
Abstract
Latent Epstein-Bar virus genomes undergo epigenetic modifications which are dependent on the respective tissue type and cellular phenotype. These define distinct viral epigenotypes corresponding with latent viral gene expression profiles. Viral Latent Membrane Proteins 1 and 2A can induce cellular DNA methyltransferases, thereby influencing the methylation status of the viral and cellular genomes. Therefore, not only the viral genomes carry epigenetic modifications, but also the cellular genomes adopt major epigenetic alterations upon EBV infection. The distinct cellular epigenotypes of EBV-infected cells differ from the epigenotypes of their normal counterparts. In Burkitt lymphoma (BL), nasopharyngeal carcinoma (NPC) and EBV-associated gastric carcinoma (EBVaGC) significant changes in the host cell methylome with a strong tendency towards CpG island hypermethylation are observed. Hypermethylated genes unique for EBVaGC suggest the existence of an EBV-specific “epigenetic signature”. Contrary to the primary malignancies carrying latent EBV genomes, lymphoblastoid cells (LCs) established by EBV infection of peripheral B cells in vitro are characterized by a massive genome-wide demethylation and a significant decrease and redistribution of heterochromatic histone marks. Establishing complete epigenomes of the diverse EBV-associated malignancies shall clarify their similarities and differences and further clarify the contribution of EBV to the pathogenesis, especially for the epithelial malignancies, NPC and EBVaGC.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Viral epigenotypes
- Epigenetic signature
- Genome-wide demethylation
- Heterochromatic histone marks
- Host cell methylome
3.1 Introduction
Epstein-Barr virus (EBV) is one of the eight currently known human pathogenic herpesviruses, together with herpes simplex virus (HSV) -1 and -2, varicella zoster virus (VZV), cytomegalovirus (CMV ), human herpesvirus (HHV) -6, HHV-7, and HHV-8, also called Kaposi sarcoma-associated herpesvirus (KSHV) . EBV was the first human tumor virus to be discovered, by electron microscopy in cultivated cells derived from Burkitt lymphoma (BL) biopsies (Epstein et al. 1964). Shortly after, seroepidemiology linked EBV with endemic BL (Henle and Henle 1966), the stunning ability of EBV to induce B cell growth (Henle et al. 1967; Moore et al. 1967) and to immortalize and growth-transform B cells (Pope et al. 1968) was recognized, and EBV was identified as the agent causing infectious mononucleosis (Henle et al. 1968). Localizing EBV to the malignant cells of anaplastic carcinomas of the nasopharynx (NPC), but not to the plenty of infiltrating lymphocytes, installed EBV as the first epitheliotropic human tumor virus (Wolf et al. 1973), a decade before high-risk papillomaviruses were identified in cervical carcinoma biopsies (Durst et al. 1983).
Meanwhile, EBV was also detected in between 30 and 40 % of classical Hodgkin lymphoma subtypes world-wide (Weiss et al. 1987, 1989), but close to 100 % of Hodgkin lymphoma in severely immunosuppressed hosts, and in a subset of gastric carcinomas (Burke et al. 1990; Shibata et al. 1991), encompassing approximately 8–10 % of this world-wide occurring sporadic tumor. Beyond BL, NPC and gastric carcinoma, EBV was also associated with T or natural killer (NK) cell lymphomas (Harabuchi et al. 1990), leiomyosarcoma (McClain et al. 1995), and post-transplant lymphoproliferative disease (Hamilton-Dutoit et al. 1993). All in all, EBV is probably co-responsible for about 200,000 new cases of cancer per year corresponding to between 1 and 2 % of all human neoplastic disease (Munz 2014). Furthermore, ever since elevated anti-EBV antibody levels were found in patients with systemic lupus erythematosus (Evans et al. 1971) and multiple sclerosis (Sumaya et al. 1980), EBV has been incriminated as a trigger of autoimmune diseases (reviewed in Niller et al. 2009, 2011). Therefore, the discovery of EBV, more than 50 years ago, has enormously stimulated tumor virology and cancer research in general, and high hopes have been pinned to it that EBV might turn out as a Rosetta stone for understanding multistep carcinogenesis, and also for better understanding the role of viruses in immunopathological disorders (de The 1984, 1985).
Because after primary infection EBV genomes persist in host cell nuclei as circular chromatin-structured mini-chromosomes in a latent mode, EBV has served as a very prolific model system for the development of large scale sequencing technology (Baer et al. 1984), and for studying the cellular mechanisms of DNA replication (Yates et al. 1985), and transcriptional and epigenetic regulation of gene expression (Li and Minarovits 2003). Altogether, EBV is one of the most comprehensively, if not the best-studied of all viruses. Therefore, we dedicate a separate book chapter to the epigenetic alterations in EBV-associated tumors. EBV gene expression is highly dependent on epigenetic regulatory mechanisms. In diverse cell types carrying latent viral genomes, genetically identical viral genomes bear different epigenetic marks, thereby leading to specific patterns of viral latent gene expression and establishing distinct viral epigenotypes (Minarovits 2006). However, also the host cell genomes of EBV-infected cancer cells carry distinct epigenetic marks differing from each other in dependence of the respective tumor type and differing from the epigenetic profiles of their healthy counterparts (Niller et al. 2009). Before we review the current state of epigenetic alterations in selected EBV-associated lymphoid and epithelial malignancies, we give a brief description of the biology and clinical aspects of EBV infection, the viral latency gene products with an emphasis on their epigenetic impact, the establishment of latency in memory B cells, and the different epigenotypes that EBV genomes may assume.
3.2 Physiological Course of EBV Infection
EBV is the only known human member of the subfamily Gammaherpesvirinae that belongs to the genus Lymphocryptovirus, the name reflecting the viral property of homing to and “hiding” in the B lymphocyte compartment. Worldwide, primary EBV infection usually occurs very early in life with no or little symptoms so that at the age of 10 years most people have turned seropositive. In Europe and the USA, primary infection of up to 50 % of the population occurs later in life, i.e. in adolescence or early adulthood. In this case, symptomatic infection may ensue in up to 50 % of the infected youths, with the more severe cases presenting as infectious mononucleosis, also named kissing disease, typically including pharyngitis, tonsillitis, fever, malaise, lymphadenopathy. The disease is often accompanied with splenomegaly, a minority also with hepatomegaly, and characteristically with more than 50 % of atypical mononuclear cells in the blood smear which can be mistaken for leukemia, and from which the name mononucleosis is derived. In rare and extreme cases of mononucleosis, splenic rupture or fulminant hepatitis with liver failure may occur (reviewed in Longnecker et al. 2013).
Contrary to VZV, the virus causing chickenpox, the infectivity of EBV is not high enough to make airborne infection the regular way of transmission. Thus, EBV is transmitted by intimate contact, usually through saliva, mostly from asymptomatic mothers which are occasionally shedding the virus due to intermittent lytic replication from their tonsillar epithelia and their salivary glands, to their infants, or between adolescents. The virus is assumed to initially enter B cells at the surface of the tonsils or the oropharyngeal lymphoid tissue of Waldeyer’s ring, through engagement of its major envelope glycoprotein gp350/220, with the CD21 surface receptor which is also known as the CR2 receptor for the complement component C3d (Fingeroth et al. 1984). Tonsillar epithelial cells most likely serve as helpful enhancers for initial high-level virus production and occasional virus shedding later-on, as they can take up the virus through direct cell to cell contact and sustain an efficient lytic viral replication more easily than lymphoid cells (Borza and Hutt-Fletcher 2002; Hadinoto et al. 2009).
In the course of primary infection, the lytic replication cycle is triggered by the expression of two major viral immediate early genes BRLF1 and BZLF1 which in turn activate transcription of the viral early proteins whose expression leads to viral DNA replication, and the sequential expression of viral late genes which mainly code for virus structural building blocks. During an incubation time of approximately 4 weeks, the lytic replication cycle leads to a massive local virus production and shedding within the oropharyngeal lymphoid tissue, with the virus spreading systemically to the blood stream. Within lymphoid tissues, naïve, germinal center and memory B cells are infected. A high percentage of the circulating blood memory B cell pool is infected in acute mononucleosis which may, in extreme cases, affect up to 50 % of circulating memory B cells. Contrary to naïve B cells within the lymphoid tissue, naïve B cells in the blood stream remain largely uninfected (Hochberg et al. 2004). In the course of mononucleosis the immune system mounts an efficient defense through both its cellular and humoral arms. The vigorous immune response more so than the cytolytic viral damage leads to the well-known clinical symptoms of mononucleosis.
When primary infection is finally overcome, the virus remains latent in a minority of 1–10 in a million of peripheral B cells, where no viral gene expression occurs at all, or only a very restricted set of viral latency genes is occasionally expressed upon cell division (Qu and Rowe 1992; Miyashita et al. 1997; Hochberg et al. 2004). After primary infection, the latent virus persists invisibly to the immune system in the healthy host, and the lytic replication cycle is restricted to lymphoid surface or salivary gland epithelia to which the immune system has only limited access (reviewed in Longnecker et al. 2013).
3.3 EBV Latency Types and Tumors
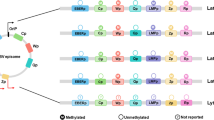
In EBV-associated tumor biopsies and EBV-infected cultured cell lines, the gene expression patterns of latent viral genomes depend largely on the host cell phenotype or stage of B cell development. Based on the activity status of the C promoter (Cp), we discern two major latency groups, Cp-on and Cp-off latency. When the major B lymphoid-specific promoter Cp is on, the full set of latency gene products, including all six EBNA (Epstein-Barr nuclear antigen) proteins, two latent membrane proteins, the multiply spliced BART transcripts, the non-translated EBER1 and -2 RNAs and three clusters of BHRF1 and BART microRNAs, are expressed. This expression pattern is commonly called latency type III. Except in BL cell lines where genetic alterations have caused permanent growth and which have undergone an in vitro switch to latency type III, cells in latency III have been immortalized by EBV, and are driven to permanent lymphoblastoid growth. When undergoing growth-transformation, the complete EBNA-transcription program is first directed by the W promoter (Wp) which becomes progressively methylated, before upstream Cp takes over the transcriptional control of the EBNA genes (Woisetschlaeger et al. 1990; Jansson et al. 1992; Tierney et al. 2000).
The astonishing ability and efficiency of EBV to immortalize and growth-transform B cells from the peripheral blood (Pope et al. 1968; Nilsson et al. 1971) in vitro has captured the interest of tumor virologists since its discovery and has also dominated EBV research and tumor virology until a decade ago. However, this ability is not to be mistaken for bona fide oncogenesis, because the specific viral contribution to growth-transformation and BL-oncogenesis appear to be rather distinct (Niller et al. 2004b). In order to identify the impact of EBV-induced transformation on the methylome of primary human B cells , Hansen et al. (2014) performed whole genome bisulfite sequencing and gene expression analysis of uninfected quiescent B-cells, B-cells activated by CD40 Ligand (CD40L) plus interleukin 4 (IL-4) for 3 weeks, and EBV-infected B cells obtained from the same donors 3 and 6 weeks after the infection. Surprisingly, compared to normal B cells, large scale hypomethylated blocks comprising two thirds of the cellular genome were induced 6 weeks after EBV infection, but not by B cell activation or 3 weeks after infection. These hypomethylated blocks significantly overlap with the positions of lamina-associated domains (LADs) and large organized chromatin lysine (K9)-modified regions (LOCKs), previously described in lung and pulmonary fibroblasts (Guelen et al. 2008; Wen et al. 2009). Comparison with the data obtained from the ENCODE project on the tier 1 lymphoblastoid cell line (LCL ) GM12878 suggested, that they may also be enriched in H3K27me3, while depleted of all transcription factors. Furthermore, these regions were associated with gene expression hypervariability, and largely corresponded to hypomethylated blocks previously described in human cancer (Hansen et al. 2011). These results suggest, that the formation of hypomethylated blocks may play a central role in the EBV induced transformation of B cells (Hansen et al. 2014). Accordingly, EBV-induced transformation led to a genome-wide decrease and redistribution of hetreochromatic marks (Hernando et al. 2014). Thus, the physiologically activated B-cell appears to be highly different from the growth-transformed B-lymphoblast. EBV-converted LCs can only expand in the absence of an anti-EBV directed immune response, i.e. if taken into in vitro culture or if the immune system is defunct, e.g. in XLP (X chromosome-linked lymphoproliferation) patients or in severely immune suppressed patients who suffer early-onset post-transplant lymphoproliferative disease (PTLD). LCLs which can be EBV-transformed in vitro from the white blood cells of healthy donors, are assumed to correspond to early onset PTLD cells. Therefore, early onset PTLD does not constitute a primarily malignant tumor, because it may be resolved, if immune control kicks back in early enough. However, it may turn monoclonal in the course and regularly become malignant, if growing too long out of immune control. In support of a molecular distinction between BL and PTLD tumors, the molecular signatures of viral and cellular protein and microRNA expression indicated that the respective contribution of EBV to both tumor types was different in each case (Navari et al. 2014).
A special latency type, called latency IIb has recently been highlighted by the secondary in vitro infection of chronic lymphocytic B cell leukemia (B-CLL) cells which exhibited EBNA2, but not LMP1 expression, which use Wp or both Wp and Cp, and which do not proliferate (Rasul et al. 2014). Secondarily EBV-infected B-CLL cells have also been found in vivo (Lewin et al. 1991). Latency IIb has also been observed in mononucleosis (Niedobitek et al. 1997; Kurth et al. 2000), in PTLD disorders (Oudejans et al. 1995; Brink et al. 1997), and in a unique case of a threefold mixed-latency type endemic BL with type IIb expression from a single chromosomally integrated viral genome (Kelly et al. 2006). Most likely latency IIb represents a transitional stage which may have got frozen on the path to full-blown growth-transformation exhibiting Cp-on latency III, through constraints imposed on viral gene expression by the cellular phenotype, e.g. secondarily infected CLL cells. Cells exhibiting latency IIb may normally get deleted by the healthy immune response, after primary infection is resolved (reviewed in Klein and Nagy 2010; Klein et al. 2013).
When Cp does not become switched on, expression of the key viral replication protein EBNA1 is governed by the Q promoter (Qp) i nstead of Cp, and the key immortalizing EBNA2 protein and the other four EBNA proteins are not expressed. Cells from EBV-infected germinal center B cells (latency I or II) or memory B cells (latency 0), and also from EBV-associated tumor biopsies, e.g. Burkitt lymphoma (latency I), Hodgkin lymphoma (latency II), NPC (latency II, +/− LMP1, +BARF1 expression) and gastric carcinoma (latency I, +/− LMP2A, +BARF1, +BARF0 expression), exhibit Cp-off expression patterns (Table 3.1). In addition, the two epithelial cancers highly express the BART, but not the BHRF1 microRNAs (Kim et al. 2007; Zhu et al. 2009; Cancer Genome Atlas Research Network 2014). Cp-off tumors constitute primarily malignant lymphomas or cancers which do not require a grossly defunct immune system in order to develop, but may arise in the face of a more or less functional or, in the case of BL or Hodgkin lymphoma, an even hyperstimulated, yet possibly imbalanced immune system, while EBV-associated epithelial cancers can, in addition, be viewed as molecular accidents by which the virus entered and succeeded to persist in a non-physiological target cell.
The fundamental difference between Cp-off and Cp-on latency types is further underscored by the fact that both NPC and EBVaGC are not characterized by a massive genome demethylation , as EBV-immortalized cells are, but they belong to the group of CIMP cancers, i.e. they exhibit a CpG Island Methylator Phenotype. The importance of immune hyperstimulation for explaining the pathogenesis of endemic BL has, to our knowledge for the first time, been brought to attention and discussed by Bornkamm, Klein, and Lenoir in 1987 (Klein 1987; Lenoir and Bornkamm 1987). The possibility of a complex temporary composite between immune suppression and hyperstimulation in the same patient, i.e. in patients with HIV, solid organ transplant, parasitic infestation or holoendemic malaria, and the occasional confusion which may arise from this complex overlay has been clarified (Niller et al. 2004b). Recently, the discussion has been re-visited under the aspect of novel epidemiological data, i.e. analysing BL incidence in equatorial Africa with a combination of both holoendemic malaria and HIV (Rickinson 2014). The disproportionate immune hyperstimulation may be provided by antigens of Plasmodium falciparum or through other parasitic infections (Araujo et al. 1996, 1999; Torgbor et al. 2014), together with hotspots of arboviral outbreaks, or by early-phase HIV-infection in the case of HIV-associated BL (Kalter et al. 1985; van den Bosch and Lloyd 2000; van den Bosch 2004). An immune response skewed from Th1 towards Th2 helper cells may further contribute to increase BL risk (Lubega 2007; van den Bosch 2012).
3.4 EBV Latency Gene Products
For a more extensive description of viral latency gene products, we refer to recent reviews (Niller et al. 2011; Tsao et al. 2015) and a textbook chapter (Longnecker et al. 2013), and for a quick overview to Table 3.1. In short, the EBER genes code for two small abundant nuclear RNAs. As a matter of routine, they serve histo-pathologists to identify EBV-infected cells by in situ hybridization. EBERs complex with the La autoantigen of systemic lupus and with the ribosomal protein L22, and bind dsRNA dependent protein kinase R, thereby blocking the pro-apoptotic interferon α dependent signaling pathway (Clarke et al. 1991). The EBERs exert tumorigenic and anti-apoptotic functions in addition to blocking dsRNA-PKR (Ruf et al. 2000). A significant amount of La-complexed EBERs can be secreted by infected cells which leads to TLR3 (Toll-like receptor) signaling in neighbouring cells. EBER-induced innate immune responses may be associated with EBV-induced autoimmune pathology (Iwakiri et al. 2009). Interestingly, although the EBERs are non-translated RNAs, both contain at least one hypothetical full length open reading frame, the significance of which has so far not been experimentally addressed (Niller et al. 2004c).
EBNA1 is the only viral transcription and replication protein required for oriP replication and nuclear maintenance through the nuclear matrix attachment region (Sugden and Warren 1989; Jankelevich et al. 1992; White et al. 2001) encompassing oriP. Within the viral genome, EBNA1 binds oriP and the latency promoter Qp, and EBNA1-binding sites within the cellular genome have been described as well (O’Neil et al. 2008; Canaan et al. 2009; Dresang et al. 2009; d’Herouel et al. 2010; Lu et al. 2010, 2011). EBNA1-bound oriP also acts as a long-distance enhancer within the viral genome for the mutually exclusive control of the viral latency promoters Cp and Qp and for additional viral promoters (Gahn and Sugden 1995; Zetterberg et al. 2004). EBV-specific epigenetic alterations may be due to viral gene products interacting with cis-acting regulatory elements and with cellular epigenetic regulators (Table 3.2). As oriP and Qp are constitutively unmethylated, EBNA1 keeps its binding sites in the viral genome free of CpG methylation (Lin et al. 2000; Salamon et al. 2000, 2001). EBNA1 may even serve as a Bookmarking protein which keeps cellular genes marked for activation throughout the cell cycle (Niller and Minarovits 2012). In gastric carcinoma cell lines, EBNA1 by itself kept the bidirectional promoter for the tumor suppressor gastrokine genes GKN1 and GKN2 methylation -free (Lu et al. 2014). Several transcriptional activators have been shown to cooperate with EBNA1 in activating cellular genes. In EBNA1 expressing B cell lymphoma cells, EBNA1 and Sp1 coactivated the antiapoptotic survivin promoter (Lu et al. 2011). The ability of EBNA1 to induce transcriptional activation may also be connected to its binding to Brd4, a bromodomain protein which preferentially interacts with acetylated chromatin (Lin et al. 2008). Further, nucleosome assembly protein NAP1 supports EBNA1 replicative and transcriptional functions (Wang and Frappier 2009). EBNA1 may play a larger chromatin modifying role than previously thought. It attracts a histone deubiquitylating complex to oriP (Sarkari et al. 2009), and interacts with multiple nuclear proteins, among them high mobility group (HMG) B2 (Jourdan et al. 2012), and HMGA proteins (Coppotelli et al. 2013).
EBNA2 is considered the main transforming viral protein (Hammerschmidt and Sugden 1989) which acts as major transactivator of promoters via indirect binding to RBP-Jκ/CBF1-recognition sequences involved in immortalizing and growth-transforming B cells in vitro (Ling et al. 1993). Besides CBF1, additional cellular transcription factors binding to sites for PU.1, AUF1, and CRE tether EBNA2 to its transcriptional targets (Johannsen et al. 1995; Fuentes-Panana et al. 2000). Specific targets of EBNA2 are viral promoters Cp, LMP2Ap and LMP1p, and promoters for B cell activation markers which push the resting B cell towards entering the cell cycle (reviewed in Gyory and Minarovits 2005). Although EBNA2 activity overlaps with Notch signaling, it governs its own extensive cellular transcriptional program and epigenetic reprogramming via recruitment of histone acetyl transferases (HAT) p300, CBP and PCAF to drive B cell proliferation (Wang et al. 2000; Zhao et al. 2011b). Further, EBNA2 interacts with human SNF-SWI-homolog chromatin modifier proteins in order to open up chromatin conformation (Wu et al. 1996).
EBNA-LP (EBNA5 in the Stockholm nomenclature) is a protein containing the repetitive units of the internal viral W-repeat . During the course of growth-transformation, it is the first viral latency protein to be expressed and it greatly enhances transformation efficiency, as it serves as a transcriptional coactivator for EBNA2 (Mannick et al. 1991). Because the W-repeat is frequently recombined during lytic viral replication, EBNA-LP exhibits a considerable size variation (Finke et al. 1987). EBNA2 coactivation through EBNA-LP is mediated by displacing the PML-nuclear body protein Sp100 from PML-NBs (Ling et al. 2005) and by displacing HDAC4 and HDAC5 from EBNA2-bound promoters (Portal et al. 2006).
Among viral latency proteins, the three members of the EBNA3 family, EBNA3A (EBNA3), EBNA3B (EBNA4), and EBNA3C (EBNA6) dominate the cytotoxic immune response against EBV (reviewed in Taylor et al. 2015). The three genes are part of the multiply spliced EBNA transcription unit, are arranged in tandem and show distant homology to each other. EBNA3C is the best-studied of the three proteins. While EBNA3A and -3C are required for EBV-mediated growth-transformation of primary B cells (Tomkinson et al. 1993), EBNA3B is not (Tomkinson and Kieff 1992). By competing with EBNA2 for promoter-bound CBF1 they modulate the transcriptional effects of EBNA2 (Johannsen et al. 1996). Like EBNA2, EBNA3C interacts also with the cellular histone acetyl transferase (HAT) p300, thereby downmodulating the EBNA2 effect (Cotter and Robertson 2000). Furthermore, EBNA3C forms a complex with HDACs 1 and 2, prothymosin α, and the corepressors mSin3A and NCoR to down-regulate transcription (Knight et al. 2003). EBNA3C-regulated genes overlap significantly with EBNA2-, EBN3A-, and EBNA3B-regulated genes (White et al. 2010; Zhao et al. 2011a; McClellan et al. 2013). The requirement for continuous EBNA3A and -3C expression for transformed B cell growth was explained with their binding of the transcriptional co-repressor CtBP, via which cell cycle checkpoint control proteins p16INK4A, p14ARF are epigenetically down-regulated through the deposition of inhibitory histone modifications on their promoters (Skalska et al. 2010; Maruo et al. 2011). Recruitment of PRC2 and deposition of the repressive histone mark H3K27me3 on the promoter of pro-apoptotic Bcl-2 family member Bim is dependent on EBNA3A and -3C, too (Paschos et al. 2012).
The latent membrane protein LMP1 is an integral membrane protein containing six transmembrane domains which can mimic a constitutively active TNF receptor and, like EBNA2, is essential for the growth-transformation of primary B cells. Its expression is governed by its terminal repeat promoter L1-TRp, while the bidirectional promoter LMP1p for LMP1 and LMP2B, is mainly active in growth-transformed B cells. In the germinal center, LMP1 signaling may replace and overlap with the CD40 signal transduction pathway (Uchida et al. 1999). Therefore, LMP1 may contribute to apoptosis resistance during the physiologically vulnerable and apoptosis-prone phase of the GC reaction, in case the contact between the B cell and the T helper cell is lost. However, LMP1 is not only expressed on EBV-transformed B cells and Hodgkin lymphoma (HL) cells, but variably also on the surface of NPC cells. In NPC cells, LMP1 induces the level of DNMTs 1, -3A and -3B leading to the epigenetic repression of several tumor suppressor genes (Tsai et al. 2002, 2006; Seo et al. 2008). In GC cells , however, and in GC-derived EBV-positive HL cells, only DNMT3A was upregulated by LMP1, while the other two DNMTs were downregulated (Leonard et al. 2011). In Hodgkin lymphoma cell lines, LMP1 induces NF-κB signaling which has broad effects on the transcriptional expression profile of the HL cell. One of the induced genes is the PRC1 component Bmi-I which contributes to the survival of HL cells through its influence on transcriptional regulation of many genes (Dutton et al. 2007). Independently of NF-κB, in GC and HL cells LMP1 induces KDM6B (JMJD3), an eraser enzyme removing the methyl groups of H3K27me3 repressive marks (Anderton et al. 2011). LMP1 induces several microRNAs, miR-155 and miR-146a (Motsch et al. 2007), miR-29b (Anastasiadou et al. 2010), and miR-10b (Li et al. 2010), thereby leading to the downregulation of several cellular genes or promoting metastasis. In NPC cells , LMP1 cooperates with LMP2A in upregulating miR-155 which on its part downregulates KDM3A (JMJD1A) expression, a histone demethylase for removing the repressive H3K9me2 mark (Du et al. 2011). In epithelial cells, LMP1 induces epithelial-mesenchymal transition (EMT) , and LMP1 expression is associated with metastasis of clinical NPC tumors (Horikawa et al. 2007).
The other latent membrane protein LMP2A containing 12 transmembrane domains mimics a constitutively active B cell receptor (Merchant et al. 2000; Mancao and Hammerschmidt 2007), and it is not as essential for the growth-transformation of primary B cells as LMP1 or EBNA2. Its expression is governed by the LMP2Ap and, in the case of the shorter splice variant in LCs, LMP2B, by the bidirectional promoter LMP1p for both LMP1 and LMP2B. The LMP2A mRNA can only be transcribed after an initial circularization of the viral genome upon infection (Laux et al. 1988). In the GC reaction , LMP2A showing strong homology to the cellular B cell receptor may replace, overlap and interfere with the B cell receptor signal transduction pathway. Therefore, LMP2A may contribute to apoptosis resistance during the GC reaction, in case the contact between the B cell and the antigen presenting cell is lost. In EBV-associated gastric carcinoma cells (EBVaGC) which do not express LMP1, LMP2A may induce DNMT1 or DNMT3B via STAT3 phosphorylation eventually leading to the widespread methylation of tumor suppressor genes, like PTEN (Hino et al. 2009) and a multitude of additional genes (Zhao et al. 2013b). In epithelial cells, LMP2A has transforming potential (Scholle et al. 2000; Fukuda and Longnecker 2007) and induces EMT (Kong et al. 2010).
The multiply spliced BART RNAs , also called CST (complementary strand transcripts), are transcribed in all viral latency forms, but at particularly high levels in NPC cells and also in EBVaGC (Smith 2001; Cancer Genome Atlas Research Network 2014). The BARTs contain reading frames potentially coding for proteins named BARF0, RK-BARF0, A73, and RPMS1 and introns encoding a set of 40 BART microRNAs in two clusters (Pfeffer et al. 2004). The reading frames have been expressed in vitro and their products analyzed for interactions and function. The proteins may modulate EBNA2 transcriptional regulation and modify Notch signaling pathways, if the proteins are actually expressed in vivo (Smith et al. 2000; van Beek et al. 2003; Thornburg et al. 2004). The BART microRNAs are preferentially expressed in epithelial cells (Cai et al. 2006) and in NPC and EBVaGC cell lines and tumor tissues (Kim et al. 2007; Zhu et al. 2009; Cancer Genome Atlas Research Network 2014). Many viral and cellular target genes have been identified in microRNA targetome studies (Dolken et al. 2010; Riley et al. 2012; Skalsky et al. 2012). Distinct BART microRNAs modulate viral LMP1 and LMP2A expression (Lo et al. 2007; Lung et al. 2009), expression of viral lytic genes BALF5, BRLF1, and BZLF1 (Barth et al. 2008; Jung et al. 2014), downregulate the expression of cellular pro-apoptotic genes PUMA, Bim and BID (Choy et al. 2008; Marquitz et al. 2011), genes involved in immune recognition (Nachmani et al. 2009; Dolken et al. 2010), tumor suppressor genes DICE1 and CDH1 (Lei et al. 2013; Hsu et al. 2014), and the epithelial cell-specific suppressor of metastasis NDRG1 in NPC cells (Kanda et al. 2015).
BARF1 , a transforming gene product from the viral BamHI A region which was previously counted among the lytic genes, is expressed from latent viral genomes in the EBV-associated epithelial cancers, NPC and EBVaGC (Decaussin et al. 2000; zur Hausen et al. 2000; Seto et al. 2005). BARF1 shows homology with the human colony stimulating factor 1 receptor (CSF1R ), the gene product of the c-Fms proto-oncogene (Strockbine et al. 1998) and appears to modulate the host immune response to EBV infection (reviewed in Takada 2012).
The BHRF1 cluster of four microRNAs is mostly expressed in latency type III, but not in epithelial cells (Pfeffer et al. 2004; Cai et al. 2006; Kim et al. 2007). During the early phase of B cell infection , BHRF1 microRNAs may induce apoptosis resistance and cell proliferation (Seto et al. 2010). In primary lymphomas exhibiting latency type III, miR-BHRF1-3 may contribute to the downregulation of the T cell attracting chemokine CXCL11 (Xia et al. 2008).
3.5 Establishing EBV Latency in Memory B Cells
Ground-breaking work has established the blood memory B cell as the site of permanent EBV latency in vivo (Miyashita et al. 1997; Babcock et al. 1998). However, there is some uncertainty on which path permanent viral latency is established. The traditional view is certainly that upon EBV infection of naïve cells in the tonsils, the expanded and growth-transformed infected B cells undergo a germinal center passage, upon which they end up as memory B cells (Fig. 3.1). According to this model, the viral latency gene expression pattern would change in concordance with the B cell phenotype or the developmental stage on its path through the germinal center reaction. An implication of this model is that the EBV-transformed lymphoblast is the direct precursor of the BL cell. Specifically, a switch of the cellular phenotype accompanied by a switch in the expression profile from the extensive latency III pattern of growth-transformed lymphoblasts to the restricted latency I or 0 patterns of GC or memory cells, respectively, is required by this model (Hawkins et al. 2013), reviewed in (Rickinson 2014). Although such a latency switch has not been observed in vivo, a phenotypic switch of LCs, not accompanied by a latency type switch, could be engineered in vitro through overexpression of c-Myc in conditionally EBNA2-expressing cells (Polack et al. 1996; Pajic et al. 2001). Partial latency type switches could be reconstructed in vitro by addition of IL-21 or by coculture of LCs with CD4+ T cells secreting soluble CD40L (Kis et al. 2010; Nagy et al. 2012). Furthermore, EBV superinfection of BL cells exhibiting a viral latency type I of their endogenous viral genomes silenced EBNA2 expression of the incoming viral genomes after an initial phase of Wp-on or Cp-on latency (Hughes et al. 2011, 2012).

Establishment of EBV latency in memory B cells of the peripheral blood. The traditional model (below, brown arrows) posits that upon EBV infection of naïve B cells in the tonsils, they become growth-transformed cells exhibiting a viral latency type III gene expression pattern, then some of them undergo a germinal center passage, where they switch from latency type III to latency type I or II, either by a promoter switch from Cp to Qp or by deletion of the EBNA2 reading frame, upon which they end up as memory B cells (Thorley-Lawson 2001). The alternative model (above, blue arrows) posits that upon EBV infection of memory B cells in the tonsils, they circularize the incoming viral genome, don’t become growth-transformed, and remain in latency types 0, I, or II. All naïve or memory B cells which express the highly immunogenic latency type III pattern are extinguished by the cellular immune response. Generally, EBV-infected cells exhibiting latency III tend to avoid GC reactions. However, in cases of immune hyperstimulation, EBV-infected memory B cells may regularly enter or re-enter GC reactions (Araujo et al. 1999) where molecular accidents, like the Burkitt-translocation may occur (Niller et al. 2003; Rossi and Bonetti 2004)
Another view, first proposed more than 10 years ago, posits that the EBV transformed LC is fundamentally different from the physiologically antigen-activated B cell which undergoes a GC reaction (Fig. 3.1). In this model, the LC, whose epigenome was recently described by Hansen et al. (2014) is not the direct precursor of the BL cell, because EBV-transformed LCs exhibiting a latency class III expression pattern are mostly extinguished by the vigorous immune response which is mounted in the course of mononucleosis or also during asymptomatic primary infection (Niller et al. 2003, 2004a, b). Thus, a prior LC stage is not required for BL oncogenesis. Here, BL oncogenesis depends on c-Myc binding to its recognition site in the locus control region (LCR) of the viral genome which expresses latency I or II functions, but not on prior EBNA2 expression. A minority of BLs which exhibit a Wp-directed gene expression pattern close to latency type III, but without expressing EBNA2 (Kelly et al. 2002), then likely rose from a secondary latency type I to III switch in a pre-existing incipient BL tumor, made possible by bouts of immune suppression due to severe malaria tropica (Whittle et al. 1984). EBNA1 expression in this subset of BL tumors is governed by Wp instead of Cp or Qp, which may signify that an imminent EBV-driven growth-transformation was at its onset abrogated by deletion of EBNA2, before fully growth-transformed cells arose which would have been exterminated by the immune response Kelly et al. 2002). EBNA2 suppression in BL cells is then a tertiary event, achieved either through deletion of the EBNA2 reading frame in the minority of cases, or through extirpation of all BL cells that arrived at the highly immunogenic LC phenotype in the majority of BL cases, after the patients re-gained immune control (Kelly et al. 2002; Niller et al. 2004b). Cp-on latency type III as it occurs in LCs in vitro and in early-onset PTLDs in vivo is mostly regarded as a form of viral latency (Rowe et al. 1987). Latency III, however, may also be viewed as an alternative viral activation or re-activation form alongside the lytic replication cycle, but not as a form of latency in the strict sense, for which only Cp-off latency types 0, I or II would qualify. Thus, latency type III has been also named the B cell growth-program (Thorley-Lawson 2001). This notion is supported by the observation that upon demethylating treatment of a latency type I BL cell culture, lytic cycle transcription and the growth-program were activated in distinct cell subsets (Masucci et al. 1989).
The ability of EBV to sustain two distinct paths to viral activation, both lytic cycle and growth-program, seems to be due to the architectural intricacy of the EBV genome, in contrast to the HSV and CMV genomes (for review see Niller et al. 2007). How frequently the two distinct activation paths may overlap in one single cell subset (Ma et al. 2012), or how frequently the growth program may follow upon the lytic cycle program (Kalla and Hammerschmidt 2012) in EBV-associated malignancies, remains to be established.
Upon in vitro infection of isolated peripheral blood B cells, linear EBV genomes are circularized to become covalently closed circular (ccc) extrachromosomal episomes as early as 16 h post infection. Infected G0 cells do not need to pass through G1 or to enter S phase for EBV genomes to become circularized, however, a single G0 to early-G1 transition is sufficient for circularization (Hurley and Thorley-Lawson 1988). Interestingly, in already activated or cycling cells, incoming EBV was subject to a linear chromosomal integration, but not to circularization (Hurley et al. 1991), reviewed in (Morissette and Flamand 2010). Circularization occurs, before EBV-infected B cells express activation markers and start proliferating, and well before the viral episomes are amplified later-on in the course of growth-transformation (latency III) which would be the natural course of events in vitro (Hurley and Thorley-Lawson 1988). The possibility of establishing viral latency in vitro before cellular activation and proliferation take place reflects on the in vivo situation during primary infection. During mononucleosis, several distinct patterns of latency gene expression are observed, including latency III (Niedobitek et al. 1997) which is the most prominent, due to the ease of in vitro culture and because growth-transformed B cells overwhelm the organism, if unchecked by the immune system. And indeed, the rare carriers of X chromosome-linked lymphoproliferative (XLP) disease , are overwhelmed by growth-transformed B cells after primary infection, due to a mutant SH2D1A gene coding for SLAM-associated protein (SAP) which is important for immune cell signaling, and mononucleosis may end deadly (Coffey et al. 1998), reviewed in (Latour and Veillette 2003). However, if kept at bay through an ever increasing immune response, infected B cells are basically able to establish EBV latency, even if they do not complete a cell cycle, or if they cycle only transiently and become resting again, and if they never reach a Cp-governed LC stage exhibiting latency III.
Correspondingly, during the systemic spread of EBV from Waldeyer’s ring to the blood stream, many naïve, as well as resting cells, memory cells and germinal center cells are directly infected, but infected naïve cells do not expand or participate in GC reactions (Kurth et al. 2000), and the expanding EBV-infected GC and memory cells in tonsils of mononucleosis patients do not participate in GC reactions either (Kurth et al. 2003). This observation was confirmed by Araujo et al. (1999) who did not find any EBV-infected cells in GC reactions in a series of excised tonsils from children in Berlin (Araujo et al. 1999). Thus, under normal immune conditions, EBV infected cells do not even seem to regularly reach the germinal center. However, in tonsils from Northeast Brazil where children suffered a high load of parasitic infections at the time (Araujo et al. 1996), a GC passage of EBV-infected cells and the expansion of EBV-positive GC clones was indeed observed. However, the hyperstimulated GC environment permitted only latency type I or occasional type II viral gene expression, but not latency type III viral gene expression (Araujo et al. 1999). Because the viral growth-program which depends on EBNA2 expression is inhibitory for AID expression which is the key enzyme of the germinal center reaction, latency III is apparently incompatible with a physiological GC reaction (Panagopoulos et al. 2004; Tobollik et al. 2006; Boccellato et al. 2007). More recently, this was supported by Roughan et al. (2010) who found that EBV-infected cells in non-IM tonsils from persistently EBV-infected carriers which expressed GC markers did not express the latency III protein EBNA2 and did not expand in physiological GC reactions which would yield 100s or 1000s of cells, but achieved only abortive reactions of less than ten cells (Roughan et al. 2010). The general “avoidance behaviour” of GC reactions by EBV-infected cells is also in accordance with the observation made in transgenic mice that LMP1 inhibits GC formation (Uchida et al. 1999). If a regular GC cell suffers a c-Myc-translocation or becomes a Reed-Sternberg cell, it is designated for apoptosis. However, if such a “crashed” B cell is EBV-infected, it may have a bigger than remote chance to survive the GC reaction and become the founder cell of a hematological malignancy, like a BL or a Hodgkin lymphoma. This is then due to the viral or cellular anti-apoptotic functions induced by the latent virus or exerted by its gene products (Niller et al. 2003; Rossi and Bonetti 2004).
Altogether, after Hansen et al. (2014) described the massive demethylation taking place during EBV-transformation of peripheral blood B cells to the growth-program (Hansen et al. 2014), the evidence for the establishment of latency through direct infection of memory B cells (Kurth et al. 2000) and for the origin of BL without a general detour through an LC stage (Niller et al. 2003, 2004a, b; Rossi and Bonetti 2004), is further solidifying (Niller et al. 2014b). A point which still needs clarification is whether the cellular epigenomes and gene expression profiles of early-onset PTLDs are actually the same as or as close to the molecular profiles of the in vitro EBV-transformed LC (Hansen et al. 2014) as is generally assumed. A complete bisulfite sequence map of early-onset PTLD genomes would be very helpful to clarify this point. Furthermore, complete methylome s of EBV-associated malignancies besides the endemic BL cell line Daudi (Kreck et al. 2013) are desiderata.
3.6 Epigenetic Regulation of EBV Latency Promoters
Cell-type specific and developmental stage-dependent viral gene expression patterns are achieved by the repertoires of viral and cellular transcription factors present in the respective cell types and by diverse epigenetic mechanisms acting on the major viral latency promoters thereby defining distinct viral epigenotypes (Minarovits 2006; Fernandez et al. 2009; Takacs et al. 2009; Arvey et al. 2013). Before the growth-program of Cp-on latency is established upon in vitro infection, Wp is governing latency III gene expression at first. However, Wp becomes gradually switched off by CpG-methylation, while Cp takes over (Tierney et al. 2000; Elliott et al. 2004). In in vitro EBV-infected tonsillar GC B cells, LMP1-induced DNMT3A binding to Wp correlates with the promoter methylation status (Leonard et al. 2011). In Cp-on latency , unmethylated Cp is located to an acetylation island, while in Cp-off latency, heterochromatic histone mark s are deposited on the methylated Cp (Fejer et al. 2008). In this case, among the EBNA proteins, solely replication protein EBNA1 is expressed from Qp. In Cp-on latency, Qp is suppressed by heterochromatic histone marks and binding of repressor proteins, but not by CpG-methylation (Zhang and Pagano 1999; Salamon et al. 2001; Fejer et al. 2008). CTCF binding closely upstream contributes to the protection of Qp from methylation which stays constitutively unmethylated in all latency types (Tempera et al. 2010). The LMP promoters are also regulated by epigenetic modifications (reviewed in Takacs et al. 2009, 2010). LMP1p activity correlates with its methylation status, but not with transcription factor occupancy (Minarovits et al. 1994; Salamon et al. 2001; Takacs et al. 2001). LMP2Ap activity again is co-regulated by its methylation status, histone marks and transcription factor occupancy (Salamon et al. 2003; Gerle et al. 2007). The EBER genes closely upstream of constitutively unmethylated oriP (Salamon et al. 2000) are also constitutively unmethylated and expressed in all latency types (Minarovits et al. 1992), but their promoters are sensitive to CpG-methylation which prevents binding of transcription factors c-Myc and ATF (Niller et al. 2003; Banati et al. 2008).
Recently, the chromatin boundary protein CCCTC-binding factor (CTCF ) has come into the focus of viral latency studies. CTCF mRNA and protein was expressed at higher levels in BL cells than in LCs, and it contributed to EBNA2 repression by downregulating Cp via its binding site between oriP and Cp (Chau et al. 2006). A knockdown of CTCF in BL cells did not lead to the activation of Cp, and a switch to latency III was not observed. However, deletion of that CTCF-binding site led to a prolonged transcription from Cp of incoming viral genomes in one superinfected EBV-positive BL cell line. Thus, CTCF was suggested to contribute, among other factors, to Cp silencing (Hughes et al. 2012). Contrary, Salamon et al. (2009) did not find a correlation between CTCF binding and Cp or Qp activity in several cell lines and, by genomic footprinting found hints for additional proteins besides CTCF playing a role in determining viral latency types (Salamon et al. 2009). CTCF binding at Qp may not serve a repressive function, but as a chromatin boundary separating methylated and unmethylated regions (Tempera et al. 2010). Holdorf et al. (2011) profiled the occupancy of CTCF, RAD21 (a cohesin component), and RNA polymerase II on the EBV episomes in Raji cells, a latency III type BL cell line. They found very prominent RNA pol II binding in the middle of oriP and at a couple of other locations which coincided with known promoter sequences. Raji is a BL cell line which does not use Wp or Cp, but a so far unknown promoter, in order to express a latency III transcriptional pattern (Woisetschlaeger et al. 1989; Walls and Perricaudet 1991). Thus, this large peak of RNA pol II binding within oriP may constitute the missing latency III promoter for the EBNA transcripts in Raji cells (Fig. 3.2). Further, they found CTCF sites not only at active and inactive latency control elements, but also at lytic promoters suggesting several distinct functions of CTCF, like repression, activation, or chromatin insulation, depending on the context of additional binding proteins. Some of the CTCF sites overlapped with cohesin binding, but not RNA pol II binding (Holdorf et al. 2011).

Binding of CTCF, cohesin, RAD21, ZNF143, c-Myc to the latent EBV genome. The EBV genome (not drawn to scale) is depicted as an episome which was circularized at the terminal repeats (TR) immediately after infection. Binding sites for CTCF, cohesin, and RAD21 (a subunit of the RNA pol II complex) were determined by Holdorf et al. (2011) in Raji cells, the binding sites for CTCF, cohesin, RAD21 and the transcriptional activator ZNF143 were determined by Arvey et al. (2012, 2013) mainly in a metaanalysis from EBV-transformed lymphoblastoid cell lines. The binding site for c-Myc at the EBER locus was determined by Niller et al. (2003) in several cell lines, among them Raji. The locus control region encompassing the EBER locus and the origin of plasmid replication (oriP) with its elements FR (family of repeats) and DS (dyad symmetry) is crucial for regulating the latency type (Tempera et al. 2011). In latency type I, the LCR loops to and activates Qp, in latency III it loops to and activates Cp, and also acts as a transcriptional enhancer for the bidirectional LMP promoter for LMP1 and LMP2B (latent membrane proteins). Rep* is a sequence adding to oriP function. Cp, the repetitive Wps, and Qp are alternative latent promoters for the EBNA (Epstein-Barr nuclear antigen) transcripts. Lytic cycle promoters around the viral genome are labeled according to the EBV mapping nomenclature
A recent metaanalysis was performed on data sets which were extracted from public databases containing total genomic chromatin data from large scale deep-seq projects that included EBV-transformed LCs. Thereby, a total of 19 CTCF binding sites was found around the EBV genome, mostly located to important regulatory regions (Arvey et al. 2012, 2013). One major role of CTCF may be to support distinct three-dimensional conformations of viral genomes depending on the latency type. In latency I, the locus control region (LCR ) of EBV containing oriP looped to Qp, while in latency III the LCR was contacting Cp. Chromatin loop formation and maintenance between the LCR and other regions of the viral genome was dependent on intact CTCF binding sites (Tempera et al. 2011). However, how the selectivity of the looping is achieved remains to be established. One possibility is certainly that cohesin may play a distinguishing role. Thus, the knockdown of cohesin subunits RAD21 and SMC1 led to the loss of an LCR-LMP-region loop in LCs (Arvey et al. 2012). This is in concordance with the immunoglobulin H locus, where cohesin binding was associated with multiple chromatin looping and recombinatorial activity (Degner et al. 2009), and with KSHV , where chromatin looping was facilitated by CTCF-cohesin interactions (Kang et al. 2011). Remarkably, the viral LCR exhibited a partial non-specific stickiness for protein (Arvey et al. 2013), probably reflecting on the nuclear matrix attachment function of the LCR (Jankelevich et al. 1992). Therefore, it is almost certain that many more factors than just CTCF and cohesin are involved in specific chromatin looping and viral 3D structure accompanying the regulation of viral latency (Fig. 3.2). For example binding of ZNF143, a most intriguing zinc-finger protein which mediates promoter looping in the cellular chromatin by directly binding to promoters and indirectly to CTCF-cohesin complexes (Bailey et al. 2015), was found just upstream of the EBERs and at a few additional locations of the viral genome, in each case colocalized with CTCF (Arvey et al. 2012). At the same location of the LCR, a binding site for c-Myc had been found in Raji cells by in vivo footprinting and chromatin immune precipitation which likely plays a crucial role in the origin of endemic BL tumors in the GC reaction (Niller et al. 2003, 2004a, b).
3.7 Epigenetic Profiles of Cellular Genomes in EBV-Associated Neoplasms
Cancers in general exhibit epigenomic profiles which are different from that of their healthy cellular counterparts (Baylin and Jones 2011) and from each other (Costello et al. 2000). As a rule, cancer cells carry stretches of hypomethylated DNA throughout their genomes, but exhibit also cancer type-associated hypermethylations at specific CpG islands which are frequently overlapping with tissue-specific regulatory elements and with developmentally regulated PRC-binding elements (Esteller 2007a, b; Baylin and Jones 2011). Thus, also EBV-associated neoplasms differ regarding their transcriptomes and, as far as partial or complete CpG-methylation maps have been established, also regarding their epigenomes. Furthermore, the EBV-infected counterparts of a specific tumor type exhibit transcriptomes and epigenetic profiles different from their EBV-negative counterparts. Lymphomas and epithelial cancers are two major groups of EBV-associated malignancies, whose epigenomes need to be individually studied in order to learn more about their individual modes of pathogenesis.
B cells transformed to the LC phenotype through EBV infection exhibit a massive hypomethylation affecting two thirds of their genome and one third of all genes in a complete genomic bisulfite sequence analysis. A total of almost 3000 small differentially methylated regions (DMRs), mostly located in the vicinity of promoters, amounting to a total of 1 MB, were also observed in LCs, approximately half of them hyper- and hypomethylated (Hansen et al. 2014). Hypomethylation of around 250 genes was observed in LCs using a 27K chip, but no hypermethylation (Hernando et al. 2013). Demethylation of most promoters associated with the transformation process was associated with binding of B cell specific transcription factors, half of them binding NF-κB (Hernando et al. 2013). Correspondingly, a major part of the epigenetic reprogramming in LCs and a set of different DLBCLs (Diffuse Large B cell Lymphomas) was due to NF-κB signaling (Vento-Tormo et al. 2014). Hypomethylated genes are involved in cellular proliferation, B cell signaling, chemotaxis, cell adhesion, immune response and inflammation (Hernando et al. 2013). An earlier study found hypomethylated genes involved in cell cycling, transcriptional regulation and the immune response (Caliskan et al. 2011). Parallel to demethylation , EBV transformation leads to a major redistribution and decrease of heterochromatic histone mark s (Hernando et al. 2014).
A focused analysis of the p16INK4A and Bim loci found heterochromatic histone marks which were deposited through PRC2 mediated by EBNA3A and -3C (Skalska et al. 2010; Paschos et al. 2012). A major finding of both Hernando et al. (2013) and Hansen et al. (2014) is the clear distinction of epigenomes between EBV-transformed LCs, on the one hand, and CD40L/IL4-activated B cells, on the other hand. The methylation profile of activated B cells was very close to that of quiescent B cells, while that of LCs had drifted far away (Hansen et al. 2014). Correspondingly, the heterochromatin profile of transformed LCs could also be achieved through infection with EBNA2- or LMP1-deleted viruses, but not through CD40L/IL4-stimulation of B cells to proliferate. Thus, the transformation trait could be clearly dissected from mere proliferation of activated B cells in those experiments (Hernando et al. 2013). This is a most intriguing observation which certainly deserves further in-depth analysis.
The epigenetic profile of BL tumors seems to considerably differ from that of LCs. So far, there is only one completed bisulfite sequence map of an EBV-positive endemic BL cell line, Daudi (Kreck et al. 2013). Sixty nine percent of all genomic CpG dinucleotides were methylated, and more than 90 % of 969 genes which are hypermethylated in mature aggressive B cell lymphomas including BL, were also silenced by methylation in Daudi cells (Martin-Subero et al. 2009b; Kreck et al. 2013). Nearly 60 % of hypermethylated genes in BL tumors were target genes of PRC2 in embryonic stem cells (Martin-Subero et al. 2009a). PRC2-targeted hypermethylation does probably not depend on EBV infection. Possibly, BLs are derived from precursor cells with stem-cell features (Martin-Subero et al. 2009b). In addition, c-Myc hyperactivity in BL tumors may contribute to their “stemness phenotype” (Takahashi et al. 2007). The overall tendency of BL is towards a high methylation profile, contrary to the hypomethylation-prone profile of LCs. A point which needs further clarification is whether primary BL tumors exhibit the same epigenetic profile as the cell line Daudi. Also in this case complete bisulfite maps would be helpful.
The epigenetic profiles of both NPC and EBV -associated GC suggest a CpG Island (CGI) Methylator Phenotype (CIMP) for both epithelial cancers (reviewed in Niller et al. 2007, 2012, 2014a, c; Kaneda et al. 2012; Lo et al. 2012). EBV gene expression patterns in NPC and GC belong mostly to latency I (or II) with a variable expression of LMP1 in the case of NPC, and with expression of BARF1, BARF0 and a variable expression of LMP2A in addition to EBNA1 and the BART microRNAs in the case of GC (Sugiura et al. 1996), reviewed in (Niller et al. 2012). Because EBV is usually monoclonal in malignant tissues, a causal role for the virus is assumed (Raab-Traub and Flynn 1986). Furthermore, a rather peculiar case of a very rapidly developing EBV-positive GC in an immune suppressed patient suggests a direct role for EBV in GC carcinogenesis (Au et al. 2005). However, the sequential order of carcinogenesis in both epithelial cancer types remains to be elucidated. Chromosomal aberrations have been found in NPC precursor lesions, i.e. in high grade dysplasia and carcinoma in situ, however, virus-infected normal epithelia have so far not been found for both NPC and GC (Kaneda et al. 2012; Lo et al. 2012).
Nevertheless, it is very likely that EBV infection is causal for a major epigenetic disruption of epithelial cells on their path to malignancy. One mechanism by which EBV causes the hypermethylation in EBVaGC, may be that LMP2A, in the cases where it is expressed, activates DNMT1 and DNMT3B expression which on their part lead to a global but non-random CGI-methylation in EBVaGC (Hino et al. 2009; Zhao et al. 2013b). How target specificity is achieved remains unclear at the moment. Candidate gene studies have reported a series of genes significantly more frequently hypermethylated in EBVaGC, but not in the other GC subtypes, e.g. CDH1 (Wu et al. 2000; Sudo et al. 2004), p16INK4A, PTEN, RASSF1A, GSTP1, MGMT, MINT2 (Kang et al. 2002), p14ARF, p15INK4B, p16INK4A (Chong et al. 2003), p14ARF, p16INK4A (Sakuma et al. 2004), p73 (Ushiku et al. 2007), HOXA10 (Kang et al. 2008). EBVaGC and microsatellite instability were mutually exclusive (Chang et al. 2003), which was reflected in constitutively unmethylated MLH1 and MSH2 genes in EBVaGC (Kang et al. 2002; Vo et al. 2002). An epigenetic field of cancerization, like in Helicobacter pylori-associated GC (Ushijima 2007), was not observed in the surrounding mucosa of EBVaGC (Enomoto et al. 2007; Ushiku et al. 2007). Further tumor suppressor genes WNT5A (Liu et al. 2013) and SSTR1 (Zhao et al. 2013a) were found to be hypermethylated in EBV-positive GCs (reviewed in Niller et al. 2009, 2014a). F urther, hypermethylated promoters of p16INK4A, FHIT, CRBP1, WWOX, and DLC1, HOXA11 have recently been reported or confirmed (Cui et al. 2015; He et al. 2015).
Candidate gene approaches were extended through genome wide screens. A remarkable genome-wide methylation analysis of 51 GC samples yielded three different GC epigenotypes , two low- and high-methylation epigenotypes in EBV-negative GCs, and one very high-methylation epigenotype of EBV-positive GCs. In the EBV-infected subtype, a surplus of 270 genes was methylated, the DNA repair gene MLH1 was regularly unmethylated, and, contrary to the EBV-negative hypermethylated subtype, PRC target genes of embryonic stem cells were not enriched. Furthermore, EBV infection of an EBV-negative GC cell line led to an increase of DNA methylation which resembled the methylation patterns of the EBV-infected very high-methylation GC epigenotype. Thus, in addition to the methylation mechanisms of other GC subtypes which may mostly be due to long-lasting Helicobacter pylori infection, additional mechanisms of aberrant methylation are assumed to act in the EBV-infected epigenotype. Hypermethylation could so far not simply be attributed to one of the latent gene products (Matsusaka et al. 2011). Consistent with the results of Matsusaka et al. (2011), a comparison of cultured GC cells yielded 886 genes which became hypermethylated in EBV-infected subclones, but not in uninfected subclones. Many of those genes are involved in tumor suppression pathways (Zhao et al. 2013b).
Two recent genome-wide studies of genetic and epigenetic alterations, one on 98 GC samples of different subtypes in comparison with 31 normal tissue samples (Wang et al. 2014), and the other one on 295 primary gastric adenocarcinomas including 9 % of EBVaGC (Cancer Genome Atlas Research Network 2014), confirmed the very high genomic CIMP status of EBVaGC (Toyota et al. 1999; Matsusaka et al. 2011). EBVaGC turns out to exhibit the highest CIMP status of all tumor types studied so far by The Cancer Genome Atlas Research Network (TCGA, reviewed in Li et al. 2014b). A list of 526 genes epigenetically silenced in EBVaGC (Cancer Genome Atlas Research Network 2014) and a list of 90 genes epigenetically silenced in different subtypes of GC (Wang et al. 2014) is available in the Supplementary Material of the respective papers. Interestingly, there is an intersection of 36 genes between the two lists of TCGA and Wang et al. (2014), an intersection of eleven genes between TCGA and Matsusaka et al. (2011), and only one gene between Wang et al. (2014) and Matsusaka et al. (2011), but no single gene is present on all three lists.
The epigenetic analysis of undifferentiated NPC is somewhat lagging behind compared to that of EBVaGC. Like EBVaGC, also NPC belongs to the group of CIMP-cancers . In the cases of NPC, where LMP1 is expressed, LMP1 may induce the expression of all three DNMTs and therefore play a role in the frequent hypermethylation of tumor suppressor genes in the tumor tissue (Tsai et al. 2002, 2006), reviewed in (Niller et al. 2009, 2012, 2014a, c; Lo et al. 2012). A comparative analysis of the cellular methylation status and LMP1 expression between NPC tissues and normal nasopharyngeal tissue showed that LMP1 expression correlated with the degree of methylation of CGIs at ten pre-selected tumor suppressor genes (Challouf et al. 2012).
Candidate gene approaches have identified a number of genes frequently hypermethylated in NPC tissue in comparison with healthy nasopharyngeal mucosal tissue, e.g. p16INK4A (Lo et al. 1996), CDH1 and CTNNB1 (Zheng et al. 1999), RASSF1A (Lo et al. 2001), EDNRB (Lo et al. 2002), TSLC1 (Hui et al. 2003), ZMYND10/BLU (Qiu et al. 2004), CHFR (Cheung et al. 2005), GADD45G (Ying et al. 2005), PCDH10 (Ying et al. 2006), LTF (Yi et al. 2006; Zhou et al. 2008), DLC1 (Peng et al. 2006), RASAL1 (Jin et al. 2007a), ADAMTS18 (Jin et al. 2007b), CCNA1 (Yanatatsaneejit et al. 2008), ADAMTS9 (Lung et al. 2008), DLEC1 (Ayadi et al. 2008), IRF8 (Lee et al. 2008), OPCML (Cui et al. 2008), BRD7 (Liu et al. 2008), ZNF382 (Cheng et al. 2010), PCDH8 (He et al. 2012), RRAD (Mo et al. 2012). CGI hypermethylation may have practical consequences for diagnostics and therapy (reviewed in Niller et al. 2009, 2012, c; Li et al. 2011, 2014b; Lo et al. 2012). Tumor suppressor or marker genes frequently methylated in NPCs may allow the use of methylation sensitive PCR (MSP) for the early detection of NPCs in high risk groups besides high levels of anti-EBV-IgA in serum samples, e.g. RASSF1A, p16INK4A, WIF1, CHFR and RIZ1 (Hutajulu et al. 2011), CACNA2D3 (Wong et al. 2013), CDK10 (You et al. 2013), ASS1 (Lan et al. 2014), CYB5R2 (Xiao et al. 2014). In addition, epigenetic alterations acquired by tumor cells during chemotherapy may play a role in the development of drug resistance and may allow predictions on the prognosis of EBVaGC. Hypermethylation of RARB2 (Kwong et al. 2002; Seo et al. 2008), CRBP1 and CRBP4 (Kwong et al. 2005b), and RARRES/TIG1 (Sriuranpong et al. 2004; Kwong et al. 2005a) prevent retinoic acid-mediated growth inhibition. High levels of H3K27me3 (Cai et al. 2011) and EZH2 (Hwang et al. 2012) may indicate the overall level of tumor suppressor gene silencing. Hypermethylation at DLC1, CHFR, and PEG10, and hypomethylation at ABCC5, ERBB2, and GSTP1 may predict taxol resistance of NPC tumors (Zhang et al. 2012).
Beyond the candidate gene analyses, genome-wide methylation analyses were performed on several NPC tumor samples and cell lines in comparison with normal tissue and non-transformed nasopharyngeal cell lines by chromatin immunoprecipitation combined with promoter microarray hybridization (MeDIP-chip ) (Li et al. 2014a). In this study, an overall high methylation level of CGIs was found, and many previously reported genes were confirmed as methylated in NPC tumors and cell lines . Two newly found genes, PAX5 and SLIT2 , were selected and again confirmed as methylated in tumors and cell lines. Several genes from the Wnt pathway, i.e. SFRP, DACT, and DKK family members were selected and examined for their methylation status and tumor suppressor function in NPC tumors and cell lines. Several members of these gene groups were indeed more frequently hypermethylated in NPC derived material than in normal nasopharyngeal cells and exhibited a tumor suppressor function in a colony formation assay. Remarkably, while in NPC tumors, the top affected pathway was the Wnt signaling pathway, the MAPK signaling pathway was the mostly affected pathway of NPC cell lines (Li et al. 2014a). This raises a note of caution in dealing with cell lines instead of tumor samples, because their epigenotype may have been altered in the course of long-term in vitro culture. On the other hand, if not microdissected, tumor samples are composed of distinct tissue types, e.g. cancer cells and infiltrating immune cells which make their epigenotype a composite of distinct epigenotypes.
Until very recently there was only a surprisingly small subset of four known hypermethylated gene loci which EBVaGC and NPC had in common, i.e. PTPRG, ASS1, GSTP1, and MIPOL1 (for a recent review and a detailed comparison of hypermethylated genes in EBVaGC versus NPC see (Niller et al. 2014a), and Table 3.3). However, a comprehensive 450K-chip methylome analysis of 60 NPC tumor samples in comparison with other tumor methylomes already established by The Cancer Genome Atlas (TCGA) Research consortium increased the overlap of methylated genes between EBVaGC and NPC (Dai et al. 2015). Dai et al. showed that also NPC belongs to the highest methylated of all cancers, described additional genes hypermethylated both in EBVaGC and NPC, confirmed a significant overlap of de novo methylated genes with bivalent chromatin markers H3K4me3 and H3K27me3 in all examined cancer types, and found a remarkably strong peak of hypermethylation at chromosomal position 6p21.3 for both EBVaGC and NPC, and a weaker, but significant methylation-peak at 6p21.3 also for prostate cancer (Dai et al. 2015). Chromosomal region 6p21.3 contains several genes apparently involved in tumorigenesis and importantly contains the human leukocyte antigen (HLA) genes which have earlier been shown to be strongly linked with NPC risk (Tse et al. 2009; Tang et al. 2012). The currently known intersection of hypermethylated genes in both EBVaGC and NPC is shown in Table 3.3. which illustrates the accent on chromosomal region 6p21.3. In-depth comparative analysis of complete genomic bisulfite sequence maps which have to be established yet for early-onset PTLD, NPC and EBVaGC, and the genome-wide analysis of histone marks may further reveal the overlaps and differences between EBV-associated tumors and thereby illuminate the specific role of EBV in the patho-epigenetics of both EBV-associated carcinomas. Furthermore, analysing the function of all newly described hypermethylated genes is of importance for understanding EBV-associated cancerogenesis.
3.8 Conclusion
The sequence-, promoter- or locus-specificity of the mechanisms targeting hypermethylation or hypomethylation to viral and cellular promoters in EBV-infected cells is most intriguing and needs further clarification. It should be sorted out which mechanisms are involved in the establishment of widespread hypomethylated blocks during the outgrowth of LCs, despite the expression of LMP1 and LMP2A which are capable of inducing cellular DNMTs. The mechanisms targeting hypermethylation at PRC target genes of embryonic stem cells in lymphomas, and targeting hypermethylation at non-PRC target genes in the highly methylated CIMP-cancer EBVaGC may clarify the reason for the apparently EBV-specific “epigenetic signature” . Further in-depth analyses of the overlaps and differences in the methylomes of EBV-associated malignancies and functional analyses of the hypermethylated genes are required to sort out the specific contribution of EBV in each case. Epigenetic marks and genes differentially methylated in EBV-associated neoplasms may also have a diagnostic and prognostic value. Because epigenetic changes are principally reversible, drugs which affect the epigenotype of an EBV-associated tumor may be exploited for cancer treatment.
References
Anastasiadou E, Boccellato F, Vincenti S, Rosato P, Bozzoni I, Frati L, Faggioni A, Presutti C, Trivedi P (2010) Epstein-Barr virus encoded LMP1 downregulates TCL1 oncogene through miR-29b. Oncogene 29:1316–1328
Anderton JA, Bose S, Vockerodt M, Vrzalikova K, Wei W, Kuo M, Helin K, Christensen J, Rowe M, Murray PG, Woodman CB (2011) The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s lymphoma. Oncogene 30:2037–2043
Araujo I, Foss HD, Bittencourt A, Hummel M, Demel G, Mendonca N, Herbst H, Stein H (1996) Expression of Epstein-Barr virus-gene products in Burkitt’s lymphoma in Northeast Brazil. Blood 87:5279–5286
Araujo I, Foss HD, Hummel M, Anagnostopoulos I, Barbosa HS, Bittencourt A, Stein H (1999) Frequent expansion of Epstein-Barr virus (EBV) infected cells in germinal centres of tonsils from an area with a high incidence of EBV-associated lymphoma. J Pathol 187:326–330
Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM (2012) An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 12:233–245
Arvey A, Tempera I, Lieberman PM (2013) Interpreting the Epstein-Barr Virus (EBV) epigenome using high-throughput data. Viruses 5:1042–1054
Au WY, Pang A, Chan EC, Chu KM, Shek TW, Kwong YL (2005) Epstein-barr virus-related gastric adenocarcinoma: an early secondary cancer post hemopoietic stem cell transplantation. Gastroenterology 129:2058–2063
Ayadi W, Karray-Hakim H, Khabir A, Feki L, Charfi S, Boudawara T, Ghorbel A, Daoud J, Frikha M, Busson P, Hammami A (2008) Aberrant methylation of p16, DLEC1, BLU and E-cadherin gene promoters in nasopharyngeal carcinoma biopsies from Tunisian patients. Anticancer Res 28:2161–2167
Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA (1998) EBV persistence in memory B cells in vivo. Immunity 9:395–404
Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C (1984) DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310:207–211
Bailey SD, Zhang X, Desai K, Aid M, Corradin O, Cowper-Sal Lari R, Akhtar-Zaidi B, Scacheri PC, Haibe-Kains B, Lupien M (2015) ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat Commun 2:6186
Banati F, Koroknai A, Salamon D, Takacs M, Minarovits-Kormuta S, Wolf H, Niller HH, Minarovits J (2008) CpG-methylation silences the activity of the RNA polymerase III transcribed EBER-1 promoter of Epstein-Barr virus. FEBS Lett 582:705–709
Barth S, Pfuhl T, Mamiani A, Ehses C, Roemer K, Kremmer E, Jaker C, Hock J, Meister G, Grasser FA (2008) Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic Acids Res 36:666–675
Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer 11:726–734
Boccellato F, Anastasiadou E, Rosato P, Kempkes B, Frati L, Faggioni A, Trivedi P (2007) EBNA2 interferes with the germinal center phenotype by downregulating BCL6 and TCL1 in non-Hodgkin’s lymphoma cells. J Virol 81:2274–2282
Borza CM, Hutt-Fletcher LM (2002) Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med 8:594–599
Brink AA, Dukers DF, van den Brule AJ, Oudejans JJ, Middeldorp JM, Meijer CJ, Jiwa M (1997) Presence of Epstein-Barr virus latency type III at the single cell level in post-transplantation lymphoproliferative disorders and AIDS related lymphomas. J Clin Pathol 50:911–918
Burke AP, Yen TS, Shekitka KM, Sobin LH (1990) Lymphoepithelial carcinoma of the stomach with Epstein-Barr virus demonstrated by polymerase chain reaction. Mod Pathol 3:377–380
Cai X, Schafer A, Lu S, Bilello JP, Desrosiers RC, Edwards R, Raab-Traub N, Cullen BR (2006) Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog 2:e23
Cai MY, Tong ZT, Zhu W, Wen ZZ, Rao HL, Kong LL, Guan XY, Kung HF, Zeng YX, Xie D (2011) H3K27me3 protein is a promising predictive biomarker of patients’ survival and chemoradioresistance in human nasopharyngeal carcinoma. Mol Med 17:1137–1145
Caliskan M, Cusanovich DA, Ober C, Gilad Y (2011) The effects of EBV transformation on gene expression levels and methylation profiles. Hum Mol Genet 20:1643–1652
Canaan A, Haviv I, Urban AE, Schulz VP, Hartman S, Zhang Z, Palejev D, Deisseroth AB, Lacy J, Snyder M, Gerstein M, Weissman SM (2009) EBNA1 regulates cellular gene expression by binding cellular promoters. Proc Natl Acad Sci U S A 106:22421–22426
Cancer Genome Atlas Research Network (2014) Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513:202–209
Challouf S, Ziadi S, Zaghdoudi R, Ksiaa F, Ben Gacem R, Trimeche M (2012) Patterns of aberrant DNA hypermethylation in nasopharyngeal carcinoma in Tunisian patients. Clin Chim Acta 413:795–802
Chang MS, Lee HS, Kim HS, Kim SH, Choi SI, Lee BL, Kim CW, Kim YI, Yang M, Kim WH (2003) Epstein-Barr virus and microsatellite instability in gastric carcinogenesis. J Pathol 199:447–452
Chau CM, Zhang XY, McMahon SB, Lieberman PM (2006) Regulation of Epstein-Barr virus latency type by the chromatin boundary factor CTCF. J Virol 80:5723–5732
Cheng Y, Geng H, Cheng SH, Liang P, Bai Y, Li J, Srivastava G, Ng MH, Fukagawa T, Wu X, Chan AT, Tao Q (2010) KRAB zinc finger protein ZNF382 is a proapoptotic tumor suppressor that represses multiple oncogenes and is commonly silenced in multiple carcinomas. Cancer Res 70:6516–6526
Cheung HW, Ching YP, Nicholls JM, Ling MT, Wong YC, Hui N, Cheung A, Tsao SW, Wang Q, Yeun PW, Lo KW, Jin DY, Wang X (2005) Epigenetic inactivation of CHFR in nasopharyngeal carcinoma through promoter methylation. Mol Carcinog 43:237–245
Chong JM, Sakuma K, Sudo M, Ushiku T, Uozaki H, Shibahara J, Nagai H, Funata N, Taniguchi H, Aburatani H, Fukayama M (2003) Global and non-random CpG-island methylation in gastric carcinoma associated with Epstein-Barr virus. Cancer Sci 94:76–80
Choy EY, Siu KL, Kok KH, Lung RW, Tsang CM, To KF, Kwong DL, Tsao SW, Jin DY (2008) An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med 205:2551–2560
Clarke PA, Schwemmle M, Schickinger J, Hilse K, Clemens MJ (1991) Binding of Epstein-Barr virus small RNA EBER-1 to the double-stranded RNA-activated protein kinase DAI. Nucleic Acids Res 19:243–248
Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, Cahn AP, Durham J, Heath P, Wray P, Pavitt R, Wilkinson J, Leversha M, Huckle E, Shaw-Smith CJ, Dunham A, Rhodes S, Schuster V, Porta G, Yin L, Serafini P, Sylla B, Zollo M, Franco B, Bolino A, Seri M, Lanyi A, Davis JR, Webster D, Harris A, Lenoir G, de St Basile G, Jones A, Behloradsky BH, Achatz H, Murken J, Fassler R, Sumegi J, Romeo G, Vaudin M, Ross MT, Meindl A, Bentley DR (1998) Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet 20:129–135
Coppotelli G, Mughal N, Callegari S, Sompallae R, Caja L, Luijsterburg MS, Dantuma NP, Moustakas A, Masucci MG (2013) The Epstein-Barr virus nuclear antigen-1 reprograms transcription by mimicry of high mobility group A proteins. Nucleic Acids Res 41:2950–2962
Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, Caligiuri MA, Yates A, Nishikawa R, Su Huang H, Petrelli NJ, Zhang X, O’Dorisio MS, Held WA, Cavenee WK, Plass C (2000) Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet 24:132–138
Cotter MA, Robertson ES (2000) Modulation of histone acetyltransferase activity through interaction of Epstein-Barr nuclear antigen 3C with prothymosin alpha. Mol Cell Biol 20:5722–5735
Cui Y, Ying Y, van Hasselt A, Ng KM, Yu J, Zhang Q, Jin J, Liu D, Rhim JS, Rha SY, Loyo M, Chan AT, Srivastava G, Tsao GS, Sellar GC, Sung JJ, Sidransky D, Tao Q (2008) OPCML is a broad tumor suppressor for multiple carcinomas and lymphomas with frequently epigenetic inactivation. PLoS One 3:e2990
Cui Y, Gao D, Linghu E, Zhan Q, Chen R, Brock MV, Herman JG, Guo M (2015) Epigenetic changes and functional study of HOXA11 in human gastric cancer. Epigenomics 7:201–213
Dai W, Cheung AK, Ko JM, Cheng Y, Zheng H, Ngan RK, Ng WT, Lee AW, Yau CC, Lee VH, Lung ML (2015) Comparative methylome analysis in solid tumors reveals aberrant methylation at chromosome 6p in nasopharyngeal carcinoma. Cancer Med 4:1079–1090
de The G (1984) Virus-associated lymphomas, leukaemias and immunodeficiencies in Africa. IARC Sci Publ 63:727–744
de The G (1985) The Epstein-Barr virus (EBV): a Rosetta Stone for understanding the role of viruses in immunopathological disorders and in human carcinogenesis. Biomed Pharmacother 39:49–51
Decaussin G, Sbih-Lammali F, de Turenne-Tessier M, Bouguermouh A, Ooka T (2000) Expression of BARF1 gene encoded by Epstein-Barr virus in nasopharyngeal carcinoma biopsies. Cancer Res 60:5584–5588
Degner SC, Wong TP, Jankevicius G, Feeney AJ (2009) Cutting edge: developmental stage-specific recruitment of cohesin to CTCF sites throughout immunoglobulin loci during B lymphocyte development. J Immunol 182:44–48
d’Herouel AF, Birgersdotter A, Werner M (2010) FR-like EBNA1 binding repeats in the human genome. Virology 405:524–529
Dolken L, Malterer G, Erhard F, Kothe S, Friedel CC, Suffert G, Marcinowski L, Motsch N, Barth S, Beitzinger M, Lieber D, Bailer SM, Hoffmann R, Ruzsics Z, Kremmer E, Pfeffer S, Zimmer R, Koszinowski UH, Grasser F, Meister G, Haas J (2010) Systematic analysis of viral and cellular microRNA targets in cells latently infected with human gamma-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 7:324–334
Dresang LR, Vereide DT, Sugden B (2009) Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J Virol 83:2930–2940
Du ZM, Hu LF, Wang HY, Yan LX, Zeng YX, Shao JY, Ernberg I (2011) Upregulation of MiR-155 in nasopharyngeal carcinoma is partly driven by LMP1 and LMP2A and downregulates a negative prognostic marker JMJD1A. PLoS One 6:e19137
Durst M, Gissmann L, Ikenberg H, zur Hausen H (1983) A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A 80:3812–3815
Dutton A, Woodman CB, Chukwuma MB, Last JI, Wei W, Vockerodt M, Baumforth KR, Flavell JR, Rowe M, Taylor AM, Young LS, Murray PG (2007) Bmi-1 is induced by the Epstein-Barr virus oncogene LMP1 and regulates the expression of viral target genes in Hodgkin lymphoma cells. Blood 109:2597–2603
Elliott J, Goodhew EB, Krug LT, Shakhnovsky N, Yoo L, Speck SH (2004) Variable methylation of the Epstein-Barr virus Wp EBNA gene promoter in B-lymphoblastoid cell lines. J Virol 78:14062–14065
Enomoto S, Maekita T, Tsukamoto T, Nakajima T, Nakazawa K, Tatematsu M, Ichinose M, Ushijima T (2007) Lack of association between CpG island methylator phenotype in human gastric cancers and methylation in their background non-cancerous gastric mucosae. Cancer Sci 98:1853–1861
Epstein MA, Achong BG, Barr YM (1964) Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1:702–703
Esteller M (2007a) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 8:286–298
Esteller M (2007b) Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet 16(Spec No 1):R50–R59
Evans AS, Rothfield NF, Niederman JC (1971) Raised antibody titres to E.B. virus in systemic lupus erythematosus. Lancet 1:167–168
Fejer G, Koroknai A, Banati F, Gyory I, Salamon D, Wolf H, Niller HH, Minarovits J (2008) Latency type-specific distribution of epigenetic marks at the alternative promoters Cp and Qp of Epstein-Barr virus. J Gen Virol 89:1364–1370
Fernandez AF, Rosales C, Lopez-Nieva P, Grana O, Ballestar E, Ropero S, Espada J, Melo SA, Lujambio A, Fraga MF, Pino I, Javierre B, Carmona FJ, Acquadro F, Steenbergen RD, Snijders PJ, Meijer CJ, Pineau P, Dejean A, Lloveras B, Capella G, Quer J, Buti M, Esteban JI, Allende H, Rodriguez-Frias F, Castellsague X, Minarovits J, Ponce J, Capello D, Gaidano G, Cigudosa JC, Gomez-Lopez G, Pisano DG, Valencia A, Piris MA, Bosch FX, Cahir-McFarland E, Kieff E, Esteller M (2009) The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res 19:438–451
Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearon DT (1984) Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci U S A 81:4510–4514
Finke J, Rowe M, Kallin B, Ernberg I, Rosen A, Dillner J, Klein G (1987) Monoclonal and polyclonal antibodies against Epstein-Barr virus nuclear antigen 5 (EBNA-5) detect multiple protein species in Burkitt’s lymphoma and lymphoblastoid cell lines. J Virol 61:3870–3878
Fuentes-Panana EM, Peng R, Brewer G, Tan J, Ling PD (2000) Regulation of the Epstein-Barr virus C promoter by AUF1 and the cyclic AMP/protein kinase A signaling pathway. J Virol 74:8166–8175
Fukuda M, Longnecker R (2007) Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway. J Virol 81:9299–9306
Gahn TA, Sugden B (1995) An EBNA-1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstein-Barr virus LMP gene. J Virol 69:2633–2636
Gerle B, Koroknai A, Fejer G, Bakos A, Banati F, Szenthe K, Wolf H, Niller HH, Minarovits J, Salamon D (2007) Acetylated histone H3 and H4 mark the upregulated LMP2A promoter of Epstein-Barr virus in lymphoid cells. J Virol 81:13242–13247
Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453:948–951
Gyory I, Minarovits J (2005) Epigenetic regulation of lymphoid specific gene sets. Biochem Cell Biol 83:286–295
Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA (2009) The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog 5:e1000496
Hamilton-Dutoit SJ, Raphael M, Audouin J, Diebold J, Lisse I, Pedersen C, Oksenhendler E, Marelle L, Pallesen G (1993) In situ demonstration of Epstein-Barr virus small RNAs (EBER 1) in acquired immunodeficiency syndrome-related lymphomas: correlation with tumor morphology and primary site. Blood 82:619–624
Hammerschmidt W, Sugden B (1989) Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature 340:393–397
Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, Briem E, Zhang K, Irizarry RA, Feinberg AP (2011) Increased methylation variation in epigenetic domains across cancer types. Nat Genet 43:768–775
Hansen KD, Sabunciyan S, Langmead B, Nagy N, Curley R, Klein G, Klein E, Salamon D, Feinberg AP (2014) Large-scale hypomethylated blocks associated with Epstein-Barr virus-induced B-cell immortalization. Genome Res 24:177–184
Harabuchi Y, Yamanaka N, Kataura A, Imai S, Kinoshita T, Mizuno F, Osato T (1990) Epstein-Barr virus in nasal T-cell lymphomas in patients with lethal midline granuloma. Lancet 335:128–130
Hawkins JB, Delgado-Eckert E, Thorley-Lawson DA, Shapiro M (2013) The cycle of EBV infection explains persistence, the sizes of the infected cell populations and which come under CTL regulation. PLoS Pathog 9:e1003685
He D, Zeng Q, Ren G, Xiang T, Qian Y, Hu Q, Zhu J, Hong S, Hu G (2012) Protocadherin8 is a functional tumor suppressor frequently inactivated by promoter methylation in nasopharyngeal carcinoma. Eur J Cancer Prev 21:569–575
He D, Zhang YW, Zhang NN, Zhou L, Chen JN, Jiang Y, Shao CK (2015) Aberrant gene promoter methylation of p16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein-Barr virus-associated gastric carcinomas. Med Oncol 32:525
Henle G, Henle W (1966) Immunofluorescence in cells derived from Burkitt’s lymphoma. J Bacteriol 91:1248–1256
Henle W, Diehl V, Kohn G, Zur Hausen H, Henle G (1967) Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science 157:1064–1065
Henle G, Henle W, Diehl V (1968) Relation of Burkitt’s tumor-associated herpes-type virus to infectious mononucleosis. Proc Natl Acad Sci U S A 59:94–101
Hernando H, Shannon-Lowe C, Islam AB, Al-Shahrour F, Rodriguez-Ubreva J, Rodriguez-Cortez VC, Javierre BM, Mangas C, Fernandez AF, Parra M, Delecluse HJ, Esteller M, Lopez-Granados E, Fraga MF, Lopez-Bigas N, Ballestar E (2013) The B cell transcription program mediates hypomethylation and overexpression of key genes in Epstein-Barr virus-associated proliferative conversion. Genome Biol 14:R3
Hernando H, Islam AB, Rodriguez-Ubreva J, Forne I, Ciudad L, Imhof A, Shannon-Lowe C, Ballestar E (2014) Epstein-Barr virus-mediated transformation of B cells induces global chromatin changes independent to the acquisition of proliferation. Nucleic Acids Res 42:249–263
Hino R, Uozaki H, Murakami N, Ushiku T, Shinozaki A, Ishikawa S, Morikawa T, Nakaya T, Sakatani T, Takada K, Fukayama M (2009) Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res 69:2766–2774
Hochberg D, Souza T, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA (2004) Acute infection with Epstein-Barr virus targets and overwhelms the peripheral memory B-cell compartment with resting, latently infected cells. J Virol 78:5194–5204
Holdorf MM, Cooper SB, Yamamoto KR, Miranda JJ (2011) Occupancy of chromatin organizers in the Epstein-Barr virus genome. Virology 415:1–5
Horikawa T, Yang J, Kondo S, Yoshizaki T, Joab I, Furukawa M, Pagano JS (2007) Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res 67:1970–1978
Hsu CY, Yi YH, Chang KP, Chang YS, Chen SJ, Chen HC (2014) The Epstein-Barr virus-encoded microRNA MiR-BART9 promotes tumor metastasis by targeting E-cadherin in nasopharyngeal carcinoma. PLoS Pathog 10:e1003974
Hughes DJ, Dickerson CA, Shaner MS, Sample CE, Sample JT (2011) trans-Repression of protein expression dependent on the Epstein-Barr virus promoter Wp during latency. J Virol 85:11435–11447
Hughes DJ, Marendy EM, Dickerson CA, Yetming KD, Sample CE, Sample JT (2012) Contributions of CTCF and DNA methyltransferases DNMT1 and DNMT3B to Epstein-Barr virus restricted latency. J Virol 86:1034–1045
Hui AB, Lo KW, Kwong J, Lam EC, Chan SY, Chow LS, Chan AS, Teo PM, Huang DP (2003) Epigenetic inactivation of TSLC1 gene in nasopharyngeal carcinoma. Mol Carcinog 38:170–178
Hurley EA, Thorley-Lawson DA (1988) B cell activation and the establishment of Epstein-Barr virus latency. J Exp Med 168:2059–2075
Hurley EA, Agger S, McNeil JA, Lawrence JB, Calendar A, Lenoir G, Thorley-Lawson DA (1991) When Epstein-Barr virus persistently infects B-cell lines, it frequently integrates. J Virol 65:1245–1254
Hutajulu SH, Indrasari SR, Indrawati LP, Harijadi A, Duin S, Haryana SM, Steenbergen RD, Greijer AE, Middeldorp JM (2011) Epigenetic markers for early detection of nasopharyngeal carcinoma in a high risk population. Mol Cancer 10:48
Hwang CF, Huang HY, Chen CH, Chien CY, Hsu YC, Li CF, Fang FM (2012) Enhancer of zeste homolog 2 overexpression in nasopharyngeal carcinoma: an independent poor prognosticator that enhances cell growth. Int J Radiat Oncol Biol Phys 82:597–604
Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T, Imai S, Fujieda M, Kawa K, Takada K (2009) Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J Exp Med 206:2091–2099
Jankelevich S, Kolman JL, Bodnar JW, Miller G (1992) A nuclear matrix attachment region organizes the Epstein-Barr viral plasmid in Raji cells into a single DNA domain. EMBO J 11:1165–1176
Jansson A, Masucci M, Rymo L (1992) Methylation of discrete sites within the enhancer region regulates the activity of the Epstein-Barr virus BamHI W promoter in Burkitt lymphoma lines. J Virol 66:62–69
Jin H, Wang X, Ying J, Wong AH, Cui Y, Srivastava G, Shen ZY, Li EM, Zhang Q, Jin J, Kupzig S, Chan AT, Cullen PJ, Tao Q (2007a) Epigenetic silencing of a Ca(2+)-regulated Ras GTPase-activating protein RASAL defines a new mechanism of Ras activation in human cancers. Proc Natl Acad Sci U S A 104:12353–12358
Jin H, Wang X, Ying J, Wong AH, Li H, Lee KY, Srivastava G, Chan AT, Yeo W, Ma BB, Putti TC, Lung ML, Shen ZY, Xu LY, Langford C, Tao Q (2007b) Epigenetic identification of ADAMTS18 as a novel 16q23.1 tumor suppressor frequently silenced in esophageal, nasopharyngeal and multiple other carcinomas. Oncogene 26:7490–7498
Johannsen E, Koh E, Mosialos G, Tong X, Kieff E, Grossman SR (1995) Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by J kappa and PU.1. J Virol 69:253–262
Johannsen E, Miller CL, Grossman SR, Kieff E (1996) EBNA-2 and EBNA-3C extensively and mutually exclusively associate with RBPJkappa in Epstein-Barr virus-transformed B lymphocytes. J Virol 70:4179–4183
Jourdan N, Jobart-Malfait A, Dos Reis G, Quignon F, Piolot T, Klein C, Tramier M, Coppey-Moisan M, Marechal V (2012) Live-cell imaging reveals multiple interactions between Epstein-Barr virus nuclear antigen 1 and cellular chromatin during interphase and mitosis. J Virol 86:5314–5329
Jung YJ, Choi H, Kim H, Lee SK (2014) MicroRNA miR-BART20-5p stabilizes Epstein-Barr virus latency by directly targeting BZLF1 and BRLF1. J Virol 88:9027–9037
Kalla M, Hammerschmidt W (2012) Human B cells on their route to latent infection – early but transient expression of lytic genes of Epstein-Barr virus. Eur J Cell Biol 91:65–69
Kalter SP, Riggs SA, Cabanillas F, Butler JJ, Hagemeister FB, Mansell PW, Newell GR, Velasquez WS, Salvador P, Barlogie B, Rios A, Hersh EM (1985) Aggressive Non-Hodgkin’s lymphomas in immunocompromised homosexual males. Blood 66:655–659
Kanda T, Miyata M, Kano M, Kondo S, Yoshizaki T, Iizasa H (2015) Clustered microRNAs of the Epstein-Barr virus cooperatively downregulate an epithelial cell-specific metastasis suppressor. J Virol 89:2684–2697
Kaneda A, Matsusaka K, Aburatani H, Fukayama M (2012) Epstein-Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res 72:3445–3450
Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, Ro JY (2002) Epstein-Barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol 160:787–794
Kang GH, Lee S, Cho NY, Gandamihardja T, Long TI, Weisenberger DJ, Campan M, Laird PW (2008) DNA methylation profiles of gastric carcinoma characterized by quantitative DNA methylation analysis. Lab Invest 88:161–170
Kang H, Wiedmer A, Yuan Y, Robertson E, Lieberman PM (2011) Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog 7:e1002140
Kelly G, Bell A, Rickinson A (2002) Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat Med 8:1098–1104
Kelly GL, Milner AE, Baldwin GS, Bell AI, Rickinson AB (2006) Three restricted forms of Epstein-Barr virus latency counteracting apoptosis in c-myc-expressing Burkitt lymphoma cells. Proc Natl Acad Sci U S A 103:14935–14940
Kim DN, Chae HS, Oh ST, Kang JH, Park CH, Park WS, Takada K, Lee JM, Lee WK, Lee SK (2007) Expression of viral microRNAs in Epstein-Barr virus-associated gastric carcinoma. J Virol 81:1033–1036
Kis LL, Salamon D, Persson EK, Nagy N, Scheeren FA, Spits H, Klein G, Klein E (2010) IL-21 imposes a type II EBV gene expression on type III and type I B cells by the repression of C- and activation of LMP-1-promoter. Proc Natl Acad Sci U S A 107:872–877
Klein G (1987) In defense of the “old” Burkitt lymphoma scenario. In: Klein G (ed) Advances in viral oncology. Raven Press, New York, pp 207–211
Klein E, Nagy N (2010) Restricted expression of EBV encoded proteins in in vitro infected CLL cells. Semin Cancer Biol 20:410–415
Klein E, Nagy N, Rasul AE (2013) EBV genome carrying B lymphocytes that express the nuclear protein EBNA-2 but not LMP-1: type IIb latency. Oncoimmunology 2:e23035
Knight JS, Lan K, Subramanian C, Robertson ES (2003) Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J Virol 77:4261–4272
Kong QL, Hu LJ, Cao JY, Huang YJ, Xu LH, Liang Y, Xiong D, Guan S, Guo BH, Mai HQ, Chen QY, Zhang X, Li MZ, Shao JY, Qian CN, Xia YF, Song LB, Zeng YX, Zeng MS (2010) Epstein-Barr virus-encoded LMP2A induces an epithelial-mesenchymal transition and increases the number of side population stem-like cancer cells in nasopharyngeal carcinoma. PLoS Pathog 6:e1000940
Kreck B, Richter J, Ammerpohl O, Barann M, Esser D, Petersen BS, Vater I, Murga Penas EM, Bormann Chung CA, Seisenberger S, Lee Boyd V, Smallwood S, Drexler HG, Macleod RA, Hummel M, Krueger F, Hasler R, Schreiber S, Rosenstiel P, Franke A, Siebert R (2013) Base-pair resolution DNA methylome of the EBV-positive Endemic Burkitt lymphoma cell line DAUDI determined by SOLiD bisulfite-sequencing. Leukemia 27:1751–1753
Kurth J, Spieker T, Wustrow J, Strickler GJ, Hansmann LM, Rajewsky K, Kuppers R (2000) EBV-infected B cells in infectious mononucleosis: viral strategies for spreading in the B cell compartment and establishing latency. Immunity 13:485–495
Kurth J, Hansmann ML, Rajewsky K, Kuppers R (2003) Epstein-Barr virus-infected B cells expanding in germinal centers of infectious mononucleosis patients do not participate in the germinal center reaction. Proc Natl Acad Sci U S A 100:4730–4735
Kwong J, Lo KW, To KF, Teo PM, Johnson PJ, Huang DP (2002) Promoter hypermethylation of multiple genes in nasopharyngeal carcinoma. Clin Cancer Res 8:131–137
Kwong J, Lo KW, Chow LS, Chan FL, To KF, Huang DP (2005a) Silencing of the retinoid response gene TIG1 by promoter hypermethylation in nasopharyngeal carcinoma. Int J Cancer 113:386–392
Kwong J, Lo KW, Chow LS, To KF, Choy KW, Chan FL, Mok SC, Huang DP (2005b) Epigenetic silencing of cellular retinol-binding proteins in nasopharyngeal carcinoma. Neoplasia 7:67–74
Lan J, Tai HC, Lee SW, Chen TJ, Huang HY, Li CF (2014) Deficiency in expression and epigenetic DNA Methylation of ASS1 gene in nasopharyngeal carcinoma: negative prognostic impact and therapeutic relevance. Tumour Biol 35:161–169
Latour S, Veillette A (2003) Molecular and immunological basis of X-linked lymphoproliferative disease. Immunol Rev 192:212–224
Laux G, Perricaudet M, Farrell PJ (1988) A spliced Epstein-Barr virus gene expressed in immortalized lymphocytes is created by circularization of the linear viral genome. EMBO J 7:769–774
Lee KY, Geng H, Ng KM, Yu J, van Hasselt A, Cao Y, Zeng YX, Wong AH, Wang X, Ying J, Srivastava G, Lung ML, Wang LD, Kwok TT, Levi BZ, Chan AT, Sung JJ, Tao Q (2008) Epigenetic disruption of interferon-gamma response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene 27:5267–5276
Lei T, Yuen KS, Xu R, Tsao SW, Chen H, Li M, Kok KH, Jin DY (2013) Targeting of DICE1 tumor suppressor by Epstein-Barr virus-encoded miR-BART3* microRNA in nasopharyngeal carcinoma. Int J Cancer 133:79–87
Lenoir GM, Bornkamm G (1987) Burkitt’s Lymphoma, a human cancer model for the study of the multistep development of cancer: proposal for a new scenario. In: Klein G (ed) Advances in viral oncology. Raven Press, New York, pp 173–206
Leonard S, Wei W, Anderton J, Vockerodt M, Rowe M, Murray PG, Woodman CB (2011) Epigenetic and transcriptional changes which follow Epstein-Barr virus infection of germinal center B cells and their relevance to the pathogenesis of Hodgkin’s lymphoma. J Virol 85:9568–9577
Lewin N, Minarovits J, Weber G, Ehlin-Henriksson B, Wen T, Mellstedt H, Klein G, Klein E (1991) Clonality and methylation status of the Epstein-Barr virus (EBV) genomes in in vivo-infected EBV-carrying chronic lymphocytic leukemia (CLL) cell lines. Int J Cancer 48:62–66
Li H, Minarovits J (2003) Host cell-dependent expression of latent Epstein-Barr virus genomes: regulation by DNA methylation. Adv Cancer Res 89:133–156
Li G, Wu Z, Peng Y, Liu X, Lu J, Wang L, Pan Q, He ML, Li XP (2010) MicroRNA-10b induced by Epstein-Barr virus-encoded latent membrane protein-1 promotes the metastasis of human nasopharyngeal carcinoma cells. Cancer Lett 299:29–36
Li LL, Shu XS, Wang ZH, Cao Y, Tao Q (2011) Epigenetic disruption of cell signaling in nasopharyngeal carcinoma. Chin J Cancer 30:231–239
Li L, Zhang Y, Fan Y, Sun K, Su X, Du Z, Tsao SW, Loh TK, Sun H, Chan AT, Zeng YX, Chan WY, Chan FK, Tao Q (2014a) Characterization of the nasopharyngeal carcinoma methylome identifies aberrant disruption of key signaling pathways and methylated tumor suppressor genes. Epigenomics 7:155–173
Li L, Zhang Y, Guo BB, Chan FK, Tao Q (2014b) Oncogenic induction of cellular high CpG methylation by Epstein-Barr virus in malignant epithelial cells. Chin J Cancer 33:604–608
Lin IG, Tomzynski TJ, Ou Q, Hsieh CL (2000) Modulation of DNA binding protein affinity directly affects target site demethylation. Mol Cell Biol 20:2343–2349
Lin A, Wang S, Nguyen T, Shire K, Frappier L (2008) The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J Virol 82:12009–12019
Ling PD, Rawlins DR, Hayward SD (1993) The Epstein-Barr virus immortalizing protein EBNA-2 is targeted to DNA by a cellular enhancer-binding protein. Proc Natl Acad Sci U S A 90:9237–9241
Ling PD, Peng RS, Nakajima A, Yu JH, Tan J, Moses SM, Yang WH, Zhao B, Kieff E, Bloch KD, Bloch DB (2005) Mediation of Epstein-Barr virus EBNA-LP transcriptional coactivation by Sp100. EMBO J 24:3565–3575
Liu H, Zhang L, Niu Z, Zhou M, Peng C, Li X, Deng T, Shi L, Tan Y, Li G (2008) Promoter methylation inhibits BRD7 expression in human nasopharyngeal carcinoma cells. BMC Cancer 8:253
Liu X, Wang Y, Wang X, Sun Z, Li L, Tao Q, Luo B (2013) Epigenetic silencing of WNT5A in Epstein-Barr virus-associated gastric carcinoma. Arch Virol 158:123–132
Lo KW, Cheung ST, Leung SF, van Hasselt A, Tsang YS, Mak KF, Chung YF, Woo JK, Lee JC, Huang DP (1996) Hypermethylation of the p16 gene in nasopharyngeal carcinoma. Cancer Res 56:2721–2725
Lo KW, Kwong J, Hui AB, Chan SY, To KF, Chan AS, Chow LS, Teo PM, Johnson PJ, Huang DP (2001) High frequency of promoter hypermethylation of RASSF1A in nasopharyngeal carcinoma. Cancer Res 61:3877–3881
Lo KW, Tsang YS, Kwong J, To KF, Teo PM, Huang DP (2002) Promoter hypermethylation of the EDNRB gene in nasopharyngeal carcinoma. Int J Cancer 98:651–655
Lo AK, To KF, Lo KW, Lung RW, Hui JW, Liao G, Hayward SD (2007) Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc Natl Acad Sci U S A 104:16164–16169
Lo KW, Chung GT, To KF (2012) Deciphering the molecular genetic basis of NPC through molecular, cytogenetic, and epigenetic approaches. Semin Cancer Biol 22:79–86
Lo KW, Chung GT, To K (2013) Acquired genetic and epigenetic alterations in nasopharyngeal carcinoma. In: Busson P (ed) Nasopharyngeal carcinoma. Springer, New York, pp 61–81
Longnecker R, Kieff E, Cohen JI (2013) Epstein Barr virus. In: Knipe DM, Howley PM (eds) Fields virology, 6th edn. Lippincott Williams & Wilkins, Philadelphia, pp 1898–1959
Lu F, Wikramasinghe P, Norseen J, Tsai K, Wang P, Showe L, Davuluri RV, Lieberman PM (2010) Genome-wide analysis of host-chromosome binding sites for Epstein-Barr Virus Nuclear Antigen 1 (EBNA1). Virol J 7:262
Lu J, Murakami M, Verma SC, Cai Q, Haldar S, Kaul R, Wasik MA, Middeldorp J, Robertson ES (2011) Epstein-Barr Virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 410:64–75
Lu F, Tempera I, Lee HT, Dewispelaere K, Lieberman PM (2014) EBNA1 binding and epigenetic regulation of gastrokine tumor suppressor genes in gastric carcinoma cells. Virol J 11:12
Lubega J (2007) T-helper 1 versus T-helper 2 lymphocyte immunodysregulation is the central factor in genesis of Burkitt lymphoma: hypothesis. Infect Agent Cancer 2:10
Lung HL, Lo PH, Xie D, Apte SS, Cheung AK, Cheng Y, Law EW, Chua D, Zeng YX, Tsao SW, Stanbridge EJ, Lung ML (2008) Characterization of a novel epigenetically-silenced, growth-suppressive gene, ADAMTS9, and its association with lymph node metastases in nasopharyngeal carcinoma. Int J Cancer 123:401–408
Lung RW, Tong JH, Sung YM, Leung PS, Ng DC, Chau SL, Chan AW, Ng EK, Lo KW, To KF (2009) Modulation of LMP2A expression by a newly identified Epstein-Barr virus-encoded microRNA miR-BART22. Neoplasia 11:1174–1184
Ma SD, Yu X, Mertz JE, Gumperz JE, Reinheim E, Zhou Y, Tang W, Burlingham WJ, Gulley ML, Kenney SC (2012) An Epstein-Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J Virol 86:7976–7987
Mancao C, Hammerschmidt W (2007) Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 110:3715–3721
Mannick JB, Cohen JI, Birkenbach M, Marchini A, Kieff E (1991) The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. J Virol 65:6826–6837
Marquitz AR, Mathur A, Nam CS, Raab-Traub N (2011) The Epstein-Barr Virus BART microRNAs target the pro-apoptotic protein Bim. Virology 412:392–400
Martin-Subero JI, Ammerpohl O, Bibikova M, Wickham-Garcia E, Agirre X, Alvarez S, Bruggemann M, Bug S, Calasanz MJ, Deckert M, Dreyling M, Du MQ, Durig J, Dyer MJ, Fan JB, Gesk S, Hansmann ML, Harder L, Hartmann S, Klapper W, Kuppers R, Montesinos-Rongen M, Nagel I, Pott C, Richter J, Roman-Gomez J, Seifert M, Stein H, Suela J, Trumper L, Vater I, Prosper F, Haferlach C, Cruz Cigudosa J, Siebert R (2009a) A comprehensive microarray-based DNA methylation study of 367 hematological neoplasms. PLoS One 4:e6986
Martin-Subero JI, Kreuz M, Bibikova M, Bentink S, Ammerpohl O, Wickham-Garcia E, Rosolowski M, Richter J, Lopez-Serra L, Ballestar E, Berger H, Agirre X, Bernd HW, Calvanese V, Cogliatti SB, Drexler HG, Fan JB, Fraga MF, Hansmann ML, Hummel M, Klapper W, Korn B, Kuppers R, Macleod RA, Moller P, Ott G, Pott C, Prosper F, Rosenwald A, Schwaenen C, Schubeler D, Seifert M, Sturzenhofecker B, Weber M, Wessendorf S, Loeffler M, Trumper L, Stein H, Spang R, Esteller M, Barker D, Hasenclever D, Siebert R, Molecular Mechanisms in Malignant Lymphomas Network Project of the Deutsche, K (2009b) New insights into the biology and origin of mature aggressive B-cell lymphomas by combined epigenomic, genomic, and transcriptional profiling. Blood 113:2488–2497
Maruo S, Zhao B, Johannsen E, Kieff E, Zou J, Takada K (2011) Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc Natl Acad Sci U S A 108:1919–1924
Masucci MG, Contreras-Salazar B, Ragnar E, Falk K, Minarovits J, Ernberg I, Klein G (1989) 5-Azacytidine up regulates the expression of Epstein-Barr virus nuclear antigen 2 (EBNA-2) through EBNA-6 and latent membrane protein in the Burkitt’s lymphoma line rael. J Virol 63:3135–3141
Matsusaka K, Kaneda A, Nagae G, Ushiku T, Kikuchi Y, Hino R, Uozaki H, Seto Y, Takada K, Aburatani H, Fukayama M (2011) Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res 71:7187–7197
McClain KL, Leach CT, Jenson HB, Joshi VV, Pollock BH, Parmley RT, DiCarlo FJ, Chadwick EG, Murphy SB (1995) Association of Epstein-Barr virus with leiomyosarcomas in children with AIDS. N Engl J Med 332:12–18
McClellan MJ, Wood CD, Ojeniyi O, Cooper TJ, Kanhere A, Arvey A, Webb HM, Palermo RD, Harth-Hertle ML, Kempkes B, Jenner RG, West MJ (2013) Modulation of enhancer looping and differential gene targeting by Epstein-Barr virus transcription factors directs cellular reprogramming. PLoS Pathog 9:e1003636
Merchant M, Caldwell RG, Longnecker R (2000) The LMP2A ITAM is essential for providing B cells with development and survival signals in vivo. J Virol 74:9115–9124
Minarovits J (2006) Epigenotypes of latent herpesvirus genomes. Curr Top Microbiol Immunol 310:61–80
Minarovits J, Hu LF, Marcsek Z, Minarovits-Kormuta S, Klein G, Ernberg I (1992) RNA polymerase III-transcribed EBER 1 and 2 transcription units are expressed and hypomethylated in the major Epstein-Barr virus-carrying cell types. J Gen Virol 73(Pt 7):1687–1692
Minarovits J, Hu LF, Imai S, Harabuchi Y, Kataura A, Minarovits-Kormuta S, Osato T, Klein G (1994) Clonality, expression and methylation patterns of the Epstein-Barr virus genomes in lethal midline granulomas classified as peripheral angiocentric T cell lymphomas. J Gen Virol 75(Pt 1):77–84
Miyashita EM, Yang B, Babcock GJ, Thorley-Lawson DA (1997) Identification of the site of Epstein-Barr virus persistence in vivo as a resting B cell. J Virol 71:4882–4891
Mo Y, Midorikawa K, Zhang Z, Zhou X, Ma N, Huang G, Hiraku Y, Oikawa S, Murata M (2012) Promoter hypermethylation of Ras-related GTPase gene RRAD inactivates a tumor suppressor function in nasopharyngeal carcinoma. Cancer Lett 323:147–154
Moore GE, Gerner RE, Franklin HA (1967) Culture of normal human leukocytes. JAMA 199:519–524
Morissette G, Flamand L (2010) Herpesviruses and chromosomal integration. J Virol 84:12100–12109
Motsch N, Pfuhl T, Mrazek J, Barth S, Grasser FA (2007) Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) induces the expression of the cellular microRNA miR-146a. RNA Biol 4:131–137
Munz C (2014) Dendritic cells during Epstein Barr virus infection. Front Microbiol 5:308
Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O (2009) Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5:376–385
Nagy N, Adori M, Rasul A, Heuts F, Salamon D, Ujvari D, Madapura HS, Leveau B, Klein G, Klein E (2012) Soluble factors produced by activated CD4+ T cells modulate EBV latency. Proc Natl Acad Sci U S A 109:1512–1517
Navari M, Fuligni F, Laginestra MA, Etebari M, Ambrosio MR, Sapienza MR, Rossi M, De Falco G, Gibellini D, Tripodo C, Pileri SA, Leoncini L, Piccaluga PP (2014) Molecular signature of Epstein Barr virus-positive Burkitt lymphoma and post-transplant lymphoproliferative disorder suggest different roles for Epstein Barr virus. Front Microbiol 5:728
Niedobitek G, Agathanggelou A, Herbst H, Whitehead L, Wright DH, Young LS (1997) Epstein-Barr virus (EBV) infection in infectious mononucleosis: virus latency, replication and phenotype of EBV-infected cells. J Pathol 182:151–159
Niller HH, Minarovits J (2012) Similarities between the Epstein-Barr virus (EBV) nuclear protein EBNA1 and the pioneer transcription factor FoxA: is EBNA1 a “bookmarking” oncoprotein that alters the host cell epigenotype? Pathogens 1:37–51
Niller HH, Salamon D, Ilg K, Koroknai A, Banati F, Bauml G, Rucker O, Schwarzmann F, Wolf H, Minarovits J (2003) The in vivo binding site for oncoprotein c-Myc in the promoter for Epstein-Barr virus (EBV) encoding RNA (EBER) 1 suggests a specific role for EBV in lymphomagenesis. Med Sci Monit 9:HY1–HY9
Niller HH, Salamon D, Banati F, Schwarzmann F, Wolf H, Minarovits J (2004a) The LCR of EBV makes Burkitt’s lymphoma endemic. Trends Microbiol 12:495–499
Niller HH, Salamon D, Ilg K, Koroknai A, Banati F, Schwarzmann F, Wolf H, Minarovits J (2004b) EBV-associated neoplasms: alternative pathogenetic pathways. Med Hypotheses 62:387–391
Niller HH, Salamon D, Rahmann S, Ilg K, Koroknai A, Banati F, Schwarzmann F, Wolf H, Minarovits J (2004c) A 30 kb region of the Epstein-Barr virus genome is colinear with the rearranged human immunoglobulin gene loci: implications for a “ping-pong evolution” model for persisting viruses and their hosts. A review. Acta Microbiol Immunol Hung 51:469–484
Niller HH, Wolf H, Minarovits J (2007) Epstein-Barr virus. In: Minarovits J, Gonczol E, Valyi-Nagy T (eds) Latency strategies of herpesviruses. Springer, New York, pp 154–191
Niller HH, Wolf H, Minarovits J (2009) Epigenetic dysregulation of the host cell genome in Epstein-Barr virus-associated neoplasia. Semin Cancer Biol 19:158–164
Niller HH, Wolf H, Ay E, Minarovits J (2011) Epigenetic dysregulation of Epstein-Barr virus latency and development of autoimmune disease. Adv Exp Med Biol 711:82–102
Niller HH, Banati F, Ay E, Minarovits J (2012) Epigenetic changes in virus-associated neoplasms. In: Minarovits J, Niller HH (eds) Patho-epigenetics of disease. Springer, New York, pp 179–225
Niller HH, Banati F, Minarovits J (2014a) Epigenetic alterations in nasopharyngeal carcinoma and Epstein-Barr virus (EBV) associated gastric carcinoma: a lesson in contrasts. J Nasopharyng Carcinoma 1:e9
Niller HH, Szenthe K, Minarovits J (2014b) Epstein-Barr virus-host cell interactions: an epigenetic dialog? Front Genet 5:367
Niller HH, Tarnai Z, Decsi G, Zsedenyi A, Banati F, Minarovits J (2014c) Role of epigenetics in EBV regulation and pathogenesis. Future Microbiol 9:747–756
Nilsson K, Klein G, Henle W, Henle G (1971) The establishment of lymphoblastoid lines from adult and fetal human lymphoid tissue and its dependence on EBV. Int J Cancer 8:443–450
O’Neil JD, Owen TJ, Wood VH, Date KL, Valentine R, Chukwuma MB, Arrand JR, Dawson CW, Young LS (2008) Epstein-Barr virus-encoded EBNA1 modulates the AP-1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J Gen Virol 89:2833–2842
Oudejans JJ, Jiwa M, van den Brule AJ, Grasser FA, Horstman A, Vos W, Kluin PM, van der Valk P, Walboomers JM, Meijer CJ (1995) Detection of heterogeneous Epstein-Barr virus gene expression patterns within individual post-transplantation lymphoproliferative disorders. Am J Pathol 147:923–933
Pajic A, Polack A, Staege MS, Spitkovsky D, Baier B, Bornkamm GW, Laux G (2001) Elevated expression of c-myc in lymphoblastoid cells does not support an Epstein-Barr virus latency III-to-I switch. J Gen Virol 82:3051–3055
Panagopoulos D, Victoratos P, Alexiou M, Kollias G, Mosialos G (2004) Comparative analysis of signal transduction by CD40 and the Epstein-Barr virus oncoprotein LMP1 in vivo. J Virol 78:13253–13261
Paschos K, Parker GA, Watanatanasup E, White RE, Allday MJ (2012) BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res 40:7233–7246
Peng D, Ren CP, Yi HM, Zhou L, Yang XY, Li H, Yao KT (2006) Genetic and epigenetic alterations of DLC-1, a candidate tumor suppressor gene, in nasopharyngeal carcinoma. Acta Biochim Biophys Sin (Shanghai) 38:349–355
Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, John B, Enright AJ, Marks D, Sander C, Tuschl T (2004) Identification of virus-encoded microRNAs. Science 304:734–736
Polack A, Hortnagel K, Pajic A, Christoph B, Baier B, Falk M, Mautner J, Geltinger C, Bornkamm GW, Kempkes B (1996) c-myc activation renders proliferation of Epstein-Barr virus (EBV)-transformed cells independent of EBV nuclear antigen 2 and latent membrane protein 1. Proc Natl Acad Sci U S A 93:10411–10416
Pope JH, Horne MK, Scott W (1968) Transformation of foetal human keukocytes in vitro by filtrates of a human leukaemic cell line containing herpes-like virus. Int J Cancer 3:857–866
Portal D, Rosendorff A, Kieff E (2006) Epstein-Barr nuclear antigen leader protein coactivates transcription through interaction with histone deacetylase 4. Proc Natl Acad Sci U S A 103:19278–19283
Qiu GH, Tan LK, Loh KS, Lim CY, Srivastava G, Tsai ST, Tsao SW, Tao Q (2004) The candidate tumor suppressor gene BLU, located at the commonly deleted region 3p21.3, is an E2F-regulated, stress-responsive gene and inactivated by both epigenetic and genetic mechanisms in nasopharyngeal carcinoma. Oncogene 23:4793–4806
Qu L, Rowe DT (1992) Epstein-Barr virus latent gene expression in uncultured peripheral blood lymphocytes. J Virol 66:3715–3724
Raab-Traub N, Flynn K (1986) The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell 47:883–889
Rasul E, Salamon D, Nagy N, Leveau B, Banati F, Szenthe K, Koroknai A, Minarovits J, Klein G, Klein E (2014) The MEC1 and MEC2 lines represent two CLL subclones in different stages of progression towards prolymphocytic leukemia. PLoS One 9:e106008
Rickinson AB (2014) Co-infections, inflammation and oncogenesis: future directions for EBV research. Semin Cancer Biol 26:99–115
Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA (2012) EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J 31:2207–2221
Rossi G, Bonetti F (2004) EBV and Burkitt’s lymphoma. N Engl J Med 350:2621
Roughan JE, Torgbor C, Thorley-Lawson DA (2010) Germinal center B cells latently infected with Epstein-Barr virus proliferate extensively but do not increase in number. J Virol 84:1158–1168
Rowe M, Rowe DT, Gregory CD, Young LS, Farrell PJ, Rupani H, Rickinson AB (1987) Differences in B cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt’s lymphoma cells. EMBO J 6:2743–2751
Ruf IK, Rhyne PW, Yang C, Cleveland JL, Sample JT (2000) Epstein-Barr virus small RNAs potentiate tumorigenicity of Burkitt lymphoma cells independently of an effect on apoptosis. J Virol 74:10223–10228
Sakuma K, Chong JM, Sudo M, Ushiku T, Inoue Y, Shibahara J, Uozaki H, Nagai H, Fukayama M (2004) High-density methylation of p14ARF and p16INK4A in Epstein-Barr virus-associated gastric carcinoma. Int J Cancer 112:273–278
Salamon D, Takacs M, Myohanen S, Marcsek Z, Berencsi G, Minarovits J (2000) De novo DNA methylation at nonrandom founder sites 5′ from an unmethylated minimal origin of DNA replication in latent Epstein-Barr virus genomes. Biol Chem 381:95–105
Salamon D, Takacs M, Ujvari D, Uhlig J, Wolf H, Minarovits J, Niller HH (2001) Protein-DNA binding and CpG methylation at nucleotide resolution of latency-associated promoters Qp, Cp, and LMP1p of Epstein-Barr virus. J Virol 75:2584–2596
Salamon D, Takacs M, Schwarzmann F, Wolf H, Minarovits J, Niller HH (2003) High-resolution methylation analysis and in vivo protein-DNA binding at the promoter of the viral oncogene LMP2A in B cell lines carrying latent Epstein-Barr virus genomes. Virus Genes 27:57–66
Salamon D, Banati F, Koroknai A, Ravasz M, Szenthe K, Bathori Z, Bakos A, Niller HH, Wolf H, Minarovits J (2009) Binding of CCCTC-binding factor in vivo to the region located between Rep* and C-promoter of Epstein-Barr virus is unaffected by CpG methylation and does not correlate with Cp activity. J Gen Virol 90:1183–1189
Sarkari F, Sanchez-Alcaraz T, Wang S, Holowaty MN, Sheng Y, Frappier L (2009) EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog 5:e1000624
Scholle F, Bendt KM, Raab-Traub N (2000) Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J Virol 74:10681–10689
Seo SY, Kim EO, Jang KL (2008) Epstein-Barr virus latent membrane protein 1 suppresses the growth-inhibitory effect of retinoic acid by inhibiting retinoic acid receptor-beta2 expression via DNA methylation. Cancer Lett 270:66–76
Seto E, Yang L, Middeldorp J, Sheen TS, Chen JY, Fukayama M, Eizuru Y, Ooka T, Takada K (2005) Epstein-Barr virus (EBV)-encoded BARF1 gene is expressed in nasopharyngeal carcinoma and EBV-associated gastric carcinoma tissues in the absence of lytic gene expression. J Med Virol 76:82–88
Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W (2010) Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog 6:e1001063
Shibata D, Tokunaga M, Uemura Y, Sato E, Tanaka S, Weiss LM (1991) Association of Epstein-Barr virus with undifferentiated gastric carcinomas with intense lymphoid infiltration. Lymphoepithelioma-like carcinoma. Am J Pathol 139:469–474
Skalska L, White RE, Franz M, Ruhmann M, Allday MJ (2010) Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog 6:e1000951
Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, Nusbaum JD, Feederle R, Delecluse HJ, Luftig MA, Tuschl T, Ohler U, Cullen BR (2012) The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog 8:e1002484
Smith P (2001) Epstein-Barr virus complementary strand transcripts (CSTs/BARTs) and cancer. Semin Cancer Biol 11:469–476
Smith PR, de Jesus O, Turner D, Hollyoake M, Karstegl CE, Griffin BE, Karran L, Wang Y, Hayward SD, Farrell PJ (2000) Structure and coding content of CST (BART) family RNAs of Epstein-Barr virus. J Virol 74:3082–3092
Sriuranpong V, Mutirangura A, Gillespie JW, Patel V, Amornphimoltham P, Molinolo AA, Kerekhanjanarong V, Supanakorn S, Supiyaphun P, Rangdaeng S, Voravud N, Gutkind JS (2004) Global gene expression profile of nasopharyngeal carcinoma by laser capture microdissection and complementary DNA microarrays. Clin Cancer Res 10:4944–4958
Strockbine LD, Cohen JI, Farrah T, Lyman SD, Wagener F, DuBose RF, Armitage RJ, Spriggs MK (1998) The Epstein-Barr virus BARF1 gene encodes a novel, soluble colony-stimulating factor-1 receptor. J Virol 72:4015–4021
Sudo M, Chong JM, Sakuma K, Ushiku T, Uozaki H, Nagai H, Funata N, Matsumoto Y, Fukayama M (2004) Promoter hypermethylation of E-cadherin and its abnormal expression in Epstein-Barr virus-associated gastric carcinoma. Int J Cancer 109:194–199
Sugden B, Warren N (1989) A promoter of Epstein-Barr virus that can function during latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA replication during latent infection. J Virol 63:2644–2649
Sugiura M, Imai S, Tokunaga M, Koizumi S, Uchizawa M, Okamoto K, Osato T (1996) Transcriptional analysis of Epstein-Barr virus gene expression in EBV-positive gastric carcinoma: unique viral latency in the tumour cells. Br J Cancer 74:625–631
Sumaya CV, Myers LW, Ellison GW (1980) Epstein-Barr virus antibodies in multiple sclerosis. Arch Neurol 37:94–96
Takacs M, Salamon D, Myohanen S, Li H, Segesdi J, Ujvari D, Uhlig J, Niller HH, Wolf H, Berencsi G, Minarovits J (2001) Epigenetics of latent Epstein-Barr virus genomes: high resolution methylation analysis of the bidirectional promoter region of latent membrane protein 1 and 2B genes. Biol Chem 382:699–705
Takacs M, Segesdi J, Banati F, Koroknai A, Wolf H, Niller HH, Minarovits J (2009) The importance of epigenetic alterations in the development of Epstein-Barr virus-related lymphomas. Mediterr J Hematol Infect Dis 1:e2009012
Takacs M, Banati F, Koroknai A, Segesdi J, Salamon D, Wolf H, Niller HH, Minarovits J (2010) Epigenetic regulation of latent Epstein-Barr virus promoters. Biochim Biophys Acta 1799:228–235
Takada K (2012) Role of EBER and BARF1 in nasopharyngeal carcinoma (NPC) tumorigenesis. Semin Cancer Biol 22:162–165
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Tang M, Lautenberger JA, Gao X, Sezgin E, Hendrickson SL, Troyer JL, David VA, Guan L, McIntosh CE, Guo X, Zheng Y, Liao J, Deng H, Malasky M, Kessing B, Winkler CA, Carrington M, De The G, Zeng Y, O’Brien SJ (2012) The principal genetic determinants for nasopharyngeal carcinoma in China involve the HLA class I antigen recognition groove. PLoS Genet 8:e1003103
Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD (2015) The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol 33:787–821
Tempera I, Wiedmer A, Dheekollu J, Lieberman PM (2010) CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog 6:e1001048
Tempera I, Klichinsky M, Lieberman PM (2011) EBV latency types adopt alternative chromatin conformations. PLoS Pathog 7:e1002180
Thorley-Lawson DA (2001) Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol 1:75–82
Thornburg NJ, Kusano S, Raab-Traub N (2004) Identification of Epstein-Barr virus RK-BARF0-interacting proteins and characterization of expression pattern. J Virol 78:12848–12856
Tierney RJ, Kirby HE, Nagra JK, Desmond J, Bell AI, Rickinson AB (2000) Methylation of transcription factor binding sites in the Epstein-Barr virus latent cycle promoter Wp coincides with promoter down-regulation during virus-induced B-cell transformation. J Virol 74:10468–10479
Tobollik S, Meyer L, Buettner M, Klemmer S, Kempkes B, Kremmer E, Niedobitek G, Jungnickel B (2006) Epstein-Barr virus nuclear antigen 2 inhibits AID expression during EBV-driven B-cell growth. Blood 108:3859–3864
Tomkinson B, Kieff E (1992) Use of second-site homologous recombination to demonstrate that Epstein-Barr virus nuclear protein 3B is not important for lymphocyte infection or growth transformation in vitro. J Virol 66:2893–2903
Tomkinson B, Robertson E, Kieff E (1993) Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J Virol 67:2014–2025
Torgbor C, Awuah P, Deitsch K, Kalantari P, Duca KA, Thorley-Lawson DA (2014) A multifactorial role for P. falciparum malaria in endemic Burkitt’s lymphoma pathogenesis. PLoS Pathog 10:e1004170
Toyota M, Ahuja N, Suzuki H, Itoh F, Ohe-Toyota M, Imai K, Baylin SB, Issa JP (1999) Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res 59:5438–5442
Tsai CN, Tsai CL, Tse KP, Chang HY, Chang YS (2002) The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc Natl Acad Sci U S A 99:10084–10089
Tsai CL, Li HP, Lu YJ, Hsueh C, Liang Y, Chen CL, Tsao SW, Tse KP, Yu JS, Chang YS (2006) Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res 66:11668–11676
Tsao SW, Tsang CM, To KF, Lo KW (2015) The role of Epstein-Barr virus in epithelial malignancies. J Pathol 235:323–333
Tse KP, Su WH, Chang KP, Tsang NM, Yu CJ, Tang P, See LC, Hsueh C, Yang ML, Hao SP, Li HY, Wang MH, Liao LP, Chen LC, Lin SR, Jorgensen TJ, Chang YS, Shugart YY (2009) Genome-wide association study reveals multiple nasopharyngeal carcinoma-associated loci within the HLA region at chromosome 6p21.3. Am J Hum Genet 85:194–203
Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N, Kikutani H (1999) Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science 286:300–303
Ushijima T (2007) Epigenetic field for cancerization. J Biochem Mol Biol 40:142–150
Ushiku T, Chong JM, Uozaki H, Hino R, Chang MS, Sudo M, Rani BR, Sakuma K, Nagai H, Fukayama M (2007) p73 gene promoter methylation in Epstein-Barr virus-associated gastric carcinoma. Int J Cancer 120:60–66
van Beek J, Brink AA, Vervoort MB, van Zijp MJ, Meijer CJ, van den Brule AJ, Middeldorp JM (2003) In vivo transcription of the Epstein-Barr virus (EBV) BamHI-A region without associated in vivo BARF0 protein expression in multiple EBV-associated disorders. J Gen Virol 84:2647–2659
van den Bosch CA (2004) Is endemic Burkitt’s lymphoma an alliance between three infections and a tumour promoter? Lancet Oncol 5:738–746
van den Bosch C (2012) A role for RNA viruses in the pathogenesis of Burkitt’s lymphoma: the need for reappraisal. Adv Hematol 2012:494758
van den Bosch C, Lloyd G (2000) Chikungunya fever as a risk factor for endemic Burkitt’s lymphoma in Malawi. Trans R Soc Trop Med Hyg 94:704–705
Vento-Tormo R, Rodriguez-Ubreva J, Lisio LD, Islam AB, Urquiza JM, Hernando H, Lopez-Bigas N, Shannon-Lowe C, Martinez N, Montes-Moreno S, Piris MA, Ballestar E (2014) NF-kappaB directly mediates epigenetic deregulation of common microRNAs in Epstein-Barr virus-mediated transformation of B-cells and in lymphomas. Nucleic Acids Res 42:11025–11039
Vo QN, Geradts J, Gulley ML, Boudreau DA, Bravo JC, Schneider BG (2002) Epstein-Barr virus in gastric adenocarcinomas: association with ethnicity and CDKN2A promoter methylation. J Clin Pathol 55:669–675
Walls D, Perricaudet M (1991) Novel downstream elements upregulate transcription initiated from an Epstein-Barr virus latent promoter. EMBO J 10:143–151
Wang S, Frappier L (2009) Nucleosome assembly proteins bind to Epstein-Barr virus nuclear antigen 1 and affect its functions in DNA replication and transcriptional activation. J Virol 83:11704–11714
Wang L, Grossman SR, Kieff E (2000) Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc Natl Acad Sci U S A 97:430–435
Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, Tsui WY, Ho SL, Chan AK, Man JL, Foglizzo V, Ng MK, Chan AS, Ching YP, Cheng GH, Xie T, Fernandez J, Li VS, Clevers H, Rejto PA, Mao M, Leung SY (2014) Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet 46:573–582
Weiss LM, Strickler JG, Warnke RA, Purtilo DT, Sklar J (1987) Epstein-Barr viral DNA in tissues of Hodgkin’s disease. Am J Pathol 129:86–91
Weiss LM, Movahed LA, Warnke RA, Sklar J (1989) Detection of Epstein-Barr viral genomes in Reed-Sternberg cells of Hodgkin’s disease. N Engl J Med 320:502–506
Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP (2009) Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet 41:246–250
White RE, Wade-Martins R, James MR (2001) Sequences adjacent to oriP improve the persistence of Epstein-Barr virus-based episomes in B cells. J Virol 75:11249–11252
White RE, Groves IJ, Turro E, Yee J, Kremmer E, Allday MJ (2010) Extensive co-operation between the Epstein-Barr virus EBNA3 proteins in the manipulation of host gene expression and epigenetic chromatin modification. PLoS One 5:e13979
Whittle HC, Brown J, Marsh K, Greenwood BM, Seidelin P, Tighe H, Wedderburn L (1984) T-cell control of Epstein-Barr virus-infected B cells is lost during P. falciparum malaria. Nature 312:449–450
Woisetschlaeger M, Strominger L, Speck SH (1989) Mutually exclusive use of viral promoters in Epstein-Barr virus latently infected lymphocytes. Proc Natl Acad Sci U S A 86:6498–6502
Woisetschlaeger M, Yandava CN, Furmanski LA, Strominger JL, Speck SH (1990) Promoter switching in Epstein-Barr virus during the initial stages of infection of B lymphocytes. Proc Natl Acad Sci U S A 87:1725–1729
Wolf H, zur Hausen H, Becker V (1973) EB viral genomes in epithelial nasopharyngeal carcinoma cells. Nat New Biol 244:245–247
Wong AM, Kong KL, Chen L, Liu M, Wong AM, Zhu C, Tsang JW, Guan XY (2013) Characterization of CACNA2D3 as a putative tumor suppressor gene in the development and progression of nasopharyngeal carcinoma. Int J Cancer 133:2284–2295
Wu DY, Kalpana GV, Goff SP, Schubach WH (1996) Epstein-Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF-SWI complex, hSNF5/Ini1. J Virol 70:6020–6028
Wu MS, Shun CT, Wu CC, Hsu TY, Lin MT, Chang MC, Wang HP, Lin JT (2000) Epstein-Barr virus-associated gastric carcinomas: relation to H. pylori infection and genetic alterations. Gastroenterology 118:1031–1038
Xia T, O’Hara A, Araujo I, Barreto J, Carvalho E, Sapucaia JB, Ramos JC, Luz E, Pedroso C, Manrique M, Toomey NL, Brites C, Dittmer DP, Harrington WJ Jr (2008) EBV microRNAs in primary lymphomas and targeting of CXCL-11 by ebv-mir-BHRF1-3. Cancer Res 68:1436–1442
Xiao X, Zhao W, Tian F, Zhou X, Zhang J, Huang T, Hou B, Du C, Wang S, Mo Y, Yu N, Zhou S, You J, Zhang Z, Huang G, Zeng X (2014) Cytochrome b5 reductase 2 is a novel candidate tumor suppressor gene frequently inactivated by promoter hypermethylation in human nasopharyngeal carcinoma. Tumour Biol 35:3755–3763
Yanatatsaneejit P, Chalermchai T, Kerekhanjanarong V, Shotelersuk K, Supiyaphun P, Mutirangura A, Sriuranpong V (2008) Promoter hypermethylation of CCNA1, RARRES1, and HRASLS3 in nasopharyngeal carcinoma. Oral Oncol 44:400–406
Yates JL, Warren N, Sugden B (1985) Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 313:812–815
Yi HM, Li H, Peng D, Zhang HJ, Wang L, Zhao M, Yao KT, Ren CP (2006) Genetic and epigenetic alterations of LTF at 3p21.3 in nasopharyngeal carcinoma. Oncol Res 16:261–272
Ying J, Srivastava G, Hsieh WS, Gao Z, Murray P, Liao SK, Ambinder R, Tao Q (2005) The stress-responsive gene GADD45G is a functional tumor suppressor, with its response to environmental stresses frequently disrupted epigenetically in multiple tumors. Clin Cancer Res 11:6442–6449
Ying J, Li H, Seng TJ, Langford C, Srivastava G, Tsao SW, Putti T, Murray P, Chan AT, Tao Q (2006) Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene 25:1070–1080
You Y, Yang W, Wang Z, Zhu H, Li H, Lin C, Ran Y (2013) Promoter hypermethylation contributes to the frequent suppression of the CDK10 gene in human nasopharyngeal carcinomas. Cell Oncol (Dordr) 36:323–331
Zetterberg H, Borestrom C, Nilsson T, Rymo L (2004) Multiple EBNA1-binding sites within oriPl are required for EBNA1-dependent transactivation of the Epstein-Barr virus C promoter. Int J Oncol 25:693–696
Zhang L, Pagano JS (1999) Interferon regulatory factor 2 represses the Epstein-Barr virus BamHI Q latency promoter in type III latency. Mol Cell Biol 19:3216–3223
Zhang X, Li W, Li H, Ma Y, He G, Tan G (2012) Genomic methylation profiling combined with gene expression microarray reveals the aberrant methylation mechanism involved in nasopharyngeal carcinoma taxol resistance. Anticancer Drugs 23:856–864
Zhao B, Mar JC, Maruo S, Lee S, Gewurz BE, Johannsen E, Holton K, Rubio R, Takada K, Quackenbush J, Kieff E (2011a) Epstein-Barr virus nuclear antigen 3C regulated genes in lymphoblastoid cell lines. Proc Natl Acad Sci U S A 108:337–342
Zhao B, Zou J, Wang H, Johannsen E, Peng CW, Quackenbush J, Mar JC, Morton CC, Freedman ML, Blacklow SC, Aster JC, Bernstein BE, Kieff E (2011b) Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc Natl Acad Sci U S A 108:14902–14907
Zhao J, Liang Q, Cheung KF, Kang W, Lung RW, Tong JH, To KF, Sung JJ, Yu J (2013b) Genome-wide identification of Epstein-Barr virus-driven promoter methylation profiles of human genes in gastric cancer cells. Cancer 119:304–312
Zheng Z, Pan J, Chu B, Wong YC, Cheung AL, Tsao SW (1999) Downregulation and abnormal expression of E-cadherin and beta-catenin in nasopharyngeal carcinoma: close association with advanced disease stage and lymph node metastasis. Hum Pathol 30:458–466
Zhou Y, Zeng Z, Zhang W, Xiong W, Wu M, Tan Y, Yi W, Xiao L, Li X, Huang C, Cao L, Tang K, Li X, Shen S, Li G (2008) Lactotransferrin: a candidate tumor suppressor-Deficient expression in human nasopharyngeal carcinoma and inhibition of NPC cell proliferation by modulating the mitogen-activated protein kinase pathway. Int J Cancer 123:2065–2072
Zhu JY, Pfuhl T, Motsch N, Barth S, Nicholls J, Grasser F, Meister G (2009) Identification of novel Epstein-Barr virus microRNA genes from nasopharyngeal carcinomas. J Virol 83:3333–3341
zur Hausen A, Brink AA, Craanen ME, Middeldorp JM, Meijer CJ, van den Brule AJ (2000) Unique transcription pattern of Epstein-Barr virus (EBV) in EBV-carrying gastric adenocarcinomas: expression of the transforming BARF1 gene. Cancer Res 60:2745–2748
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Niller, H.H., Banati, F., Salamon, D., Minarovits, J. (2016). Epigenetic Alterations in Epstein-Barr Virus-Associated Diseases. In: Minarovits, J., Niller, H. (eds) Patho-Epigenetics of Infectious Disease. Advances in Experimental Medicine and Biology, vol 879. Springer, Cham. https://doi.org/10.1007/978-3-319-24738-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-24738-0_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24736-6
Online ISBN: 978-3-319-24738-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)