtRF1 recognizes any of the stop codons used in mitochondria [190, 192, 193]. In human mitochondria, the AGG and AGA codons may not function as stop codons. Instead, they promote a −1 frameshift that creates a standard TGA stop codon that can be decodified by mtRF1a [24]. Nonetheless, an analysis from all the vertebrate genomes showed that a −1 frameshift (or even a −2 frameshift) could not originate a canonical TGA stop in every ORF ending in AGG or AGA [188].
-
ICT1 (immature colon carcinoma transcript-1) is widely distributed in mitochondria from all eukaryotic phyla [188]. ICT1 is a codon-independent release factor that lost the CR domain. In addition, it is an integral component of the mitoribosome and a crucial component for its assembly [194]. ICT1 is the eukaryotic ortholog of bacterial ArfB. This protein is a rescue factor of stalled ribosomes in prematurely truncated mRNAs and is also part of the bacterial ribosome [195, 196]. ICT1’s role in mitochondrial translation is still not completely understood. The position of ICT1 in the mitoribosome is incompatible with the mechanism used by ArfB [193, 195]. In fact, the ICT1 integrated to the mitoribosome has no release factor activity [192]. This protein can rescue ribosomal complexes not only at the ends of mRNAs, but also in the middle of mRNAs, and even can rescue ribosomes depleted of mRNAs [192, 194]. ICT1 might terminate translation of ORFs ending in AGG and AGA since these codons are unassigned in mammalian mitochondria, and mitoribosomes stalled at AGG/AGA codons might be recognized by ICT1 [192, 193].
-
C12orf65 is a release factor that probably derived from ICT1. It has a wide phylogenetic distribution and is only absent in viridiplantae [188]. Contrary to ICT1, C12orf65 is a mitochondrial soluble matrix protein that does not exhibit ribosomal-dependent peptidyl hydrolase activity. However, ICT1 overexpression partially complements C12orf65’s absence, indicating that both proteins must have some overlapping functions [197].
-
mtRF2s lack experimental data about their function or mitochondrial localization. mtRF2 has a narrow phylogenetic distribution, consistently found in land plants, red algae, dictyosteliida and some stramenopiles (brown algae, oomycetes and Blastocystis). It has been lost at least five times during eukaryotic evolution, in concordance with the reassignment of the UGA codon to Trp [188].
The final step of mitochondrial translation consists of recycling of the mitoribosomes. Once the nascent chain is released, the ribosome recycling factor 1 (RRF1mt) and the specialized EF-G2mt (see above) separate ribosome subunits to allow new cycles of translation [181, 198]. In addition, IF3mt may attach to the ribosomal SSU to prevent futile association of the mitoribosome until an initiation complex is formed [199].
9 Mitochondrial Translation Is Coupled to Protein Assembly
The majority of proteins encoded by mitochondrial DNA are subunits of the respiratory chain complexes and the ATP synthase. These proteins are usually hydrophobic, with two or more transmembrane stretches. Thus, it is expected that mitochondrial translation machinery is physically coupled to the mitochondrial inner membrane. Mitochondrial and cytoplasmic subunits have to assemble and acquire the necessary prosthetic groups in coordination to make active enzymes. Indeed, major progress in the field has come from beaker’s yeast S. cerevisiae. Defects in the coordination of mitochondrial-encoded subunit synthesis and assembly are proposed to affect the cell physiology. When cytochrome c oxidase is not assembled, then Cox1 synthesis in mitochondria is downregulated [200, 201]. Cox1 is part of the central core of the enzyme and has 12 transmembrane stretches. Downregulation of Cox1 synthesis may prevent generation of pro-oxidant species, because the poorly assembled heme a present in Cox1 has peroxidase activity [202]. When the ATPase F1 sector is not assembled, then translation of the ATP8/ATP6 transcript is downregulated [203, 204]. This prevents accumulation on the membrane of Atp6–Atp9 rings that could interfere with the membrane potential [205, 206]. Translation of the mitochondrial COB mRNA, coding for the cytochrome b subunit from the bc1 complex is also tightly linked to enzyme assembly [207]. Coordination of mitochondrial translation and assembly is an intricate process that requires many factors, many of them specific for each mitochondrial-coded protein [159, 203, 208, 209] (Fig. 3). In general, initiation of mitochondrial mRNA translation is achieved by translational activators (discussed above), which assist in the positioning of the ribosome on the initiator AUG. Translational activators are themselves associated with the inner membrane and with other translational activators to tether translation initiation to the localization of assembly of nascent peptides [155, 156]. Some of these mRNA-specific activators act exclusively on the 5′-UTR of the target mRNA and are no longer required in downstream events after translation initiation. Some other translational activators have dual functions. Together with specific chaperones, they physically interact with the newly synthesized peptide and are key players in the coordination of translation and assembly. In general, it is proposed that once the respiratory complex proceeds to assembly, these chaperones/translational activators are released from the assembly intermediary and recycled for new rounds of translation [159, 208]. Equivalent processes were described for C. reinhardtii photosynthetic complexes in chloroplasts [165].

General model for the coupling of protein synthesis and membrane assembly in yeast mitochondria. Translational activators assist the ribosome in localization of the AUG start codon through recognition of specific sequences within the 5′-UTR of each mRNA Cbs1/Cbs2/Cbp3/Cbp6 (cytochrome b synthesis) [207, 210]; Pet309/Mss51 (Cox1 synthesis) [157, 200, 201, 211]; Pet111 (Cox2 synthesis) [212]; Pet494/Pet122/Pet54 (Cox3 synthesis) [213–215], Atp22 (Atp6 and Atp8 synthesis) [204]; Aep1, Aep2 (Atp9 synthesis) [216, 217]. Some of these activators play a second role in coordination of translation/assembly. They physically interact with newly made peptides (mtSubunits) and with additional chaperones to form assembly intermediaries. This is the case for Cbp3/Cbp6 [207] and Mss51 [201]. Once the respiratory complexes assemble with cytosolic, imported subunits (CytSubunits), then the chaperones/translational activators are released and recycled. Translational activators are now ready for new rounds of translation
In human mitochondria, the scenario is not as clear as with yeast. However, some orthologs of the yeast translational activators and chaperones are present in humans, and some of them may have similar roles [164, 209, 218, 219].
10 Concluding Remarks
Mitochondrial translation evolution is an exciting field in biology. Many efforts have been made to understand mitochondrial translation by studying yeast, mammals, plant and trypanosomatid species, and to a lesser degree apicomplexans, nematodes and Drosophila. Since the mitochondrial translation machinery has extensively diverged among eukaryotes, it is necessary to study it in many disparate groups of eukaryotes in order to understand its evolution. To date, many questions remain open in this field. For instance: (1) What are the role and mechanism of action in the translation of mitochondrial-encoded small and large non-coding RNAs discovered a few years ago [220–222]? (2) How do mRNA polyadenylation and edition regulate stability, editing and translation in different species? (3) Are there specialized ribosomes devoted to translating a specific mRNA in mitochondria? If this is the case, how are populations of each type of ribosome regulated in different conditions or cell types? (4) What are the mechanism and regulation of the recently discovered “programmed translational bypass”? This process was first discovered in the yeast Magnusiomyces capitatus [223, 224]. In this organism, almost all protein-coding genes in mtDNA have insertions of 27–55 nucleotides, called byps. Translation of these insertions would lead to frameshifts and premature termination of translation, so a precise mechanism to bypass these elements is necessary. Is translation bypass present in different eukaryotic groups? (5) What is the role of the recently discovered mitochondrial RNA granules in translation [127]? Are they present exclusively on mammalian mitochondria, or do they have a broader prevalence in eukaryotes? (6) Since Shine-Dalgarno sequences are absent in many eukaryotic lineages, how is the start codon AUG recognized by the initiation complex among different phyla? (7) Are the translation and assembly of nascent peptides prevalent processes in eukaryotes? The field of mitochondrial evolution awaits answers to these exiting questions.
References
Hori H, Osawa S. Origin and evolution of organisms as deduced from 5S ribosomal RNA sequences. Mol Biol Evol. 1987;4:445–72.
Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–81.
Degli Esposti M. Bioenergetic evolution in proteobacteria and mitochondria. Genome Biol Evol. 2014;6:3238–3251. doi:10.1093/gbe/evu257.
Millar AH, Heazlewood JL, Kristensen BK, Braun HP, Moller IM. The plant mitochondrial proteome. Trends Plant Sci. 2005; 10:36-43. doi:10.1016/j.tplants.2004.12.002.
Premsler T, Zahedi RP, Lewandrowski U, Sickmann A. Recent advances in yeast organelle and membrane proteomics. Proteomics. 2009;9:4731–43. doi:10.1002/pmic.200900201.
Szklarczyk R, Huynen MA. Mosaic origin of the mitochondrial proteome. Proteomics. 2010;10:4012–24. doi:10.1002/pmic.201000329.
Gawryluk RM, Chisholm KA, Pinto DM, Gray MW. Compositional complexity of the mitochondrial proteome of a unicellular eukaryote (Acanthamoeba castellanii, supergroup Amoebozoa) rivals that of animals, fungi, and plants. J Proteomics. 2014;109:400–16. doi:10.1016/j.jprot.2014.07.005.
Hikosaka K, Kita K, Tanabe K. Diversity of mitochondrial genome structure in the Phylum Apicomplexa. Mol Biochem Parasitol. 2013;188:26–33. doi:10.1016/j.molbiopara.2013.02.006.
Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–80. doi:10.1093/nar/27.8.1767.
Gualberto JM, Mileshina D, Wallet C, Niazi AK, Weber-Lotfi F, Dietrich A. The plant mitochondrial genome: dynamics and maintenance. Biochimie. 2014;100:107–20. doi:10.1016/j.biochi.2013.09.016.
Burger G, Gray MW, Forget L, Lang BF. Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol Evol. 2013;5:418–38. doi:10.1093/gbe/evt008.
Bohnert M, Pfanner N, van der Laan M. Mitochondrial machineries for insertion of membrane proteins. Curr Opin Struct Biol. 2015;33:92–102.
Stuart K, Feagin JE. Mitochondrial DNA of kinetoplastids. Int Rev Cytol. 1992;141:65–88.
Burger G, Forget L, Zhu Y, Gray MW, Lang BF. Unique mitochondrial genome architecture in unicellular relatives of animals. Proc Natl Acad Sci USA. 2003;100:892–7. doi:10.1073/pnas.0336115100.
Campbell WH, Gowri G. Codon usage in higher plants, green algae, and cyanobacteria. Plant Physiol. 1990;92:1–11.
Knight RD, Landweber LF, Yarus M. How mitochondria rede-fine the code. J Mol Evol. 2001;53:299–313.
Bezerra AR, Guimaraes AR, Santos MA. Non-standard genetic codes define new concepts for protein engineering. Life (Basel). 2015;5:1610–28.
Inagaki Y, Ehara M, Watanabe KI, Hayashi-Ishimaru Y, Ohama T. Directionally evolving genetic code: the UGA codon from stop to tryptophan in mitochondria. J Mol Evol. 1998;47:378–84.
Hayashi-Ishimaru Y, Ohama T, Kawatsu Y, Nakamura K, Osawa S. UAG is a sense codon in several chlorophycean mitochondria. Curr Genet. 1996;30:29–33.
Jacob JE, Vanholme B, Van Leeuwen T, Gheysen G. A unique genetic code change in the mitochondrial genome of the parasitic nematode Radopholus similis. BMC Res Notes. 2009;2:192.
Nedelcu AM, Lee RW, Lemieux C, Gray MW, Burger G. The complete mitochondrial DNA sequence of Scenedesmus obliquus reflects an intermediate stage in the evolution of the green algal mitochondrial genome. Genome Res. 2000;10:819–31.
Xu W, Xing T, Zhao M, Yin X, Xia G, Wang M. Synonymous codon usage bias in plant mitochondrial genes is associated with intron number and mirrors species evolution. PLoS One. 2015;10:e0131508.
Osawa S, Ohama T, Jukes TH, Watanabe K. Evolution of the mitochondrial genetic code. I. Origin of AGR serine and stop codons in metazoan mitochondria. J Mol Evol. 1989;29:202–7.
Temperley R, Richter R, Dennerlein S, Lightowlers RN, Chrzanowska-Lightowlers ZM. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science. 2010;327:301. doi:10.1126/science.1180674.
Clark-Walker GD, Weiller GF. The structure of the small mito-chondrial DNA of Kluyveromyces thermotolerans is likely to reflect the ancestral gene order in fungi. J Mol Evol. 1994;38:593–601.
Desjardins P, Morais R. Nucleotide sequence and evolution of coding and noncoding regions of a quail mitochondrial genome. J Mol Evol. 1991;32:153–61.
Yokobori S, Ueda T, Feldmaier-Fuchs G, Paabo S, Ueshima R, Kondow A, Nishikawa K, Watanabe K. Complete DNA sequence of the mitochondrial genome of the ascidian Halocynthia roretzi (Chordata, Urochordata). Genetics. 1999;153:1851–62.
Foury F, Roganti T, Lecrenier N, Purnelle B. The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett. 1998;440:325–31.
Jacobs HT, Elliott DJ, Math VB, Farquharson A. Nucleotide sequence and gene organization of sea urchin mitochondrial DNA. J Mol Biol. 1988;202:185–217.
Ziaie Z, Suyama Y. The cytochrome oxidase subunit I gene of Tetrahymena: a 57 amino acid NH2-terminal extension and a 108 amino acid insert. Curr Genet. 1987;12:357–68.
Pritchard AE, Seilhamer JJ, Mahalingam R, Sable CL, Venuti SE, Cummings DJ. Nucleotide sequence of the mitochondrial genome of Paramecium. Nucleic Acids Res. 1990;18:173–80.
Suzuki T, Miyauchi K, Suzuki T, Yokobori S, Shigi N, Kondow A, Takeuchi N, Yamagishi A, Watanabe K. Taurine-containing uridine modifications in tRNA anticodons are required to decipher non-universal genetic codes in ascidian mitochondria. J Biol Chem. 2011;286:35494–8.
Moriya J, Yokogawa T, Wakita K, Ueda T, Nishikawa K, Crain PF, Hashizume T, Pomerantz SC, McCloskey JA, Kawai G, et al. A novel modified nucleoside found at the first position of the anticodon of methionine tRNA from bovine liver mitochondria. Biochemistry. 1994;33:2234–9.
Takemoto C, Spremulli LL, Benkowski LA, Ueda T, Yokogawa T, Watanabe K. Unconventional decoding of the AUA codon as me-thionine by mitochondrial tRNAMet with the anticodon f5CAU as revealed with a mitochondrial in vitro translation system. Nucleic Acids Res. 2009;37:1616–27.
Osawa S, Jukes TH. Evolution of the genetic code as affected by anticodon content. Trends Genet. 1988;4:191–8.
Osawa S, Ohama T, Jukes TH, Watanabe K, Yokoyama S. Evo-lution of the mitochondrial genetic code. II. Reassignment of codon AUA from isoleucine to methionine. J Mol Evol. 1989;29:373–80.
Sengupta S, Yang X, Higgs PG. The mechanisms of codon reassignments in mitochondrial genetic codes. J Mol Evol. 2007;64:662–88.
Silva RM, Miranda I, Moura G, Santos MA. Yeast as a model organism for studying the evolution of non-standard genetic codes. Brief Funct Genomic Proteomic. 2004;3:35–46.
Miranda I, Silva R, Santos MA. Evolution of the genetic code in yeasts. Yeast. 2006;23:203–13.
Schultz DW, Yarus M. Transfer RNA mutation and the malleability of the genetic code. J Mol Biol. 1994;235:1377–80.
Schultz DW, Yarus M. On malleability in the genetic code. J Mol Evol. 1996;42:597–601.
Jia W, Higgs PG. Codon usage in mitochondrial genomes: distinguishing context-dependent mutation from translational selection. Mol Biol Evol. 2008;25:339–51.
Abascal F, Posada D, Knight RD, Zardoya R. Parallel evolution of the genetic code in arthropod mitochondrial genomes. PLoS Biol. 2006;4:e127.
Chin JW. Expanding and reprogramming the genetic code of cells and animals. Annu Rev Biochem. 2014;83:379–408.
Hancock K, Hajduk SL. Sequence of Trypanosoma brucei tRNA genes encoding cytosolic tRNAs. Nucleic Acids Res. 1992;20:2602.
Turmel M, Lemieux C, Burger G, Lang BF, Otis C, Plante I, Gray MW. The complete mitochondrial DNA sequences of Nephroselmis olivacea and Pedinomonas minor. Two radically different evolutionary patterns within green algae. Plant Cell. 1999;11:1717–30.
Martin RP, Schneller JM, Stahl AJ, Dirheimer G. Import of nuclear deoxyribonucleic acid coded lysine-accepting transfer ribonucleic acid (anticodon C-U-U) into yeast mitochondria. Biochemistry. 1979;18:4600–5.
Kamenski P, Kolesnikova O, Jubenot V, Entelis N, Krasheninnikov IA, Martin RP, Tarassov I. Evidence for an adaptation mechanism of mitochondrial translation via tRNA import from the cytosol. Mol Cell. 2007;26:625–37.
Rubio MA, Rinehart JJ, Krett B, Duvezin-Caubet S, Reichert AS, Soll D, Alfonzo JD. Mammalian mitochondria have the innate ability to import tRNAs by a mechanism distinct from protein import. Proc Natl Acad Sci USA. 2008;105:9186–91.
Vinogradova E, Salinas T, Cognat V, Remacle C, Marechal-Drouard L. Steady-state levels of imported tRNAs in Chlamydomonas mitochondria are correlated with both cytosolic and mitochondrial codon usages. Nucleic Acids Res. 2009;37:1521–8.
Salinas-Giege T, Giege R, Giege P. tRNA biology in mitochondria. Int J Mol Sci. 2015;16:4518–59. doi:10.3390/ijms16034518.
Watanabe Y, Suematsu T, Ohtsuki T. Losing the stem-loop structure from metazoan mitochondrial tRNAs and co-evolution of interacting factors. Front Genet. 2014;5:109.
Giege R, Juhling F, Putz J, Stadler P, Sauter C, Florentz C. Structure of transfer RNAs: similarity and variability. Wiley Interdiscip Rev RNA. 2012;3:37–61. doi:10.1002/wrna.103.
Suzuki T, Suzuki T. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2014;42:7346–57. doi:10.1093/nar/gku390.
Machnicka MA, Olchowik A, Grosjean H, Bujnicki JM. Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 2014;11:1619–29. doi:10.4161/15476286.2014.992273.
Weber F, Dietrich A, Weil JH, Marechal-Drouard L. A potato mitochondrial isoleucine tRNA is coded for by a mitochondrial gene possessing a methionine anticodon. Nucleic Acids Res. 1990;18:5027–30.
Hazle T, Bonen L. Comparative analysis of sequences preceding protein-coding mitochondrial genes in flowering plants. Mol Biol Evol. 2007;24:1101–12.
Christian BE, Spremulli LL. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta. 2012;1819:1035–54. doi:10.1016/j.bbagrm.2011.11.009.
Aphasizhev R, Aphasizheva I. Mitochondrial RNA editing in trypanosomes: small RNAs in control. Biochimie. 2014;100:125–31. doi:10.1016/j.biochi.2014.01.003.
Takenaka M, Zehrmann A, Verbitskiy D, Hartel B, Brennicke A. RNA editing in plants and its evolution. Annu Rev Genet. 2013;47:335–52.
Norbury CJ. Cytoplasmic RNA: a case of the tail wagging the dog. Nat Rev Mol Cell Biol. 2013;14:643–53.
Tian B, Manley JL. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 2013;38:312–20.
Silva IJ, Saramago M, Dressaire C, Domingues S, Viegas SC, Arraiano CM. Importance and key events of prokaryotic RNA decay: the ultimate fate of an RNA molecule. Wiley Interdiscip Rev RNA. 2011;2:818–36.
Bandyra KJ, Luisi BF. Licensing and due process in the turnover of bacterial RNA. RNA Biol. 2013;10:627–35.
Temperley RJ, Wydro M, Lightowlers RN, Chrzanowska-Lightowlers ZM. Human mitochondrial mRNAs–like members of all families, similar but different. Biochim Biophys Acta. 2010;1797:1081–5.
Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, Filipovska A, Mattick JS. The human mitochondrial transcriptome. Cell. 2011;146:645–58.
Bernt M, Braband A, Schierwater B, Stadler PF. Genetic aspects of mitochondrial genome evolution. Mol Phylogenet Evol. 2012;69:328–38.
Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–9.
Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–4.
Slomovic S, Laufer D, Geiger D, Schuster G. Polyadenylation and degradation of human mitochondrial RNA: the prokaryotic past leaves its mark. Mol Cell Biol. 2005;25:6427–35. doi:10.1128/MCB.25.15.6427-6435.2005.
Gagliardi D, Stepien PP, Temperley RJ, Lightowlers RN, Chrzanowska-Lightowlers ZM. Messenger RNA stability in mitochondria: different means to an end. Trends Genet. 2004;20:260–7. doi:10.1016/j.tig.2004.04.006.
Etheridge RD, Aphasizheva I, Gershon PD, Aphasizhev R. 3’ adenylation determines mRNA abundance and monitors completion of RNA editing in T. brucei mitochondria. EMBO J. 2008;27:1596–608. doi:10.1038/emboj.2008.87.
Schuster G, Stern D. RNA polyadenylation and decay in mitochondria and chloroplasts. Prog Mol Biol Transl Sci. 2009;85:393–422. doi:10.1016/S0079-6603(08)00810-6.
Dziembowski A, Piwowarski J, Hoser R, Minczuk M, Dmochowska A, Siep M, van der Spek H, Grivell L, Stepien PP. The yeast mitochondrial degradosome. Its composition, interplay between RNA helicase and RNase activities and the role in mitochondrial RNA metabolism. J Biol Chem. 2003;278:1603–1611.
Turk EM, Das V, Seibert RD, Andrulis ED. The mitochondrial RNA landscape of Saccharomyces cerevisiae. PLoS One. 2013;8:e78105.
Schafer B, Hansen M, Lang BF. Transcription and RNA-processing in fission yeast mitochondria. RNA. 2005;11:785–95. doi:10.1261/rna.7252205.
Kolondra A, Labedzka-Dmoch K, Wenda JM, Drzewicka K, Golik P. The transcriptome of Candida albicans mitochondria and the evolution of organellar transcription units in yeasts. BMC Genomics. 2015;16:827.
Butow RA, Zhu H, Perlman P, Conrad-Webb H. The role of a conserved dodecamer sequence in yeast mitochondrial gene expression. Genome. 1989;31:757–60.
Zhu H, Conrad-Webb H, Liao XS, Perlman PS, Butow RA. Functional expression of a yeast mitochondrial intron-encoded protein requires RNA processing at a conserved dodecamer sequence at the 3’ end of the gene. Mol Cell Biol. 1989;9:1507–12.
Chateigner-Boutin AL, Small I. Organellar RNA editing. Wiley Interdiscip Rev RNA. 2011;2:493–506.
Takenaka M, Verbitskiy D, Zehrmann A, Hartel B, Bayer-Csaszar E, Glass F, Brennicke A. RNA editing in plant mitochondria-connecting RNA target sequences and acting proteins. Mitochondrion. 2014;19 Pt B:191–197.
Read LK, Lukes J, Hashimi H. Trypanosome RNA editing: the complexity of getting U in and taking U out. Wiley Interdiscip Rev RNA. 2016;7:33–51.
Lavrov DV, Adamski M, Chevaldonne P, Adamska M. Extensive mitochondrial mRNA editing and unusual mitochondrial genome organization in calcaronean sponges. Curr Biol. 2016;26:86–92.
Gray MW. Evolutionary origin of RNA editing. Biochemistry. 2012;51:5235–42.
Knoop V. Plant mitochondrial genome peculiarities evolving in the earliest vascular plant lineages. J Syst Evol. 2013;51:1–12.
Rudinger M, Funk HT, Rensing SA, Maier UG, Knoop V. RNA editing: only eleven sites are present in the Physcomitrella patens mitochondrial transcriptome and a universal nomenclature proposal. Mol Genet Genomics. 2009;281:473–81.
Bentolila S, Oh J, Hanson MR, Bukowski R. Comprehensive high-resolution analysis of the role of an Arabidopsis gene family in RNA editing. PLoS Genet. 2013;9:e1003584.
Grewe F, Herres S, Viehover P, Polsakiewicz M, Weisshaar B, Knoop V. A unique transcriptome: 1782 positions of RNA editing alter 1406 codon identities in mitochondrial mRNAs of the lycophyte Isoetes engelmannii. Nucleic Acids Res. 2011;39:2890–902.
Hecht J, Grewe F, Knoop V. Extreme RNA editing in coding islands and abundant microsatellites in repeat sequences of Selaginella moellendorffii mitochondria: the root of frequent plant mtDNA re-combination in early tracheophytes. Genome Biol Evol. 2011;3:344–58.
Benne R, Van den Burg J, Brakenhoff JP, Sloof P, Van Boom JH, Tromp MC. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell. 1986;46:819–26.
Blum B, Bakalara N, Simpson L. A model for RNA editing in kinetoplastid mitochondria: “guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell. 1990;60:189–98.
Liu M, Spremulli L. Interaction of mammalian mitochondrial ribosomes with the inner membrane. J Biol Chem. 2000;275:29400–6. doi:10.1074/jbc.M002173200.
Pfeffer S, Woellhaf MW, Herrmann JM, Forster F. Organization of the mitochondrial translation machinery studied in situ by cryoelectron tomography. Nat Commun. 2015;6:6019. doi:10.1038/ncomms7019.
Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–44. doi:10.1016/j.cell.2009.08.005.
Tzagoloff A, Myers AM. Genetics of mitochondrial biogenesis. Annu Rev Biochem. 1986;55:249–85. doi:10.1146/annurev.bi.55.070186.001341.
Bogenhagen DF, Martin DW, Koller A. Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metab. 2014;19:618–29. doi:10.1016/j.cmet.2014.03.013.
De Silva D, Tu YT, Amunts A, Fontanesi F, Barrientos A. Mitochondrial ribosome assembly in health and disease. Cell Cycle. 2015;14:2226–50. doi:10.1080/15384101.2015.1053672.
Kitakawa M, Isono K. The mitochondrial ribosomes. Biochimie. 1991;73:813–25. doi:10.1016/0300-9084(91)90061-5.
Ramulu HG, Groussin M, Talla E, Planel R, Daubin V, Brochier-Armanet C. Ribosomal proteins: toward a next generation standard for prokaryotic systematics? Mol Phylogenet Evol. 2014;75:103–17. doi:10.1016/j.ympev.2014.02.013.
Desmond E, Brochier-Armanet C, Forterre P, Gribaldo S. On the last common ancestor and early evolution of eukaryotes: reconstructing the history of mitochondrial ribosomes. Res Microbiol. 2011;162:53–70. doi:10.1016/j.resmic.2010.10.004.
van der Sluis EO, Bauerschmitt H, Becker T, Mielke T, Frauenfeld J, Berninghausen O, Neupert W, Herrmann JM, Beckmann R. Parallel structural evolution of mitochondrial ribosomes and OXPHOS complexes. Genome Biol Evol. 2015;7:1235–51. doi:10.1093/gbe/evv061evv061.
Smits P, Smeitink JA, van den Heuvel LP, Huynen MA, Ettema TJ. Reconstructing the evolution of the mitochondrial ribosomal proteome. Nucleic Acids Res. 2007;35:4686–703. doi:10.1093/nar/gkm441.
Brown WM, George M Jr, Wilson AC. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA. 1979;76:1967–71.
Haag-Liautard C, Coffey N, Houle D, Lynch M, Charlesworth B, Keightley PD. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 2008;6:e204.
Neiman M, Taylor DR. The causes of mutation accumulation in mitochondrial genomes. Proc Biol Sci. 2009;276:1201–9. doi:10.1098/rspb.2008.1758.
Lukes J, Archibald JM, Keeling PJ, Doolittle WF, Gray MW. How a neutral evolutionary ratchet can build cellular complexity. IUBMB Life. 2011;63:528–37.
Barreto FS, Burton RS. Evidence for compensatory evolution of ribosomal proteins in response to rapid divergence of mitochondrial rRNA. Mol Biol Evol. 2013;30:310–4. doi:10.1093/molbev/mss228.
Rand DM, Haney RA, Fry AJ. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol Evol. 2004;19:645–53. doi:10.1016/j.tree.2004.10.003.
Berry S. Endosymbiosis and the design of eukaryotic electron transport. Biochim Biophys Acta. 2003;1606:57–72. doi:S0005272803000847[pii].
Gabaldon T, Rainey D, Huynen MA. Tracing the evolution of a large protein complex in the eukaryotes, NADH:ubiquinone oxidoreductase (Complex I). J Mol Biol. 2005;348:857–70. doi:10.1016/j.jmb.2005.02.067.
Yamaguchi K, Subramanian AR. The plastid ribosomal pro-teins. Identification of all the proteins in the 50 S subunit of an orga-nelle ribosome (chloroplast). J Biol Chem. 2000;275:28466–82. doi:10.1074/jbc.M005012200.
Yamaguchi K, von Knoblauch K, Subramanian AR. The plastid ribosomal proteins. Identification of all the proteins in the 30 S sub-unit of an organelle ribosome (chloroplast). J Biol Chem. 2000;275:28455–65. doi:10.1074/jbc.M004350200.
Spirina O, Bykhovskaya Y, Kajava AV, O’Brien TW, Nierlich DP, Mougey EB, Sylvester JE, Graack HR, Wittmann-Liebold B, Fischel-Ghodsian N. Heart-specific splice-variant of a human mitochondrial ribosomal protein (mRNA processing; tissue specific splicing). Gene. 2000;261:229–34. doi:10.1016/S0378-1119(00)00504-7.
Mogk A, Haslberger T, Tessarz P, Bukau B. Common and specific mechanisms of AAA+ proteins involved in protein quality control. Biochem Soc Trans. 2008;36:120–5. doi:10.1042/BST0360120.
Daubner GM, Clery A, Allain FH. RRM-RNA recognition: NMR or crystallography and new findings. Curr Opin Struct Biol. 2013;23:100–8. doi:10.1016/j.sbi.2012.11.006.
Kaur J, Stuart RA. Truncation of the Mrp20 protein reveals new ribosome-assembly subcomplex in mitochondria. EMBO Rep. 2011;12:950–5. doi:10.1038/embor.2011.133.
Ryden-Aulin M, Shaoping Z, Kylsten P, Isaksson LA. Ribo-some activity and modification of 16S RNA are influenced by deletion of ribosomal protein S20. Mol Microbiol. 1993;7:983–92.
Tobin C, Mandava CS, Ehrenberg M, Andersson DI, Sanyal S. Ribosomes lacking protein S20 are defective in mRNA binding and subunit association. J Mol Biol. 2010;397:767–76. doi:10.1016/j.jmb.2010.02.004.
Sorensen MA, Fricke J, Pedersen S. Ribosomal protein S1 is required for translation of most, if not all, natural mRNAs in Escherichia coli in vivo. J Mol Biol. 1998;280:561–9. doi:10.1006/jmbi.1998.1909.
O’Brien TW. Evolution of a protein-rich mitochondrial ribo-some: implications for human genetic disease. Gene. 2002;286:73–9. doi:S0378111901008083 [pii].
Amunts A, Brown A, Toots J, Scheres SH, Ramakrishnan V. Ribosome. The structure of the human mitochondrial ribosome. Science. 2015;348:95–8. doi:10.1126/science.aaa1193.
Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D, Ban N. Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science. 2015;348:303–8. doi:10.1126/science.aaa3872
Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell. 2003;115:97–108. doi:S0092867403007621 [pii].
Schneider A, Ebert D. Covariation of mitochondrial genome size with gene lengths: evidence for gene length reduction during mitochondrial evolution. J Mol Evol. 2004;59:90–6.
Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014;505:515–9. doi:10.1038/nature12890.
Simpson L, Thiemann OH, Savill NJ, Alfonzo JD, Maslov DA. Evolution of RNA editing in trypanosome mitochondria. Proc Natl Acad Sci USA. 2000;97:6986–93. doi:97/13/6986 [pii].
Antonicka H, Shoubridge EA. Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep. 2015;S2211–1247:00055–8. doi:10.1016/j.celrep.2015.01.030.
Gualerzi CO, Pon CL. Initiation of mRNA translation in bacteria: structural and dynamic aspects. Cell Mol Life Sci. 2015;72:4341–67.
Atkinson GC, Kuzmenko A, Kamenski P, Vysokikh MY, Lakunina V, Tankov S, Smirnova E, Soosaar A, Tenson T, Hauryliuk V. Evolutionary and genetic analyses of mitochondrial translation initiation factors identify the missing mitochondrial IF3 in S. cerevisiae. Nucleic Acids Res. 2012;40:6122–34.
Hinttala R, Sasarman F, Nishimura T, Antonicka H, Brunel-Guitton C, Schwartzentruber J, Fahiminiya S, Majewski J, Faubert D, Ostergaard E, Smeitink JA, Shoubridge EA. An N-terminal formyl methionine on COX 1 is required for the assembly of cytochrome c oxidase. Hum Mol Genet. 2015;24:4103–13.
Tucker EJ, Hershman SG, Kohrer C, Belcher-Timme CA, Patel J, Goldberger OA, Christodoulou J, Silberstein JM, McKenzie M, Ryan MT, Compton AG, Jaffe JD, Carr SA, Calvo SE, RajBhandary UL, Thorburn DR, Mootha VK. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 2011;14:428–34.
Li Y, Holmes WB, Appling DR, RajBhandary UL. Initiation of protein synthesis in Saccharomyces cerevisiae mitochondria without formylation of the initiator tRNA. J Bacteriol. 2000;182:2886–92.
Lee C, Tibbetts AS, Kramer G, Appling DR. Yeast AEP3p is an accessory factor in initiation of mitochondrial translation. J Biol Chem. 2009;284:34116–25.
Marzi S, Knight W, Brandi L, Caserta E, Soboleva N, Hill WE, Gualerzi CO, Lodmell JS. Ribosomal localization of translation initiation factor IF2. RNA. 2003;9:958–69.
Moreno JM, Sorensen HP, Mortensen KK, Sperling-Petersen HU. Macromolecular mimicry in translation initiation: a model for the initiation factor IF2 on the ribosome. IUBMB Life. 2000;50:347–54.
Spencer AC, Spremulli LL. Interaction of mitochondrial initiation factor 2 with mitochondrial fMet-tRNA. Nucleic Acids Res. 2004;32:5464–70.
Tibbetts AS, Oesterlin L, Chan SY, Kramer G, Hardesty B, Appling DR. Mammalian mitochondrial initiation factor 2 supports yeast mitochondrial translation without formylated initiator tRNA. J Biol Chem. 2003;278:31774–80.
Koc EC, Burkhart W, Blackburn K, Koc H, Moseley A, Spremulli LL. Identification of four proteins from the small subunit of the mammalian mitochondrial ribosome using a proteomics approach. Protein Sci. 2001;10:471–81.
Koc EC, Burkhart W, Blackburn K, Moyer MB, Schlatzer DM, Moseley A, Spremulli LL. The large subunit of the mammalian mitochondrial ribosome. Analysis of the complement of ribosomal proteins present. J Biol Chem. 2001;276:43958–69.
Spremulli LL, Coursey A, Navratil T, Hunter SE. Initiation and elongation factors in mammalian mitochondrial protein biosynthesis. Prog Nucleic Acid Res Mol Biol. 2004;77:211–61.
Laursen BS, Siwanowicz I, Larigauderie G, Hedegaard J, Ito K, Nakamura Y, Kenney JM, Mortensen KK, Sperling-Petersen HU. Characterization of mutations in the GTP-binding domain of IF2 resulting in cold-sensitive growth of Escherichia coli. J Mol Biol. 2003;326:543–51.
Gaur R, Grasso D, Datta PP, Krishna PD, Das G, Spencer A, Agrawal RK, Spremulli L, Varshney U. A single mammalian mito-chondrial translation initiation factor functionally replaces two bacterial factors. Mol Cell. 2008;29:180–90.
Roll-Mecak A, Cao C, Dever TE, Burley SK. X-Ray structures of the universal translation initiation factor IF2/eIF5B: conformational changes on GDP and GTP binding. Cell. 2000;103:781–92.
Guenneugues M, Caserta E, Brandi L, Spurio R, Meunier S, Pon CL, Boelens R, Gualerzi CO. Mapping the fMet-tRNA(f)(Met) binding site of initiation factor IF2. Embo J. 2000;19:5233–40.
Misselwitz R, Welfle K, Krafft C, Welfle H, Brandi L, Caserta E, Gualerzi CO. The fMet-tRNA binding domain of translational initiation factor IF2: role and environment of its two Cys residues. FEBS Lett. 1999;459:332–6.
Wienk H, Tomaselli S, Bernard C, Spurio R, Picone D, Gualerzi CO, Boelens R. Solution structure of the C1-subdomain of Bacillus stearothermophilus translation initiation factor IF2. Protein Sci. 2005;14:2461–8.
Grunberg-Manago M, Dessen P, Pantaloni D, Godefroy-Colburn T, Wolfe AD, Dondon J. Light-scattering studies showing the effect of initiation factors on the reversible dissociation of Escherichia coli ribosomes. J Mol Biol. 1975;94:461–78.
Yassin AS, Haque ME, Datta PP, Elmore K, Banavali NK, Spremulli LL, Agrawal RK. Insertion domain within mammalian mitochondrial translation initiation factor 2 serves the role of eubacterial initiation factor 1. Proc Natl Acad Sci USA. 2011;108:3918–23.
Koc EC, Spremulli LL. Identification of mammalian mitochondrial translational initiation factor 3 and examination of its role in initiation complex formation with natural mRNAs. J Biol Chem. 2002;277:35541–9.
Dallas A, Noller HF. Interaction of translation initiation factor 3 with the 30S ribosomal subunit. Mol Cell. 2001;8:855–64.
Pioletti M, Schlunzen F, Harms J, Zarivach R, Gluhmann M, Avila H, Bashan A, Bartels H, Auerbach T, Jacobi C, Hartsch T, Yonath A, Franceschi F. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. Embo J. 2001;20:1829–39.
Sette M, Spurio R, van Tilborg P, Gualerzi CO, Boelens R. Identification of the ribosome binding sites of translation initiation factor IF3 by multidimensional heteronuclear NMR spectroscopy. RNA. 1999;5:82–92.
Dieckmann CL, Staples RR. Regulation of mitochondrial gene expression in Saccharomyces cerevisiae. Int Rev Cytol. 1994;152:145–81.
Christian BE, Spremulli LL. Preferential selection of the 5’-terminal start codon on leaderless mRNAs by mammalian mitochondrial ribosomes. J Biol Chem. 2010;285:28379–86.
Fox TD. Genetics of mitochondrial translation. In: Hershey JWB, Matthews MB, Sonenberg N, editors. Translational control. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1996. p. 733–58.
Herrmann JM, Woellhaf MW, Bonnefoy N. Control of protein synthesis in yeast mitochondria: the concept of translational activators. Biochim Biophys Acta. 2013;1833:286–94.
Zamudio-Ochoa A, Camacho-Villasana Y, Garcia-Guerrero AE, Perez-Martinez X. The Pet309 pentatricopeptide repeat motifs mediate efficient binding to the mitochondrial COX1 transcript in yeast. RNA Biol. 2014;11:953–67.
Naithani S, Saracco SA, Butler CA, Fox TD. Interactions among COX1, COX2, and COX3 mRNA-specific translational activator proteins on the inner surface of the mitochondrial inner membrane of Saccharomyces cerevisiae. Mol Biol Cell. 2003;14:324–33.
Ott M, Amunts A, Brown A. Organization and regulation of mitochondrial protein synthesis. Annu Rev Biochem. 2016; In press. doi:10.1146/annurev-biochem-060815-014334.
Sanchirico ME, Fox TD, Mason TL. Accumulation of mitochondrially synthesized Saccharomyces cerevisiae Cox2p and Cox3p depends on targeting information in untranslated portions of their mRNAs. Embo J. 1998;17:5796–804.
Coffin JW, Dhillon R, Ritzel RG, Nargang FE. The Neurospora crassa cya-5 nuclear gene encodes a protein with a region of homology to the Saccharomyces cerevisiae PET309 protein and is required in a post-transcriptional step for the expression of the mitochondrially encoded COXI protein. Curr Genet. 1997;32:273–80.
Costanzo MC, Bonnefoy N, Williams EH, Clark-Walker GD, Fox TD. Highly diverged homologs of Saccharomyces cerevisiae mitochondrial mRNA-specific translational activators have orthologous functions in other budding yeasts. Genetics. 2000;154:999–1012.
Kuhl I, Dujeancourt L, Gaisne M, Herbert CJ, Bonnefoy N. A genome wide study in fission yeast reveals nine PPR proteins that regulate mitochondrial gene expression. Nucleic Acids Res. 2011;39:8029–41.
Szklarczyk R, Wanschers BF, Cuypers TD, Esseling JJ, Riemersma M, van den Brand MA, Gloerich J, Lasonder E, van den Heuvel LP, Nijtmans LG, Huynen MA. Iterative orthology predic-tion uncovers new mitochondrial proteins and identifies C12orf62 as the human ortholog of COX14, a protein involved in the assembly of cytochrome c oxidase. Genome Biol. 2012;13:R12.
Choquet Y, Wollman FA. Translational regulations as specific traits of chloroplast gene expression. FEBS Lett. 2002;529:39–42.
Lefebvre-Legendre L, Choquet Y, Kuras R, Loubery S, Douchi D, Goldschmidt-Clermont M. A nucleus-encoded chloroplast protein regulated by iron availability governs expression of the photosystem I subunit PsaA in Chlamydomonas reinhardtii. Plant Physiol. 2015;167:1527–40.
Prikryl J, Rojas M, Schuster G, Barkan A. Mechanism of RNA stabilization and translational activation by a pentatricopeptide repeat protein. Proc Natl Acad Sci USA. 2011;108:415–20.
Weraarpachai W, Sasarman F, Nishimura T, Antonicka H, Aure K, Rotig A, Lombes A, Shoubridge EA. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am J Hum Genet. 2011;90:142–51.
Williams EH, Butler CA, Bonnefoy N, Fox TD. Translation initiation in Saccharomyces cerevisiae mitochondria: functional interactions among mitochondrial ribosomal protein Rsm28p, initiation factor 2, methionyl-tRNA-formyltransferase and novel protein Rmd9p. Genetics. 2007;175:1117–26.
Arita M, Suematsu T, Osanai A, Inaba T, Kamiya H, Kita K, Sisido M, Watanabe Y, Ohtsuki T. An evolutionary ‘intermediate state’ of mitochondrial translation systems found in Trichinella species of parasitic nematodes: co-evolution of tRNA and EF-Tu. Nucleic Acids Res. 2006;34:5291–9.
Ohtsuki T, Watanabe Y, Takemoto C, Kawai G, Ueda T, Kita K, Kojima S, Kaziro Y, Nyborg J, Watanabe K. An “elongated” translation elongation factor Tu for truncated tRNAs in nematode mitochondria. J Biol Chem. 2001;276:21571–7.
Sakurai M, Watanabe Y, Watanabe K, Ohtsuki T. A protein ex-tension to shorten RNA: elongated elongation factor-Tu recognizes the D-arm of T-armless tRNAs in nematode mitochondria. Biochem J. 2006;399:249–56. doi:10.1042/BJ20060781.
Suematsu T, Sato A, Sakurai M, Watanabe K, Ohtsuki T. A unique tRNA recognition mechanism of Caenorhabditis elegans mitochondrial EF-Tu2. Nucleic Acids Res. 2005;33:4683–91.
Cristodero M, Mani J, Oeljeklaus S, Aeberhard L, Hashimi H, Ramrath DJ, Lukes J, Warscheid B, Schneider A. Mitochondrial translation factors of Trypanosoma brucei: elongation factor-Tu has a unique subdomain that is essential for its function. Mol Microbiol. 2013;90:744–55.
Sharma MR, Booth TM, Simpson L, Maslov DA, Agrawal RK. Structure of a mitochondrial ribosome with minimal RNA. Proc Natl Acad Sci USA. 2009;106:9637–42.
Esseiva AC, Naguleswaran A, Hemphill A, Schneider A. Mito-chondrial tRNA import in Toxoplasma gondii. J Biol Chem. 2004;279:42363–8.
Pino P, Aeby E, Foth BJ, Sheiner L, Soldati T, Schneider A, Soldati-Favre D. Mitochondrial translation in absence of local tRNA aminoacylation and methionyl tRNA Met formylation in Apicomplexa. Mol Microbiol. 2010;76:706–18.
Gaillardin C, Duchateau-Nguyen G, Tekaia F, Llorente B, Casaregola S, Toffano-Nioche C, Aigle M, Artiguenave F, Blandin G, Bolotin-Fukuhara M, Bon E, Brottier P, de Montigny J, Dujon B, Durrens P, Lepingle A, Malpertuy A, Neuveglise C, Ozier-Kalogeropoulos O, Potier S, Saurin W, Termier M, Wesolowski-Louvel M, Wincker P, Souciet J, Weissenbach J. Genomic exploration of the hemiascomycetous yeasts: 21. Comparative functional classification of genes. FEBS Lett. 2000;487:134–49. doi:S0014579300022924 [pii].
Rosenthal LP, Bodley JW. Purification and characterization of Saccharomyces cerevisiae mitochondrial elongation factor Tu. J Biol Chem. 1987;262:10955–9.
Chiron S, Suleau A, Bonnefoy N. Mitochondrial translation: elongation factor tu is essential in fission yeast and depends on an exchange factor conserved in humans but not in budding yeast. Genetics. 2005;169:1891–901. doi:10.1534/genetics.104.037473.
Atkinson GC, Baldauf SL. Evolution of elongation factor G and the origins of mitochondrial and chloroplast forms. Mol Biol Evol. 2011;28:1281–92. doi:10.1093/molbev/msq316.
Suematsu T, Watanabe O, Kita K, Yokobori S, Watanabe Y. Arabidopsis thaliana mitochondrial EF-G1 functions in two different translation steps. J Biochem. 2013;155:107–14. doi:10.1093/jb/mvt105.
Tsuboi M, Morita H, Nozaki Y, Akama K, Ueda T, Ito K, Nierhaus KH, Takeuchi N. EF-G2mt is an exclusive recycling factor in mammalian mitochondrial protein synthesis. Mol Cell. 2009;35:502–10.
Gupta A, Mir SS, Jackson KE, Lim EE, Shah P, Sinha A, Siddiqi MI, Ralph SA, Habib S. Recycling factors for ribosome disassembly in the apicoplast and mitochondrion of Plasmodium falciparum. Mol Microbiol. 2013;88:891–905. doi:10.1111/mmi.12230.
Johnson RA, McFadden GI, Goodman CD. Characterization of two malaria parasite organelle translation elongation factor G proteins: the likely targets of the anti-malarial fusidic acid. PLoS One. 2011;6:e20633. doi:10.1371/journal.pone.0020633.
Scolnick E, Tompkins R, Caskey T, Nirenberg M. Release factors differing in specificity for terminator codons. Proc Natl Acad Sci USA. 1968;61:768–74.
Klaholz BP. Molecular recognition and catalysis in translation termination complexes. Trends Biochem Sci. 2011;36:282–92.
Duarte I, Nabuurs SB, Magno R, Huynen M. Evolution and diversification of the organellar release factor family. Mol Biol Evol. 2012;29:3497–512. doi:10.1093/molbev/mss157.
Nozaki Y, Matsunaga N, Ishizawa T, Ueda T, Takeuchi N. HMRF1L is a human mitochondrial translation release factor involved in the decoding of the termination codons UAA and UAG. Genes Cells. 2008;13:429–38. doi:10.1111/j.1365-2443.2008.01181.x.
Soleimanpour-Lichaei HR, Kuhl I, Gaisne M, Passos JF, Wydro M, Rorbach J, Temperley R, Bonnefoy N, Tate W, Lightowlers R, Chrzanowska-Lightowlers Z. mtRF1a is a human mitochondrial translation release factor decoding the major termination codons UAA and UAG. Mol Cell. 2007;27:745–57. doi:10.1016/j.molcel.2007.06.031.
Young DJ, Edgar CD, Murphy J, Fredebohm J, Poole ES, Tate WP. Bioinformatic, structural, and functional analyses support release factor-like MTRF1 as a protein able to decode nonstandard stop codons beginning with adenine in vertebrate mitochondria. RNA. 2010;16:1146–55.
Akabane S, Ueda T, Nierhaus KH, Takeuchi N. Ribosome rescue and translation termination at non-standard stop codons by ICT1 in mammalian mitochondria. PLoS Genet. 2014;10:e1004616. doi:10.1371/journal.pgen.1004616.
Lind C, Sund J, Aqvist J. Codonreading specificities of mitochondrial release factors and translation termination at non-standard stop codons. Nat Commun. 2013;4:2940. doi:10.1038/ncomms3940.
Richter R, Rorbach J, Pajak A, Smith PM, Wessels HJ, Huynen MA, Smeitink JA, Lightowlers RN, Chrzanowska-Lightowlers ZM. A functional peptidyl-tRNA hydrolase, ICT1, has been recruited into the human mitochondrial ribosome. EMBO J. 2010;29:1116–25. doi:10.1038/emboj.2010.14.
Gagnon MG, Seetharaman SV, Bulkley D, Steitz TA. Structur-al basis for the rescue of stalled ribosomes: structure of YaeJ bound to the ribosome. Science. 2012;335:1370–2. doi:10.1126/science.1217443.
Handa Y, Inaho N, Nameki N. YaeJ is a novel ribosome-associated protein in Escherichia coli that can hydrolyze peptidyl-tRNA on stalled ribosomes. Nucleic Acids Res. 2010;39:1739–48. doi:10.1093/nar/gkq1097.
Antonicka H, Ostergaard E, Sasarman F, Weraarpachai W, Wibrand F, Pedersen AM, Rodenburg RJ, van der Knaap MS, Smeitink JA, Chrzanowska-Lightowlers ZM, Shoubridge EA. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet. 2011;87:115–22.
Rorbach J, Richter R, Wessels HJ, Wydro M, Pekalski M, Farhoud M, Kuhl I, Gaisne M, Bonnefoy N, Smeitink JA, Lightowlers RN, Chrzanowska-Lightowlers ZM. The human mitochondrial ribosome recycling factor is essential for cell viability. Nucleic Acids Res. 2008;36:5787–99. doi:10.1093/nar/gkn576.
Christian BE, Spremulli LL. Evidence for an active role of IF3mt in the initiation of translation in mammalian mitochondria. Biochemistry. 2009;48:3269–78. doi:10.1021/bi8023493.
Barrientos A, Zambrano A, Tzagoloff A. Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. Embo J. 2004;23:3472–82.
Perez-Martinez X, Broadley SA, Fox TD. Mss51p promotes mitochondrial Cox1p synthesis and interacts with newly synthesized Cox1p. Embo J. 2003;22:5951–61.
Khalimonchuk O, Bird A, Winge DR. Evidence for a pro-oxidant intermediate in the assembly of cytochrome oxidase. J Biol Chem. 2007;282:17442–9.
Rak M, Su CH, Xu JT, Azpiroz R, Singh AM, Tzagoloff A. Regulation of mitochondrial translation of the ATP8/ATP6 mRNA by Smt1p. Mol Biol Cell. 2016.
Rak M, Tzagoloff A. F1-dependent translation of mitochondrially encoded Atp6p and Atp8p subunits of yeast ATP synthase. Proc Natl Acad Sci USA. 2009;106:18509–14.
Godard F, Tetaud E, Duvezin-Caubet S, di Rago JP. A genetic screen targeted on the FO component of mitochondrial ATP synthase in Saccharomyces cerevisiae. J Biol Chem. 2011;286:18181–9.
Rak M, Gokova S, Tzagoloff A. Modular assembly of yeast mitochondrial ATP synthase. Embo J. 2011;30:920–30.
Gruschke S, Kehrein K, Rompler K, Grone K, Israel L, Imhof A, Herrmann JM, Ott M. Cbp3-Cbp6 interacts with the yeast mitochondrial ribosomal tunnel exit and promotes cytochrome b synthesis and assembly. J Cell Biol. 2011;193:1101–14.
Mick DU, Fox TD, Rehling P. Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation. Nat Rev Mol Cell Biol. 2011;12:14–20.
Dennerlein S, Rehling P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J Cell Sci. 2015;128:833–7.
Rodel G. Translational activator proteins required for cytochrome b synthesis in Saccharomyces cerevisiae. Curr Genet. 1997;31:375–9.
Manthey GM, McEwen JE. The product of the nuclear gene PET309 is required for translation of mature mRNA and stability or production of intron-containing RNAs derived from the mitochondrial COX1 locus of Saccharomyces cerevisiae. Embo J. 1995;14:4031–43.
Mulero JJ, Fox TD. PET111 acts in the 5’-leader of the Saccha-romyces cerevisiae mitochondrial COX2 mRNA to promote its translation. Genetics. 1993;133:509–16.
Costanzo MC, Fox TD. Product of Saccharomyces cerevisiae nuclear gene PET494 activates translation of a specific mitochondrial mRNA. Mol Cell Biol. 1986;6:3694–703.
Costanzo MC, Seaver EC, Fox TD. At least two nuclear gene products are specifically required for translation of a single yeast mitochondrial mRNA. Embo J. 1986;5:3637–41.
Costanzo MC, Fox TD. Specific translational activation by nuclear gene products occurs in the 5’ untranslated leader of a yeast mitochondrial mRNA. Proc Nat Acad Sci USA. 1988;85:2677–81.
Finnegan PM, Payne MJ, Keramidaris E, Lukins HB. Characterization of a yeast nuclear gene, AEP2, required for accumulation of mitochondrial mRNA encoding subunit 9 of the ATP synthase. Curr Genet. 1991;20:53–61.
Payne MJ, Finnegan PM, Smooker PM, Lukins HB. Characterization of a second nuclear gene, AEP1, required for expression of the mitochondrial OLI1 gene in Saccharomyces cerevisiae. Curr Genet. 1993;24:126–35.
Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta. 2012;1817:851–62.
Wanschers BF, Szklarczyk R, van den Brand MA, Jonckheere A, Suijskens J, Smeets R, Rodenburg RJ, Stephan K, Helland IB, Elkamil A, Rootwelt T, Ott M, van den Heuvel L, Nijtmans LG, Huynen MA. A mutation in the human CBP4 ortholog UQCC3 im-pairs complex III assembly, activity and cytochrome b stability. Hum Mol Genet. 2014;23:6356–65.
Duarte FV, Palmeira CM, Rolo AP. The role of microRNAs in mitochondria: small players acting wide. Genes (Basel). 2014;5:865–86.
Rackham O, Shearwood AM, Mercer TR, Davies SM, Mattick JS, Filipovska A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA. 2011;17:2085–93.
Richter-Dennerlein R, Dennerlein S, Rehling P. Integrating mitochondrial translation into the cellular context. Nat Rev Mol Cell Biol. 2015;16:586–92.
Lang BF, Jakubkova M, Hegedusova E, Daoud R, Forget L, Brejova B, Vinar T, Kosa P, Fricova D, Nebohacova M, Griac P, Tomaska L, Burger G, Nosek J. Massive programmed translational jumping in mitochondria. Proc Natl Acad Sci USA. 2014;111:5926–31.
Nosek J, Tomaska L, Burger G, Lang BF. Programmed translational bypassing elements in mitochondria: structure, mobility, and evolutionary origin. Trends Genet. 2015;31:187–94.
Watanabe Y, Tsurui H, Ueda T, Furushima R, Takamiya S, Kita K, Nishikawa K, Watanabe K. Primary and higher order structures of nematode (Ascaris suum) mitochondrial tRNAs lacking either the T or D stem. J Biol Chem. 1994;269:22902–6.
Acknowledgments
This work was supported by a research grant from Consejo Nacional de Ciencia y Tecnología (47514 to XP-M) and fellowships 255917 (to AE. G-G), 298954 (to AZ-O) and 250726 (to RG-V); Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT), UNAM (IN204414 to XP-M). AE.G-G and RG-V are students of the Programade Doctoradoen Ciencias Biomédicas, UNAM.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
García-Guerrero, A.E., Zamudio-Ochoa, A., Camacho-Villasana, Y., García-Villegas, R., Reyes-Prieto, A., Pérez-Martínez, X. (2016). Evolution of Translation in Mitochondria. In: Hernández, G., Jagus, R. (eds) Evolution of the Protein Synthesis Machinery and Its Regulation. Springer, Cham. https://doi.org/10.1007/978-3-319-39468-8_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-39468-8_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39467-1
Online ISBN: 978-3-319-39468-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)