
Abstract
Skeletal muscle possesses remarkable ability to change its size and force-producing capacity in response to physiological stimuli. Impairment of the cellular processes that govern these attributes also affects muscle mass and function in pathological conditions. Myostatin, a member of the TGF-β family, has been identified as a key regulator of muscle development, and adaptation in adulthood. In muscle, myostatin binds to its type I (ALK4/5) and type II (ActRIIA/B) receptors to initiate Smad2/3 signalling and the regulation of target genes that co-ordinate the balance between protein synthesis and degradation. Interestingly, evidence is emerging that other TGF-β proteins act in concert with myostatin to regulate the growth and remodelling of skeletal muscle. Consequently, dysregulation of TGF-β proteins and their associated signalling components is increasingly being implicated in muscle wasting associated with chronic illness, ageing, and inactivity. The growing understanding of TGF-β biology in muscle, and its potential to advance the development of therapeutics for muscle-related conditions is reviewed here.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
5.1 Introduction
Skeletal muscle is a highly adaptive tissue, capable of responding to imposed physiological stimuli, to tune its performance as a contractile tissue, a metabolic regulator, and a secretor of factors that influence other organs. Finely regulated processes controlling protein synthesis and degradation enable changes in muscle mass and function as demands require.
The adaptive properties of skeletal muscle also make it susceptible to loss of mass and functional capacity, when protein turnover is adversely affected in pathological contexts. Significant wasting of skeletal muscle is associated with many diseases, including (but not limited to) advanced progression of certain cancers, sepsis, organ failure, HIV/AIDS, anorexia, disuse, advanced ageing and neuromuscular disorders (Ciciliot et al. 2013; Dam et al. 2014; Jung et al. 2014; Mondello et al. 2015; Puthucheary et al. 2013; Tang et al. 2002; Visvanathan and Chapman 2009; Wall et al. 2014). As the debilitating effects of muscle wasting reduce quality of life and survival, placing significant burden on our healthcare system, it is necessary to understand the mechanisms that regulate muscle attributes, and use this information to advance the development of muscle-directed therapeutics.
Increasingly, muscle wasting observed in chronic illness is being linked with perturbed regulation of the transforming growth factor-β (TGF-β) signalling network. The TGF-β ligand family comprises 34 structurally-related proteins (Cusella-De Angelis et al. 1994; Wu and Hill 2009) broadly classified as: TGF-β isoforms, growth differentiation factors (GDFs), bone morphogenetic proteins (BMPs), activins and inhibins (Massague 1998). Other members include Nodal, lefty and anti-Müllerian hormone. Myostatin (also known as GDF-8) has been identified as a critical regulator of muscle development and homeostasis (Mcpherron et al. 1997), although subsequent studies have begun to identify other TGF-β proteins, such as TGF-β1, activins and several BMPs, as also having crucial roles in muscle. This review considers the significance of signalling regulated by the TGF-β network as a regulator of muscle attributes in relation to musculoskeletal disorders.
5.2 The TGF-β Signalling Network in Overview
In the human body, the vast majority of cell types express at least one TGF-β family ligand and their cognate receptors (Blobe et al. 2000). Together, TGF-β proteins and their signalling components exert physiological control over proliferation, differentiation, apoptosis, adhesion and extracellular matrix deposition, thereby controlling embryogenesis, organogenesis and adult tissue homeostasis (Buijs et al. 2007; Chang et al. 2002; Massague 1990; Shi and Massague 2003). While this review focuses on myostatin and other TGF-β family proteins with emerging roles in the biology and disease of skeletal muscle, it is beneficial to review the processes that control ligand synthesis and activity.
5.2.1 Ligand Synthesis and Secretion
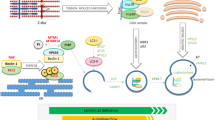
Like most TGF-β family proteins, myostatin is synthesised as a precursor protein comprising an N-terminal prodomain and a C-terminal mature domain (Fig. 5.1). During biosynthesis, hydrophobic residues at the N-terminus of the prodomain bind to residues in the mature domain, an interaction that is critical for correct protein formation (Walton et al. 2010). Two monomers are linked covalently at a site within the mature domain (Husken-Hindi et al. 1994), and non-covalently between the two prodomains. Following complex assembly, proprotein convertases cleave between the pro- and mature domains (Dubois et al. 1995; Gentry et al. 1988), resulting in a covalently bound mature protein non-covalently associated to its prodomain dimer. From herein, TGF-β family proteins are referred to as to the ‘mature dimer’ and the resultant prodomain dimer is described as the ‘prodomain’. Following synthesis, the mature dimer and prodomain complex is secreted from the cell, and in many instances, localised to the extracellular matrix (ECM).

Synthesis of TGF-β proteins. (a) TGF-β proteins are synthesised as precursor proteins consisting of an N-terminal prodomain and a C-terminal mature domain. Prodomains form tight contacts within their mature domains, which facilitate folding and dimerisation. (b) Dimeric precursors are cleaved between the pro- and mature domains by proprotein convertases, such as furin, to form (c) a complex comprising prodomain non-covalently associated to the mature TGF-β dimer
Some TGF-β proteins, such as myostatin, activins and TGF-β isoforms, bind to the ECM via specific interactions (Ramirez and Rifkin 2009; Rifkin 2005; Sengle et al. 2008a). The ECM network of glycoproteins plays essential roles in the structure, survival, migration and proliferation of cells (Assoian et al. 1983; Brunner et al. 1989; Gentry and Nash 1990), and can also act as an important intermediate reservoir for many members of the family prior to their activation (Fig. 5.2) (Sengle et al. 2008a). ECM components vary between organs as they are temporally and spatially expressed and secreted by proximal tissues, but the three major proteins comprising the ECM are collagen, fibronectin and fibrillin (Vakonakis and Campbell 2007). Interestingly, TGF-β proteins are also critical regulators of ECM production, regulating collagen synthesis and deposition by fibroblasts (Akhurst and Hata 2012).

Localisation of TGF-β proteins to the extracellular matrix. The prodomains target TGF-β proteins to the ECM in the vicinity of target cells. (a) Some TGF-βs, such as BMP-7 bind directly to fibrillin microfibrils in the ECM, (b) while others, such as activin A, bind fibrillin-associated proteins, such as perlecan. (c) The TGF-β isoforms bind to LTBPs, which then bind to fibrillin in the ECM
The prodomains of the TGF-β isoforms bind the ECM via an intermediate association with latent TGF-β-binding proteins (LTBPs). Prodomain binding to LTBPs promotes the formation of large latent complexes (LLCs). The C-terminal region of LTBPs binds to the N-terminal region of fibrillin, targeting LLCs to the ECM, an interaction that is essential for normal TGF-β signalling (Ramirez et al. 2008). Other TGF-β proteins, including BMP-2, -4, -7, -10 and GDF-5 can bind directly to fibrillin via their prodomains (Sengle et al. 2008a), whereas activin prodomains can bind heparin sulphate proteoglycans, such as perlecan, in the ECM (Li et al. 2010).
5.2.2 Prodomains Control TGF-β Ligand Activity and Localisation in the Extracellular Matrix
Prodomains can modulate the signalling potential of their associated TGF-β proteins. For the majority of TGF-β family members, the interaction between the mature protein and their associated prodomains is weak, allowing them to be readily displaced in the presence of their target receptors (Sengle et al. 2008b; Walton et al. 2009). However, myostatin, GDF-11, TGF-β1, TGF-β2, TGF-β3, BMP-10 and human GDF-9 bind their prodomains with high affinity and are secreted in a latent form (Simpson et al. 2012). The prodomains confer latency by shielding the receptor-binding epitopes (Bottinger et al. 1996; De Crescenzo et al. 2001; Ge et al. 2006; Hill et al. 2002; Thies et al. 2001).
The latency of some TGF-β proteins provides an additional barrier in activity regulation. Myostatin is expressed in skeletal muscle and is a potent negative regulator of muscle mass, therefore an additional level of regulation is necessary to constrain its effects. The BMP-1/tolloid family of metalloproteinases is hypothesised to activate latent myostatin, by cleaving after the aspartate-76 residue within the myostatin prodomain and releasing mature active myostatin.(Lee 2008; Wolfman et al. 2003). Other latent TGF-βs can be activated by thrombospondin-1 and integrins, which alter the conformation of the prodomain enabling the release of the mature active proteins (Fig. 5.3) (Annes et al. 2004; Ribeiro et al. 1999).

TGF-β activation. (a) The TGF-β isoforms, myostatin and GDF-11 bind their prodomains with high affinity, with the prodomains shielding the receptor-binding epitopes. In order for these ligands to signal, they must be liberated from their prodomains via an activation mechanism, enabling them to bind to their receptors. (b) For the other TGF-β proteins, such as activin A, they are secreted in an ‘active’ form. These ligands have a higher affinity for their signalling receptors than their prodomains and do not require an activation step
5.2.3 TGF-β Family Members Target Specific Receptor Complexes and Transcription Factors
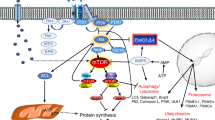
The biological effects of myostatin are initiated upon engagement of the ligand with two specific transmembrane serine/threonine kinase receptors (Fig. 5.4). Binding of mature myostatin to the extracellular domain of a type II activin receptor (ActRII) leads to recruitment, phosphorylation and activation of a type I activin receptor (ActRI), also known as an activin receptor-like kinase (ALK) (Ten Dijke and Arthur 2007). Type II and type I transmembrane receptors are classified by their structural and functional properties. There are five mammalian type II receptors: ActRIIA, ActRIIB, TβRII, BMPRII and AMHRII, and seven type I receptors: activin receptor-like kinases (ALK) 1-7 (Shimasaki et al. 2004). Predominantly, the type II receptors facilitate ligand engagement, whereas the type I receptors determine the signalling specificity by phosphorylating specific Smad proteins that modulate intracellular signalling and regulate target gene transcription (Ten Dijke and Arthur 2007).

TGF-β signalling pathway. TGF-β signalling is divided into two main intracellular pathways (the TGF-β and BMP pathways) according to the type I and II receptors the ligands target and the intracellular Smads they activate. The binding of a growth factor to a type II receptor leads to recruitment of a type I receptor. The TGF-β subgroup phosphorylate type I receptors. In turn, type I receptors phosphorylate Smads 2 or 3. Conversely, activation by the BMP subgroup leads to phosphorylation of Smads 1, 5 or 8. All Smads form complexes with Smad4, and translocate to the nucleus to regulate gene transcription with co-activators, co-repressors and additional transcription factors. Smad6 and 7 are inhibitory Smads that prevent Smads forming complexes with Smad4, and also bind to type I receptors
Myostatin can signal through the type II receptors ActRIIA/B and BMPRII, and the type I receptors ALK4 and ALK5 (Sidis et al. 2006; Tsuchida et al. 2004). Activation of ALK4 and ALK5 receptors proceeds to phosphorylate intracellular Smad2 or 3 transcription factors (Wrighton et al. 2009b), which then bind to Smad4, and the resulting Smad complex translocates to the nucleus to regulate gene transcription (Massague and Gomis 2006; Wrighton et al. 2009a). Other TGF-β proteins that initiate Smad2/3 signalling include the TGF-β isoforms, activins and Nodal. Notably, in parallel with this ActRII-Smad2/3 signalling pathway, the TGF-β network supports signalling mediated by other ligands that activate ALKs1, 2, 3 and 6 to phosphorylate Smad1/5/8. BMPs, other GDFs and AMH can employ this axis via their binding to discrete type II receptors (Massague and Gomis 2006).
As there are only a handful of type I and type II receptors for the large number of TGF-β proteins, several TGF-β proteins can utilise the same receptor complexes. For example, activin A, activin B, BMP-4, BMP-7 and GDF-5 can bind ActRIIA/B (Greenwald et al. 2003; Harrison et al. 2004; Tsuchida et al. 2004). Similarly, BMP-2, BMP-4, BMP-7, BMP-15 and GDF-9 bind BMPRII (Shi et al. 2000). In some circumstances, more than one ligand can utilise the same receptor complex; both myostatin and activin A can initiate signalling via ActRIIA/B and ALK4 (Tsuchida et al. 2004). The interplay of numerous ligands and receptor combinations and the interaction of downstream Smad signalling with other pathways varies by cell type and context, thus creating a system for highly varied and nuanced effects of the TGF-β network.
5.2.4 Extracellular Regulation of TGF-β Signalling
Further regulation of TGF-β signalling is facilitated by extracellular, membrane-bound and intracellular proteins. Some members of the TGF-β family also require accessory receptors, such as betaglycan, endoglin and cripto, to enhance their interaction with signalling receptors. For example, betaglycan acts as a co-receptor for the TGF-β isoforms, most notably TGF-β2, and inhibin (Cheifetz et al. 1990; Lewis et al. 2000). Within cells, inhibitory Smad proteins 6 and 7 govern TGF-β signalling through a negative feedback loop. Smad6 and 7 bind to type I receptors, to inhibit the phosphorylation of receptor-regulated Smads (Gazzerro and Canalis 2006). In addition, Ski binds to Smad4 to prevent the formation of Smad complexes, as well as retaining existing Smad complexes bound to DNA to prevent binding of new complexes (Luo 2003).
Follistatin is an extracellular protein found in circulating and membrane-bound forms, and exhibits high affinity binding for activins, but also bind myostatin, GDF-11 and some BMPs (Lee and Mcpherron 2001; Nakamura et al. 1990; Tsuchida 2006). In the case of activins, follistatin neutralises activity by shielding approximately a third of the residues on the mature ligand, including both the type I and type II receptor-binding sites (Harrington et al. 2006). Follistatin-related gene (FLRG) is an inhibitor of TGF-β signalling that also contains a follistatin-binding domain, which binds similar ligands as follistatin (Tsuchida et al. 2000). There are also specific extracellular BMP antagonists, such as noggin, that preferentially bind certain BMP family members and reduce their access to signalling receptors (Gazzerro and Canalis 2006; Shimasaki et al. 2004). Collectively, these extra-cellular proteins confer an additional layer of regulation to control the bioactivity of the TGF-β family of ligands.
5.3 The Role of TGF-β Signalling in Skeletal Muscle Development and Homeostasis
The formation of skeletal muscle (myogenesis) commences early during embryonic development. Pluripotent muscle progenitor cells originating from the somites give rise to myoblasts, the precursor cells of skeletal muscle fibres (Kubota et al. 1989; Mauro 1961). The fusion of myoblasts produces multi-nucleated muscle fibres, which, remarkably, can be maintained for life. In parallel, a population of progenitor cells persist as ‘satellite cells’, which reside in proximity with muscle fibres as a reservoir of cells for repair of muscle fibres. Proliferation and recruitment of the resident satellite cells occurs when damaged muscle fibres must be repaired (or regenerated). Post-natal adaptation of muscle mass occurs largely via changes in the size of existing muscle fibres, although in some instances recruitment of satellite cell may also provide a means of contributing additional nuclei to existing fibres during episodes of growth.
Myostatin, in particular, has been defined as a potent negative regulator of skeletal muscle mass (Mcpherron et al. 1997). Myostatin knockout mice (Mstn -/-) exhibit dramatically increased muscle mass as a result of increased fibre numbers being formed during development, and increased fibre size in adulthood. The significance of myostatin in skeletal muscle is evident from its conservation across species, as loss of myostatin during development has been associated with a ‘hypermuscular’ phenotype in rodents (mice, rats), dogs, birds (chickens, turkeys), sheep, pigs, horses and humans. Although other members of the TGF-β family have also been found to exert unique effects upon muscle development and post-natal adaptation (Kollias and Mcdermott 2008; Lee et al. 2005), myostatin remains arguably the most dominant regulator of skeletal muscle phenotype in development.
5.3.1 Skeletal Muscle Development
Cells of somatic origin are directed down a program of myogenesis commencing early in embryonic development according to episodes of determination, differentiation and maturation. Determination begins with the formation of pluripotent muscle progenitor cells (Mauro 1961) that give rise to myoblasts, the precursor cells of skeletal muscle fibres (Kubota et al. 1989). Myoblasts completing an active proliferative phase typically undergo differentiation, which entails irreversible withdrawal from the cell cycle, and the expression of early muscle-specific genes, including the MyoD and MEF2 family of transcription factors (Joulia-Ekaza and Cabello 2006). These myogenic factors support myoblast commitment and fusion, and the subsequent expression of late muscle-specific genes (Acharyya et al. 2004; Megeney et al. 1996; Wheeler et al. 1999). As myoblasts fuse into post-mitotic syncytial structures spanning from tendon to tendon, the specialised proteins required to assemble the excitation-contraction apparatus are synthesised, and support the maturation of muscle fibres. For most species, the number of skeletal muscle fibres is determined before birth and maintained largely until approaching senescence.
5.3.2 Myostatin as a Regulator of Myogenesis
During embryonic myogenesis, myostatin signalling initiates the Smad2/3 complex to translocate to the nucleus and block production of the muscle transcription factor MyoD. Inhibition of MyoD prevents myoblast proliferation, differentiation and fusion through cell cycle arrest (i.e., myoblasts accumulate in the G1 and G2 phase of the cell cycle) (Allen and Unterman 2007; Langley et al. 2002; Rios et al. 2002; Thomas et al. 2000; Zhu et al. 2004). This coincides with up-regulation of p21, a potent inhibitor of the cyclin-Cdk complexes (Thomas et al. 2000) that support cell cycle progression. Cyclin-Cdk complex inhibition results in reduced phosphorylation of Rb, a tumour suppressor protein that inhibits cell growth. Hypo-phosphorylated Rb inhibits cell cycle progression by binding and repressing the activity of the E2F family of transcription factors (Lam and La Thangue 1994), thereby preventing their transcription of S-phase-specific genes (La Thangue 1996).
5.3.3 Myostatin as a Regulator of Muscle Fibre Size
In mature muscle fibres, the maintenance of size is dependent on the balance between protein synthesis and protein degradation. While these processes are paramount to maintaining muscle health and homeostasis, the interaction of signalling pathways that influence protein turnover in muscle continues to be expanded upon. As with the process of muscle fibre formation, myostatin appears to play a prominent role in governing the processes controlling muscle fibre size.
Stimulation of the canonical Smad2/3 pathway by myostatin suppresses the transcription and activity of MyoD and the myogenic regulatory factor myogenin to limit protein synthesis. While these effects serve to maintain satellite cells in a quiescent state (Langley et al. 2002; Liu et al. 2001), the same process limits the growth of muscle fibres by restricting expression of muscle proteins. Although myostatin has been shown to activate the canonical Smad pathway in muscle, crosstalk with other non-Smad pathways also contributes to the regulation of protein turnover in muscle fibres (Kollias and Mcdermott 2008; Mcpherron et al. 1997) (Fig. 5.5).

Increased activation of the Smad2/3 signalling pathway results in muscle wasting. Heightened activation of the Smad2/3 signalling pathway, as when myostatin or activin A are overexpressed, leads to inactivation of Akt and additional dephosphorylation of the transcription factor FoxO3. Dephosphorylated FoxO3 can translocate to the nucleus and induce the expression of the muscle-specific ubiquitin ligases, atrogin-1 and MuRF-1. These ligases ubiquitinate myofibrillar proteins, such as myosin, targeting them for degradation via the ubiquitin-proteasome system, resulting in muscle wasting. Akt is also a central mediator of protein synthesis, where its phosphorylation by PI3K activates mTOR-mediated phosphorylation of S6 ribosomal protein to increase protein translation
The IGF-1/PI3K/Akt signalling cascade is a regulator of processes associated with cell cycle proliferation and cell survival, and in muscle, is also a key governor of protein synthesis and glucose metabolism (Chen et al. 2014; Morissette et al. 2009; Trendelenburg et al. 2009; Zdychova and Komers 2005). Phosphorylation of Akt results in increased protein synthesis through activation of mTOR, and subsequently 4E-BP1 and S6K (Bodine et al. 2001; Laplante and Sabatini 2012). mTOR is a key contributor to skeletal muscle growth and as such, can be regulated through a variety of signalling targets. In vitro and in vivo studies have demonstrated that the absence of myostatin results in increased Akt activity and therefore elevated mTOR signalling (Lipina et al. 2010). Prevention of muscle atrophy also occurs downstream of Akt through inhibition of apoptotic factors such as Bax, and the Forkhead box O (FoxO) transcription factor family (Fig. 5.5) (Hribal et al. 2003). During the differentiation of myoblasts, as myostatin expression increases, FoxOs are dephosphorylated and enter the nucleus, where they promote transcription of the muscle-specific E3-ubiquitin ligases, atrogin-1 and MuRF-1 (Zhao et al. 2007). These muscle-specific ligases mark muscle proteins for degradation via the ubiquitin-proteasome pathway. During homeostasis, the protein synthesis and degradation pathways are in balance to maintain muscle mass (Lokireddy et al. 2011; Sartori et al. 2009). In the Mstn -/- mouse, the IGF-1/PI3K/Akt pathway is de-repressed, increasing protein synthesis, while FoxO proteins remain in the cytoplasm in the phosphorylated form, unable to initiate transcription of the catabolic E3-ubiquitin ligases (Mcpherron et al. 1997). Thus, elevated expression of myostatin promotes increased atrogin-1 and MuRF-1 transcription, while myostatin inhibition has the opposite effect.
While the Smad/Akt/FoxO axis is a major regulator of muscle size, other cellular pathways intersect with this network, adding an additional layer of control to the system. Crosstalk exists between the Smad pathway and the mitogen-activated protein kinase (MAPK) pathways of the extracellular signal-related kinase 1/2 (ERK1/2), p38 and JNK (Hanafusa et al. 1999; Mulder 2000; Philip et al. 2005). TGF-β proteins can bind to MAPK receptors in a cell-specific and context-dependent manner, but MAPKs that are not activated by TGF-βs are capable of regulating Smad proteins and Smad complexes (Javelaud and Mauviel 2005; Rahimi and Leof 2007). To date, the role of ERK1/2 in skeletal muscle mass remains incompletely defined. Further research in this area could yield new insights into muscle regulation in the future.
5.3.4 Other TGF-β Proteins as Regulators of Muscle Development and Maturation
Although myostatin is well characterised as a negative regulator of muscle, other members of the TGF-β are also essential in guiding myogenesis and supporting muscle homeostasis. For instance, TGF-β1 expressed during myogenesis inhibits the expression of muscle-specific mRNA and proteins in vitro (Massague et al. 1986), and inhibits differentiation of myoblasts (Cusella-De Angelis et al. 1994; Massague et al. 1986; Olson et al. 1986). TGF-β1 has also been shown to inhibit skeletal muscle satellite cell differentiation, proliferation and fusion in vitro, suggesting a role in regulating muscle regeneration (Allen and Boxhorn 1987; Li et al. 2004).
Other members of the TGF-β family are also implicated in muscle homeostasis. The broad spectrum TGF-β antagonist, follistatin, binds several TGF-β family members in addition to myostatin, including activins, GDF-11, and BMP-2, -4, -6, and -7 (Glister et al. 2004; Schneyer et al. 2008; Sidis et al. 2006). Transgenic mice over expressing follistatin exhibit dramatically increased muscle mass as a product of increased muscle fibre number and size(Lee and Mcpherron 2001). Similarly, transgenic overexpression of a dominant negative ActRIIB in mice, which binds similar TGF-β proteins as follistatin, also yields a hypermuscular phenotype (Lee and Mcpherron 2001). Importantly, when follistatin or the dominant negative ActRIIB were overexpressed in Mstn -/- mice, which already exhibit twice the muscle mass of wild type mice, a further increase in muscle mass was still observed (Lee 2007; Winbanks et al. 2012). As this additional increase occurred in the absence of myostatin, at least one other TGF-β protein capable of binding follistatin and ActRIIB must contribute to the negative regulation of muscle mass under homeostatic conditions. Recent studies have subsequently revealed that activin A and B contribute to the negative regulation of muscle mass (Chen et al. 2015).
Historically, research into the control of muscle homeostasis has focussed on the Smad2/3 signalling arm of the TGF-β family. This has largely left the role of the Smad1/5/8 pathway in skeletal muscle unchartered. However, recent reports have begun to show that the Smad1/5/8 axis is a positive regulator of muscle mass (Sartori et al. 2013; Winbanks et al. 2013). Increases in muscle mass and Smad1/5 phosphorylation were observed when BMP-7, or a constitutively active ALK3, were overexpressed in muscle (Winbanks et al. 2013), with growth mediated by increased mTOR activity. Interestingly, while blockade of Smad2/3 signalling results in muscle hypertrophy, overexpression of Smad6, an inhibitor of Smad1/5/8 signalling, had no effect on endogenous muscle mass (Lee 2007; Lee and Mcpherron 2001; Lee et al. 2005; Winbanks et al. 2013). Collectively, the studies to date suggest that the Smad2/3 arm of the TGF-β signalling pathway is prominent in regulating muscle mass, but that the parallel opposing arm of Smad1/5/8 is required for sustaining or promoting muscle hypertrophy. This latter aspect is noted from observations that muscles up-regulate expression of BMPs and their signalling receptors to protect against muscle atrophy in models of denervation. Inhibiting BMP signalling in this context exacerbates muscle wasting (Sartori et al. 2013; Winbanks et al. 2013). Mechanistically, the BMP-Smad1/5/8 signalling axis was shown to negatively regulate the transcription of E3 ubiquitin ligases that drive proteasome-mediated protein breakdown (Sartori et al. 2013; Winbanks et al. 2013). Thus, the BMP-Smad1/5/8 signalling arm of the TGF-β network exerts positive effects on protein synthesis and degradation in muscle depending on the context. These findings suggest multiple TGF-β family members contribute simultaneously to the establishment of skeletal muscle attributes.
5.4 The Role of TGF-β Signalling in Post-natal Muscle Regeneration
The regeneration of muscle fibres after damage recapitulates many of the events involved in myogenesis during embryonic development. Myostatin tightly governs the activity of transcription factors involved with lineage progression within both satellite cells and myoblasts, and by doing so is a key regulator in muscle regeneration following injury (Cornelison et al. 2000; Langley et al. 2002; Mccroskery et al. 2003).
To better dissect the role of myostatin during regeneration, several injury models have been employed. Following injection with myotoxic agents, the muscles of 24-month-old senescent Mstn -/- mice regenerate similarly to their wild-type counterparts (Wagner et al. 2005). However, while wild-type muscles eventually established a similar histological profile, injured Mstn -/- muscles were able to attain larger myofibre diameters at an earlier phase of regeneration than was possible for wild-type muscles (Wagner et al. 2005). These findings provide evidence for myostatin playing a more complex role in muscle regeneration than solely governing lineage control within muscle itself, and highlight the possibility of non-muscle factors contributing to this regenerative phenotype.
Using a dry-ice injury-regeneration model in Mstn -/- and wild-type bovine muscle, it was demonstrated that myostatin can govern chemokine secretion following injury (Iwasaki et al. 2013). The injured muscles of Mstn -/- cows did not parallel the level of elevation of chemokines (CXCL1, CXCL2, CXCL6 and CCL2) found in wild-type counterparts, as measured by protein level and transcript abundance: a result that is mirrored in vitro. Whether these chemokines are typically all of muscle origin in an in vivo setting is yet to be established. These findings, however, highlight that a complete picture of myostatin and TGF-β signalling in muscle regeneration is still being pieced together. Similarly control of muscle size can be heavily influenced by the inflammatory response, which also plays a major role during muscle regeneration.
Inflammatory cells feature heavily in the regenerative process, and are also implicated in pro-fibrotic signalling, throughout which TGF-β is a major coordinator of fibrotic deposition (Bernasconi et al. 1999; Gosselin et al. 2004). After injury, migration of macrophages and fibroblasts increase the production of ECM components (Serrano and Munoz-Canoves 2010). TGF-β, in conjunction with other signalling molecules, including tumour necrosis factor-alpha (TNF-α), promotes the production of ECM components collagen and elastin (Gosselin and Martinez 2004; Gosselin et al. 2004). ECM components provide a support framework within the damaged muscle, which is then degraded during the regeneration process (Serrano and Munoz-Canoves 2010). While external factors can modulate TGF-β signalling during regeneration, myostatin expression is significantly down-regulated in satellite cells, enabling lineage progression and myofibre re-population (Cornelison et al. 2000). This tight regulation of TGF-β signalling is important in controlling muscle remodelling following trauma through injury or disease.
5.5 TGF-β Signalling in Skeletal Muscle Disease
The majority of the 34 TGF-β proteins play critical and overlapping roles in organogenesis and post-natal tissue homeostasis. Intriguingly, these potent signalling proteins only utilise a very small number of receptors. The canonical signalling pathway uses five type II receptors (TβRII, ActRII, ActRIIB, BMPRII and AMHRII) and seven type I receptors (ALKs 1-7) to finely regulate the inputs of the distinct TGF-β proteins. These receptors can interact with multiple TGF-β proteins. Therefore, the expression and activity of ligands is tightly regulated, and this strict control extends to all their associated components including signalling receptors, inhibitors and the downstream molecules that transduce their signals to the nucleus for gene expression.
Owing to these ligands’ secreted nature, altered expression of specific TGF-β family proteins in disease settings can exert deleterious effects in either the tissue of origin or at distal sites. Given the diverse biological roles of TGF-β family proteins, it is not surprising that disruption within the signalling network has detrimental consequences for cell function. As examples, many mutations in TGF-β proteins or their associated signalling components have been identified in human disease (Table 5.1). Though mutations within the TGF-β signalling network often result in developmental disorders, vascular diseases and cancer (Gordon and Blobe 2008), ongoing research is revealing that altered TGF-β network signalling also exerts detrimental effects on tissue function, including in skeletal muscle. Consequently, manipulation of TGF-β signalling may offer the means to target TGF-β actions in muscle to improve specific disease states.
5.5.1 Muscular Dystrophy
Collectively, muscular dystrophies are a group of disorders characterised by progressive loss of functional muscle mass and an accumulation of non-functional fibrotic tissue. A large array of genetic mutations have been identified within dystrophic muscle fibres (Cohn and Campbell 2000), predominantly within genes encoding for proteins that make up the dystrophin-associated protein complex (DAPC) (Tsuchida 2006). The DAPC is a highly specialised and regulated network of scaffolding proteins anchoring the outside of the muscle fibre to the inside, which protects muscle fibres from injury under typical mechanical loading (Campbell and Kahl 1989; Ervasti et al. 1990). Proteins of the DAPC not only provide physical links, but also act to transmit signals regarding membrane stability and force transduction (Constantin 2014).
As the most common heritable neuromuscular disorder, Duchenne muscular dystrophy (DMD) is a severe and debilitating condition that typically affects young males, confining them to a wheelchair early in their youth (Engvall and Wewer 2003). In DMD, mutations in the dystrophin gene prevent the production of functional dystrophin protein (Hoffman et al. 1987; Jacobs et al. 1981; Koenig et al. 1987). Dystrophin is a pivotal member of the DAPC acting both as a large, central tether and as a marshalling molecule for many associated proteins that assemble in its proximity (Constantin 2014). The mutations that cause the loss of dystrophin, or similarly important structural proteins from the DAPC, weaken the overall integrity of muscle fibres. These more fragile fibres are susceptible to contraction-induced injury and subject to bouts of ongoing degeneration and regeneration. Repeated cycles of breakdown and repair are hypothesised to compromise the regenerative process over time (Jiang et al. 2014). Similarly, muscles comprised of structurally impaired fibres are vulnerable to infiltration by adipose, fibrotic and inflammatory cells, which further impair the muscles’ regenerative capacity (Acharyya et al. 2007; Andreetta et al. 2006; Jacobs et al. 1981).
During muscle regeneration after injury, TGF-β1 is increased to regulate a short inflammatory response in damaged muscles, enabling the removal of cellular debris from sites of new fibre formation (Serrano and Munoz-Canoves 2010). In dystrophic muscle, the repeated cycles of regeneration leaves the muscles persistently exposed to TGF-β1, inflammatory cells and their signalling molecules (Cohn et al. 2007). This aberrant signalling promotes the production of collagen and other ECM proteins, exacerbating fibrotic accumulation (Sabatelli et al. 2012; Zanotti et al. 2010). As TGF-β signalling is often associated with facilitating ongoing fibrotic infiltration, TGF-β inhibition has been proposed as potential therapeutic strategy in the dystrophic setting (Chen et al. 2005; Zanotti et al. 2010).
Several studies have investigated the potential outcomes of inhibiting the TGF-β signalling pathway in DMD. Administration of neutralising antibodies against TGF-β significantly resolved fibrosis in treated mdx mice, a model for DMD, when compared to their untreated littermates (Andreetta et al. 2006). While no change was observed regarding the ongoing cycles of degeneration and regeneration, there was an increase in inflammatory CD4+ lymphocytes, highlighting a potential hurdle when considering immunomodulation accompanying long-term treatment (Andreetta et al. 2006). As an alternate approach, the angiotensin II type 1 receptor blocker losartan, a widely-used FDA-approved drug for hypertension, has been demonstrated to inhibit TGF-β effects when administered to mdx mice, broadly replicating the effects of TGF-β inhibition by neutralising antibodies (Cohn et al. 2007). Losartan administration to mdx mice was reported to enhance limb grip strength compared to untreated mdx mice (Cohn et al. 2007), and reduce cardiac, diaphragm and skeletal muscle fibrosis (Spurney et al. 2011). Though effects on fibrotic signature have been significant, improvements in cardiac and limb muscle function to date appear relatively modest. These findings suggest that targeting TGF-β signalling may be beneficial in ameliorating fibrosis associated with dystrophy, but offers modest potential to enhance muscle strength.
Consequently, interventions that target other TGF-β members may be required. In this regard, several alternate strategies have been pursued to inhibit the actions of myostatin and other related TGF-β family members that repress muscle mass. These include treatment with a soluble form of the ActRIIB receptor, which comprises the extracellular domain fused to an IgG-Fc domain (Morine et al. 2010; Pistilli et al. 2011), myostatin-neutralising antibodies (Bogdanovich et al. 2002), and the administration of a myostatin prodomain fused to an IgG-Fc domain (Bogdanovich et al. 2005). Increased expression of soluble follistatin (as an inhibitor of myostatin and activins) via gene delivery using adeno-associated virus-based vectors (AAV) has also been pursued (Rodino-Klapac et al. 2009). Promising early studies in mice have supported clinical evaluation of viral vector-mediated administration of follistatin to treat Becker muscular dystrophy and sporadic inclusion body myositis (Mendell et al. 2015). Each of these intervention approaches has been demonstrated to increase muscle mass and functional capacity to an extent, thus supporting the suggestion that strategic manipulation of TGF-β network activity in dystrophic muscles may hold promise for maintaining or restoring the functionality of musculature in this setting.
5.5.2 Marfan Syndrome
Marfan syndrome (MFS) is an autosomal dominant disease resulting from mutations in the fibrillin-1 gene, causing loss of this key protein in the ECM and a crucial regulator of TGF-β signalling (Dietz et al. 1991; Hollister et al. 1990; Kaartinen and Warburton 2003; Neptune et al. 2003). Patients with MFS exhibit a range of clinical symptoms, including defects in the musculoskeletal, cardiovascular and ocular systems. Altered ECM integrity contributes to reduced ability of the aortic wall to withstand intraluminal pressure, which can result in the formation of an aortic aneurysm, a leading cause of morbidity in patients with MFS (Judge and Dietz 2005). Although the most visible manifestation of the disease is the increase in bone growth that results in hyper-flexible joints and malformations of digits, limbs and the chest wall, respiratory muscle dysfunction, poor skeletal muscle development and clinical weakness are also observed (Behan et al. 2003).
The mutations in fibrillin-1 associated with MFS render the fibrillin-1 protein sensitive to proteolysis and degradation (Ramirez and Dietz 2007; Ramirez and Rifkin 2009; Reinhardt et al. 1997). As fibrillin-1 is required for TGF-β localisation via LTBPs (Kaartinen and Warburton 2003; Neptune et al. 2003), the lack of ECM structural integrity in patients with MFS enhances the level of active TGF-β that is released from the matrix (Kaartinen and Warburton 2003; Neptune et al. 2003). TGF-β protein levels that are significantly elevated in the sera of patients with MFS are hypothesised to contribute to MFS aetiology by disrupting signalling during tissue development and adaptation (Hillebrand et al. 2014; Neptune et al. 2003). Mouse studies have also identified TGF-β signalling to be a major contributor to the prolapsing of the mitral valve, a pathology common in MFS (Ng et al. 2004). The dysregulation of TGF-β that can manifest pathologies, such as those found in MFS, highlights the importance of regulating TGF-β signalling through ligand release and activation within the ECM production, as well as ligand production (Ramirez and Dietz 2007).
Administration of a TGF-β neutralising antibody or losartan to fibrillin-1-deficient mice modelling MFS can restore muscle fibre diameter (reduced in the diseased state) to wild-type dimensions, enhance the regenerative capacity of muscles, and slow the progression of aortic pathology (Cohn et al. 2007; Habashi et al. 2006). However, while murine studies show promise for the drug, the use of losartan in individuals with MFS has yielded mixed results. Depending on the study, clinical use of losartan has had modest effects on normalising circulating TGF-β levels (Franken et al. 2014; Groenink et al. 2013; Lacro et al. 2013, 2014; Pees et al. 2013). Moreover, there has been some discrepancy in the apparent effects of losartan treatment on measures of aorta morphology (Franken et al. 2014; Groenink et al. 2013; Lacro et al. 2013, 2014; Pees et al. 2013). Collectively, these findings serve to provide a unique insight into treating MFS via TGF-β inhibition, highlighting that this disease may be more nuanced than originally anticipated and that researchers will require an even greater understanding of the disease aetiology. Whether TGF-β inhibition through losartan therapy will be clinically effective for all patients with MFS remains to be conclusively determined. While the mismatch between preclinical and clinical trial results could emphasise the innate differences that exist between the physiology of mouse and man, the findings are indicative of a field moving forward in their understanding of the complex relationship between TGF-β regulation and disease pathology.
5.5.3 Muscle Wasting Associated with Disuse
Muscle atrophy is a major problem in conditions of disuse, such as limb casting, immobilisation, or patients undergoing intensive care unit hospitalisation. In patients confined to extended bed rest, significant muscle atrophy leads to a poorer prognosis for recovery, increasing their hospital stay duration, and can also severely impair the successfulness of long-term rehabilitation post discharge.
Animal studies of limb immobilisation have demonstrated that ensuing disuse atrophy of muscles is a product of simultaneous reductions in protein synthesis and increases in protein degradation (Booth and Seider 1979). This contrasts with denervation-induced muscle atrophy, where both protein synthesis and degradation increase, though protein breakdown exceeds protein synthesis (Argadine et al. 2009).
Recently, Zhang et al. provided an important link between mechanical load-sensing via the dystrophin-associated protein complex and the protein synthesis/degradation pathways in unloaded muscles, which implicated the TGF-β network (Zhang et al. 2014). In wild-type mice, plantaris muscles comprising fast-twitch muscle fibres and soleus muscles comprising slow twitch muscle fibres lost differing amounts of mass and exhibited differing expression of dystrophin mRNA over the course of unloading (Zhang et al. 2014). Comparisons of unloading atrophy in wild-type and dystrophin-deficient mice revealed relative sparing of some dystrophin-deficient muscles, accompanied with reduced expression of the E3 ubiquitin ligases MuRF-1 and atrogin-1, responsible for controlling ubiquitin/proteasome degradation. These observations point to a role of the dystrophin-associated protein complex as a regulator of protein degradation pathways in unloaded muscles. Additionally, wild-type mice exhibited up-regulation of TGF-β1 and increased phosphorylation of Smad3 subsequent to unloading, while these proteins, which were expressed at a greater basal rate in mdx mice, were reduced after the unloading period. As the TGF-β1/Smad3 pathway influences protein synthesis and degradation processes, these findings suggest that regulation of TGF-β network signalling downstream of the dystrophin-associated protein complex contributes to changes in muscle mass with unloading. As a comparison, examination of denervated muscles has determined that myostatin transcription is increased in neurogenic atrophy (Baumann et al. 2003). Subsequent studies have identified a peak in myostatin protein level anywhere between 3-28 days post-denervation, where Smad2 phosphorylation is at its highest level around 3 days (Shao et al. 2007; Zhang et al. 2006).
Collectively, these studies implicate increased myostatin and Smad2/3 signalling as contributing to muscle atrophy in mouse models of disuse or denervation. The involvement of myostatin in disuse atrophy in humans remains unclear. While some studies have reported unchanged myostatin mRNA levels following unloading (De Boer et al. 2007; Jones et al. 2004), increased myostatin transcripts have been observed in reduced limb use associated with chronic osteoarthritis (Reardon et al. 2001). Whether myostatin is elevated temporally or only in discrete models of disease is yet to be identified. As myostatin levels can be controlled independently of transcription, further investigation may need to pursue protein detection techniques to analyse myostatin activity in sera or in muscle itself.
5.5.4 Sarcopenia
Sarcopenia is defined as the loss of muscle mass and functional capacity specifically associated with advancing age (Roubenoff 2000). Hallmarks of sarcopenia include muscle fibre atrophy, reduced muscle fibre number, diminished satellite cell activity, and progressive deterioration of pre-synaptic structures of the neuromuscular junction. Rodent studies of sarcopenia have identified gene signatures associated with oxidative stress and motoneuron degeneration as early indicators of sarcopenia, but at the signalling level, there is also evidence that the TGF-β network plays a role in ageing-associated muscle wasting. Muscle biopsies from human septuagenarians have been noted to exhibit increased myostatin expression relative to the muscles of young adults (Leger et al. 2008). Aged mice and humans also demonstrate elevated levels of TGF-β1 protein with an accompanying elevation in Smad3 phosphorylation (Carlson et al. 2009), the sera of which can inhibit the myogenic potential of satellite cells in vitro (Carlson et al. 2009). These findings point to mechanisms by which elevated TGF-β1 and myostatin may contribute to the suppressed regenerative capacity of aged muscle.
Seeking to prevent or overcome sarcopenic muscle atrophy, other studies have identified the inhibition of myostatin-Smad2/3 signalling as a potential therapeutic target. Mice lacking myostatin have been reported to exhibit reduced susceptibility to ageing-associated atrophy of fast twitch muscle fibres compared with wild-type mice. Treatment of aged mice with neutralising antibodies that target myostatin has also been shown to preserve muscle mass and force-producing capacity, and reduce markers of apoptosis (Murphy et al. 2010). Similar effects have been achieved using recombinant viral vectors to deliver myostatin prodomain constructs to old animals (Collins-Hooper et al. 2014) or via the administration of soluble ActRIIB receptors as ligand traps (Chiu et al. 2013). In aged mice, attenuation of TGF-β signalling using a receptor kinase inhibitor has also proven beneficial for enhancing muscle regeneration after experimental injury (Carlson et al. 2009). Paralleling the benefits of losartan treatment in mouse models of muscular dystrophy, administering losartan to aged mice confers improvements in muscle mass and function and reduced fibrosis (Burks et al. 2011). Combined, these studies support further investigation into blocking TGF-β signalling as a preventative or restorative intervention for sarcopenia.
5.5.5 Cachexia
As many as 80 % of all patients contending with the progression of advanced cancer exhibit symptoms of cachexia, a debilitating syndrome characterised by the loss of body mass and fatigue (Tisdale 2009). The loss of muscle mass is a leading contributing factor for increased morbidity and mortality among cancer patients, accounting for nearly 30 % of cancer-related deaths (Anker et al. 2003; Tisdale 2002, 2009). Perturbation of the autoimmune, neuromuscular and inflammatory systems has also been implicated in the manifestation of cachexia (Jespersen et al. 2006; Tan and Fearon 2008). The degree of weight loss experienced by patients varies by cancer type, with cachexia occurring especially frequently and being particularly severe in association with pancreatic and gastric cancers (Dewys 1986). Although a common side effect of cancer and chemotherapy is decreased appetite (Theologides 1979), cachexia is a hypercatabolic state that cannot be reversed by hyper-caloric food intake (Bosaeus et al. 2001). Importantly, cachexia is not unique to patients with cancer, as patients with other forms of chronic illness can present with similar degrees of wasting. A particularly striking example is that of cardiac cachexia diagnosed in patients with chronic heart failure. Severe loss of muscle mass in these individuals has similar implications as in patients with cancer, with significant weight loss representing a contributing factor to death in a significant percentage of patients with heart disease (Glass and Roubenoff 2010; Wigmore et al. 1997).
Our understanding of the aetiology of cancer cachexia has greatly improved in recent years, with the current model recognising the condition as a product of the combined insult of tumour- and host-derived factors (Fearon et al. 2013; Morley et al. 2006). Increased expression and excess signalling of the pro-inflammatory cytokines TNF-α, IL-6 and IL-1β are heavily implicated in the manifestation of cachexia (Iwase et al. 2004; Kuroda et al. 2007; Moses et al. 2009; Oliff et al. 1987; Scott et al. 1996), but in tandem with these pro-cachectic factors, a growing body of work has identified specific TGF-β family proteins as dominant players in pathogenesis of cachexia (Zhou et al. 2010).
TGF-β family members that have been associated with cancer cachexia include TGF-β1 (Ikushima and Miyazono 2010), activin A (Pirisi et al. 2000; Wildi et al. 2001), BMP-4 (Thawani et al. 2010), GDF-15 (Bauskin et al. 2006) and myostatin (Lokireddy et al. 2012). Several studies have also established direct effects of specific TGF-β proteins as cachectic factors. Administration of TGF-β1 into mice xenografted with human breast cancers induced severe weight loss and fibrosis (Zugmaier et al. 1991). Elevated levels of activin A and activin B induced in mice as a consequence of inhibin knock-out promotes rapid loss of muscle and fat mass that is ultimately fatal (Matzuk et al. 1992, 1994). Systemic overexpression of myostatin or activin A in mice by implantation of genetically engineered Chinese hamster ovary cells also promotes extensive wasting of lean and fat mass (Zhou et al. 2010; Zimmers et al. 2002). Importantly, both activin and myostatin have also been shown to induce significant muscle wasting if over expressed in healthy mice in the absence of other tumour-derived factors (Chen et al. 2014). These findings illustrate that excessive levels of specific TGF-β family ligands can be considered as direct contributors to the development of cachexia, and thus may also serve as sensitive biomarkers of patient prognosis.
The growing interest in the TGF-β network as a factor in the etiology of cachexia has spurred considerable effort to evaluate interventions that might modulate the actions of excessive TGF-β ligand levels. SB431542, a small molecule inhibitor of TGF-β signalling via selective antagonism of ALK4/5/7 (Laping et al. 2002), has proven capable of reducing the tumourigenic effects of TGF-β (Halder et al. 2005; Watt et al. 2010). Treatment mice bearing activin-expressing tumours with antibodies against activin A was effective at reversing cachexia and prolonging survival (Zhou et al. 2010). Antibodies designed to neutralise myostatin have also demonstrated efficacy in protecting mice bearing cachexia-inducing carcinomas (Murphy et al. 2011). Arguably, the most potent effects have been observed in studies evaluating the administration of soluble ActRIIB receptors as ‘ligand traps’ for circulating TGF-β family members (Zhou et al. 2010). Promising results in mouse models have motivated the clinical evaluation of soluble ActRIIA and ActRIIB designs (Attie et al. 2013).
The use of modified receptors, however, has raised concerns about the potential off-target effects as they still retain their capacity to bind multiple ligands (Massague 1998), which could disrupt normal endogenous signalling. One such concern is the potential disruption of BMP-9 and BMP-10, which operate as regulators of angiogenesis (Allen and Unterman 2007; Vallese et al. 2013; Zhang and Bradley 1996). Concerns over adverse events have recently tempered enthusiasm for continued development of soluble ActRIIB receptors for long-term. However, the exploration of TGF-β network antagonism as a treatment for diseases associated with muscle wasting is still a field of in its infancy. Continued study will hopefully yield useful interventions for long-term patient benefits (Attie et al. 2013).
One alternative to soluble ActRII-based interventions currently showing promise is the use of antibodies directed against the endogenous ActRIIB receptor (Lach-Trifilieff et al. 2014). This intervention has been demonstrated to increase muscle mass in both animals and human subjects with conditions of muscle wasting. However, given the previously reported side-effect risks of using soluble ActRIIB receptors (Attie et al. 2013), it may also eventuate that targeting the endogenous ActRIIB receptor at the systemic level may present similar challenges. Consequently, ligand-specific inhibitors may prove the most effective strategy to mitigate muscle wasting with minimal risk of off-target effects. These may take the form of antibodies (Yaden et al. 2014) or proteins that mimic the prodomain of specific TGF-β ligands (Chen et al. 2015). The latter approach was recently shown to effectively prevent activin-induced muscle wasting in mouse models (Chen et al. 2015). Ongoing research should help to identify the most promising approaches for modulating the TGF-β network to combat muscle wasting.
5.6 Concluding Remarks
The complexity of the TGF-β network provides the means for this system to finely regulate a variety of different cellular processes in many different cell types and organ systems, including skeletal muscle. Considerable effort has provided us with an appreciation for the role of the TGF-β network as a regulator of skeletal muscle development and adaptation, and when perturbed, as a contributor to muscle disease. However, it is clear that our understanding of the significance of the TGF-β network in muscle will be enhanced with continued examination of TGF-β family members and their signalling mechanisms. Novel approaches to modulating the TGF-β network are increasingly showing promise as prospective interventions for muscle disease. Though hurdles remain to be faced in realising the widespread clinical use of these approaches, continued dedication to better understanding the TGF-β network in muscle will hopefully aid the development of invaluable new therapeutics, and help us to more completely define the mechanisms that govern the attributes of skeletal muscle in health and disease.
References
Acharyya S, Ladner KJ, Nelsen LL et al (2004) Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest 114:370–378
Acharyya S, Villalta SA, Bakkar N et al (2007) Interplay of IKK/NF-kappa B signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest 117:889–901
Akhurst RJ, Hata A (2012) Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov 11:790–811
Allen RE, Boxhorn LK (1987) Inhibition of skeletal muscle satellite cell differentiation by transforming growth factor-beta. J Cell Physiol 133:567–572
Allen DL, Unterman TG (2007) Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol 292:C188–C199
Andreetta F, Bernasconi P, Baggi F et al (2006) Immunomodulation of TGF-beta1 in mdx mouse inhibits connective tissue proliferation in diaphragm but increases inflammatory response: Implications for antifibrotic therapy. J Neuroimmunol 175:77–86
Anker SD, Negassa A, Coats AJ et al (2003) Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting-enzyme inhibitors: an observational study. Lancet 361:1077–1083
Annes JP, Chen Y, Munger JS et al (2004) Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol 165:723–734
Argadine HM, Hellyer NJ, Mantilla CB et al (2009) The effect of denervation on protein synthesis and degradation in adult rat diaphragm muscle. J Appl Physiol 107:438–444
Assoian RK, Komoriya A, Meyers CA et al (1983) Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem 258:7155–7160
Attie KM, Borgstein NG, Yang Y et al (2013) A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerve 47:416–423
Baldwin RL, Friess H, Yokoyama M et al (1996) Attenuated ALK5 receptor expression in human pancreatic cancer: correlation with resistance to growth inhibition. Int J Cancer 67:283–288
Baran W, Szepietowski JC, Mazur G et al (2007) TGF-beta(1) gene polymorphism in psoriasis vulgaris. Cytokine 38:8–11
Baumann AP, Ibebunjo C, Grasser WA et al (2003) Myostatin expression in age and denervation-induced skeletal muscle atrophy. J Musculoskelet Neuronal Interact 3:8–16
Bauskin AR, Brown DA, Kuffner T et al (2006) Role of macrophage inhibitory cytokine-1 in tumorigenesis and diagnosis of cancer. Cancer Res 66:4983–4986
Behan WM, Longman C, Petty RK et al (2003) Muscle fibrillin deficiency in Marfan’s syndrome myopathy. J Neurol Neurosurg Psychiatry 74:633–638
Belville C, Josso N, Picard JY (1999) Persistence of Mullerian derivatives in males. Am J Med Genet 89:218–223
Berg JN, Gallione CJ, Stenzel TT et al (1997) The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 61:60–67
Bernasconi P, Di Blasi C, Mora M et al (1999) Transforming growth factor-beta 1 and fibrosis in congenital muscular dystrophies. Neuromusc Disord 9:28–33
Blobe GC, Schiemann WP, Lodish HF (2000) Role of transforming growth factor beta in human disease. N Engl J Med 342:1350–1358
Bodine SC, Stitt TN, Gonzalez M et al (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3:1014–1019
Bogdanovich S, Krag TO, Barton ER et al (2002) Functional improvement of dystrophic muscle by myostatin blockade. Nature 420:418–421
Bogdanovich S, Perkins KJ, Krag TO et al (2005) Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J 19:543–549
Booth FW, Seider MJ (1979) Early change in skeletal muscle protein synthesis after limb immobilization of rats. J Appl Physiol 47:974–977
Border WA, Noble NA (1994) Transforming growth factor beta in tissue fibrosis. N Engl J Med 331:1286–1292
Bosaeus I, Daneryd P, Svanberg E et al (2001) Dietary intake and resting energy expenditure in relation to weight loss in unselected cancer patients. Int J Cancer 93:380–383
Bottinger EP, Factor VM, Tsang ML et al (1996) The recombinant proregion of transforming growth factor beta1 (latency-associated peptide) inhibits active transforming growth factor beta1 in transgenic mice. Proc Natl Acad Sci U S A 93:5877–5882
Brown DJ, Kim TB, Petty EM et al (2002) Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the gene encoding noggin. Am J Hum Genet 71:618–624
Brunner AM, Marquardt H, Malacko AR et al (1989) Site-directed mutagenesis of cysteine residues in the pro region of the transforming growth factor beta 1 precursor. Expression and characterization of mutant proteins. J Biol Chem 264:13660–13664
Buijs JT, Henriquez NV, Van Overveld PG et al (2007) TGF-beta and BMP7 interactions in tumour progression and bone metastasis. Clin Exp Metastasis 24:609–617
Burks TN, Andres-Mateos E, Marx R et al. (2011) Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci Transl Med 3:82ra37
Campbell KP, Kahl SD (1989) Association of dystrophin and an integral membrane glycoprotein. Nature 338:259–262
Carlson ME, Conboy MJ, Hsu M et al (2009) Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell 8:676–689
Chang H, Brown CW, Matzuk MM (2002) Genetic analysis of the mammalian transforming growth factor-beta superfamily. Endocr Rev 23:787–823
Cheifetz S, Hernandez H, Laiho M et al (1990) Distinct transforming growth factor-beta (TGF-beta) receptor subsets as determinants of cellular responsiveness to three TGF-beta isoforms. J Biol Chem 265:20533–20538
Chen YW, Nagaraju K, Bakay M et al (2005) Early onset of inflammation and later involvement of TGF beta in Duchenne muscular dystrophy. Neurology 65:826–834
Chen H, Brady Ridgway J, Sai T et al (2013) Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc Natl Acad Sci U S A 110:11887–11892
Chen JL, Walton KL, Winbanks CE et al (2014) Elevated expression of activins promotes muscle wasting and cachexia. FASEB J 28:1711–1723
Chen JL, Walton KL, Al-Musawi SL et al (2015) Development of novel activin-targeted therapeutics. Mol Ther 23:434–444
Chiu CS, Peekhaus N, Weber H et al (2013) Increased muscle force production and bone mineral density in ActRIIB-Fc-treated mature rodents. J Gerontol A Biol Sci Med Sci 68:1181–1192
Ciciliot S, Rossi AC, Dyar KA et al (2013) Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol 45:2191–2199
Clement JH, Raida M, Sanger J et al (2005) Bone morphogenetic protein 2 (BMP-2) induces in vitro invasion and in vivo hormone independent growth of breast carcinoma cells. Int J Oncol 27:401–407
Cohn RD, Campbell KP (2000) Molecular basis of muscular dystrophies. Muscle Nerve 23:1456–1471
Cohn RD, Van Erp C, Habashi JP et al (2007) Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med 13:204–210
Collins-Hooper H, Sartori R, Macharia R et al. (2014) Propeptide-mediated inhibition of myostatin increases muscle mass through inhibiting proteolytic pathways in aged mice. J Gerontol A Biol Sci Med Sci 69(9):1049–1059
Constantin B (2014) Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta-Biomembr 1838:635–642
Cornelison DD, Olwin BB, Rudnicki MA et al (2000) MyoD(-/-) satellite cells in single-fiber culture are differentiation defective and MRF4 deficient. Dev Biol 224:122–137
Cusella-De Angelis MG, Molinari S, Le Donne A et al (1994) Differential response of embryonic and fetal myoblasts to TGF beta: a possible regulatory mechanism of skeletal muscle histogenesis. Development 120:925–933
D’abronzo FH, Swearingen B, Klibanski A et al (1999) Mutational analysis of activin/transforming growth factor-beta type I and type II receptor kinases in human pituitary tumors. J Clin Endocrinol Metab 84:1716–1721
Dam TT, Peters KW, Fragala M et al (2014) An evidence-based comparison of operational criteria for the presence of sarcopenia. J Gerontol A Biol Sci Med Sci 69:584–590
De Boer MD, Selby A, Atherton P et al (2007) The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J Physiol 585:241–251
De Crescenzo G, Grothe S, Zwaagstra J et al (2001) Real-time monitoring of the interactions of transforming growth factor-beta (TGF-beta) isoforms with latency-associated protein and the ectodomains of the TGF-beta type II and III receptors reveals different kinetic models and stoichiometries of binding. J Biol Chem 276:29632–29643
Dean JC (2007) Marfan syndrome: clinical diagnosis and management. Eur J Hum Genet 15:724–733
Dewys WD (1986) Weight-loss and nutritional abnormalities in cancer-patients – incidence, severity and significance. Clin Oncol 5:251–261
Dietz HC, Cutting GR, Pyeritz RE et al (1991) Marfan-syndrome caused by a recurrent denovo missense mutation in the fibrillin gene. Nature 352:337–339
Dubois CM, Laprise MH, Blanchette F et al (1995) Processing of transforming growth factor beta 1 precursor by human furin convertase. J Biol Chem 270:10618–10624
Engvall E, Wewer UM (2003) The new frontier in muscular dystrophy research: booster genes. FASEB J 17:1579–1584
Eppert K, Scherer SW, Ozcelik H et al (1996) MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell 86:543–552
Ervasti JM, Ohlendieck K, Kahl SD et al (1990) Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345:315–319
Fearon K, Arends J, Baracos V (2013) Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 10:90–99
Franken R, Radonic T, Den Hartog AW et al (2014) The revised role of TGF-beta in aortic aneurysms in Marfan syndrome. Neth Heart J
Gazzerro E, Canalis E (2006) Bone morphogenetic proteins and their antagonists. Rev Endocr Metab Disord 7:51–65
Ge GX, Hopkins DR, Greenspan DS (2006) GDF11 forms a bone morphogenetic protein 1-activated latent complex that can modulate nerve growth factor-induced differentiation of PC12 cells. FASEB J 20:A515
Gentry LE, Nash BW (1990) The pro domain of pre-pro-transforming growth factor beta 1 when independently expressed is a functional binding protein for the mature growth factor. Biochemistry 29:6851–6857
Gentry LE, Lioubin MN, Purchio AF et al (1988) Molecular events in the processing of recombinant type 1 pre-pro-transforming growth factor beta to the mature polypeptide. Mol Cell Biol 8:4162–4168
Glass D, Roubenoff R (2010) Recent advances in the biology and therapy of muscle wasting. Ann N Y Acad Sci 1211:25–36
Glister C, Kemp CF, Knight PG (2004) Bone morphogenetic protein (BMP) ligands and receptors in bovine ovarian follicle cells: actions of BMP-4, -6 and -7 on granulosa cells and differential modulation of Smad-1 phosphorylation by follistatin. Reproduction 127:239–254
Goggins M, Shekher M, Turnacioglu K et al (1998) Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res 58:5329–5332
Gong Y, Krakow D, Marcelino J et al (1999) Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet 21:302–304
Gordon KJ, Blobe GC (2008) Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta 1782:197–228
Gosselin LE, Martinez DA (2004) Impact of TNF-alpha blockade on TGF-beta 1 and type I collagen mRNA expression in dystrophic muscle. Muscle Nerve 30:244–246
Gosselin LE, Williams JE, Deering M et al (2004) Localization and early time course of TGF-beta 1 mRNA expression in dystrophic muscle. Muscle Nerve 30:645–653
Grady WM, Myeroff LL, Swinler SE et al (1999) Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res 59:320–324
Greenwald J, Groppe J, Gray P et al (2003) The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol Cell 11:605–617
Groenink M, Den Hartog AW, Franken R et al (2013) Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J 34:3491–3500
Groppe JC, Shore EM, Kaplan FS (2007) Functional modeling of the ACVR1 (R206H) mutation in FOP. Clin Orthop Relat Res 462:87–92
Habashi JP, Judge DP, Holm TM et al (2006) Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312:117–121
Hahn SA, Schutte M, Hoque AT et al (1996) DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271:350–353
Halder SK, Beauchamp RD, Datta PK (2005) A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia 7:509–521
Hanafusa H, Ninomiya-Tsuji J, Masuyama N et al (1999) Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J Biol Chem 274:27161–27167
Harrington AE, Morris-Triggs SA, Ruotolo BT et al (2006) Structural basis for the inhibition of activin signalling by follistatin. EMBO J 25:1035–1045
Harrison CA, Gray PC, Fischer WH et al (2004) An activin mutant with disrupted ALK4 binding blocks signaling via type II receptors. J Biol Chem 279:28036–28044
Harrison CA, Al-Musawi SL, Walton KL (2011) Prodomains regulate the synthesis, extracellular localisation and activity of TGF-beta superfamily ligands. Growth Factors 29:174–186
Hill JJ, Davies MV, Pearson AA et al (2002) The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem 277:40735–40741
Hillebrand M, Millot N, Sheikhzadeh S et al (2014) Total serum transforming growth factor-beta 1 is elevated in the entire spectrum of genetic aortic syndromes. Clin Cardiol 37:672–679
Hoffman EP, Brown RH, Kunkel LM (1987) Dystrophin – the protein product of the duchenne muscular-dystrophy locus. Cell 51:919–928
Hollister DW, Godfrey M, Sakai LY et al (1990) Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan-syndrome. N Engl J Med 323:152–159
Howe JR, Roth S, Ringold JC et al (1998) Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 280:1086–1088
Howe JR, Bair JL, Sayed MG et al (2001) Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 28:184–187
Hribal ML, Nakae J, Kitamura T et al (2003) Regulation of insulin-like growth factor-dependent myoblast differentiation by Foxo forkhead transcription factors. J Cell Biol 162:535–541
Husken-Hindi P, Tsuchida K, Park M et al (1994) Monomeric activin A retains high receptor binding affinity but exhibits low biological activity. J Biol Chem 269:19380–19384
Ikushima H, Miyazono K (2010) TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer 10:415–424
Iwasaki S, Miyak M, Hayashi S et al (2013) Effect of myostatin on chemokine expression in regenerating skeletal muscle cells. Cells Tissues Organs 198:66–74
Iwase S, Murakami T, Saito Y et al (2004) Steep elevation of blood interleukin-6 (IL-6) associated only with late stages of cachexia in cancer patients. Eur Cytokine Netw 15:312–316
Jacobs PA, Hunt PA, Mayer M et al (1981) Duchenne muscular-dystrophy (DMD) in a female with an X-autosome translocation – further evidence that the dmd locus is at Xp21. Am J Hum Genet 33:513–518
Javelaud D, Mauviel A (2005) Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene 24:5742–5750
Jespersen J, Kjaer M, Schjerling P (2006) The possible role of myostatin in skeletal muscle atrophy and cachexia. Scand J Med Sci Sports 16:74–82
Jiang CH, Wen YF, Kuroda K et al (2014) Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Dis Model Mech 7:997–1004
Jones SW, Hill RJ, Krasney PA et al (2004) Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J 18:1025–1027
Joulia-Ekaza D, Cabello G (2006) Myostatin regulation of muscle development: molecular basis, natural mutations, physiopathological aspects. Exp Cell Res 312:2401–2414
Judge DP, Dietz HC (2005) Marfan’s syndrome. Lancet 366:1965–1976
Jung B, Nougaret S, Conseil M et al (2014) Sepsis is associated with a preferential diaphragmatic atrophy a critically Ill patient study using tridimensional computed tomography. Anesthesiology 120:1182–1191
Kaartinen V, Warburton D (2003) Fibrillin controls TGF-beta activation. Nat Genet 33:331–332
Kinoshita A, Saito T, Tomita H et al (2000) Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet 26:19–20
Knaus PI, Lindemann D, Decoteau JF et al (1996) A dominant inhibitory mutant of the type II transforming growth factor beta receptor in the malignant progression of a cutaneous T-cell lymphoma. Mol Cell Biol 16:3480–3489
Koenig M, Hoffman EP, Bertelson CJ et al (1987) Complete cloning of the Duchenne muscular-dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50:509–517
Kollias HD, Mcdermott JC (2008) Transforming growth factor-beta and myostatin signaling in skeletal muscle. J Appl Physiol 104:579–587
Kubota T, Zhang Q, Wrana JL et al (1989) Multiple forms of SppI (secreted phosphoprotein, osteopontin) synthesized by normal and transformed rat bone cell populations: regulation by TGF-beta. Biochem Biophys Res Commun 162:1453–1459
Kuroda K, Nakashima J, Kanao K et al (2007) Interleukin 6 is associated with cachexia in patients with prostate cancer. Urology 69:113–117
La Thangue NB (1996) E2F and the molecular mechanisms of early cell-cycle control. Biochem Soc Trans 24:54–59
Lach-Trifilieff E, Minetti GC, Sheppard K et al (2014) An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol 34:606–618
Lacro RV, Guey LT, Dietz HC et al. (2013) Characteristics of children and young adults with Marfan syndrome and aortic root dilation in a randomized trial comparing atenolol and Losartan therapy. Am Heart J 165:828–835
Lacro RV, Dietz HC, Sleeper LA et al (2014) Atenolol versus Losartan in children and young adults with Marfan’s syndrome. N Engl J Med 371:2061–2071
Lam EW, La Thangue NB (1994) DP and E2F proteins: coordinating transcription with cell cycle progression. Curr Opin Cell Biol 6:859–866
Lane KB, Machado RD, Pauciulo MW et al (2000) Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 26:81–84
Langley B, Thomas M, Bishop A et al (2002) Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem 277:49831–49840
Laping NJ, Grygielko E, Mathur A et al (2002) Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol Pharmacol 62:58–64
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293
Lee SJ (2007) Quadrupling muscle mass in mice by targeting TGF-beta signaling pathways. PLoS One 2, e789
Lee SJ (2008) Genetic analysis of the role of proteolysis in the activation of latent myostatin. PLoS One 3, e1628
Lee SJ, Mcpherron AC (2001) Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A 98:9306–9311
Lee SJ, Reed LA, Davies MV et al (2005) Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci U S A 102:18117–18122
Leger B, Derave W, De Bock K et al (2008) Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of akt phosphorylation. Rejuvenation Res 11:163–175
Lehmann K, Seemann P, Stricker S et al (2003) Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc Natl Acad Sci U S A 100:12277–12282
Lehmann K, Seemann P, Silan F et al (2007) A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet 81:388–396
Lewis KA, Gray PC, Blount AL et al (2000) Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 404:411–414
Li B, Khanna A, Sharma V et al (1999) TGF-beta1 DNA polymorphisms, protein levels, and blood pressure. Hypertension 33:271–275
Li Y, Foster W, Deasy BM et al (2004) Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: a key event in muscle fibrogenesis. Am J Pathol 164:1007–1019
Li S, Shimono C, Norioka N et al (2010) Activin A binds to perlecan through its pro-region that has heparin/heparan sulfate binding activity. J Biol Chem 285:36645–36655
Lipina C, Kendall H, Mcpherron AC et al (2010) Mechanisms involved in the enhancement of mammalian target of rapamycin signalling and hypertrophy in skeletal muscle of myostatin-deficient mice. FEBS Lett 584:2403–2408
Liu D, Black BL, Derynck R (2001) TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev 15:2950–2966
Loeys BL, Chen J, Neptune ER et al (2005) A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 37:275–281
Lokireddy S, Mcfarlane C, Ge X et al (2011) Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting. Mol Endocrinol 25:1936–1949
Lokireddy S, Wijesoma IW, Bonala S et al (2012) Myostatin is a novel tumoral factor that induces cancer cachexia. Biochem J 446:23–36
Loscalzo J (2001) Genetic clues to the cause of primary pulmonary hypertension. N Engl J Med 345:367–371
Luo K (2003) Negative regulation of BMP signaling by the ski oncoprotein. J Bone Joint Surg Am 85-A(Suppl 3):39–43
Maehara Y, Kakeji Y, Kabashima A et al (1999) Role of transforming growth factor-beta 1 in invasion and metastasis in gastric carcinoma. J Clin Oncol 17:607–614
Massague J (1990) The transforming growth factor-beta family. Annu Rev Cell Biol 6:597–641
Massague J (1998) TGF-beta signal transduction. Annu Rev Biochem 67:753–791
Massague J, Gomis RR (2006) The logic of TGFbeta signaling. FEBS Lett 580:2811–2820
Massague J, Cheifetz S, Endo T et al (1986) Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc Natl Acad Sci U S A 83:8206–8210
Matyas G, Arnold E, Carrel T et al (2006) Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome-related disorders. Hum Mutat 27:760–769
Matzuk MM, Finegold MJ, Su JG et al (1992) Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature 360:313–319
Matzuk MM, Finegold MJ, Mather JP et al (1994) Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci U S A 91:8817–8821
Mauro A (1961) Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9:493–495
Mcallister KA, Grogg KM, Johnson DW et al (1994) Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 8:345–351
Mccaffrey TA, Du B, Consigli S et al (1997) Genomic instability in the type II TGF-beta1 receptor gene in atherosclerotic and restenotic vascular cells. J Clin Invest 100:2182–2188
Mccroskery S, Thomas M, Maxwell L et al (2003) Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol 162:1135–1147
Mcpherron AC, Lawler AM, Lee SJ (1997) Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387:83–90
Megeney LA, Kablar B, Garrett K et al (1996) MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev 10:1173–1183
Mendell JR, Sahenk Z, Malik V et al (2015) A phase 1/2a follistatin gene therapy trial for Becker muscular dystrophy. Mol Ther 23:192–201
Mondello P, Mian M, Aloisi C et al (2015) Cancer cachexia syndrome: pathogenesis, diagnosis, and new therapeutic options. Nutr Cancer 67:12–26
Morine KJ, Bish LT, Selsby JT et al (2010) Activin IIB receptor blockade attenuates dystrophic pathology in a mouse model of Duchenne muscular dystrophy. Muscle Nerve 42:722–730
Morissette MR, Cook SA, Buranasombati C et al (2009) Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol 297:C1124–C1132
Morley JE, Thomas DR, Wilson MM (2006) Cachexia: pathophysiology and clinical relevance. Am J Clin Nutr 83:735–743
Moses AG, Maingay J, Sangster K et al (2009) Pro-inflammatory cytokine release by peripheral blood mononuclear cells from patients with advanced pancreatic cancer: relationship to acute phase response and survival. Oncol Rep 21:1091–1095
Mulder KM (2000) Role of Ras and Mapks in TGFbeta signaling. Cytokine Growth Factor Rev 11:23–35
Murphy KT, Koopman R, Naim T et al (2010) Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J 24:4433–4442
Murphy KT, Chee A, Gleeson BG et al (2011) Antibody-directed myostatin inhibition enhances muscle mass and function in tumor-bearing mice. Am J Physiol Regul Integr Comp Physiol 301:R716–R726
Myeroff LL, Parsons R, Kim SJ et al (1995) A transforming growth factor beta receptor type II gene mutation common in colon and gastric but rare in endometrial cancers with microsatellite instability. Cancer Res 55:5545–5547
Nakamura T, Takio K, Eto Y et al (1990) Activin-binding protein from rat ovary is follistatin. Science 247:836–838
Neptune ER, Frischmeyer PA, Arking DE et al (2003) Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 33:407–411
Ng CM, Cheng A, Myers LA et al (2004) TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest 114:1586–1592
Oliff A, Defeo-Jones D, Boyer M et al (1987) Tumors secreting human TNF/cachectin induce cachexia in mice. Cell 50:555–563
Olson EN, Sternberg E, Hu JS et al (1986) Regulation of myogenic differentiation by type beta transforming growth factor. J Cell Biol 103:1799–1805
Pannu H, Avidan N, Tran-Fadulu V et al (2006) Genetic basis of thoracic aortic aneurysms and dissections: potential relevance to abdominal aortic aneurysms. Ann N Y Acad Sci 1085:242–255
Pees C, Laccone F, Hagl M et al (2013) Usefulness of Losartan on the size of the ascending aorta in an unselected cohort of children, adolescents, and young adults with Marfan syndrome. Am J Cardiol 112:1477–1483
Philip B, Lu Z, Gao Y (2005) Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal 17:365–375
Pirisi M, Fabris C, Luisi S et al (2000) Evaluation of circulating activin-A as a serum marker of hepatocellular carcinoma. Cancer Detect Prev 24:150–155
Pistilli EE, Bogdanovich S, Goncalves MD et al (2011) Targeting the activin type IIB receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am J Pathol 178:1287–1297
Puthucheary ZA, Rawal J, Mcphail M et al (2013) Acute skeletal muscle wasting in critical illness. JAMA 310:1591–1600
Rahimi RA, Leof EB (2007) TGF-beta signaling: a tale of two responses. J Cell Biochem 102:593–608
Ramirez F, Dietz HC (2007) Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev 17:252–258
Ramirez F, Rifkin DB (2009) Extracellular microfibrils: contextual platforms for TGFbeta and BMP signaling. Curr Opin Cell Biol 21:616–622
Ramirez F, Carta L, Lee-Arteaga S et al (2008) Fibrillin-rich microfibrils – structural and instructive determinants of mammalian development and physiology. Connect Tissue Res 49:1–6
Reardon KA, Davis J, Kapsa RM et al (2001) Myostatin, insulin-like growth factor-1, and leukemia inhibitory factor mRNAs are upregulated in chronic human disuse muscle atrophy. Muscle Nerve 24:893–899
Reinhardt DP, Ono RN, Sakai LY (1997) Calcium stabilizes fibrillin-1 against proteolytic degradation. J Biol Chem 272:1231–1236
Ribeiro SM, Poczatek M, Schultz-Cherry S et al (1999) The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-beta. J Biol Chem 274:13586–13593
Rifkin DB (2005) Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem 280:7409–7412
Riggins GJ, Kinzler KW, Vogelstein B et al (1997) Frequency of Smad gene mutations in human cancers. Cancer Res 57:2578–2580
Rios R, Carneiro I, Arce VM et al (2002) Myostatin is an inhibitor of myogenic differentiation. Am J Physiol Cell Physiol 282:C993–C999
Rodino-Klapac LR, Haidet AM, Kota J et al (2009) Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve 39:283–296
Roubenoff R (2000) Sarcopenia and its implications for the elderly. Eur J Clin Nutr 54(Suppl 3):S40–S47
Sabatelli P, Gualandi F, Gara SK et al (2012) Expression of collagen VI alpha 5 and alpha 6 chains in human muscle and in Duchenne muscular dystrophy-related muscle fibrosis. Matrix Biol 31:187–196
Salam MT, Gauderman WJ, Mcconnell R et al (2007) Transforming growth factor- 1 C-509 T polymorphism, oxidant stress, and early-onset childhood asthma. Am J Respir Crit Care Med 176:1192–1199
Sartori R, Milan G, Patron M et al (2009) Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 296:C1248–C1257
Sartori R, Schirwis E, Blaauw B et al (2013) BMP signaling controls muscle mass. Nat Genet 45:1309–1318
Schneyer AL, Sidis Y, Gulati A et al (2008) Differential antagonism of activin, myostatin and growth and differentiation factor 11 by wild-type and mutant follistatin. Endocrinology 149:4589–4595
Scott HR, Mcmillan DC, Crilly A et al (1996) The relationship between weight loss and interleukin 6 in non-small-cell lung cancer. Br J Cancer 73:1560–1562
Sengle G, Charbonneau NL, Ono RN et al (2008a) Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J Biol Chem 283:13874–13888
Sengle G, Ono RN, Lyons KM et al (2008b) A new model for growth factor activation: type II receptors compete with the prodomain for BMP-7. J Mol Biol 381:1025–1039
Serrano AL, Munoz-Canoves P (2010) Regulation and dysregulation of fibrosis in skeletal muscle. Exp Cell Res 316:3050–3058
Shao CX, Liu M, Wu X et al (2007) Time-dependent expression of myostatin RNA transcript and protein in gastrocnemius muscle of mice after sciatic nerve resection. Microsurgery 27:487–493
Shi Y, Massague J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113:685–700
Shi YQ, Katsev S, Cai C et al (2000) BMP signaling is required for heart formation in vertebrates. Dev Biol 224:226–237
Shimasaki S, Moore RK, Otsuka F et al (2004) The bone morphogenetic protein system in mammalian reproduction. Endocr Rev 25:72–101
Shore EM, Xu M, Feldman GJ et al (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38:525–527
Shovlin CL, Hughes JM, Scott J et al (1997) Characterization of endoglin and identification of novel mutations in hereditary hemorrhagic telangiectasia. Am J Hum Genet 61:68–79
Sidis Y, Mukherjee A, Keutmann H et al (2006) Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 147:3586–3597
Simpson CM, Stanton PG, Walton KL et al (2012) Activation of latent human GDF9 by a single residue change (Gly 391 Arg) in the mature domain. Endocrinology 153:1301–1310
Slayton RL, Williams L, Murray JC et al (2003) Genetic association studies of cleft lip and/or palate with hypodontia outside the cleft region. Cleft Palate Craniofac J 40:274–279
Spurney CF, Sali A, Guerron AD et al (2011) Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient Mdx mice. J Cardiovasc Pharmacol Ther 16:87–95
Tan BH, Fearon KC (2008) Cachexia: prevalence and impact in medicine. Curr Opin Clin Nutr Metab Care 11:400–407
Tanabe A, Taketani S, Endo-Ichikawa Y et al (2000) Analysis of the candidate genes responsible for non-syndromic cleft lip and palate in Japanese people. Clin Sci 99:105–111
Tang AM, Forrester J, Spiegelman D et al (2002) Weight loss and survival in HIV-positive patients in the era of highly active antiretroviral therapy. J Acquir Immune Defic Syndr 31:230–236
Ten Dijke P, Arthur HM (2007) Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol 8:857–869
Thawani JP, Wang AC, Than KD et al (2010) Bone morphogenetic proteins and cancer: review of the literature. Neurosurgery 66:233–246, discussion 246
Theologides A (1979) Cancer cachexia. Cancer 43:2004–2012
Thiagalingam S, Lengauer C, Leach FS et al (1996) Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet 13:343–346
Thies RS, Chen T, Davies MV et al (2001) GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors 18:251–259
Thomas JT, Lin K, Nandedkar M et al (1996) A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat Genet 12:315–317
Thomas JT, Kilpatrick MW, Lin K et al (1997) Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat Genet 17:58–64
Thomas M, Langley B, Berry C et al (2000) Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem 275:40235–40243
Thys M, Schrauwen I, Vanderstraeten K et al (2007) The coding polymorphism T263I in TGF-beta1 is associated with otosclerosis in two independent populations. Hum Mol Genet 16:2021–2030
Tisdale MJ (2002) Cachexia in cancer patients. Nat Rev Cancer 2:862–871
Tisdale MJ (2009) Mechanisms of cancer cachexia. Physiol Rev 89:381–410
Trendelenburg AU, Meyer A, Rohner D et al (2009) Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol 296:C1258–C1270
Tsuchida K (2006) The role of myostatin and bone morphogenetic proteins in muscular disorders. Expert Opin Biol Ther 6:147–154
Tsuchida K, Arai KY, Kuramoto Y et al (2000) Identification and characterization of a novel follistatin-like protein as a binding protein for the TGF-beta family. J Biol Chem 275:40788–40796
Tsuchida K, Nakatani M, Matsuzaki T et al (2004) Novel factors in regulation of activin signaling. Mol Cell Endocrinol 225:1–8
Vakonakis I, Campbell ID (2007) Extracellular matrix: from atomic resolution to ultrastructure. Curr Opin Cell Biol 19:578–583
Vallese D, Negroni E, Duguez S et al (2013) The Rag2(-)Il2rb(-)Dmd(-) mouse: a novel dystrophic and immunodeficient model to assess innovating therapeutic strategies for muscular dystrophies. Mol Ther 21:1950–1957
Villanueva A, Garcia C, Paules AB et al (1998) Disruption of the antiproliferative TGF-beta signaling pathways in human pancreatic cancer cells. Oncogene 17:1969–1978
Visvanathan R, Chapman IM (2009) Undernutrition and anorexia in the older person. Gastroenterol Clin North Am 38:39–409
Wagner KR, Liu X, Chang X et al (2005) Muscle regeneration in the prolonged absence of myostatin. Proc Natl Acad Sci U S A 102:2519–2524
Wall BT, Dirks ML, Snijders T et al (2014) Substantial skeletal muscle loss occurs during only 5 days of disuse. Acta Physiol (Oxf) 210:600–611
Walton KL, Makanji Y, Wilce MC et al (2009) A common biosynthetic pathway governs the dimerization and secretion of inhibin and related transforming growth factor beta (TGFbeta) ligands. J Biol Chem 284:9311–9320
Walton KL, Makanji Y, Chen J et al. (2010) Two distinct regions of latency associated peptide coordinate stability of the latent TGF-{beta}1 complex. J Biol Chem 285(22):17029–17037
Watt KI, Jaspers RT, Atherton P et al (2010) SB431542 treatment promotes the hypertrophy of skeletal muscle fibers but decreases specific force. Muscle Nerve 41:624–629
Wheeler MT, Snyder EC, Patterson MN et al (1999) An E-box within the MHC IIB gene is bound by MyoD and is required for gene expression in fast muscle. Am J Physiol 276:C1069–C1078
Wigmore SJ, Plester CE, Richardson RA et al (1997) Changes in nutritional status associated with unresectable pancreatic cancer. Br J Cancer 75:106–109
Wildi S, Kleeff J, Maruyama H et al (2001) Overexpression of activin A in stage IV colorectal cancer. Gut 49:409–417
Winbanks CE, Weeks KL, Thomson RE et al (2012) Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol 197:997–1008
Winbanks CE, Chen JL, Qian H et al (2013) The bone morphogenetic protein axis is a positive regulator of skeletal muscle mass. J Cell Biol 203:345–357
Wolfman NM, Mcpherron AC, Pappano WN et al (2003) Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci U S A 100:15842–15846
Wrighton KH, Lin X, Feng XH (2009a) Phospho-control of TGF-beta superfamily signaling. Cell Res 19:8–20
Wrighton KH, Lin X, Yu PB et al (2009b) Transforming growth factor {beta} Can stimulate Smad1 phosphorylation independently of bone morphogenic protein receptors. J Biol Chem 284:9755–9763
Wu MY, Hill CS (2009) Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell 16:329–343
Yaden BC, Wang YX, Wilson JM et al (2014) Inhibition of activin A ameliorates skeletal muscle injury and rescues contractile properties by inducing efficient remodeling in female mice. Am J Pathol 184:1152–1166
Yamada Y, Miyauchi A, Goto J et al (1998) Association of a polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J Bone Miner Res 13:1569–1576
Zanotti S, Gibertini S, Mora M (2010) Altered production of extra-cellular matrix components by muscle-derived Duchenne muscular dystrophy fibroblasts before and after TGF-beta 1 treatment. Cell Tissue Res 339:397–410
Zdychova J, Komers R (2005) Emerging role of Akt kinase/protein kinase B signaling in pathophysiology of diabetes and its complications. Physiol Res 54:1–16
Zhang H, Bradley A (1996) Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development 122:2977–2986
Zhang DL, Liu M, Ding F et al (2006) Expression of myostatin RNA transcript and protein in gastrocnemius muscle of rats after sciatic nerve resection. J Muscle Res Cell Motil 27:37–44
Zhang P, Li WJ, Liu HJ et al (2014) Dystrophin involved in the susceptibility of slow muscles to hindlimb unloading via concomitant activation of TGF-beta 1/Smad3 signaling and ubiquitin-proteasome degradation in mice. Cell Biochem Biophys 70:1057–1067
Zhao J, Brault JJ, Schild A et al (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6:472–483
Zhou XP, Woodford-Richens K, Lehtonen R et al (2001) Germline mutations in BMPR1A/ALK3 cause a subset of cases of juvenile polyposis syndrome and of Cowden and Bannayan-Riley-Ruvalcaba syndromes. Am J Hum Genet 69:704–711
Zhou X, Wang JL, Lu J et al (2010) Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142:531–543
Zhu X, Topouzis S, Liang LF et al (2004) Myostatin signaling through Smad2, Smad3 and Smad4 is regulated by the inhibitory Smad7 by a negative feedback mechanism. Cytokine 26:262–272
Zimmers TA, Davies MV, Koniaris LG et al (2002) Induction of cachexia in mice by systemically administered myostatin. Science 296:1486–1488
Zugmaier G, Paik S, Wilding G et al (1991) Transforming growth factor beta 1 induces cachexia and systemic fibrosis without an antitumor effect in nude mice. Cancer Res 51:3590–3594
Acknowledgements
This work was supported by grant funding (526648, 566820, 1006488, 1078907) from the National Health and Medical Research Council (NHMRC, Australia). JLC is supported by a postdoctoral fellowship from the Cancer Council Victoria. KLW is supported by an Early Career Seed from the Victorian Cancer Agency. PG is supported by a Career Development Fellowship (1046782) from the NHMRC. JLC and TDC were previously supported by Australian Postgraduate Awards. PG was previously supported by a Senior Research Fellowship, sponsored by Pfizer Australia. CAH was supported by a Career Development Fellowship (1013533) from the NHMRC. Hudson Institute of Medical Research and Baker IDI Heart and Diabetes Institute are supported in part by the Operational Infrastructure Support Program of the Victorian Government.
Conflict of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Chen, J.L., Colgan, T.D., Walton, K.L., Gregorevic, P., Harrison, C.A. (2016). The TGF-β Signalling Network in Muscle Development, Adaptation and Disease. In: White, J., Smythe, G. (eds) Growth Factors and Cytokines in Skeletal Muscle Development, Growth, Regeneration and Disease. Advances in Experimental Medicine and Biology, vol 900. Springer, Cham. https://doi.org/10.1007/978-3-319-27511-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-27511-6_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-27509-3
Online ISBN: 978-3-319-27511-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)