
Abstract
Alpha-1 antitrypsin deficiency (AATD) is a common genetic disorder characterized by a mutation in the SERPINA1 gene that codes for the alpha-1 antitrypsin protein. A homozygous mutation leads to the production of the Z protein, which takes an abnormal configuration that prevents its secretion from the hepatocyte, leading to a concomitant deficiency of the protein in the lung. The retained mutant Z protein in the endoplasmic reticulum of the hepatocyte activates a cascade of cell injury that leads to apoptosis, fibrosis, and in some individuals, cirrhosis. Clinical manifestations and progression of disease are highly variable even amongst Pi*ZZ homozygote individuals, suggesting a role from other genetic and environmental influences. Current treatment for liver disease from AATD consists mostly of supportive care and liver transplantation if indicated, but new and promising therapeutic approaches that include gene therapy, stem cell transplantation, and stimulation of autophagy are currently under investigation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Alpha-1 antitrypsin
- Metabolic
- Genetic
- Enzyme
- Emphysema
- PAS positive
- Diastase resistant
- Cirrhosis
- Liver transplantation
- Gene therapy
- Autophagy
Patient-Level Questions
-
1.
What is alpha-1 antitrypsin (AAT) deficiency?
Alpha-1 antitrypsin is a glycoprotein produced in the liver and works by preventing neutrophil proteases from destroying host tissues during inflammation. A hereditary mutation in the gene that codes for this protein leads to the production of a modified AAT protein, which folds in a way that prevents its secretion from the liver cell. Therefore, the abnormal AAT protein accumulates in the liver cell, leading to cell injury and eventually, severe scarring or cirrhosis . In addition, the absence of AAT protein in the lung leads to the absence of protection against the digestive actions of enzymes such as neutrophil elastase and proteinase 3 on the tissue structure of the lung, resulting in the premature development of emphysema.
-
2.
What can be done about it?
Lung disease from AATD can be treated with intra venous replacement with the protein, leading to improvement in lung function. Unfortunately, liver disease from AATD still has no direct treatment available, other than supportive measures to prevent or treat complications of cirrhosis . In those who have cirrhosis with complications, liver transplantation may be necessary. Currently, many investigators are actively studying various strategies geared towards removal or prevention of accumulation of the mutant AAT protein in the liver.
-
3.
Is the rest of my family at risk?
By virtue of its hereditary nature, the disease may affect more than one member of the family. However, the severity of the disease and clinical manifestations vary greatly amongst individuals with the same genetic mutations, suggesting that other genetic modifiers and environmental factors may affect the clinical presentation of the disease. It would be prudent to screen family members for the disease to provide early detection and possible intervention and monitoring, if the disease is clinically significant.
-
4.
Why do some patients with AATD manifest only lung disease, whereas others manifest only liver disease?
The peak ages for the diagnosis of liver and lung disea se in AATD differ, with liver disorders usually detected during childhood or old age, whereas lung disease develops in middle age. This may give the impression that the presence of one organ involvement excludes the other, although the risk of lung and liver disease indeed seems to be quite independent of each other.
Lung disease in AATD results from deficiency of the AAT protein in the lungs, whereas liver disease results from the accumulation of the mutant protein in the liver cell, leading to liver injury and fibrosis. The involvement of one organ rather than the other may also be partially explained by the various genetic mutations that lead to AATD. Some cases of AATD involve the production of abnormal AAT proteins which predisposes to liver disease and in severe cases, lung disease, whereas other cases involve a decreased or complete absence of production of the AAT protein in the liver which can cause lung disease but not liver disease. However, even in the homozygous AATD, the clinical manifestation of the genetic mutation is highly variable, and the natural history is influenced by other genetic and environmental modifiers.
-
5.
Are there things that I should avoid because of my diagnosis of AATD?
The development and severity of emphysema in AATD is significant ly increased up to 1000-fold by cigarette smoking, so one must avoid smoking, pollution, or occupational exposure to inhaled substances that are toxic to the lungs in the setting of AATD. In terms of liver disease, there is no evidence to indicate that alcohol use exacerbates the progression of the liver disease. However, it would be prudent to avoid excessive alcohol intake, given a theoretical risk of an additive injury on top of the AATD that may cause a more rapid progression of the liver disease. An increase in the production of the mutant Z protein has been demonstrated with the use of nonsteroidal anti-inflammatory drugs (NSAIDS) in animal models of AAT liver disease, so it would also be prudent to avoid the use of NSAIDS in this patient population.
Introduction
First described by Laurell and Erikson in 1963, alpha-1 antitrypsin deficiency (AATD) was noted as an absent alpha-1 band on protein electrophoresis of the serum of a patient with a lung disorder [1]. Alpha-1 antitrypsin (AAT) is a serum glycoprotein synthesized by the liver and secreted into the blood. It can inhibit trypsin in vitro, but it physiologically functions to inhibit neutrophil elastase and several related neutrophil serine proteases released during inflammation, and prevent them from destroying structural host tissues. As such, its production is regulated by inflammation, with serum levels rising three to five times during the host response to inflammation [2]. The normal allele of the AAT gene (SERPINA1-serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 1) on chromosome 14 is the M gene, which produces the M protein and normal serum levels of AAT. Mutations in the AAT gene may present as one of over 100 variant alleles, but majority of patients with liver disease from AATD have a Glu342Lys substitution in both genes and are Pi*ZZ homozygotes (also known as ZZ; Pi :protease inhibitor). About 0.05 % of the United States population is homozygous for the Z allele, whereas 2 % of the population is heterozygous [3].
The mutant Z gene drives the production of a mutant Z protein which folds abnormally, impeding its secretion from the endoplasmic reticulum (ER) of the hepatocyte. Its accumulation in the hepatocyte ER leads to hepatocyte injury, inflammation, fibrosis, and in some patients, cirrhosis of the liver [4]. In addition, failure to excrete the protein from the liver leads to a deficiency state in the lung, with Pi*ZZ homozygotes having serum levels of only 10–15 % of normal ranges [2]. This translates to disinhibited connective tissue breakdown by the neutrophil proteases in the lung, resulting in premature emphysema [5]. Heterozygote carriers of a Z allele combined with a normal M gene, Pi*MZ, are generally healthy but appear to be more susceptible to liver disease in the presence of another injurious agent to the hepatocyte. The S variant, causing the Glu288Val substitution in the AAT gene, is common in North American and Western European populations particularly in Spain and Portugal. It may cause significant lung disease or liver disease when it is present in combination with the Z allele (Pi*SZ), but not when present in homozygous form Pi*SS [3]. Other rare mutations can yield a protein with a normal M migration on electrophoresis (M Duarte and M malton alleles), but when present in the heterozygous state with a Z allele, the protein may accumulate in the ER of the hepatocyte and cause liver disease. Such mutations can be detected by a profoundly low AAT level in peripheral blood that is below that expected for a Pi*MZ phenotype (Table 19.1) [6, 7]. The presence of null genes or other unusual alleles that do not direct the synthesis of a protein product that accumulates in the ER of the liver will not cause liver disease, but null genes with resulting undetectable serum AAT level will almost certainly result in lung disease.
Alpha-1 antitrypsin deficiency is the most common hereditary neonatal liver disease. It can lead to cirrhosis and hepatocellular carcinoma in some adults [8]. Although the classical Pi*ZZ form of AAT is found in 1 in 1600 to 1 in 2000 live births in most populations [9], prospective screening studies in Sweden report that only about 10 % of affected individuals develop clinically significant liver disease by the time they reach the fourth decade of life [10, 11], suggesting that other genetic and/or environmental factors may play a role in the phenotypic expression of the disease.
Pathophysiology of the AAT ZZ Liver Disease
The mutant Z protein is transcribed and translated in the hepatocyte, then t ransported into the endoplasmic reticulum, where it undergoes folding into its final conformation in preparation for secretion. Unfortunately, the mutant protein folds inefficiently due to the amino acid substitution. Its abnormal configuration impairs its secretion from the ER, and these proteins are directed mostly into a variety of proteolytic pathways referred to as “ER-associated degradation (ERAD),” but some survive and take various conformations, including the linkage of large groups of Z protein “polymers” by noncovalent bonds that allow them to become visible on light microscopy as “globules” [12]. Although the actual degradation mechanism remains to be elucidated, calnexin, and ER manosidase have been identified to be possible points of control [13]. Pi*ZZ homozygous patients with less efficient ERAD mechanisms are more susceptible to development of liver disease than those with more efficient mechanisms [14]. Autophagy is another important proteolytic pathway that is upregulated by the accumulation of Z protein polymers, leading to the degradation of abnormal proteins by specialized vacuoles. Increasing autophagy in animal models has led to a reduction in the accumulation of the Z protein polymers and consequent liver injury [15, 16]. Hepatocytes with large burden of polymerized Z proteins undergo apoptosis, initiating a chronic process of cell injury that leads to cell death, fibrosis, and eventually in some individuals, cirrhosis . However, it is well recognized that individuals with the same Pi*ZZ genotype demonstrate variable extents of clinical liver injury, invoking the probable roles of environmental and genetic modifiers that alter the rate and severity of the clinical manifestation of the disease.
Clinical Presentation and Natural History
As is seen in other liver diseases that affect neonates, persistent jaundice is the most common presenting symptom at 4–8 weeks of age in those who manifest the disease. Accompanying symptoms may include poor feeding, poor weight gain, and pruritus. Some babies may have hepatomegaly, and the laboratory derangements may demonstrate either a hepatitis picture with elevated serum transaminases, or cholestasis with elevated alkaline phosphatase [17]. Occasionally, neonates present with bleeding from the gastrointestinal tract or from the umbilical stump, or easy bruising [18].
Liver disease from AATD may be diagnosed later in childhood, presenting with asymptomatic hepatomegaly or elevated transaminases, or with jaundice that may occur during the course of an unrelated illness. It can also remain further undetected until the adolescence or adulthood, where it can present with complications of cirrhosis and portal hypertension, such as splenomegaly and hypersplenism, gastrointestinal bleeding from varices, ascites, or hepatic encephalopathy [17]. The diagnosis should be considered in adults who present with chronic liver disease or cirrhosis of unknown etiology.
The natural history of liver disease due to AATD is remarkably variable. Most infants that manifested jaundice early in life recover and become asymptomatic by age 1 year, despite the few who would require liver transplantation. Majority of these children will remain without symptoms as they grow older. In the only prospective study of AATD diagnosed by screening 200,000 neonates in Sweden in the 1970s, 127 infants were diagnosed with the classic form of AATD. Eleven percent of these infants had prolonged jaundice, 6 % had hepatomegaly with or without elevated serum transaminases, and approximately 43 % had elevated serum transaminases alone. Five of the twenty neonates with clinical evidence of liver disease died of cirrhosis in early childhood. Follow-up of the remaining population at age 18 years showed that only 12 % of the ZZ phenotype had elevated serum transaminases, although there were no clinical signs of liver disease, while 10 % of the SZ phenotype had similar findings. Therefore, although 17 % of the population had clinical evidence of liver disease in the first 18 years of life, almost 90 % of those who reached adulthood had normal liver enzymes, although biopsies were not performed to confirm the absence of liver injury [11].
Lung destruction in the form of emphysema caused by AATD usually manifests later in the third decade. The incidence of liver disease in patients who were diagnosed with emphysema from AATD is not clearly defined, although one small series reported elevated liver chemistry tests in 50 % [19].
Diagnosis
The diagnosis of AATD should be considered in an individual who presents with symptoms of chronic liver disease or demonstrates elevated liver chemistry tests, particularly those who do not have an obvious etiology. The disease is characterized by the presence of altered AAT proteins that accumulates in the liver, such that circulating serum AAT levels are expected to be low and the serum AAT phenotype determination by isoelectric-focusing electrophoresis would reveal the altered protein type. While screening for the disease is typically performed with the AAT level measurement, it is important to remember that these levels may be falsely elevated to normal or near-normal ranges in affected patients by a concomitant inflammatory host response. On the other hand, patients with advanced cirrhosis and compromised synthetic function of the liver may have low serum protein levels, including AAT levels. Therefore, both serum concentrations and phenotype determination should be obtained when the diagnosis is seriously considered.
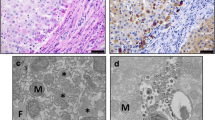
Histologically, accumulation of the abnormal AAT molecules in the liver is represented by the presence of periodic acid-Schiff-positive, diastase-resistant globules in the endoplasmic reticulum of hepatocytes (Fig. 19.1). These inclusions are eosinophilic, round to oval, 1–40 μm in diameter located prominently in periportal hepatocytes, and less so in Kupffer cells and biliary epithelial cells [20]. However, it is important to note that the globules are not present in all hepatocytes, or can be small or “dust-like” in small infants, or totally absent in neonatal livers [21].

Liver histology from a patient with ZZ alpha-1 antitrypsin deficiency, demonstrating eosinophilic periodic acid-Schiff-positive, diastase-resistant globules in the cytoplasm of the hepatocytes (Courtesy of John Hart, MD)
The lung manifestation of AATD is rarely seen in childhood or adolescence, but becomes more common when the individual reaches the mid to late 30 years of life. The development and severity of emphysema in AATD is significantly increased up to 1000-fold by cigarette smoking [22]. Alpha-1 antitrypsin deficiency has been associated with vasculitic diseases [23], and the c losest association has been demonstrated in a genome-wide sequence analysis that demonstrated the association of anti-proteinase 3 antineutrophil cytoplasmic antibody (ANCA) with the gene encoding AAT (SERPINA1) [24]. It also has been associated with panniculitis, which can respond to augmentation therapy [25, 26].
Management
Whereas the management of lung disease from AATD focuses most ly on avoidance of smoking and pollution and augmentation therapy, these strategies do not have any impact on the liver. Referral to a pulmonologist to evaluate and manage any lung manifestation would be important, as regular monitoring of lung function would be essential as the risk of emphysema increases with age. Avoidance of first-hand or second-hand smoking as well as pollution must be stressed [27]. Augmentation therapy or exogenous AAT replacement, if indicated, can benefit lung disease.
Currently, the management of liver disease due to AATD consists mainly of supportive measures, as there is no available specific therapy targeted to the liver. The use of long-term ursodiol in children with milder liver disease from AAT has been associated with improvement in liver chemistry tests, but a true benefit on histologic disease or the natural history of the disease remains to be seen [28]. When cirrhosis of the liver is suspected or established, screening for hepatocellular carcinoma and for esophageal varices should be performed, and preventive measures against some complications such as variceal bleeding or malnutrition can be instituted. In the setting of advanced fibrosis or cirrhosis, abstinence from alcohol is necessary to avoid a more rapid decompensation. When hepatic decompensation or hepatocellular carcinoma has developed, liver transplantation may be necessary. Liver transplantation in children and adults is associated with excellent survival rates of 90 and 83 % in 5 years, respectively [29]. Animal models of AAT liver disease have demonstrated a unique toxicity of nonsteroidal anti-inflammatory drugs to the Pi*ZZ liver by increasing the production of the mutant Z protein [30], so it would be prudent to avoid the use of NSAIDS in this patient population.
Various directed therapies are currently under investigation for AATD. Similar to other genetic diseases, gene therapy for AAT have been demonstrated to succeed in mouse models, including one model where inhibition of the synthesis of the mutant Z protein by 80 % and generation of the normal M AAT protein were achieved with an exogenous mRNA incorporated into recombinant adeno-associated virus vectors, allowing prevention of liver disease and lung disease at the same time [31]. This therapy appears promising with its dual benefits, provided its efficacy can be duplicated in humans and safety concerns can be overcome. The use of stem cells as a delivery tool has also been explored in mouse models, where gene transfer via lentivirally transduced hematopoietic stem cells has led to sustained human AAT expression [32]. Induced pluripotent stem cells (IPSC) have been used to correct the point mutation in individuals with AATD, and these corrected IPSC’s can differentiate into hepatocyte-like cells in vitro with demonstrated ability to function when transplanted into the liver of a mouse [33].
Studies on small molecules that may alter the folding process of the Z protein to prevent retention or polymerization and allow secretion or degradation have also been undertaken. A drug called 4-phenylbutyrate was able to increase the secretion of the mutant Z protein in cell culture and animal models [34], but unfortunately did not show this benefit in humans [35]. Other drugs that have been studied in animal models include rapamycin and carbamazepine, which are aimed at inducing autophagic degration of the mutant Z protein in the endoplasmic reticulum and preventing its accumulation. These interventions have led to reduced fibrosis and reduced levels of markers of liver injury [16, 28]. Known toxicities from these drugs in humans are a significant concern, however, so a clinical trial of carbamazepine to treat AATD in humans is currently being conducted at doses that are much lower than those given t o animal models. At the present time, use of carbamazepine and rapamycin in AATD remain experimental.
Conclusions
Homozygous AATD is a common metabolic liver disease that affects children and adults with a highly variable penetrance and natural history that are influenced by other genetic and environmental modifiers. Accumulation of the mutant Z protein in the hepatocyte ER activates an intracellular injury cascade of cell injury that leads to apoptotic cell death, fibrosis, and eventually in some individuals, cirrhosis. Currently, there is no specific treatment for liver disease from AATD other than supportive measures, and liver transplantation if indicated, but promising avenues utilizing gene therapy , stem cell transplantation, and stimulation of autophagy are currently under active investigation.
References
Laurell C, Eriksson S. The electrophoretic alpha 1 globulin pattern of serum in alpha 1 antitrypsin deficiency. Scan J Clin Lab Invest. 1963;15:132–40.
Perlmutter DH. Alpha-1 antitrypsin deficiency. In: Suchy F, Sokol R, Balisteri W, editors. Liver disease in children. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2007.
Nelson DR, Teckman J, Di Bisceglie AM, Brenner DA. Diagnosis and management of patients with alpha1-antitrypsin (A1AT) deficiency. Clin Gastroenterol Hepatol. 2012;10(6):575–80.
Lindblad D, Blomenkamp K, Teckman J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology. 2007;46(4):1228–35.
Eriksson S. Discovery of alpha 1-antitrypsin deficiency. Lung. 1990;168(Suppl):523–9.
Lomas DA, Elliott PR, Sidhar SK, Foreman RC, Finch JT, Cox DW, et al. alpha 1-Antitrypsin Mmalton (Phe52-deleted) forms loop-sheet polymers in vivo. Evidence for the C sheet mechanism of polymerization. J Biol Chem. 1995;270(28):16864–70.
Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53 → Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem. 1993;268(21):15333–5.
Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. N Engl J Med. 1986;314(12):736–9.
Sveger T. The natural history of liver disease in alpha 1-antitrypsin deficient children. Acta Paediatr Scand. 1988;77(6):847–51.
Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294(24):1316–21.
Sveger T, Eriksson S. The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology. 1995;22(2):514–7.
Lomas DA, Mahadeva R. Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest. 2002;110(11):1585–90.
Qu D, Teckman JH, Omura S, Perlmutter DH. Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J Biol Chem. 1996;271(37):22791–5.
Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, Perlmutter DH. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc Natl Acad Sci U S A. 1994;91(19):9014–8.
Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329(5988):229–32.
Kaushal S, Annamali M, Blomenkamp K, Rudnick D, Halloran D, Brunt EM, et al. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp Biol Med. 2010;235(6):700–9.
Perlmutter DH. Alpha-1-antitrypsin deficiency: diagnosis and treatment. Clin Liver Dis. 2004;8(4):839–59. viii–ix.
Hope PL, Hall MA, Millward-Sadler GH, Normand IC. Alpha-1-antitrypsin deficiency presenting as a bleeding diathesis in the newborn. Arch Dis Child. 1982;57(1):68–70.
von Schonfeld J, Breuer N, Zotz R, Liedmann H, Wencker M, Beste M, et al. Liver function in patients with pulmonary emphysema due to severe alpha-1-antitrypsin deficiency (Pi ZZ). Digestion. 1996;57(3):165–9.
Yunis EJ, Agostini Jr RM, Glew RH. Fine structural observations of the liver in alpha-1-antitrypsin deficiency. Am J Pathol. 1976;82(2):265–86.
Teckman JH, Jain A. Advances in alpha-1-antitrypsin deficiency liver disease. Curr Gastroenterol Rep. 2014;16(1):367.
Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med. 2009;360(26):2749–57.
Morris H, Morgan MD, Wood AM, Smith SW, Ekeowa UI, Herrmann K, et al. ANCA-associated vasculitis is linked to carriage of the Z allele of alpha(1) antitrypsin and its polymers. Ann Rheum Dis. 2011;70(10):1851–6.
Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. 2012;367(3):214–23.
Lyon MJ. Metabolic panniculitis: alpha-1 antitrypsin deficiency panniculitis and pancreatic panniculitis. Dermatol Ther. 2010;23(4):368–74.
Valverde R, Rosales B, Ortiz-de Frutos FJ, Rodriguez-Peralto JL, Ortiz-Romero PL. Alpha-1-antitrypsin deficiency panniculitis. Dermatol Clin. 2008;26(4):447–51. vi.
Turner AM. Alpha-1 antitrypsin deficiency: new developments in augmentation and other therapies. BioDrugs. 2013;27(6):547–58.
Lykavieris P, Ducot B, Lachaux A, Dabadie A, Broue P, Sarles J, et al. Liver disease associated with ZZ alpha1-antitrypsin deficiency and ursodeoxycholic acid therapy in children. J Pediatr Gastroenterol Nutr. 2008;47(5):623–9.
Kemmer N, Kaiser T, Zacharias V, Neff GW. Alpha-1-antitrypsin deficiency: outcomes after liver transplantation. Transplant Proc. 2008;40(5):1492–4.
Rudnick DA, Shikapwashya O, Blomenkamp K, Teckman JH. Indomethacin increases liver damage in a murine model of liver injury from alpha-1-antitrypsin deficiency. Hepatology. 2006;44(4):976–82.
Mueller C, Tang Q, Gruntman A, Blomenkamp K, Teckman J, Song L, et al. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol Ther. 2012;20(3):590–600.
Wilson AA, Kwok LW, Hovav AH, Ohle SJ, Little FF, Fine A, et al. Sustained expression of alpha1-antitrypsin after transplantation of manipulated hematopoietic stem cells. Am J Respir Cell Mol Biol. 2008;39(2):133–41.
Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, et al. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478(7369):391–4.
Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97(4):1796–801.
Teckman JH. Lack of effect of oral 4-phenylbutyrate on serum alpha-1-antitrypsin in patients with alpha-1-antitrypsin deficiency: a preliminary study. J Pediatr Gastroenterol Nutr. 2004;39(1):34–7.
Wise RA. Alpha-1 antitrypsin deficiency. In: Porter R, editor. Merck manual of diagnosis and therapy. Whitehouse Station, NJ: Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc.; 2014 [updated June 2014; cited 2015 January 2, 2015]. http://www.merckmanuals.com/professional/pulmonary_disorders/chronic_obstructive_pulmonary_disease_and_related_disorders/alpha-1_antitrypsin_deficiency.html. Accessed 2 Jan 2015.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Te, H.S. (2017). Metabolic and Genetic Liver Diseases: Alpha-1 Anti-trypsin Deficiency. In: Saeian, K., Shaker, R. (eds) Liver Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-30103-7_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-30103-7_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-30101-3
Online ISBN: 978-3-319-30103-7
eBook Packages: MedicineMedicine (R0)