
Abstract
The two clinically validated and Food and Drug Administration approved lung cancer predictive biomarkers (epidermal growth factor receptor mutations and anaplastic lymphoma kinase (ALK) translocations) occur in only about 20 % of lung adenocarcinomas and acquired resistance develops to first generation drugs. Several other oncogenic drivers for lung adenocarcinoma have emerged as potentially druggable targets with new predictive biomarkers. Oncologists are requesting testing for ROS1 translocations which predict susceptibility to crizotinib, already approved for ALK positive lung cancers. Other potential biomarkers which are currently undergoing clinical trials are RET, MET, HER2 and BRAF. Detection of these biomarkers includes fluorescent in situ hybridization and/or reverse transcriptase polymerase chain reaction (ROS1, RET, HER2), mutation analysis (BRAF) and immunohistochemistry (MET). Screening by immunohistochemistry may be useful for some biomarkers (ROS1, BRAF). Targeted next generation sequencing techniques may be useful as well. These five biomarkers are under consideration for inclusion in revised lung cancer biomarker guidelines by the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- ROS1
- RET
- MET
- HER2
- BRAF
- Multikinase inhibitors
- Fluorescent in situ hybridization
- Crizotinib
- Reverse transcriptase polymerase chain reaction
- Immunohistochemistry
1 Introduction
Two predictive biomarkers for personalized therapy of non-small cell lung cancers (NSCLC) have been well validated in clinical trials and approved by the Federal Drug Administration (FDA) : epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) translocations [1]. These two biomarkers have been the subject of the first lung cancer biomarkers guidelines from the College of American Pathologists (CAP), International Association for the Study of Lung Cancer (IASLC) and Association for Molecular Pathology (AMP) [2] as well as the CAP Lung Cancer Biomarker Reporting Template [3].
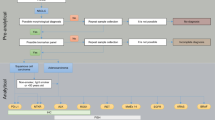
The frequency of EGFR mutations found in non-small cell lung cancers (NSCLC), more specifically in adenocarcinomas, ranges from about 15 % of whites and Hispanics to about 19 % of African Americans to about 30 % of Asian patients [4–7]. ALK translocations creating fusion genes occur in about 4–5 % of adenocarcinomas [8–12]. Lung cancers that initially respond to first generation EGFR TKIs or to crizotinib eventually develop drug resistance and relapse, typically within a year [13–19]. Since about 80 % of adenocarcinomas lack EGFR mutations or ALK translocations and since lung cancers with these abnormalities develop acquired resistance to current therapies, there has been a robust search for additional oncogenic drivers in lung cancers that might be actionable. Investigations have not yet discovered drugs that target KRAS, the most frequent oncogenic driver in lung adenocarcinomas, occurring in about 30 % of cases [1, 2]. Oncogenic drivers have not yet been identified in a substantial number of lung adenocarcinomas and, of the additional drivers that have been identified, investigations of several are sufficiently advanced that they are being considered for revisions to the CAP/IASLC/AMP lung cancer biomarker guidelines and CAP lung cancer biomarker reporting template (See Fig. 1).

Diagram showing the actionable and potentially actionable biomarkers in lung adenocarcinoma. KRAS mutation is the most common oncogenic driver, but no drugs specifically targeted to KRAS mutation are yet available. EGFR mutation and ALK translocation are clinically validated as predictive biomarkers for FDA approved TKI therapy. Emerging as biomarkers currently in clinical trials at this time are ROS1, RET, MET, Her2 and BRAF
2 ROS1
Chromosomal rearrangements of the receptor tyrosine kinase gene c-ros oncogene 1 (ROS1) are found in approximately 1–2 % of lung cancers with adenocarcinoma histology, or about 2000–4000 new cases of ROS1 positive lung cancer each year in the United States [20–24]. ROS1 has considerable amino acid homology with ALK [25]. In 2012, Bergethon et al. [20] reported sensitivity of a ROS1 positive lung cancer cell line and ROS1 transfected cell lines to the small molecule multikinase inhibitor crizotinib. They also reported a near complete response of a ROS1 positive lung cancer to crizotinib in a single patient enrolled in an expansion cohort of an early phase study [20]. In an expansion of the PROFILE 1001 study, Shaw et al. [26] reported one complete response, six partial responses and four stable disease in thirteen patients with ROS1 positive lung cancers at 8 weeks of treatment with crizotinib. These observations indicating ROS1 positive lung adenocarcinomas might respond to crizotinib, a drug that already had FDA approval for treatment of ALK positive lung cancers, produced requests for ROS1 biomarker testing by medical oncologists for lung adenocarcinomas. Typically, this has been as part of an algorithm or after adenocarcinomas were reported negative for EGFR and ALK. As a result, ROS1 has moved to the forefront of new biomarkers for lung cancer.
Similar to ALK rearrangements, ROS1 rearrangements with any of several fusion partners result in oncogenic kinase activation and the resultant oncogenic fusion kinase is susceptible to the multikinase inhibitor crizotinib [24, 27–31]. ROS1 positive adenocarcinomas share histologic and demographic features with ALK positive adenocarcinomas. ROS1 translocations tend to occur in adenocarcinomas with solid, papillary, cribriform or signet ring cell histologic patterns, tend to produce mucin and tend to arise in patients who are younger and never smokers. There are many exceptions to these general tendencies. As with other oncogenic drivers identified in lung adenocarcinomas, ROS1 translocation most often excludes the presence of other oncogenic drivers in the same tumor [20, 21, 28, 32–36].
Like ALK rearrangements, ROS1 rearrangements can be detected by a break-apart fluorescent in situ hybridization (FISH) probe that is not dependent on the specific fusion partner [20, 22, 37, 38]. Specific fusion partners are detected by reverse transcriptase polymerase chain reaction (RT-PCR) , including CD74-ROS1, SDC4-ROS1, EZR-ROS1, SLC34A22-ROS1 and FIG-ROS1 [20, 21, 29, 31, 34, 38–41]. Immunohistochemistry (IHC) can be used to screen for ROS1 positivity which can then be confirmed by FISH. IHC is performed on formalin-fixed, paraffin-embedded sections using clone D4D6 from Cell Signaling Technology. As a screening tool, IHC is reported to be highly sensitive (100 %) for ROS1 positive lung cancers confirmed by FISH and/or RT-PCR with strong diffuse staining. False positive immunostaining is reported to occur in some ROS1 negative lung cancers with considerable variability depending on the study [35, 37, 38, 42].
As with ALK positive adenocarcinomas, acquired resistance to crizotinib has been observed in ROS1 positive adenocarcinomas . Acquired resistance of a ROS1 positive lung cancer to crizotinib has been reported with a proposed mechanism of EGFR pathway activation [43] and, in another case, due to a mutation in CD74-ROS1 [44]. Therefore, similar to the situation with other oncogenic drivers of lung cancers, new drugs are under investigation for inhibiting ROS1. Davare et al. [45] reported preclinical studies which demonstrated that foretinib is a potent ROS1 inhibitor.
3 RET
The rearranged during transfection (RET) gene encodes for the RET receptor tyrosine kinase. Chromosomal rearrangements of the RET gene result in an oncogenic fusion kinase in about 1–2 % of lung cancers with adenocarcinoma histology. The majority are KIF5B-RET fusion genes with a lesser number of CCDC6-RET, NCOA4 and TRIM33 fusion genes reported [27, 34, 46–55]. Preclinical studies have reported that RET-positive lung cancer cell lines are sensitive to the multikinase inhibitors vandetanib, sunitinib, and sorafenib [56, 57]. One patient with RET positive advanced adenocarcinoma has been reported to respond to vandetanib [58]. Preliminary results from a phase II trial of the multikinase inhibitor cabozantinib were partial responses in two of three patients and stable disease in the third patient [54]. Therefore, oncologists may order RET tests for lung adenocarcinomas for possible enrollment of a patient in a clinical trial or RET may be detected in a lung cancer using next generation sequencing techniques.
Translocations of RET which result in oncogenic fusion kinases in lung adenocarcinomas have a tendency to occur in the same demographic and histologic groups as the reported tendencies for oncogenic fusion kinases from ROS1 and ALK translocations. Patients tend to be younger and never smokers and the adenocarcinomas tend to have solid, papillary and lepidic patterns and more often produce mucin. As with ALK and ROS1 positive adenocarcinomas, there are many exceptions to these general histologic and demographic tendencies for RET positive adenocarcinomas. Also, identification of a RET translocation usually excludes the presence of other oncogenic drivers such as EGFR, ALK and ROS1 in the same cancer [34, 48, 52, 54, 55].
RET translocations may be detected by FISH, by RT-PCR or by next generation sequencing [34, 48, 52, 54, 55, 59]. Immunohistochemistry for RET has had variable results and, currently, is not popular for identification of RET positive lung adenocarcinomas [52, 59].
4 MET
The MNNG-HOS transforming (MET) gene encodes a receptor tyrosine kinase and binding of its ligand hepatocyte growth factor (HGF) causes a conformational change in the MET receptor that facilitates receptor activation. MET can be activated in lung cancers by amplification and/or overexpression [60–67]. About 18 % of cases of acquired resistance to EGFR TKIs are associated with overexpression and/or amplification of MET or HGF, but prevalence of MET amplification in NSCLC patients who have not received treatment is 1–7 % [68].
Onartuzumab (MetMAb) is a recombinant, humanized, monovalent monoclonal antibody that targets MET [69]. In a phase II study patients with previously treated NSCLC were evaluated for therapy with onartuzumab plus erlotinib versus placebo plus erlotinib [70]. Patient lung cancer samples were classified as positive for MET expression or negative for MET expression by IHC using a cut-off of 50 % of malignant cells with moderate and/or strong staining intensity for classification as MET positive. The combination of onartuzumab and erlotinib resulted in improved progression free survival (PFS) and overall survival (OS) compared to placebo plus erlotinib in MET positive cases whereas the opposite was true in MET negative cases. Therefore, this IHC test provides the biomarker for MET treatment in this setting and is being considered as a companion diagnostic for onartuzumab in combination with erlotinib for treatment of lung cancer [71]. The phase II study is being followed by the MetLung phase III study [72].
ARQ 197 or tivantinib is a TKI that inhibits MET. The MARQUEE (Met Inhibitor ARQ 197 plus Erlotinib vs. Erlotinib plus placebo in NSCLC) phase III trial of tavantinib plus erlotinib in previously treated patients with locally advanced or metastatic non-squamous NSCLC was stopped not meet its primary endpoint of improved overall survival [73, 74]. Cabozantinib and ficlatuzumab, an anti-HGF monoclonal antibody, have undergone investigation in clinical trials for lung cancer combined with EGFR TKIs as well [75]. None of these drugs is currently approved for lung cancer therapy.
5 HER 2
HER2/ERBB2/NEU is a receptor tyrosine kinase of the epidermal growth factor family. Amplification or overexpression of HER2 is well known as a biomarker that predicts breast cancer response to targeted therapies. HER2 activation in lung cancer is associated with mutations, mostly insertions in exon 20, which are independent of HER2 gene amplification. These mutations are not seen in breast cancer. HER2 mutations are found in 2 % of lung adenocarcinomas. HER2 mutations are more prevalent in lung adenocarcinomas from patients who are never smokers and perhaps are more common in Asians and women. Adenocarcinomas with HER2 mutations generally lack other oncogenic drivers such as EGFR, ALK and KRAS [76–83].
Clinical trials in patients with NSCLC that have HER2 mutations have shown promising early results for therapy with afatinib [83, 84], trastuzumab [83], dacomitinib [85, 86] and neratinib plus temsirolimus [87, 88]. Therefore, detection of HER2 mutations is a potential biomarker for a small subset of lung adenocarcinomas.
HER2 expression in lung cancers by IHC has not yet proven to be a successful biomarker for selecting patients for therapy [89]. HER2 gene amplification is found in approximately 2 % of NSCLCs identified by FISH using the criteria for HER2 amplification in breast cancer [90]. Grob et al. [91] detected HER2 amplification by FISH in 3 % of NSCLC, overwhelmingly adenocarcinomas, with high-level amplification in 2 %. They also reported that HER2 amplification in lung cancer may be heterogeneous, thus impacting the outcomes of trastuzumab or other HER2 therapies based on HER2 amplification. HER2 amplification also sometimes plays a role in acquired resistance to EGFR TKIs in lung cancer patients who initially respond to these therapies [92].
6 BRAF
The BRAF gene encodes for a nonreceptor serine/threonine kinase that is activated downstream of the Ras protein. About 50 % of melanomas have BRAF mutations which activate the BRAF kinase and increase phosphorylation of downstream targets, particularly MEK, and about 80–90 % are V600E mutations. The FDA has approved vemurafenib for the treatment of BRAF V600E mutation-positive, inoperable or metastatic melanoma [93, 94] and approved the cobas 4800 BRAF V600 Mutation Test as the companion diagnostic for the biomarker [95]. IHC using the primary mouse monoclonal antibody VE1, specific for BRAF p.V600E has been studied as a screening tool for the BRAF V600E mutation [96–98]. Dabrafenib, a mutant-BRAF kinase inhibitor [99], and trametinib, a MEK inhibitor [100], have also been approved for treatment of BRAF V600E positive unresectable or metastatic melanoma.
BRAF mutations occur in about 1–5 % of lung cancers. In contrast to melanomas, V600E mutations account for 50–60 % of these mutations and non-V600E mutations account for the remainder. With few exceptions, BRAF positive lung cancers are adenocarcinomas and, in some series, patients are more likely to be current or former smokers [101–105]. Marchetti et al. [103] reported that V600E mutations occurred more frequently in women and never smokers and were associated with micropapillary pattern whereas non-V600E mutations occurred in smokers.
Cases have been reported of BRAF V600E mutated lung adenocarcinomas which responded to vemurafenib [106–108], whereas a BRAF G469L mutated lung adenocarcinoma did not [109] which anecdotally suggests that BRAF V600E mutation is a predictive biomarker for therapy of lung adenocarcinoma with vemurafenib. Two patients with BRAF V600E mutated lung NSCLC, at least one an adenocarcinoma, are reported to have had a partial responses to dabrafenib [110, 111]. In these cases, patients have developed acquired resistance similar to what is observed with targeted therapies with the other biomarkers. Clinical trials with vemurafenib [94], dabrafenib [99] and trametinib [100] will hopefully validate these therapies for BRAF V600E mutated lung NSCLC.
Testing for BRAF V600 mutations can be done by Sanger sequencing and various molecular techniques. As previously noted, the cobas 4800 BRAF V600 Mutation Test has been approved by the FDA as the companion diagnostic forBRAF V600E testing for vemurafenib therapy in melanoma [95]. BRAF V600 mutations can be detected with targeted next generation sequencing [112, 113]. IHC using the aforementioned VE1 antibody has also been reported as a successful screening tool for BRAF V600E mutation in lung adenocarcinomas [114, 115].
References
Cagle PT, Allen TC (2012) Lung cancer genotype-based therapy and predictive biomarkers: present and future. Arch Pathol Lab Med 136(12):1482–1491
Lindeman NI, Cagle PT, Beasley MB et al (2013) Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the college of american pathologists, international association for the study of lung cancer, and association for molecular pathology. Arch Pathol Lab Med 137(6):828–860
Cagle PT, Sholl LM, Lindeman NI et al (2014) Template for reporting results of biomarker testing of specimens from patients with non-small cell carcinoma of the lung. Arch Pathol Lab Med 138(2):171–174
Mitsudomi T, Yatabe Y (2007) Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci 98(12):1817–1824
Suda K, Tomizawa K, Mitsudomi T (2010) Biological and clinical significance of KRAS mutations in lung cancer: an oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev 29(1):49–60
Reinersman JM, Johnson ML, Riely GJ et al (2011) Frequency of EGFR and KRAS mutations in lung adenocarcinomas in African Americans. J Thorac Oncol 6(1):28–31
Zhang W, McQuitty EB, Olsen R et al (2014) EGFR mutations in US Hispanic versus non-Hispanic white patients with lung adenocarcinoma. Arch Pathol Lab Med 138(4):543–545
Shaw AT, Yeap BY, Mino-Kenudson M et al (2009) Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 27(26):4247–4253
Shaw AT, Yeap BY, Solomon BJ et al (2011) Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 12(11):1004–1012
Rodig SJ, Mino-Kenudson M, Dacic S et al (2009) Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res 15(16):5216–5223
Atherly AJ, Camidge DR (2012) The cost-effectiveness of screening lung cancer patients for targeted drug sensitivity markers. Br J Cancer 106(6):1100–1106
Gaughan EM, Costa DB (2011) Genotype-driven therapies for non-small cell lung cancer: focus on EGFR, KRAS and ALK gene abnormalities. Ther Adv Med Oncol 3(3):113–125
Engelman JA, Janne PA (2008) Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 14(10):2895–2899
Jackman D, Pao W, Riely GJ et al (2010) Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol 28(2):357–360
Yano S (2010) Studies for mechanism of drug resistance to EGFR-TKI. Gan To Kagaku Ryoho 37(8):1463–1466
Choi YL, Soda M, Yamashita Y et al (2010) EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 363(18):1734–1739
Doebele RC, Pilling AB, Aisner DL et al (2012) Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 18(5):1472–1482
Katayama R, Khan TM, Benes C et al (2011) Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A 108(18):7535–7540
Katayama R, Shaw AT, Khan TM et al (2012) Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med 4(120):120ra17
Bergethon K, Shaw AT, Ou SH et al (2012) ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 30(8):863–870
Janne PA, Meyerson M (2012) ROS1 rearrangements in lung cancer: a new genomic subset of lung adenocarcinoma. J Clin Oncol 30(8):878–879
Ou SH, Tan J, Yen Y, Soo RA (2012) ROS1 as a ‘druggable’ receptor tyrosine kinase: lessons learned from inhibiting the ALK pathway. Expert Rev Anticancer Ther 12(4):447–456
Rimkunas VM, Crosby K, Kelly M et al (2012) Analysis of receptor tyrosine kinase ROS1 positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res 18(16):4449–4457
Yasuda H, de Figueiredo-Pontes LL, Kobayashi S, Costa DB (2012) Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer. J Thorac Oncol 7(7):1086–1090
Chin LP, Soo RA, Soong R, Ou SH (2012) Targeting ROS1 with anaplastic lymphoma kinase inhibitors: a promising therapeutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J Thorac Oncol 7(11):1625–1630
Shaw AT, Camidge DR, Engelman JA (2012) Clinical activity of crizotinib in advanced non-small cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. J Clin Oncol 30(15 suppl):7508
Takeuchi K, Soda M, Togashi Y et al (2012) RET, ROS1 and ALK fusions in lung cancer. Nat Med 18(3):378–381
Forde PM, Rudin CM (2012) Crizotinib in the treatment of non-small-cell lung cancer. Expert Opin Pharmacother 13(8):1195–1201
Davies KD, Doebele RC (2013) Molecular pathways: ROS1 fusion proteins in cancer. Clin Cancer Res 19(15):4040–4045
Shaw AT, Hsu PP, Awad MM, Engelman JA (2013) Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer 13(11):772–787
Heigener DF, Reck M (2014) Crizotinib. Recent Results Cancer Res 201:197–205
Go H, Kim DW, Kim D et al (2013) Clinicopathologic analysis of ROS1-rearranged non-small-cell lung cancer and proposal of a diagnostic algorithm. J Thorac Oncol 8(11):1445–1450
Kim HR, Lim SM, Kim HJ et al (2013) The frequency and impact of ROS1 rearrangement on clinical outcomes in never smokers with lung adenocarcinoma. Ann Oncol 24(9):2364–2370
Pan Y, Zhang Y, Li Y et al (2014) ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: a comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung Cancer 84(2):121–126
Warth A, Muley T, Dienemann H et al (2014) ROS1 expression and translocations in non-small cell lung cancer: clinicopathological analysis of 1478 cases. Histopathology 65:187–194. doi:10.1111/his.12379
Yoshida A, Kohno T, Tsuta K et al (2013) ROS1-rearranged lung cancer: a clinicopathologic and molecular study of 15 surgical cases. Am J Surg Pathol 37(4):554–562
Sholl LM, Sun H, Butaney M et al (2013) ROS1 immunohistochemistry for detection of ROS1-rearranged lung adenocarcinomas. Am J Surg Pathol 37(9):1441–1449
Yoshida A, Tsuta K, Wakai S et al (2014) Immunohistochemical detection of ROS1 is useful for identifying ROS1 rearrangements in lung cancers. Mod Pathol 27(5):711–720
Matsuura S, Shinmura K, Kamo T et al (2013) CD74-ROS1 fusion transcripts in resected non-small cell lung carcinoma. Oncol Rep 30(4):1675–1680
Davies KD, Le AT, Theodoro MF et al (2012) Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res 18(17):4570–4579
Arai Y, Totoki Y, Takahashi H et al (2013) Mouse model for ROS1-rearranged lung cancer. PLoS One 8(2), e56010
Mescam-Mancini L, Lantuejoul S, Moro-Sibilot D et al (2014) On the relevance of a testing algorithm for the detection of ROS1-rearranged lung adenocarcinomas. Lung Cancer 83(2):168–173
Davies KD, Mahale S, Astling DP et al (2013) Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS One 8(12), e82236
Awad MM, Katayama R, McTigue M et al (2013) Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med 368(25):2395–2401
Davare MA, Saborowski A, Eide CA et al (2013) Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc Natl Acad Sci U S A 110(48):19519–19524
Gainor JF, Shaw AT (2013) Novel targets in non-small cell lung cancer: ROS1 and RET fusions. Oncologist 18(7):865–875
Ju YS, Lee WC, Shin JY et al (2012) A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res 22(3):436–445
Kohno T, Ichikawa H, Totoki Y et al (2012) KIF5B-RET fusions in lung adenocarcinoma. Nat Med 18(3):375–377
Li F, Feng Y, Fang R et al (2012) Identification of RET gene fusion by exon array analyses in “pan-negative” lung cancer from never smokers. Cell Res 22(5):928–931
Lipson D, Capelletti M, Yelensky R et al (2012) Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med 18(3):382–384
Suehara Y, Arcila M, Wang L et al (2012) Identification of KIF5B-RET and GOPC-ROS1 fusions in lung adenocarcinomas through a comprehensive mRNA-based screen for tyrosine kinase fusions. Clin Cancer Res 18(24):6599–6608
Tsuta K, Kohno T, Yoshida A et al (2014) RET-rearranged non-small-cell lung carcinoma: a clinicopathological and molecular analysis. Br J Cancer 110(6):1571–1578
Yokota K, Sasaki H, Okuda K et al (2012) KIF5B/RET fusion gene in surgically-treated adenocarcinoma of the lung. Oncol Rep 28(4):1187–1192
Drilon A, Wang L, Hasanovic A et al (2013) Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 3(6):630–635
Wang R, Hu H, Pan Y et al (2012) RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol 30(35):4352–4359
Chao BH, Briesewitz R, Villalona-Calero MA (2012) RET fusion genes in non-small-cell lung cancer. J Clin Oncol 30(35):4439–4441
Matsubara D, Kanai Y, Ishikawa S et al (2012) Identification of CCDC6-RET fusion in the human lung adenocarcinoma cell line, LC-2/ad. J Thorac Oncol 7(12):1872–1876
Gautschi O, Zander T, Keller FA et al (2013) A patient with lung adenocarcinoma and RET fusion treated with vandetanib. J Thorac Oncol 8(5):e43–e44
Sasaki H, Shimizu S, Tani Y et al (2012) RET expression and detection of KIF5B/RET gene rearrangements in Japanese lung cancer. Cancer Med 1(1):68–75
Bean J, Brennan C, Shih JY et al (2007) MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 104(52):20932–20937
Cappuzzo F, Janne PA, Skokan M et al (2009) (2009) MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol 20(2):298–304
Cappuzzo F, Marchetti A, Skokan M et al (2009) Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 27(10):1667–1674
Chen HJ, Mok TS, Chen ZH et al (2009) Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non-small cell lung cancer. Pathol Oncol Res 15(4):651–658
Engelman JA, Zejnullahu K, Mitsudomi T et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316(5827):1039–1043
Kong-Beltran M, Seshagiri S, Zha J et al (2006) Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res 66(1):283–289
Kubo T, Yamamoto H, Lockwood WW et al (2009) MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer 124(8):1778–1784
Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T (2009) Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol 4(1):5–11
Sierra JR, Tsao MS (2011) c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol 3(1 suppl):S21–S35
Surati M, Patel P, Peterson A, Salgia R (2011) Role of MetMAb (OA-5D5) in c-MET active lung malignancies. Expert Opin Biol Ther 11(12):1655–1662
Spigel DR, Ervin TJ, Ramlau RA et al (2013) Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 31(32):4105–4114
Koeppen H, Yu W, Zha J et al (2014) Biomarker analyses from a placebo-controlled phase II study evaluating erlotinib {+/-} onartuzumab in advanced non-small-cell lung cancer: MET expression levels are predictive of patient benefit. Clin Cancer Res. doi:10.1158/1078-0432.CCR-13-1836
Spigel DR, Edelman MJ, Mok T et al (2012) Treatment rationale study design for the MetLung trial: a randomized, double-blind phase III study of onartuzumab (MetMAb) in combination with erlotinib versus erlotinib alone in patients who have received standard chemotherapy for stage IIIB or IV met-positive non-small-cell lung cancer. Clin Lung Cancer 13(6):500–504
Scagliotti GV, Novello S, Schiller JH et al (2012) Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer 13(5):391–395
Sequist LV, von Pawel J, Garmey EG et al (2011) Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol 29(24):3307–3315
Robinson KW, Sandler AB (2013) The role of MET receptor tyrosine kinase in non-small cell lung cancer and clinical development of targeted anti-MET agents. Oncologist 18(2):115–122
Buttitta F, Barassi F, Fresu G et al (2006) Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int J Cancer 119(11):2586–2591
Shigematsu H, Takahashi T, Nomura M et al (2005) Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res 65(5):1642–1646
Stephens P, Hunter C, Bignell G et al (2004) Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431(7008):525–526
Arcila ME, Chaft JE, Nafa K et al (2012) Prevalence, clinicopathologic associations and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res 18(18):4910–4918
Davies H, Hunter C, Smith R et al (2005) Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res 65(17):7591–7595
Sasaki H, Shimizu S, Endo K et al (2006) EGFR and erbB2 mutation status in Japanese lung cancer patients. Int J Cancer 118(1):180–184
Sonobe M, Manabe T, Wada H, Tanaka F (2006) Lung adenocarcinoma harboring mutations in the ERBB2 kinase domain. J Mol Diagn 8(3):351–356
Mazieres J, Peters S, Lepage B et al (2013) Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol 31(16):1997–2003
De Greve J, Teugels E, Geers C et al (2012) Clinical activity of afatinib (BIBW 2992) in patients with lung adenocarcinoma with mutations in the kinase domain of HER2/neu. Lung Cancer 76(1):123–127
Kris M, Goldberg Z, Janne PA, Kim D, Martins R, Mok TSK (2012) Dacomitinib (PF-00299804), an irreversible pan-HER tyrosine kinase inhibitor (TKI), for first-line treatment of EGFR-mutant or HER2-mutant or -amplified lung cancers. Ann Oncol 23:1228
Reckamp KL, Giaccone G, Camidge DR et al (2014) A phase 2 trial of dacomitinib (PF-00299804), an oral, irreversible pan-HER (human epidermal growth factor receptor) inhibitor, in patients with advanced non-small cell lung cancer after failure of prior chemotherapy and erlotinib. Cancer 120(8):1145–1154
Gandhi L, Bahleda R, Cleary JM, Hollebecque A, Kwak EL, Pandya S (2011) Two dimensional phase I study of neratinib (NER) combined with temsirolimus (TEM) in patients (pts) with solid tumors. J Clin Oncol 29:3027
Gandhi L, Bahleda R, Tolaney SM et al (2014) Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol 32(2):68–75
Clamon G, Herndon J, Kern J et al (2005) Lack of trastuzumab activity in nonsmall cell lung carcinoma with overexpression of erb-B2: 39810: a phase II trial of cancer and leukemia group B. Cancer 103(8):1670–1675
Heinmoller P, Gross C, Beyser K et al (2003) HER2 status in non-small cell lung cancer: results from patient screening for enrollment to a phase II study of herceptin. Clin Cancer Res 9(14):5238–5243
Grob TJ, Kannengiesser I, Tsourlakis MC et al (2012) Heterogeneity of ERBB2 amplification in adenocarcinoma, squamous cell carcinoma and large cell undifferentiated carcinoma of the lung. Mod Pathol 25(12):1566–1573
Takezawa K, Pirazzoli V, Arcila ME et al (2012) HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov 2(10):922–933
Chapman PB, Hauschild A, Robert C et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26):2507–2516
Garbe C, Abusaif S, Eigentler TK (2014) Vemurafenib. Recent Results Cancer Res 201:215–225
Anderson S, Bloom KJ, Vallera DU et al (2012) Multisite analytic performance studies of a real-time polymerase chain reaction assay for the detection of BRAF V600E mutations in formalin-fixed, paraffin-embedded tissue specimens of malignant melanoma. Arch Pathol Lab Med 136(11):1385–1391
Capper D, Berghoff AS, Magerle M et al (2012) Immunohistochemical testing of BRAF V600E status in 1,120 tumor tissue samples of patients with brain metastases. Acta Neuropathol 123(2):223–233
Capper D, Preusser M, Habel A et al (2011) Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol 122(1):11–19
Marin C, Beauchet A, Capper D et al (2014) Detection of BRAF p.V600E mutations in melanoma by immunohistochemistry has a good interobserver reproducibility. Arch Pathol Lab Med 138(1):71–75
Ballantyne AD, Garnock-Jones KP (2013) Dabrafenib: first global approval. Drugs 73(12):1367–1376
Wright CJ, McCormack PL (2013) Trametinib: first global approval. Drugs 73(11):1245–1254
Brustugun OT, Khattak AM, Tromborg AK et al (2014) BRAF-mutations in non-small cell lung cancer. Lung Cancer 84(1):36–38
Kinno T, Tsuta K, Shiraishi K et al (2014) Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 25(1):138–142
Marchetti A, Felicioni L, Malatesta S et al (2011) Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol 29(26):3574–3579
Paik PK, Arcila ME, Fara M et al (2011) Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 29(15):2046–2051
Yousem SA, Nikiforova M, Nikiforov Y (2008) The histopathology of BRAF-V600E-mutated lung adenocarcinoma. Am J Surg Pathol 32(9):1317–1321
Gautschi O, Pauli C, Strobel K et al (2012) A patient with BRAF V600E lung adenocarcinoma responding to vemurafenib. J Thorac Oncol 7(10):e23–e24
Peters S, Michielin O, Zimmermann S (2013) Dramatic response induced by vemurafenib in a BRAF V600E-mutated lung adenocarcinoma. J Clin Oncol 31(20):e341–e344
Robinson SD, O’Shaughnessy JA, Lance Cowey C, Konduri K (2014) BRAF V600E-mutated lung adenocarcinoma with metastases to the brain responding to treatment with vemurafenib. Lung Cancer 85:326–330. doi:10.1016/j.lungcan.2014.05.009
Gautschi O, Peters S, Zoete V et al (2013) Lung adenocarcinoma with BRAF G469L mutation refractory to vemurafenib. Lung Cancer 82(2):365–367
Falchook GS, Long GV, Kurzrock R et al (2012) Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 379(9829):1893–1901
Rudin CM, Hong K, Streit M (2013) Molecular characterization of acquired resistance to the BRAF inhibitor dabrafenib in a patient with BRAF-mutant non-small-cell lung cancer. J Thorac Oncol 8(5):e41–e42
McCourt CM, McArt DG, Mills K et al (2013) Validation of next generation sequencing technologies in comparison to current diagnostic gold standards for BRAF, EGFR and KRAS mutational analysis. PLoS One 8(7), e69604
Tuononen K, Maki-Nevala S, Sarhadi VK et al (2013) Comparison of targeted next-generation sequencing (NGS) and real-time PCR in the detection of EGFR, KRAS, and BRAF mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma-superiority of NGS. Genes Chromosomes Cancer 52(5):503–511
Ilie M, Long E, Hofman V et al (2013) Diagnostic value of immunohistochemistry for the detection of the BRAFV600E mutation in primary lung adenocarcinoma Caucasian patients. Ann Oncol 24(3):742–748
Sasaki H, Shimizu S, Tani Y et al (2013) Usefulness of immunohistochemistry for the detection of the BRAF V600E mutation in Japanese lung adenocarcinoma. Lung Cancer 82(1):51–54
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Cagle, P.T., Raparia, K., Portier, B.P. (2016). Emerging Biomarkers in Personalized Therapy of Lung Cancer. In: Ahmad, A., Gadgeel, S. (eds) Lung Cancer and Personalized Medicine: Novel Therapies and Clinical Management. Advances in Experimental Medicine and Biology, vol 890. Springer, Cham. https://doi.org/10.1007/978-3-319-24932-2_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-24932-2_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24931-5
Online ISBN: 978-3-319-24932-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)