
Abstract
In chemotherapy a fine balance between therapeutic and toxic effects needs to be found for each patient, adapting standard combination protocols each time. Nanotherapeutics has been introduced into clinical practice for treating tumors with the aim of improving the therapeutic outcome of conventional therapies and of alleviating their toxicity and overcoming multidrug resistance.
Photodynamic therapy (PDT) is a clinically approved, minimally invasive procedure emerging in cancer treatment. It involves the administration of a photosensitizer (PS) which, under light irradiation and in the presence of molecular oxygen, produces cytotoxic species. Unfortunately, most PSs lack specificity for tumor cells and are poorly soluble in aqueous media, where they can form aggregates with low photoactivity. Nanotechnological approaches in PDT (nanoPDT) can offer a valid option to deliver PSs in the body and to solve at least some of these issues. Currently, polymeric nanoparticles (NPs) are emerging as nanoPDT system because their features (size, surface properties, and release rate) can be readily manipulated by selecting appropriate materials in a vast range of possible candidates commercially available and by synthesizing novel tailor-made materials. Delivery of PSs through NPs offers a great opportunity to overcome PDT drawbacks based on the concept that a nanocarrier can drive therapeutic concentrations of PS to the tumor cells without generating any harmful effect in non-target tissues. Furthermore, carriers for nanoPDT can surmount solubility issues and the tendency of PS to aggregate, which can severely affect photophysical, chemical, and biological properties. Finally, multimodal NPs carrying different drugs/bioactive species with complementary mechanisms of cancer cell killing and incorporating an imaging agent can be developed.
In the following, we describe the principles of PDT use in cancer and the pillars of rational design of nanoPDT carriers dictated by tumor and PS features. Then we illustrate the main nanoPDT systems demonstrating potential in preclinical models together with emerging concepts for their advanced design.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Cancer treatment is currently based on a combination of surgery, radiotherapy, chemotherapy, and, more recently, immunotherapy. Each treatment modality bears advantages and drawbacks and needs to be established depending on tumor location, stage of tumor growth, and presence of metastasis. Chemotherapy is one of the principal modes of treatment for cancer. The main Achilles’ heel in a chemotherapeutic regimen lies in poor selectivity of the treatment that generates severe side effects, contributing to decreased patient compliance and quality of life. Indeed, most chemotherapeutics are administered by the intravenous route, distribute in the whole body according to the physical chemical features (which drive interactions with plasma proteins), and reach healthy organs as well as diseased tissue. A fine balance between therapeutic and toxic effects needs to be found for each patient, adapting standard combination protocols each time. Nevertheless, the effectiveness of chemotherapy is limited by intrinsic or acquired drug resistance [1, 2].
In the past 20 years, nanotherapeutics has been introduced in the clinical practice for treating tumors with the aim of improving the therapeutic outcome of conventional pharmacological therapies and alleviating their toxicity, as well as overcoming multidrug resistance (MDR) [3–9]. By providing a protective housing for the drug, nanoscale delivery systems can in theory offer the advantages of drug protection from degradation and efficient control of pharmacokinetics and accumulation in tumor tissue, thus limiting drug interaction with healthy cells and, as a consequence, side effects. The delivery of chemotherapeutics through nanocarriers has been mainly focused on the intravenous route to reach remote sites in the body through the blood system [10]. By exploiting the presence of the dysfunctional endothelium of the tumor capillary wall and the absence of effective lymphatic drainage in solid tumors, nanocarriers can extravasate from the blood circulation and can reach the solid tumor interstitium [11–13]. This mechanism, referred as the Enhanced Permeability and Retention (EPR) effect, is the main determinant in passive targeting [14, 15]. Nanocarrier decoration with ligands that specifically recognize peculiar elements of tumors (receptors on endothelial cells of blood vessels, extracellular matrix, cancer cells) or with magnetically sensitive materials can ameliorate drug specificity, allowing its effective accumulation in a solid tumor, an approach known as active targeting [16, 17]. Nanocarriers can also be designed with exquisite responsiveness to the tumor environment (pH, temperature, redox potential) or external stimuli (light, magnetic field, ultrasound, temperature), which can, in theory, trigger drug release only at tumor level [18–25].
In the attempt to find alternative treatment modalities for cancer, photodynamic therapy (PDT) has emerged as an adjuvant therapy to target neoplastic lesions selectively [26, 27]. PDT consists in the administration (local or systemic) of a photosensitizer (PS) which accumulates in different tissue/cells and, under application of light with a specific wavelength and in the presence of molecular oxygen, produces highly reactive oxygen species (ROS), mainly singlet oxygen (1O2), finally inducing cell death and tumor regression. Selectivity is achieved partly by the accumulation of the PS in the malignant cells/tissue and partly by restricting the application of the incident light to the tumor area. PS is minimally toxic in the non-irradiated zones, although a such phototoxicity and photosensitivity can occur when PS shows a tropism for organs exposed to daylight (skin, eye) or is topically administered, as in the case of skin cancer. PDT has been approved as a primary treatment option for certain neoplastic conditions including inoperable esophageal tumors, head and neck cancers, and microinvasive endo-bronchial non-small cell lung carcinoma [28]. PDT is also being investigated in preclinical and clinical studies for other cancer types including colon, breast, prostate, and ovarian [28–30].
There are several technical difficulties in the application of PDT in cancer, partly shared by most clinically relevant chemotherapeutics. First is the difficulty in preparing pharmaceutical formulations that enable PS parenteral administration because most existing PSs are hydrophobic, aggregate easily under physiological condition, and somewhat lose their photophysical properties. Second is the selective accumulation in diseased tissues, which is often not high enough for clinical use. A third aspect is related to light-activation of PS that generally occurs at a wavelength where radiation is poorly penetrating and unable to reach deep tissues.
Nanotechnological approaches in PDT (nanoPDT) can offer a valid option to deliver a PS and to solve at least part of these issues. Currently, several nanosized carriers made of different materials, such as lipids, polymers, metals, and inorganic materials, have been proposed in nanoPDT, each type of system highlighting pros and cons [31–34]. This review focuses on polymer-based nanoparticles (NPs) specifically designed for cancer PDT. The main advantages of polymeric NPs lie in the ability to manipulate carrier properties readily by selecting polymer type and mode of carrier assembly [35, 36]. In fact, advances in polymer chemistry make it possible to produce an almost infinite number of sophisticated structures and to engineer these structures in light of a strictly defined biological rationale. As a consequence, not only are those features which affect the distribution of drug doses in the body and interaction with target cells controlled, but also spatio-temporal release of the delivered drug is predetermined.
This review covers current trends and novel concepts in the design of passively, actively, and physically targeted NPs proposed in cancer PDT, focusing on those tested in preclinical studies.
2 Photodynamic Therapy in Cancer Treatment
2.1 Principles of a PDT Treatment
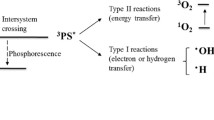
PDT is based on photochemical processes between light and an exogenous PS localized at disease level. These components, well tolerated singly by the cells, generate oxygen-based molecular species exerting a number of effects at cell and tissue level. Mechanistically, a photodynamic reaction consists in exciting PS molecules with light of appropriate wavelength, usually visible (VIS) or near-infrared (NIR), preferentially at PS maximum absorption. PS passes from the ground state to the excited state and can at this stage decay to the ground state with concomitant emission of light in the form of fluorescence. The excited PS may also undergo intersystem crossing to form a relatively more stable and long-lived excited triplet state which can either decay to the ground state or transfer electrons/energy to the surroundings through (1) electron transfer to organic molecules and molecular oxygen in cell microenvironment to form radicals finally giving hydrogen peroxide (H2O2) and hydroxyl/oxygen radicals (Type I process) or (2) transfer of energy to molecular oxygen leading to the formation of 1O2 which initiates oxidation of susceptible substrates (Type II process).
Both Type I and Type II reactions can occur simultaneously and competitively, and the ratio between these processes depends on the type of PS used, and on the concentrations of substrate and oxygen. Type II reaction, however, appears to play a central role in cytotoxicity, because of the highly efficient interaction of the 1O2 species with various biomolecules [27]. 1O2 has a lifetime of less than 3.5 μs in an aqueous environment and can diffuse only 0.01–0.02 μm during this period. Nevertheless, it should be taken into account that 1O2 senses the inherent heterogeneity of cell environment and its lifetime can consequently be affected [37]. A natural consequence is that the initial extent of the damage is limited to the site of concentration of the PS [38]. This is usually the mitochondria, plasma membrane, Golgi apparatus, lysosomes, endosomes, and endoplasmic reticulum. The nucleus and nuclear membrane are usually spared and DNA damage is rare. Net ionic charge (from −4 to +4), hydrophobicity, and the degree of asymmetry of PS are reported to play a role in cell uptake and intracellular localization.
2.1.1 Generalities on PS
Although an enormous number of chemical structures have been found to act as PSs, only a handful have proceeded to clinical trial and even fewer are commercially available [39]. PSs are generally classified as porphyrinoids and non-porphyrinoids [40]. Within porphyrinoid-based PSs, first, second, and third generation PSs are reported.
The first generation agent hematoporphyrin derivative (HpD) represents the foundation and the reference for novel PSs. Purified HpD is commercialized as porfimer sodium (Photofrin®), a lyophilized concentrated form of monomeric and oligomeric hematoporphyrin derivatives. Photofrin® is characterized by an absorption band at 630 nm (corresponding to a penetration of about 5–10 mm), a low molar extinction coefficient which in turn demands large amounts of Photofrin® and light to obtain adequate tumor eradication, and a long half-life of 452 h, leading to long-lasting photosensitivity. The time delay between drug administration and the time needed to maximize the tumor to normal cell uptake within the target tissue determines the correct delay for light application. Photofrin®-mediated PDT involves intravenous administration of PS followed by irradiation (100–200 J/cm2 of red light) 24–48 h later. During this period, Photofrin® is cleared from a number of tissues and remains concentrated at the target site [41].
Second generation PSs have been developed with the aim of alleviating certain problems associated with first-generation molecules such as prolonged skin photosensitization and suboptimal tissue penetration. 5-Aminolevulinic acid (5-ALA) is a prodrug enzymatically converted to the active PS protoporphyrin IX (PpIX) during the biosynthesis of heme. 5-ALA (Levulan®) is now approved for the topical treatment of actinic keratosis (AK) and is in clinical trials for other types of cancer [42]. Because of its poor ability to cross the skin, lipophilic derivatives have been proposed such as 5-methyl-aminolevulinate (Metvix®) and hexyl ester of 5-ALA (Hexvix®) [43].
From the porphyrin family, meta-tetra(hydroxyphenyl)porphyrin (m-THPP) and 5,10,15,20-tetrakis(4-sulfanatophenyl)-21H,23H-porphyrin (TPPS4) are the main second generation PDT sensitizers. m-THPP, although being 25–30 times more potent than HpD in tumor photonecrosis when irradiated at 648 nm, causes severe skin phototoxicity [40, 44].
Various chemical modifications of the tetrapyrrolic ring of the porphyrins characterize the different groups of the second-generation PSs [45, 46]. They have high absorption coefficients/ 1O2 quantum yields and absorption peaks in the IR (660–700 nm) or NIR (700–850 nm) regions. The serum half-life of these compounds is short and tissue accumulation is improved and occurs quickly (within 1–6 h after injection). Thus, the treatment can be carried out on the same day as the administration of the drug. In addition, the risk of burns by accidental sun exposure is low because clearance from normal tissues is rapid. Finally, toxicity to skin and internal organs in the absence of light (so-called ‘dark’ toxicity) is absent or minimal.
The chlorin family includes benzoporphyrin derivative monoacid ring A (BPD-MA, Verteporfin, Visudyne®), meta-tetra(hydroxyphenyl)chlorin (m-THPC, Foscan®), tin ethyl etiopurpurin (SnET2, Rostaporfin, Purlytin™), and N-aspartyl chlorin e6 (NPe6, Talaporfin, Ls11) which is derived from chlorophyll a. When compared to porphyrins, the structure of chlorins differs by two extra hydrogens in one pyrrole ring. This structural change leads to a bathochromic shift in the absorption band (640–700 nm) and gives ε max ~ 40,000 M−1 cm−1. Pheophorbides also have two extra hydrogens in one pyrrole unit and can be derived from chlorophyll. 2-(1-Hexyloxyethyl)-2-devinyl pyropheophorbide A (HPPH, Photochlor®) absorbs at 665 nm with ε max ~ 47,000 M−1 cm−1.
The joining of four benzene or naphthalene rings to the β-pyrrolic positions of porphyrins and the substitution of the methylene-bridge carbons with nitrogen produce phthalocyanine and naphthalocyanines, respectively. The presence of Al(III), Zn(II), Si(IV), Ru(II), and other diamagnetic metal ions with axial ligands gives hexacoordination and guarantees a satisfactory yield of 1O2 generation, thus decreasing the tendency to form PS self-aggregates and inducing high photodynamic efficiency and reduced phototoxic side effects [46].
Non-porphyrin derivatives, including hypericin, hypocrellins, methylene blue (MB), toluidine Blue, and merocyanine 540, are other potential PSs for cancer PDT [40].
Currently, research efforts are focusing on the development of third generation PSs, characterized by a higher specificity to target cells and minimal accumulation in healthy tissues. The basic approach consists in the conjugation of a PS with a targeting component, such as an antibody directed against the tumor antigens, to promote the localization and the accumulation of the drug at the diseased site [47, 48]. As discussed in the following, the most advanced strategy to ameliorate PS therapeutic outcomes relies in their delivery through engineered nanosystems.
2.1.2 Light Sources
Besides the type of PS, the selection of a light source plays a central role in achieving effective PS excitation in the bioenvironment. PS maximum absorption range, disease location, size of the area to treat, and cost are the main determinants to identify an appropriate illuminating system. Furthermore, the clinical efficacy of PDT is dependent on dosimetry: total light dose, light exposure time, light delivery mode (single vs fractionated or even metronomic), and fluence rate (intensity of light delivery) [49].
The effective excitation light magnitude is determined by the combination of optical absorption and scattering properties of the tissue. Absorption is largely because of endogenous tissue chromophores such as hemoglobin, myoglobin, and cytochromes. On the other hand, the optical scattering of a tissue depends on wavelength. For the spectral range of 450–1750 nm, tissue scattering is, in general, more prevalent than absorption, although, for the range of 450–600 nm, melanin and hemoglobin provide significant absorption, and water plays a similar role for wavelengths >1350 nm. Therefore, the optimal optical window for PDT, and for optical imaging, is in the NIR spectral region (600–1300 nm), where the scattering and absorption by tissue are minimized and, therefore, the longest penetration depth can be achieved. Within this optical window, the longer the wavelength, the deeper the penetration depth. However, light up to only approximately 800 nm can extensively generate 1O2, because longer wavelengths have insufficient energy to initiate a photodynamic reaction [50]. In fact, optical penetration depth of 780-nm light was found to be 3.62 mm in a mammary carcinoma and 2.82 mm in a lung carcinoma [51]. Currently approved PSs absorb in the visible spectral regions below 700 nm, where light penetration into the skin is only a few millimeters, clinically limiting PDT to treat topical lesions. Thus, a PDT treatment is also generally carried out with red light at higher penetration for PSs, which have maximum absorption in the blue region of the absorption spectrum.
Laser systems widely used for treating dermatological conditions allow the selection of a wavelength with a maximal effective tissue penetration of approximately 10 mm, and have been used in combination with all types of PSs [52]. The laser beams can be launched into an optical fiber applicator, enabling light to be delivered directly into internal tumors. These techniques are relatively expensive, require specialized supporting staff, and are space-consuming. It is likely that such systems will eventually be replaced by laser diode arrays, which are very convenient because they can be easily handled, require only a single phase supply, and are also relatively inexpensive. Because they are monochromatic, the choice of laser wavelength becomes crucial as it must be matched with the often narrow absorption band of the PS, with the result that one laser can only be used in combination with one (or a limited number of) PS(s). Lasers at present are the only possible light source to treat malignancies located in sites that can only be reached with optical fibers.
Several PDT treatments use filtered output high power lamps such as Tungsten Filament Quartz Halogen Lamps, Xenon Arc Lamps, and Metal Halide Lamps, especially in clinical settings. In fact, lamps can provide a broad range of wavelengths at reduced fluence rates to avoid thermal effects, not necessarily producing a dramatic increase in the time required for the treatment. A combination of narrowband, longpass, and shortpass filters is often required to select the irradiation wavelength within 10 nm to cut high-power UV radiation and IR emission (causing an undesired increase in the temperature). Because of their broad emission, lamps can be used in combination with several PSs with different absorption maxima within the emission spectrum of the lamp. Moreover, lamps normally also excite the region where photoproducts absorb, thus being responsible for some additional PDT effects. Because of their characteristics, lamps are well suited for treatment of accessible lesions, especially for larger skin lesions (with or without the use of liquid light guides). Moreover, compared to lasers, such sources offer the advantage of being less expensive and easier to handle [49].
Naturally, most of the light sources for PDT application have been developed to optimize the output near the absorption wavelengths of the main PSs. Thus, the tendency of regulatory agencies, such as the US Food and Drug Administration, has been to approve the PS and the light source to be used for its optical excitation.
2.2 Mechanisms of Cancer Cell Death
A PDT treatment is a two-stage process where a PS is administered in the body locally or by intravenous injection. After a certain period, PS accumulates in cancer cells and is activated by application of light at the level of diseased area where biological effects occur (Fig. 1a). Accumulation in solid tumors is especially critical after intravenous administration and largely related to PS physical-chemical features. PSs probably interact with tumors via low-density lipoprotein (LDL) receptors. Hydrophobic compounds and their aggregates bind to LDL whereas hydrophilic species bind to albumin and globulins [53]. Because cancer cells have elevated levels of LDL receptors, endocytosis of LDL–PS complex is preferred by malignant cells [54, 55]. PS solubility is the main determinant affecting its distribution and location inside tumor cells. Accumulation of PS in the cell organelles also depends on the charge of the sensitizer. Cationic compounds collect in mitochondria, whereas anionic species are found in lysosomes [53]. Dye sensitizers with one or two anionic charges localize in the perinuclear region, vesicles of the cell, and lysosomes, providing multiple sites of PS accumulation [56, 57]. It should be noted that subcellular localization may change with incubation time because PS may relocate to other organelles after illumination.

Treatment of a solid tumor by PDT. (A) Steps of an intravenous photodynamic treatment. (B) In tumor PDT, PS absorbs light and an electron moves to the first short-lived excited singlet state. This is followed by intersystem crossing, in which the excited electron changes its spin and produces a longer-lived triplet state. The PS triplet transfers energy to ground state triplet oxygen, which produces reactive 1O2 that can (1) directly kill tumor cells by induction of necrosis and/or apoptosis, (2) cause destruction of tumor vasculature, and (3) produce an acute inflammatory response attracting leukocytes. Adapted from [59]
Although PDT can induce many cellular and molecular signaling pathway events, the final effect is the induction of cell death through the activation of three main cell death pathways: apoptosis, necrosis, and autophagy [27, 38, 58]. The mode and the extent of cell death is related to different elements, including the concentration, the physiochemical properties and subcellular location of the PS, the concentration of oxygen, the wavelength and intensity of the light, and the cell type. For instance, it is recognized that lower doses of PDT lead to more apoptotic cells, whereas higher doses lead to proportionately more necrotic cells [59]. After PDT, cancer cells usually develop a cytoprotective mechanism to limit cytotoxic effects and detoxify from ROS, such as the production of antioxidant molecules (e.g., some amino acids, glutathione, vitamin E) and of enzymes.
Other distinct mechanisms contribute to the reduction or disappearance of tumors after PDT treatment (Fig. 1b). In fact, PDT is also able to damage the tumor-associated vasculature, leading to tumor death via lack of oxygen and nutrients [60]. The higher sensitivity of endothelial cells compared to the other proliferating tumor cells is produced by a greater PS accumulation in the endothelial cells, where biological response occurs at sub-lethal doses of PDT [61, 62]. Although microvascular damage after PDT contributes to greater tumor response, reduction in oxygen during treatment can limit tumor control by inducing the production of proangiogenic markers, creating a favorable environment for tumor recurrence [63, 64].
PDT frequently provokes a strong inflammatory reaction observed as localized edema at the target site caused by oxidative stress. PDT-induced inflammation is orchestrated by innate immune system. The acute inflammation and release of cytokines and stress response proteins induced in the tumor can lead to an invasion of leukocytes contributing both to tumor destruction and to stimulation of the immune system to recognize and destroy tumor cells [59]. Nevertheless, numerous studies have linked PDT to the adaptive immune response. The precise mechanism leading to potentiation vs suppression of adaptive immunity exerted by PDT is unclear as yet; nevertheless, it seems as though the effect of PDT on the immune system is dependent on the PS type, the treatment regimen, and the area treated. Furthermore, recent findings suggest that clinical antitumor PDT can increase antitumor immunity [65, 66].
The relative importance of each mechanism for the overall tumor response is yet to be defined and requires further research. It is clear, however, that the combination of all these components in PDT is required for optimum long-term tumor regression, especially in tumors that may have metastasized.
Finally, two general approaches may increase the antitumor effectiveness of PDT; (1) sensitization of tumor cells to PDT and (2) interference with cytoprotective molecular responses triggered by PDT in surviving tumor or stromal cells [27].
2.3 Clinical PDT for Cancer
PDT has been utilized for preneoplastic and neoplastic diseases in a wide variety of organ systems, including skin, genitourinary, esophagus, prostate, bile duct, pancreas, head and neck, and brain [28]. Several medicines have been approved or are currently in clinical trials (Table 1). At present, 68 open clinical trials on cancer PDT are ongoing.
Successful results for PDT of non-hyperkeratotic actinic keratosis have been achieved with systemically administered porfimer sodium as well as topically applied ALA and methyl-ALA (MAL). Fifty-one randomized clinical trials (RCTs) that reported the use of PDT in the treatment of actinic keratosis have been identified ([67] and clinicaltrial.gov) and aggregated data indicate better rates of complete response and better cosmetic results with PDT than with the other treatments.
Several RCTs on superficial and nodular basal cell carcinoma have been reported, comparing ALA-PDT with surgical excision, cryotherapy, or placebo [68]. In particular, for superficial basal cell carcinoma, the outcome after PDT appears similar to surgery or cryotherapy, whereas for nodular (deep) basal cell carcinoma, PDT is less effective than surgery for lesion clearance. Finally, PDT can substantially reduce the size of large squamous-cell carcinoma tumors, reducing morbidity and increasing overall curative response [69].
In the field of head and neck cancer, thousands of patients have been treated with PDT [28, 70] by systemic delivery; in particular, Foscan® was approved in Europe in 2001 for the palliative treatment of patients with advanced head and neck cancer who have exhausted other treatment options. Furthermore, various formulations of porfimer sodium, ALA, and temoporfin are currently undergoing intensive clinical investigation as an adjunctive treatment for brain tumors, such as glioblastoma multiforme, anaplastic astrocytoma, malignant ependymomas or meningiomas, melanoma, lung cancer, brain metastasis, and recurrent pituitary adenomas [71].
PDT is increasingly being used to treat cancers of the airways and other tumors in the thoracic cavity, especially non-small cell lung carcinoma [72, 73]. Different RCTs based on talaporfin or porfimer sodium-mediated PDT showed good results and complete response rate in patients with early stage lung cancer or for whom surgery is not feasible.
In gastroenterology, endoscopically accessible premalignant or malignant lesions located within the esophagus, the stomach, the bile duct, or the colorectum with a high surgical risk have become suitable targets of endoscopic PDT [74]. Photofrin®-PDT has been approved for obstructing esophageal cancer, early-stage esophageal cancer, and Barrett’s esophagus in several countries, as an alternative to esophagectomy because these are superficial and large mucosal areas that are easily accessible for light. Recent pilot studies have demonstrated that endoscopic Photofrin®-PDT is also effective in the palliative treatment of cholangiocarcinoma [34, 75], for early duodenal and ampullary cancers, and for advanced adenomas.
Because of advances in light applicators, the interstitial PDT is now becoming a practical option for solid lesions, including those in parenchymal organs such as the liver and pancreas [76, 77]. Talaporfin-mediated PDT may have efficacy in treating hepatocellular carcinoma, whereas Foscan® looked promising in the treatment of pancreatic cancer [78]. In the case of prostate cancer, Foscan® represented a viable minimally-invasive alternative to surgery or radiotherapy, reducing the risk of the post-surgical side effects of incontinence and impotence [79]. Bladder cancer tends to be a superficial condition, and for this reason it is proposed that a superficial treatment with ALA or its ester derivatives by intravesical instillation may be a preferable mean for local therapy [80].
The last PDT application refers to the treatment of gynecological cancers [81]. For cervical intraepithelial neoplasia, PDT based on chlorine e6 (Photolon®) or hexyl-ALA offers a nonscarring alternative to cone biopsy. For vulvar intraepithelial neoplasia, use of Foscan or ALA may ameliorate the need for radical mutilating surgery. Similarly, penile intraepithelial neoplasia and anal intraepithelial neoplasia have been treated with ALA-based PDT, sometimes with complete clearance. Extramammary Paget’s disease responds to PDT with porfimer sodium or ALA.
Currently, PSs are being evaluated as intraoperative diagnostic tools both by means of photodetection (PD) and fluorescence guided resection (FGR) during PDT [82]. The most recently published trials that employed PD, FGR, and PDT provided additional encouraging results, but the initial delay in tumor progression did not translate to extended overall survival.
2.4 Combining PDT to Chemotherapy
In a clinical setting, patients treated with anticancer drugs were found to fail the experiences of single agent chemotherapy because it is limited to act on specific cancer survival pathways and showed low response rates and relapse of tumor. To improve the therapeutic potential of cancer chemotherapy, it is essential to establish alternative approaches which could provide a solution to the problems involved in single drug chemotherapy. To this end, much attention has been given to combination approaches for a better long-term prognosis and to decrease side effects associated with high doses of monotherapy. One of the prime benefits of combination therapies is the potential for providing synergistic effects. The overall therapeutic response to drug combinations is generally greater than the sum of the effects of the drugs individually [83]. The best drug combination with maximal antitumor efficacy can be calculated by multiple drug effect/combination index isobologram analysis, an effective way to demonstrate that drugs are working synergistically. The prime mechanism of synergistic effect following combinational drug treatment could act on the same or different signaling pathways to achieve more-favorable outcomes at a lower dose with equal or increased efficacy [84]. Unlike monotherapy, combination therapy can modulate different signaling pathways, maximizing the therapeutic effect while overcoming toxicity and, moreover, can decrease the likelihood that resistant cancer cells develop.
As a complementary therapeutic modality, PDT can be combined with chemotherapy to enhance therapeutic outcome. In PDT, any activity of PDT-sensitizing agents is confined to the illuminated area, thus inducing non-systemic potentiated toxicity of the combinations. This should be of special importance in elderly or debilitated patients who tolerate poorly very intensive therapeutic regimens. Moreover, considering its unique 1O2-dependent cytotoxic effects, PDT can be safely combined with other antitumor treatments without the risk of inducing cross-resistance [85]. Despite this potential, few studies on combinations of PDT with standard antitumor regimens have been published to date [83].
Photochemical internalization (PCI), a specific branch of PDT, is a novel strategy utilized for the site-specific triggered drug/gene release [86–88]. PCI was initially developed at the Norwegian Radium Hospital as a method for light-enhanced cytosolic release of membrane-impermeable molecular therapeutics entrapped in endocytic vesicles. Briefly, the drug or gene of interest colocalizes with a PS in endocytic vesicles. Light-activation of the PS results in ROS-mediated damage of the membranes of these vesicles with subsequent release of the drug or gene to cytosol. This strategy is especially useful for proteins and nucleic acids that are unable to cross biological membranes, and even with a specific delivery system these molecules are taken up by endocytosis and are sequestered in endolysosomal compartments where they are subjected to enzymatic degradation, resulting in lack of biological effect. Furthermore, PCI can facilitate endolysosomal release of anti-cancer drugs and promote their subcellular redistribution after NP uptake [89]. PCI has been demonstrated to be a feasible drug delivery technology in numerous cancer cell lines and different animal models.
2.5 Drawbacks in Cancer PDT
The efficacy of a PDT treatment depends on multiple factors related to PS photochemical and physicochemical properties (1O2 production efficiency, tissue penetration of excitation light), PS biodistribution in the body, localization in a specific compartment and dose at target tissue, as well as light parameters (light dose, fluence rate, interval between administration and light exposure). Obviously, cancer tissue characteristics (vascularization, oxygenation level) play an important role in determining the therapeutic outcome of PDT.
Each of the commercially available PSs has specific characteristics, but none of them is an ideal agent. Selectivity remains a key issue in PDT. A PDT treatment can be considered to be selective in that the toxicity to tumor tissue is induced by the local activation of the PS, whereas normal tissues not exposed to light are spared. Second generation PSs show improved selectivity and clearance rate from the body so increased therapeutic efficiency and mostly alleviated toxicity caused by post-PDT photosensitization are experienced. However, Foscan® has failed FDA approval for the treatment of head and neck cancer because of poor tumor selectivity resulting in serious skin burns arising from photosensitivity [90]. Furthermore, most second generation PSs exhibit poor solubility in aqueous media, complicating intravenous delivery into the bloodstream. The low extinction coefficients of PSs often require the administration of relatively large amounts of drug to obtain a satisfactory therapeutic response, thus demanding specific vehicles (Chremophor®, propylene glycol), which can lead to unpredictable biodistribution profiles, allergy, hypersensitivity, and toxicity [91].
Several hydrophobic PSs tend to aggregate in physiological conditions via the strong attractive interactions between π-systems of the polyaromatic macrocycles and, as a consequence, to produce singlet oxygen with very low yields [92]. Aggregation is one of the determining factors which can cause a loss of PS efficacy in vivo by decreasing its bioavailability and limiting its capacity to absorb light [93]. The interactions are affected mainly by the solvent, sample concentration, temperature, and specific interactions with biological structures. Furthermore, the absorption maximum of PSs falls at relatively short wavelengths, leading to poor tissue penetration of light. This has prompted development of alternative strategies to improve quantum yields of 1O2 such as two-photon induced excitation [82, 94]. This strategy combines the energy of two photons (in the range 780–950 nm) where tissues have maximum transparency to light but where the energy of one photon is not high enough to produce 1O2.
For systemic administration, PS location and extent of PS accumulation in the target tissue depend on post-injection time [41]. At times shorter than PS half-life, the drug predominantly stays in the vascular compartment of the tumor, whereas at longer time, PS can accumulate in extravascular sites because of interstitial diffusion. Therefore, drug-light interval may play a crucial role for the therapeutic outcome. For topical administration there is a need to promote transport through the skin and to accumulate PS in the skin target. In this case there is no need to delay light application except for drugs that need metabolic pathways to become active (such as 5-ALA).
3 Injectable Nanoparticles for Photodynamic Therapy
3.1 Nanotechnology in Cancer PDT
Nanotechnology offers a great opportunity in advancing PDT based on the concept that a PS packaged in a nanoscale-carrier can result in optimized pharmacokinetics, enhancing the treatment ability to target and kill cancer cells of diseased tissue/organ while affecting as few healthy cells as possible [95, 96].
Besides improving specificity, nanoPDT is also emerging to surmount solubility issues and the aggregation tendency of PSs, which can severely affect photophysical, chemical, and biological properties. In fact, a carrier specifically engineered for nanoPDT should provide an environment where the PS can be administered in a monomeric form and can also maintain its photochemical properties in an in vivo setting without loss or alteration of photoactivity. Furthermore, a nanocarrier engineered for the therapy of solid tumors is expected to deliver therapeutic concentrations of PS in the diseased tissue and at specific subcellular locations.
NPs can be designed to transport more than one drug/bioactive species with different mechanisms of cancer cell killing. The idea to combine two drugs with different mechanisms of action and pharmacokinetics in a nanocarrier with well-tailored properties can allow control over anticancer drug/PS biological fate and promote co-localization in the same area of the body [97]. This approach is rather recent and demonstrated that cytotoxic drugs can act in concert with PS for tumor killing providing an anticancer synergistic effect, inducing antitumor immunity and sometime reverting MDR.
Another potential application of nanotechnology that has been a research hotspot in the forefront of materials science is the combination of non-invasive PDT and photothermal therapy (PTT) [98]. PTT consists in a NIR irradiation of a photo-absorbing agent which converts electromagnetic energy to local heat producing hyperthermia and subsequently cell death. Generally, noble metal NPs such as gold nanorods coupled with a PS on their surface are used to promote the tumor accumulation and synergistic PDT/PTT. Although high therapeutic outcomes, in this strategy two different wavelength lasers are usually required to allow PDT and PTT because of the absorption mismatch of PS and photothermal agents. Thus, developing a simple and effective strategy for simultaneous PDT and PTT treatment is highly desirable. Recently, researchers have been demonstrated the efficiency of NP loading with a single PS such as chlorins or some phthalocyanines that present a strong NIR absorbance and are capable of both PDT and PTT to kill cancer cells under single wavelength irradiation [99].
Nanocarriers also serve as a multimodal platform to bind/include a great variety of molecules, such as tumor-specific surface ligands for targeted nanoPDT and/or imaging agents integrating in a single platform the unique opportunity for concurrent diagnostic and treatment of cancer tumors, so-called theranostics. Recently, several multifunctional theranostic systems have been developed for real-time imaging-guided PDT of cancer [100, 101].
The general design of a carrier for cancer nanoPDT should be planned on a rational basis in the light of specific needs dictated by (1) tumor features (location, stage, metastatization), (2) selectivity for tumor tissue, which means to accumulating the largest fraction of administered dose at tumor level (cancer cells/tumor interstitium) with little or no uptake by non-target tissue/organs, and (3) stability in the body compartments, withstanding premature disassembly of nanocarrier and release of PS before the target is reached. Rational design is perhaps the most critical step in developing a nanoPDT carrier where a multidisciplinary approach at the interface between chemistry, pharmaceutical technology, biology, and medicine should be planned. In this respect, nanocarrier interactions with the biological environment (protein interaction, blood circulation time, elimination rate, transport through mucus or epithelia, cell internalization just to cite some aspects) can be properly regulated by nanocarrier overall physical-chemical properties (size, surface charge/hydrophilicity, drug loading capacity/release rate).
3.2 Fate of Intravenously Injected Nanocarriers
In analogy to several anticancer drugs, intravenous injection remains the preferred route of PS administration to reach different body compartments. In fact, the unique properties of tumor vasculature and microenvironment result in a natural tendency of a nanocarrier bearing a drug cargo to accumulate in solid tumors referred to as passive targeting [14, 15]. Architectural defectiveness and high degree of vascular density generate abnormal “leaky” tumor vessels, aberrant branching and blind loops of twisted shape. Blood flow behavior, such as direction of blood flow, is also irregular or inconsistent in these vessels. The pore size of tumor vessels varies from 100 nm to almost 1 mm in diameter, depending on the anatomic location of the tumors and the stage of tumor growth. Moreover, solid tumors are characterized by impaired lymphatic drainage which decreases the clearance of locally resident macromolecules. The enhanced permeability and retention (EPR) effect enables nanocarriers to extravasate through these gaps into extravascular spaces and to accumulate inside tumor tissues. Nevertheless, exploiting the EPR effect is complicated by the presence of physiological elimination processes, including both renal clearance and mononuclear phagocyte system (MPS) uptake [14, 15, 102].
Filtration of particles through the glomerular capillary wall (filtration-size threshold) depends on molecular weight and allows molecules with a diameter larger than 15 nm to remain in the circulation [4]. On the other hand, nanocarriers need to escape MPS, which mediates their fast disappearance from blood circulation and accumulation in the MPS organs (liver, spleen, bone marrow). Opsonin adsorption on nanocarrier surface mediates MPS recognition and is considered a key factor in controlling nanocarrier biodistribution in the body [103, 104]. Accumulation in the liver can be of benefit for the chemotherapeutic treatment of MPS localized tumors (e.g., hepatocarcinoma or hepatic metastasis arising from digestive tract or gynecological cancers, bronchopulmonary tumors) but undesirable when trying to target other body compartments. Ideally, an injectable nanocarrier has to be small enough to avoid internalization by the MPS but large enough to avoid renal clearance (100–200 nm). Recent findings highlight that variation of nanocarrier dimension in the scale length >100 nm can heavily affect blood circulation time, whereas the role of geometry in driving in vivo biodistribution has not yet been clarified [105–107].
Although extracellular matrix itself seems not to represent an evident obstacle to NP passage, tissue neighboring tumor cells are surrounded by coagulation-derived matrix gel (fibrin gel or stromal tissues) representing a further barrier to drug transport. Nevertheless, penetration in the remote area of a solid tumor (hypoxic zones) is strictly related to size for drugs, i.e., small drugs penetrate better than a high molecular weight antibody [108].
3.3 Cancer NanoPDT: Biologically-Driven Design Rules
Pharmacokinetics and cell uptake of PSs can be modified by engineering nanocarrier properties (size, surface, shape) to target tumors more specifically, which results in major clinical implications [11, 12, 109]. The general structure of multifunctional NPs for PDT of solid tumors is represented in Fig. 2a. Their rational design relies on appropriate assembling of each building element as dictated by biological requirements.

General structure of multifunctional NPs for PDT treatment of solid tumors. (A) Main components of multifunctional NPs. (B) Stimuli-sensitive NPs. Extracellular stimuli are suitable for shell shedding (to unmask moieties promoting intracellular transport) and to deliver drug cargo in proximity of cancer cells. Intracellular stimuli can be useful to activate PS intracellularly (dequenching) at specific subcellular locations or to release drug cargo in one pulse
In order to overcome opsonization, a number of strategies have been investigated to make a nanocarrier “stealth” that is able to evade MPS and long-circulating. Coating with a hydrophilic shell can form a cloud on nanocarrier surface which repels opsonins giving decreased levels of uptake by the MPS and longevity in the blood, finally promoting nanocarrier accumulation in solid tumors through EPR mechanism [110, 111]. To overcome opsonization and rapid elimination from the bloodstream produced by MPS recognition, coating with a biomimetic shell of polyethylene glycol (PEG) is the most explored strategy to obtain biomimetic long-circulating NPs [112], although other alternative polymers are under investigation [111]. Thus, nanocarriers with hydrophobic surfaces are preferentially taken into MPS organs although long-circulating nanocarriers fulfilling size requirements (less than 100 nm) can accumulate at tumor level. By exploiting passive mechanisms, only a limited nanocarrier fraction can reach the tumor site [13].
Although the presence of a hydrophilic coating allows NP escape from MPS recognition, it decreases the rate and extent of NP uptake inside cancer cells [112, 113]. A strategy to encourage nanocarrier internalization in solid tumors lies in surface decoration with ligands recognizing typical or overexpressed receptors in the tumor microenvironment, which can promote its transport through receptor-mediated endocytosis. Different chemical motifs interacting with specific receptors of endothelial cells in defective tumor vasculature (i.e., integrin receptor) and cancer cells (folate, CD44, transferrin, EGF, and some others) can be exploited for this purpose. This approach, known as active targeting, can aid selective nanocarrier accumulation inside cancer cells while avoiding healthy cells, thus decreasing treatment toxicity. Nevertheless, it has recently been demonstrated that targeted NPs can paradoxically lose targeting ability in a biological environment because of interaction with different high-affinity proteins [103] or can confine their activity to perivascular regions of a tumor (binding site barrier) [114]. Cell cycling also plays a role in NP uptake rate and amount of NPs internalized by cells because of splitting between daughter cells when the parent cell divides [115]. Thus, proper understanding of NP properties at the biointerface is a critical issue needing future investigational efforts [116].
An added sophistication to selective delivery of drug cargo in cancer cells can be brought about by utilizing certain cues inherently characteristic of the tumor microenvironment or by applying certain stimuli to this region from outside the body (Fig. 2b) [18–20, 24]. Stimuli-sensitive nanocarriers based on tailor-made materials can indeed be designed to deliver drug payload sharply and “on demand” by undergoing structural modifications under internal or external stimuli of chemical, biochemical, and physical origin. Internal stimuli typical of solid tumors include mainly pH, temperature, and reductive conditions. In fact, compared to normal/host tissues, pH value in tumor interstitium is lower with an average value of 6.84 because of up-regulated glycolysis producing lactates and protons [22]. Furthermore, once NPs are internalized through endocytic pathways involving lysosomes, pH progressively decreases from early endosomes (pH 5–6) to more acidic late endosomes (pH 4–5) [117], which can strongly alter nanocarrier stability and release features. pH sensitiveness has been widely employed to trigger NP disassembly and drug release [118]. Certain tumor microenvironments are also characterized by mild hyperthermia (1–2 °C above healthy tissues) and some treatment modalities imply rising temperature which, together with pH sensitiveness, can be of help for triggering drug release [25]. In addition, extracellular space is considered oxidative in comparison with intracellular compartment (≈100–1000 folds), mainly in the hypoxic area of tumors, caused by different concentration levels of glutathione [118]. Finally, an array of tumor-associated enzymes, either extracellular or intracellular, can be used as biochemical trigger of drug release to attain a fine control on spatial distribution of the delivered cargo [118].
3.4 Building Carriers for NanoPDT
Polymer-based NPs are matrix-type submicron-sized particles prepared from biodegradable or non-biodegradable materials. In some cases they can be nanocapsules (NCs), where an oily or aqueous core is surrounded by a polymeric shell. The main advantage of polymeric NPs is that their features (size, surface properties, and release rate of drug cargo) can easily be tuned by selecting appropriate materials among a vast number of commercially available candidates as well as by synthesizing novel tailor-made materials [20, 36].
Depending on base material, some NPs are biodegradable/bioeliminable and can be administered by the parenteral route although others are not, and thus are useful only for local applications, assuming that no systemic NP absorption occurs. When dealing with NP entering the systemic circulation, degradability of the polymer is of utmost importance because polymer accumulation in the body above a certain molecular weight can occur. Drug biopharmaceutical properties (solubility, stability, charge, molecular weight, etc.) also guide nanocarrier design and suggest the requirements of specific release features (triggered, sustained) as well as preferential location inside or outside cancer cells.
Numerous materials of synthetic and natural origin have been used to develop PS-loaded NPs. Commonly, the well-defined structure of synthetic polymers results in well-defined and finely-tunable properties of the corresponding NPs. In comparison, natural polymers offer some advantages over synthetic polymers as they are metabolized by enzymes into innocuous side-products. They also take advantage of more than a few drug loading mechanisms including electrostatic attractions, hydrophobic interactions, and covalent bonding. Moreover, NPs from natural polymers offer various possibilities for surface modification caused by the presence of functional groups on the surface of the corresponding NPs, thus enabling conjugation with targeting moieties.
Synthetic polymers commonly employed to produce NPs for drug delivery are reported in Table 2. Polymers that are hydrophobic and insoluble in water form the core of NPs, which can be further modified by depositing one or more layers of hydrophilic polymers, surfactants, or phospholipids to give shells with tailored properties. Instead, hydrophilic polymers can form NP core or shell either by cross-linking or by electrostatic interactions with ions or hydrophilic polymers of opposite charge (nanogels). A further coating of the hydrophilic core with another hydrophilic polymer or a surfactant is possible.
Amphiphilic block copolymers (ABCs) are a class of synthetic materials obtained by the polymerization of more than one type of monomer, typically one hydrophobic and one hydrophilic, so that the resulting molecule is composed of regions with opposite affinities for an aqueous solvent [119, 120]. An advantage of ABCs is their ability to self-assemble in an aqueous environment giving nanostructures spontaneously. Either hydrophobic polymers covalently modified with hydrophilic chains or hydrophilic polymers modified with a hydrophobic moiety (which can also be the active drug) have been synthesized so far. The literature abounds with studies encompassing different functional blocks that produce, beside spontaneously formed micelles (spherical, worm-like, crew-cut), an astounding range of other nanoassemblies depending on amphiphile properties [120–125]. The versatility of these materials allows proper design of nanocarriers with specific features depending on the desired application.
Stimuli-responsive polymers, referred to as “environmentally-sensitive,” “smart,” or “intelligent” polymers, incorporate a chemical motif sharply responding to small changes in physical or chemical conditions with relatively large phase or property changes of the nanocarrier. Over the past 25 years, a huge number of chemical structures and functionalizations have been proposed for numerous biomedical uses [126]. Thus, pH-, redox potential-, and thermo-responsive materials have been applied in the cancer field to build nanocarriers with triggered drug release [24, 118].
PSs can be loaded in NPs through encapsulation, covalent linkage, or post-loading. PSs have also been exposed on NP surface in some NP types. Encapsulation relies on physical entrapment of a PS in NP core or shell based on hydrophobic or electrostatic interactions and hydrogen bond formation. PS loading in the core of NPs can contribute to achieving a sustained release rate in the biological environment and timing of drug release can be finely tuned by allocating different drugs in the core or the shell, which is especially important in combination therapies. In the post-loading method, PS is added to preformed NPs by equilibrium in solution. The latter method is simple to perform although NPs can suffer premature PS leaching, which can be a drawback for in vivo application. Covalent binding of a PS to NPs is difficult to attain and requires either attachment of a PS to monomers that are then polymerized or self-assembled in NPs or post-modification of preformed NPs. Advantages of this strategy consist in preventing PS leaching from NPs and avoiding PS aggregation in biological environments. Independent of the loading strategy, aggregation of PS inside the matrix needs to be controlled to circumvent loss of PDT efficiency.
From a therapeutic standpoint, timing of drug release is important not only to drive the administration scheme (number of administrations, frequency) but also to optimize the therapeutic outcome. For example, sustained extracellular release can be expected to amplify cell response to some chemotherapeutics and to extend activity to hypoxic zones of certain tumors, resembling a metronomic therapy (subactive doses for longer time frames) [127], whereas responsiveness to external or internal stimuli can be useful to trigger drug release at specific subcellular levels. Nevertheless, timing of drug release can be finely tuned by allocating different drugs in the core or the shell, which is of utmost importance in drug-nucleic acid combination therapies [97, 128]. In all cases, drug amount released from NPs should be reasonably low in the circulation and regulated at tumor level to obtain the optimal therapeutic response.
It should be noted that release of PS from NPs is not considered determinant to achieve a therapeutic effect because molecular oxygen can penetrate polymer matrix and generated 1O2 can diffuse out of NPs to induce photodynamic reactions. In such cases, PDT efficiency depends on NP type (size and oxygen permeability of the matrix) [129].
In general, NPs can be prepared by top-down and bottom-up approaches, each method being useful for a specific material and its combination with others. Bottom-up approaches primarily consist in NP production from monomers or preformed polymers by techniques such as emulsification/solvent evaporation, interfacial deposition after solvent displacement, or salting-out [130–133]. By taking advantage of the unique properties of polymers, such as low melting temperature and the ability to self-aggregate in water, novel preparation methods of NPs based on melting/sonication can be set-up [134]. New approaches, including supercritical technology, electrospraying, premix membrane emulsification, and aerosol flow reactor methods are also under investigation [135]. In the top-down approach, originating from microfabrication tools, monodispersed nanostructures in a range of shapes can be obtained. Among them, particle replication in non-wetting templates (PRINT) technology, involving the use of a nanoscale molds to shape particles, has opened a new avenue to NP production in cancer therapy on an industrial scale [105]. Nevertheless, general principles of applicability of this method to several polymer types and the possibility to engineer surface properties finely are necessary in the near future. It is worth of note that surface properties of NPs are strictly dictated by the production method, which is especially critical when specific targeting elements have to be exposed [136].
The unique nanoscale structure of NPs provides significant increases in surface area to volume ratio which results in notably different behavior compared to larger particles. The stability of colloids, which can be at risk during manufacturing, storage, and shipping, remains a very challenging issue during pharmaceutical product development. To obtain stable NPs, the freeze-drying process is the most useful method for avoiding undesirable changes upon storage. The removal of water from drug-loaded NPs by freeze-drying may be fundamental to avoid the hydrolytic degradation of biodegradable matrix in aqueous suspension and to prevent drug leaching [137]. However, freeze-drying can promote NP aggregation and alter their properties after redispersion in pharmaceutical vehicles. Some sugars such as trehalose, glucose, sucrose, fructose, and sorbitol may be used as cryoprotectants to minimize NP instability upon freeze-drying, preventing their aggregation and protecting them from the mechanical stress of ice crystals. Physical and chemical stability of drug-loaded NPs, including their mechanisms and corresponding characterization techniques, as well as a few common strategies to overcome stability issues, have been reviewed recently [138].
In the following sections we describe different types of nanoPDT polymeric systems, highlighting novel trends in design and specific features achieved.
4 NPs Developed for NanoPDT
4.1 Polysaccharide NPs
Polysaccharides extracted from natural sources or prepared by microorganisms represent the most diffused example of natural polymers employed in the biomedical field. Their use as biomaterials has become much more common as new biological functions are identified. The array of materials that can be investigated has also increased because of new synthetic routes that have been developed for modifying polysaccharides. Their biodegradability, processability, and bioactivity also make polysaccharides very promising natural biomaterials in nanoPDT (Table 3).
Chitosan (CS) is considered one of the most widely used biopolymers for NP preparation because of its unique structural features. CS is a cationic polysaccharide composed of randomly located units of d-glucosamine and N-acetylglucosamine. CS is insoluble in water at neutral and basic pH conditions because it contains free amino groups. In contrast, in acidic pH conditions, CS is soluble because the amino groups can be protonated. CS can be cross-linked with various cross-linking agents, such as glutaraldehyde, sodium tripolyphosphate, and geneipin, to provide a hydrated network where drug molecules can be entangled. Its properties make possible the combination with other anionic polymers to provide polyionic complexes, improving the performance of the base material [139]. CS NPs can be created by emulsion cross-linking, emulsion-solvent extraction, emulsification solvent diffusion, emulsion droplet coalescence, ionotropic gelation, complex coacervation, reverse microemulsion techniques, and self-assembly [140]. Physiologically, lysozyme is the primary degrading enzyme and CS degradation rate is dependent on the degree of acetylation and crystallinity [141].
Because of cationic surface, CS-based NPs are especially suited to entrap hydrophilic PSs with the final aim to improve their cell uptake as demonstrated for the hydrophilic meso-tetra(N-methyl-4-pyridyl) porphine tetra tosylate (TMP) [142]. NPs of 560 nm in diameter were endocytosed into HCT116 colorectal carcinoma cells and elicited a more potent photocytotoxic effect than the free drug. To improve NP specificity toward cancer cells, surface-conjugation of an antibody to DR5, a cell surface apoptosis-inducing receptor up-regulated in various types of cancer, was demonstrated to enhance uptake and cytotoxicity further.
Recently, several hydrophobically modified CS derivatives able to self-assemble in NPs with a positively-charged shell entangling hydrophilic negatively-charged PSs and a hydrophobic core accommodating poorly soluble drugs have been reported [143]. It is envisaged that this feature can allow efficient delivery of multiple drugs with different physicochemical properties. When loading chlorin E6 (Ce6) in NPs fabricated from CS modified with ursodeoxycholic acid, fluorescence quenching was observed in aqueous solution. Surprisingly, Ce6 uptake into HuCC-T1 cholangiocarcinoma cells, phototoxicity and ROS generation were enhanced compared to free Ce6 [144], suggesting Ce6 photoactivation only takes place in a biological environment.
In another example, the importance of premature drug leaching from hydrophobically modified CS NPs on in vivo performance has been demonstrated [145]. In a comparative study, Ce6 was loaded into the hydrophobically-modified glycol CS-5-beta-cholanic acid conjugate (HGC-Ce6) or conjugated to glycol CS (GC-Ce6) to form NPs with similar average diameters (300–350 nm), similar in vitro 1O2 generation, and rapid uptake in SCC-7 squamous-cell carcinoma cells. When intravenously injected into tumor-bearing mice, HGC-Ce6 did not accumulate efficiently in tumor tissue because of premature Ce6 release, although GC-Ce6 showed a prolonged circulation profile, a more efficient tumor accumulation, and high therapeutic efficacy.
Implementation of CS NPs was attempted with the aim of attaining responsiveness to a reductive tumor environment. Self-assembling NPs made of glycol CS (GC) with reducible disulfide bonds conjugated with pheophorbide A (GC-SS-PheoA) were designed [146]. As shown in Fig. 3, the photoactivity of NPs in an aqueous environment was greatly suppressed by the self-quenching effect, which enabled the PheoA-SS-CC NPs to remain photo-inactive. NPs were internalized in HT-29 human colorectal adenocarcinoma cells and dissociated instantaneously by reductive cleavage of the disulfide linkers. The following efficient dequenching process resulted in effective photodynamic activity on HT-29 cells. In subcutaneous tumor-bearing mice, NPs presented prolonged blood circulation, demonstrating enhanced tumor specific targeting behavior through the EPR effect, and superior antitumor effects compared to free PheoA.

Bioreducible chitosan NPs for switchable photoactivity of PheoAA. (A) PheoA is conjugated to glycol chitosan (GC) through reducible disulfide bonds (PheoA-ss-GC). (B) Self-quenching and dequenching of free PheoA, PheoA–NPs, and PheoA-ss-NPs in different solvents. (C) NIR images of PheoA–NPs and PheoA-ss-NPs in PBS with (+) or without (−) DTT solution. (D) Ex vivo fluorescence photon counts of tumor and organs. (E) Tumor growth of HT-29 tumor-bearing mice treated with PheoA and PheoA-ss-CNPs under irradiation. (#,*p < 0.01). Adapted from [146]
It has been demonstrated that iodine-concentrated nanoformulations can enhance the 1O2 generation efficiency because of the intraparticle heavy-atom effect that facilitates intersystem crossing of the photoexcited PS from the singlet state to the long-lived triplet state. On this basis, a CS densely conjugated with diatrizoic acid (3,5-bis(acetamido)-2,4,6-triiodobenzoic acid) as an iodine-rich hydrophobic pendant and Ce6 (GC-I-Ce6) was synthesized and used to fabricate self-assembled polymeric NPs [147]. Actual improvement in the photodynamic efficacy of MDA-MB-231 human breast cancer cells demonstrated the potential of the hybrid bioconjugate approach in therapeutic applications.
Multifunctional hybrid NPs made of a glutaraldehyde-cross-linked CS entrapping indocyanine green (ICG) and gold nanorods (AuNR) were successfully prepared and used for combined PDT/PTT with a single irradiation [148]. It was found that the hybrid NPs with a spherical size of 180 nm and a broad adsorption from 650 to 900 nm effectively entrapped ICG and protected it from rapid hydrolysis. In vivo NIR imaging and biodistribution demonstrated that ICG and AuNR could be delivered to the tumor site with high accumulation. With the irradiation by 808 nm laser, CS hybrid nanospheres were able to produce simultaneously sufficient hyperthermia and ROS to kill cancer cells at irradiation sites, resulting in complete tumor disappearance in most tumor-bearing mice.
Among polysaccharides, alginates are linear polyanionic block copolymers composed of 1-4-linked β-d-mannuronic acid and α-l-guluronic acid with recognized biocompatibility and bioresorption properties. Because of ionic interactions with divalent ions, they can form cross-linked hydrated NPs. Alginate-docusate NPs cross-linked with calcium ions and encapsulating MB through electrostatic interaction have been developed [149, 150]. It was demonstrated that these NPs facilitate charge transfer and Type I reaction which is less sensitive to environmental oxygen concentration. NPs led to an increased production of ROS under both normoxic and hypoxic conditions and were able to eliminate cancer stem cells under hypoxic conditions, an important aim of current cancer therapy [151]
4.2 Protein NPs
The presence of multiple sites able to accommodate hydrophobic drugs has pointed to human serum albumin (HSA) as an interesting option to deliver hydrophobic PSs. HSA is an abundant plasma protein that is positively-charged, acidic, and multifunctional [152]. It is produced by extraction from plasma as an amphoteric, globular protein which maintains its structure in the pH range of 4–9, is soluble in 40% ethanol (an important parameter which is of great importance to albumin production processes such as cold ethanol fractionation), and can resist denaturation when heated at 60 °C for over 10 h [140]. Albumin NPs can be prepared by pH-induced desolvation which can include cross-linking by glutaraldehyde molecules, thermal and chemical treatments under emulsification, and self-assembly. NP albumin-bound (Nab)-technology has also been applied to create Abraxane® (albumin-bound paclitaxel NPs) which is currently used in clinics to deliver paclitaxel in breast, pancreatic, and lung cancers. Some examples of nanoPDT systems based on HSA, gelatin, and apoferritin are listed in Table 3.
IR780 iodide, a NIR dye for cancer imaging, PDT, and PTT were loaded into HSA NPs through protein self-assembly [153]. Compared to free IR-780, the solubility of HSA-IR780 NPs was greatly increased (1,000-fold), although a 10-fold decreased toxicity was observed. As illustrated in Fig. 4, both PTT and PDT could be observed in HSA-IR780 NPs, as determined by increased temperature and enhanced generation of 1O2 after laser irradiation at a wavelength of 808 nm. In vivo studies also showed a great tumor inhibition in mice bearing a subcutaneous xenograft of CT26 colon adenocarcinoma cells.

HSA-IR780 NPs with NIR irradiation for antitumor therapy. (A) Illustration of the concept of multifunctional NPs. (B) Heating curves of water and HSA-IR780 NPs solutions. (C) Fluorescence intensity of 1O2 sensor green (SOSG) combined with HSA-IR780 NPs solutions at different concentrations exposed to 808 nm laser irradiation (1 W cm−2). (D) NIR images and (E) IR thermal images of tumor-bearing mice intravenously administered with HSA-IR780 NPs and saline. Mice were exposed to an 808-nm laser (1 W cm−2) at 24 h post-injection. (F) Tumor growth of mice bearing CT26 tumor after various treatments as indicated. **P < 0.01, compared to the NPs with laser group. Adapted from [153]
Gelatin is another natural polymer tested for PS delivery. Gelatin is the result of acid or base catalyzed hydrolysis of collagen and its physiochemical properties depend upon the hydrolysis method employed. One of the most important properties of gelatin is its ability to form a thermally reversible gel in water under a variety of pH, temperature, and/or solute conditions [154]. This property is readily utilized by the largest part of the gelatin-based NP encapsulation methods. Gelatin NPs can be created by the water-in-oil emulsification process, the desolvation process, and the two-step desolvation process [140]. The presence of free amino groups on their surface is advantageous for surface modification with target molecules [155].
NPs formulated from biodegradable and natural gelatin were investigated for their potential to enable efficient delivery and enhanced efficacy of a well-known photodynamic agent, Hypocrellin B (HB) [156]. The HB-loaded PEG-conjugated gelatin NPs (HB-PEG-GNP), prepared by a modified two-step desolvation method, exhibited near-spherical shape, with particle size around 300 nm, and demonstrated characteristic optical properties for PDT. NPs tested for cell uptake on Daltons’ Lymphoma Ascites (DLA) cells demonstrated dose-dependent phototoxicity upon visible light treatment, and induced mitochondrial damage leading to apoptotic cell death. Biodistribution measurements in solid tumor-bearing mice revealed that NPs reduce liver uptake and increase tumor uptake with time. In vivo PDT studies showed markedly significant regression for HB-PEG-GNP treated mice in contrast to those treated with free HB. In a subsequent study, polylactic acid (PLA) was added to PEGylated gelatin with the aim of better controlling release features of an HB derivative (cyclohexane-1,2-diamino hypocrellin B, CHA2HB) [157]. PS release was observed in normal conditions, whereas enzyme assistance resulted in a relatively fast release because of partial disintegration of CHA2HB-loaded PEG-GEL/PLA NPs. In vitro experiments indicated that NPs were efficiently taken up not only by Dalton’s lymphoma cells but also by MCF-7 human breast adenocarcinoma and AGS human gastric sarcoma. Interestingly, PDT effectiveness was different for the different cell type studied and induced both apoptotic and necrotic cell death as a result of photoirradiation.
Another protein, apoferritin, has been proposed as a natural nanocage for PSs. Taking advantage of the fact that apoferritin nanocages can be disassociated into subunits at low pH (2.0) and the subunits reconstitute in a high pH (8.5) environment, a novel encapsulation approach has been proposed. As a model, MB was successfully encapsulated in apoferritin via a dissociation-reassembly process controlled by pH [158]. The resulting MB-containing apoferritin nanocages showed a positive effect on 1O2 production, and cytotoxic effects on MCF-7 human breast adenocarcinoma cells when irradiated at the appropriate wavelength.
4.3 Polyester NPs
In the last 30 years, particular attention has been focused on nanocarriers based on biodegradable polyesters such as poly(ε-caprolactone) (PCL), PLA from d- and/or l-lactic acid monomers (PLLA, PDLLA) and copolymers of lactic acid with glycolic acid (PLGA) because of their better safety profile (degradation products are water and carbon dioxide). Their use has been approved by the regulatory agencies in implantable devices and in injectable products (implants, microspheres). As core-forming polymers, polyesters are appropriate to form NPs with sustained delivery features of the incorporated drug. Indeed, polymer molecular weight and crystallinity, along with the presence of more or less hydrophobic monomers, are the key properties to control both encapsulation efficiency and biodegradation rate, which in turn allows a fine tuning of drug delivery rate [159, 160]. A molecule entrapped in a PLA or PLGA matrix is protected from inactivation occurring in the biological environment and is slowly released in the milieu as a function of diffusion through matrix micropores and degradation of the polymer itself. A drug burst followed by a slow diffusion phase and a fast erosion phase in the time-window of months is observed. By regulating polymer features in term of monomer composition and molecular weight, a large variety of materials with different degradability can be obtained.
Biodegradable polymeric NPs have received tremendous attention for delivering PSs because of their excellent biodegradability, capacity of high drug loading, the possibility of controlling drug release rate, and the existence of a large variety of derivatives (Table 4).
The simplest type of polyester-based NPs developed for PDT application consists in PLGA or PCL hydrophobic NPs entrapping hydrophobic PSs. Early studies highlighted that PDT response in both in vitro and in vivo cancer models is strictly related to NP size [161, 162], cellular internalization pathways [163, 164], and timing/dosing of light exposure [165]. The time interval between NP administration and light irradiation is a determinant for therapeutic efficacy, because it is related to the time needed for NPs to biodistribute in the body. As an example, PLGA NPs delivering SL052, a hypocrellin-based photosensitizer, induced a higher tumor cure rates in syngenic C3H/HeN mice bearing a subcutaneous xenograft of SCC-7 squamous carcinoma cells with a drug-light interval of 4 h compared to 1 h [165]. When injecting zinc(II) phthalocyanine (ZnPc)-loaded PLGA NPs intratumorally into mice bearing an Ehrlich’s Ascites Carcinoma, good PDT outcome was observed in term of tumor growth and survival compared to free ZnPc [166].
PEG is a hydrophilic polymer which can be chemically conjugated to polyester forming ABCs able to provide a vast variety of nanostructures (micelles, NPs, polymersomes, filomicelles) and to entrap hydrophobic and hydrophilic drugs [167]. The opportunity to tune the length of the single chains, the chemical composition of polyester segments, and the arrangement of PEG-polyester segments (diblock, triblock, star-shape) has allowed the building of a wide range of nanocarriers designed with specific delivery requirements. As far as spontaneous self-assembly is concerned, hydrophilic/lipophilic balance, copolymer molecular weight, and properties of the core (crystallinity, hydrophobicity) strongly affect critical micelle concentration, thereby controlling micelle disassembly in biological media [168]. When employing PEGylated polyesters to form core/shell NPs, the conformation of PEG on the surface is dictated by PEG molecular weight, architecture, and surface density [110, 112, 169].
PEGylated polyesters nowadays represent one of the most promising classes of copolymers to translate nanoncologicals in a clinical setting because of excellent biocompatibility and chemical versatility [170, 171]. Polymeric micelles of PDLLA-PEG (Genexol-PM) represent the first polyester system for passive targeting of taxanes approved in Korea in 2006 as a first-line therapy for metastatic breast and non-small cell lung cancer (Phase III) and are currently being evaluated in the USA in a Phase II study on metastatic pancreatic cancer.
Several types of NPs made of PEGylated polyesters, such as PEG-PCL, PEG-PLGA, and PEG-PLA, have been tested in nanoPDT as delivery system for hydrophobic PSs (Table 3) based on the concept that PEGylated NPs exhibited therapeutically favorable tissue distribution compared to non-PEGylated counterparts in delivering PSs in vivo [172–176].
Different entrapment strategies to incorporate PSs into NPs can severely affect photochemical profile. For instance, Ding et al. [177] demonstrated that PpIX loaded in the core of PEG-PLA micelle nanocarriers were monomeric, dimeric, and aggregated depending on the method of encapsulation (physical entrapment or chemical conjugation to the copolymer). The obvious consequence was different photochemical behavior in terms of 1O2 generation and PDT activity, with the highest PDT efficacy in the case of conjugates micelles. Along this line, chlorin-core star block PEG-PLA micelles loaded with SN-38 as second anticancer drug were found to improve significantly the cytotoxicity of SN-38 in HT-29 human colorectal adenocarcinoma cells after irradiation [178]. Micelles exhibited a prolonged plasma residence time in mice bearing subcutaneous xenografts of HT-29 human colon adenocarcinoma cells, as well as increased tumor accumulation which improved antitumor activity.
Dual drug delivery of ZnPc and the anticancer drug docetaxel from PEG-PCL was recently demonstrated by our group [179]. These systems showed superior antitumor activity compared to the free drugs in an orthotopic mice model of amelanotic melanoma. Similar synergic effects were demonstrated for PEG-PDLLA nanovesicles loaded with Hp/doxorubicin (DOX) against HepG2 human hepatocellular carcinoma cells through apoptotic cell death pathways [180].
The versatility of PEGylated polyesters and the ability to employ different polymer combinations allow the fabrication of hybrid NPs for theranostic applications. Multifunctional NPs based on PEG-PLGA mixed with PLGA-Ce6, and loaded with superparamagnetic iron oxide nanoparticles (SPIONs) for luminescence/magnetic resonance imaging and PDT, have been developed [181]. It was demonstrated that, depending on the amount of PLGA conjugated to the PS, NPs exhibited a different ability to produce 1O2 because of concentration-dependent aggregation of Ce6 in NPs when encountering a biological environment.
Combined PDT nanosystems have also been tested to evaluate the ability of PS in eliciting immune response against tumor. An ideal cancer treatment should not only cause tumor regression and eradication but also induce a systemic antitumor immunity controlling metastasis formation and long-term tumor resistance. Marrache et al. [182] formulated PLGA-PEG NPs loaded with ZnPc and modified on the surface with gold NPs (AuNPs) by using non-covalent interactions. For immune stimulation, the surface of the AuNPs was utilized to introduce 5′-purine-purine/T-CpG-pyrimidine-pyrimidine-3′-oligodeoxynucleotides (CpG-ODN) as a potent dendritic cell activating agent. In vitro cytotoxicity on 4T1 metastatic mouse breast carcinoma cells showed significant photocytotoxicity of NPs and the treatment of mouse bone marrow-derived dendritic cells with the PDT-killed 4T1 cell lysate highlighted the immunostimulant activity of PDT through involvement of several cytokines.
As an alternative to preparing NPs from preformed PEGylated polyesters, surface coating of PLGA NPs with PEGylated lecithin has recently been reported [183]. NPs loaded with both ICG and DOX were prepared in one step for combination of chemotherapy with PTT NPs showed excellent temperature response, faster DOX release under laser irradiation, and longer retention time in mice bearing a subcutaneous xenograft of MCF-7 human breast cancer cells compared with free ICG. NPs induced the apoptosis and death of both DOX-sensitive MCF-7 and DOX-resistant MCF-7/ADR cells and suppressed MCF-7 and MCF- 7/ADR tumor growth in vivo. Notably, no tumor recurrence was observed after only a single dose of NPs with laser irradiation.
Despite the obvious promises shown, PEGylated nanoncologicals are poorly prone to be uptaken inside cells (PEG dilemma) [176] and remain entangled in tumor matrix, forming an extracellular drug depot releasing drug cargo. To encourage internalization of PSs in cancer cells, the most diffused strategy lies in nanocarrier surface decoration with ligands, typical or overexpressed in tumor microenvironments, which can promote nanocarrier transport through receptor-mediated endocytosis. As an example, Master et al. built PEG-PCL micelles targeted to cancers overexpressing epidermal growth factor receptor (EGRF), such as head and neck cancers, and delivering silicon phthalocyanine [184, 185]. Analogously, micelles decorated with the 12-amino acid EGFR-targeting peptide GE11 (i.e., GE11-PEG-PCL) showed high uptake in targeted tumor cells and, as a consequence, a strong PDT response, depending on photoirradiation parameters. H2O2-activatable and O2-evolving NPs targeted to α,β integrin receptor exploiting a clever strategy to treat hypoxic tumor area—where PDT effect is expected to be poor—have recently been reported (Fig. 5) [186]. NPs bear MB and catalase in the aqueous core, a black hole quencher in the polymeric shell, and are functionalized with a tumor targeting peptide c(RGDfK) to be selectively taken up by α,β integrin-rich tumor cells. In the intracellular compartment, H2O2 penetrates into NP core and generate O2, which is the substrate for 1O2 production under light irradiation, through catalase activity. Following shell rupture and release of PS activate local PDT. In vivo studies in glioblastoma bearing mice showed that tumor growth is completely inhibited, and the tumor eliminated after 7 days of treatment.

H2O2-activatable and O2-evolving NPs for photodynamic therapy against hypoxic tumor cells. (A) Mechanism of H2O2-controllable release of photosensitizer and O2 to implement PDT. (B) In vitro release profiles of MB from NPs (with/without catalase) in the presence or absence of 100 μM H2O2. Insets: SEM micrographs of NPs incubated with 100 μM H2O2 (scale bars: 100 nm). (C) Confocal fluorescence images of U87-MG cells after 635 nm irradiation in the presence of 1O2 sensor green (SOSG) (b) SOSG only; (c) NPs + SOSG; (d) NAC + NPs + SOSG; (e) NPs (without catalase) + SOSG. (D) Normalized average intracellular fluorescence intensity of cells in C. (E) Change of relative tumor volume (V/V0) in U87-MG tumor-bearing mice injected with NPs. After 24 h, PDT treatment was performed on groups 4−7 by irradiating the tumor region with a 635-nm laser at a power of 100 mW cm−2 for 5 min. Adapted from [186]
Non-covalent approaches useful to modify NP surface have recently been investigated as an alternative to chemical functionalization of copolymers. On this basis, overwhelming interest has been taken in layer-by-layer (LbL) NPs engineered for cancer therapy [187]. Consecutive deposition through electrostatic interactions of ionized polymers with opposite charge onto a nanotemplate results in ultrathin multilayers of polymer chains. By dictating and controlling type, composition, number of alternating layers surrounding the core, and final layer thickness, multifunctional core-shell nanostructures can be obtained where multiple drugs and magnetic or luminescent layers can be formed. In this context, our group developed double coated NPs (dcNPs) targeted to CD44 receptor, which is overexpressed in several cancers [188, 189]. Negatively-charged DTX-loaded NPs of PLGA were sequentially decorated through electrostatic interactions with a polycationic shell of polyethyleneimine entangling negatively-charged TPPS4 and a final layer of hyaluronan (HA), a CD44 ligand. dcNPs bears TPPS4 completely aggregated and photochemically inactive at their surface and they release the active monomer after cell internalization, which is higher in MDA-MB231 breast cancer cells overexpressing CD44 receptor. This aspect is of relevance in view of in vivo application because dcNPs are expected to be nonfluorescent and non-photoactive in non-target organs, thereby strongly reducing phototoxicity of carried PS. Nevertheless, taking advantage of targeting to CD44 receptor, dcNPs can localize in tumor tissue, where the PDT component is disassembled from the dcNPs surface and becomes highly fluorescent and phototoxic. The concerted delivery of DTX and TPPS4 resulted in a higher uptake of the hydrophilic PS and tremendous improvement of single drug activity.
Modification of polyesters with pH-sensitive segments allows the building of NPs responsive to tumor acidic microenvironments. pH-responsive micelles made of poly(2-ethyl-2-oxazoline)-b-poly(d-l-lactide) entrapping m-THPC deliver PS at pH around 5 and suppresses release at pH 7.4. Nevertheless, micelles exhibited in vivo PDT activity similar to that of free m-THPC while strongly attenuating skin phototoxicity [190]. Analogously, PEG-b-poly(beta-aminoesters) entrapping PpIX displayed in vivo PDT activity and tumor specificity in mice bearing SCC-7 squamous carcinoma, causing complete tumor ablation [191].
4.4 Polyacrylamide NPs
Polyacrylamide (PAA) NPs are hydrogel-like nanostructures prepared by polymerization of a nanoemulsion template widely studied in PS delivery (Table 5). Hydrophilic PAA NPs can be prepared from biodegradable or non-biodegradable cross-linkers and decorated on the surface with targeting ligands.
The simplest strategy to load a PS inside PAA NPs is either encapsulation or post-loading, which can have a different impact on PDT efficacy depending on PS water solubility. When employed for hydrophilic PSs such as MB and its derivatives, conjugation gave PAA NPs with higher 1O2 production yield and enhanced cell mortality under PDT [192, 193]. When those NPs were surface-decorated with F3 peptide to attain specific targeting to 9L rat gliosarcoma cells, MDA-MB-435 human breast cancer cells, and F98 rat glioma cells, excellent PDT efficacy, increasing with the NP dose and irradiation time, was found [192, 193].
In the case of a hydrophobic PSs such as HPPH and its derivatives, post-loading in preformed amine functionalized polyacrylamide (AFPAA) gives the highest 1O2 production and phototoxicity in vitro compared to encapsulation and conjugation [194]. NPs, tested in a mice colon carcinoma xenograft, enabled fluorescence imaging of the tumor and produced a photodynamic response similar to that of free HPPH at an equivalent dose [194].
When developing multifunctional nanoplatforms loaded with more than one drug, confinement of multiple absorbing species in NPs can have a strong impact on in vitro and in vivo PDT outcomes. To operate in parallel, the two photoresponsive agents should not interfere with each other when in close proximity in the same polymeric scaffold. This aspect was clearly shown with HPPH and a tailor-made cyanine dye for PDT and fluorescence imaging [195]. By playing on HPPH/cyanine dye ratio, the undesirable quenching of the HPPH because of Förster Resonance Energy Transfer between the two molecules was minimized. NPs at optimal ratio resulted in an excellent tumor-imaging (NIR fluorescence) and PDT efficacy in mice bearing a subcutaneous xenograft of Colon 26 cells.
Besides 1O2, light can trigger the release of cytotoxic species such as nitric oxide (NO) exerting antitumor cooperative effects. Indeed, cytotoxic effects induced by NO [196] combined with PDT represent a very promising strategy in view of a multimodal cancer treatment because of the ability to attack biological substrates of different natures, to avoid MDR, and to improve selectivity of therapy. Finally, as NO release is independent of O2 availability, it can potentially very well complement PDT at the onset of hypoxic conditions. A nanoplatform releasing 1O2 and nitric oxide was thus prepared by the electrostatic entangling of two anionic photoactivable components (TPPS4 and a tailored nitro-aniline derivative) onto the cationic shell of NPs [197]. Photochemical characterization of the nanoplatform clearly showed that the drugs operate in parallel under the exclusive control of light, providing a combinatory effect of the two photogenerated cytotoxic species in B78H1 melanoma cells.
4.5 Pluronic Micelles
Among a variety of triblock copolymers, Pluronics (also termed poloxamers) have achieved the most noticeable interest in pharmaceutics because of their versatility and biocompatibility [198, 199]. Pluronics are commercial FDA-approved material, consisting of a central poly(propylene oxide) flanked by two poly(ethylene oxide) blocks, available in different molecular weights and block lengths and thus characterized by different hydrophilic-lipophilic balances. Pluronics form spontaneously nanosized micelles in aqueous media. Because of their amphiphilic character, these copolymers display surfactant properties including the ability to interact with hydrophobic surfaces and biological membranes. These systems avoid MPS uptake, increase drug solubility, and improve its circulation time and passive tumor targeting by EPR effects [198]. Previously thought to be “inert,” Pluronics display a unique set of biological activities and have been shown to be potent sensitizers of MDR cancer cells in vitro and in vivo [198, 200]. The key attribute for the biological activity of Pluronics is their ability to incorporate into membranes followed by subsequent translocation into the cells where they affect various cellular functions, such as mitochondrial respiration, ATP synthesis, activity of drug efflux transporters, apoptotic signal transduction, and gene expression. As a result, Pluronics cause drastic sensitization of MDR tumors to various anticancer agents, enhance drug transport across the blood brain and intestinal barriers, and cause transcriptional activation of gene expression both in vitro and in vivo [200].
The first example of Pluronic use as carriers for PDT agents was the delivery of Verteporfin® derivatives in monomeric form [201, 202]. Thereafter, Pluronic micelles have been considered a promising vehicle for the delivery of other PDT agents such as porphyrins, chlorins, phthalocyanines, chlorophylls, and xanthene derivatives [203–206]. Sobczyński and collaborators recently evaluated the influence of Pluronics on the photocytotoxicity and cytolocalization of four porphyrin-based PSs, i.e., tetraphenyl porphyrins 4-substituted on the phenyl groups with trimethylamine (TAPP), hydroxyl (THPP), sulfonate (TSPP), and carboxyl (TCPP), in WiDr colon adenocarcinoma cell line [205]. Pluronics were found to deaggregate the PSs and improve PS solubility efficiently. Moderate to profound effects on intracellular localization of the PSs and cellular sensitivity to photoinactivation were found. P123 and F127 strongly attenuated the uptake and photocytotoxicity of THPP and redirected the cellular uptake to endocytosis, while P123 stimulated translocation of TAPP from endocytic vesicles to a cytosolic and nuclear localization followed by an enhanced phototoxicity. P123 and F127 lowered the fraction of TCCP in endocytic vesicles followed by a reduced sensitivity to photoinactivation. F68 had only moderate effects on intracellular localization of the evaluated PSs with the exception of a higher endocytic accumulation of TCPP and lowered photocytotoxicity of TCPP and THPP.
To enhance 1O2 generation, chlorine e6 was encapsulated in heavy-atomic NPs based on Pluronic F127 and in vitro PDT efficacy was evaluated in MDA-MB-231 human breast-cancer cell line [207]. Pluronic F68 was used to increase the intracellular level of ALA in human cholangiocarcinoma cells, resulting in enhanced PpIX formation and phototoxicity [208]. Pluronics P123 and F127 improved the delivery of Photofrin® overcoming MDR in MCF-7/WT human breast and SKOV-3 ovarian cancer cells inducing cell apoptosis through photodynamic effects [209].
A few pieces of work have reported in vivo results with Pluronic-based nanoPDT. Park and Na [210] conjugated chlorine e6 in F127 micelles and observed higher internalization rates, tumor-specific distribution, and in vivo tumor growth inhibition after intravenous injection into mice bearing a colon tumor (CT-26) when compared with free PS. In subsequent work these authors introduced DOX in the formulations for combined photodynamics and chemotherapy overcoming drug resistance in drug-resistant cancer cells [211]. Furthermore, the natural PS chlorophyll (Chl) extracted from vegetables was encapsulated into Pluronic F68 micelles for in vivo cancer imaging and therapy. Results showed that, after intravenous injection and laser irradiation, the growth of melanoma cells and mouse xenograft (A375) were effectively inhibited by laser-triggered PTT and PDT synergistic effects.
A simple and biocompatible nanocomplex of MB in a combination of Pluronic F68 and oleic acid (named NanoMB) driven by the dual (electrostatic and hydrophobic) interactions between the ternary constituents was developed [212]. The nanocomplexed MB showed greatly enhanced cell internalization in different cancer cell lines while keeping the photosensitization efficiency as high as free MB, leading to distinctive phototoxicity toward cancer cells. When administered to human breast cancer xenograft mice by peritumoral injection, nanocomplexed MB was capable of facile penetration into the tumor followed by cancer cell accumulation. After five PDT treatments consisting in a combination of peritumorally injected nanocomplexed MB and selective laser irradiation, tumor volume was significantly decreased, demonstrating potential for adjuvant locoregional cancer treatment.
Additionally, Pluronics have been associated with graphene oxide and gold NPs for in vivo combined PDT and PTT. The complex graphene oxide sheet, Pluronic F127 and MB showed high tumor accumulation after intravenous injection into tumor-bearing mice, causing total ablation of tumor tissue exposed to NIR light [213]. A Pluronic-based nanogel was combined with both gold nanorods as a PTT agent and Ce6 as a PS for PDT. In both in vitro cell culture and in vivo tumor-bearing mice experiments in SCC-7 squamous carcinoma cells or NIH/3T3 fibroblast cells, a remarkably enhanced tumor ablation was observed with treatment by PDT (red laser) followed by PTT (NIR laser) as compared with separate treatments [214].
4.6 Other Systems
Polymer-PS conjugates can spontaneously form different nanostructures. The simplest example among them is represented by PEGylated HpD assembled into NPs and loaded with DOX to achieve a synergistic effect of chemotherapy and PDT [215]. This approach is useful to form core-shell-structured bioreducible self-quenched NPs that dissociate under intracellular reductive conditions, triggering the rapid release of PS in a photoactive form [216]. More complex concepts have been reported recently. NPs based on a star shaped 4-arm PEG functionalized with biotin as targeting unit and a chlorambucil-coumarin fluorophore were synthesized for site-specific and image guided treatment of cancer cells [217]. Telodendrimers, a novel class of hybrid amphiphilic polymers comprised of linear PEG and dendritic oligomers of PheoA and cholic acid (CA) formed NPs by self-assembly (Nanoporphyrins), which greatly increased the imaging sensitivity for tumor detection through background suppression in blood, as well as preferential accumulation and signal amplification in tumors. Nanoporphyrins also functioned as multiphase nanotransducers that can efficiently convert light to heat inside tumors for PTT, and light to 1O2 for PDT.
The Kataoka group studied polyion complex micelles of PEG-poly(l-lysine) block copolymers with an anionic dendrimer Pc (dPc) [218]. The PDT effect of DPc was two orders of magnitude higher than the free DPc with the same irradiation time. In vivo PDT efficacy of dPc-loaded micelles [219] was higher than free dPc and Photofrin®. Furthermore, the skin phototoxicity of the DPc-loaded micelles was significantly reduced after white light irradiation. Lu et al. have loaded in PEG-poly(l-lysine) micelles entrapping dPc and also DOX [220]. In vivo studies carried out in a xenograft model of breast cancer highlighted that the internalized DPc micelles showed unique PCI properties inside the cells and thereby facilitated DOX release from the endo-lysosomes to nuclei after photoirradiation, thus reversing MDR.
To implement multimodality of a cancer therapy, very promising results have been obtained by employing light-activated supramolecular nanoassemblies based on cyclodextrin branched polymers for simultaneous imaging and therapy [221–225]. In these systems, beside the photochemical independence of the two chromogenic centers, a high association constant between the two units was a key prerequisite to avoid displacement in a biological environment if their association is not sufficiently strong. Bichromophoric NPs exploiting TPE fluorescence have been obtained by the self-assembly of a NO photodonor, ZnPc, and a β-CD polymer [226]. The macromolecular assembly delivered its photoresponsive cargo of active molecules not only within the cytoplasm but also in human skin as exemplified in ex vivo experiments.
In the attempt to apply layer-by-layer fabrication technology to theranostic nanocapsules for cancer PDT combined with magnetic resonance imaging (MRI), a platform entrapping dendrimer porphyrin (DP) in the shells and SPIONs in the core was designed [227]. SPIONs-embedded polystyrene NPs were used as a template to build up multilayered nanocapsules through sequential poly(allylamine hydrochloride)/DP deposition. NCs exhibited typical superparamagnetic behavior, and cell viability study (HeLa) upon light irradiation revealed that NCs can successfully work in PS formulation for PDT.
5 Conclusions
PDT is gaining momentum as an alternative or complementary treatment of solid tumors. Potentiating PDT effects and coupling PDT to other treatment modalities are considered promising strategies to fight tumors. Polymeric NPs are supportive of this evolution, offering a wide variety of options to engineering multimodal systems useful for therapeutic, diagnostic and theranostic purposes. NP design needs to be established ab initio and should be driven by biologically-oriented design rules to accumulate drug cargo at the pharmacological target and by specific requirements to preserve/optimize photochemical properties of delivered PS. Rational combination of building elements in a single nanoplatform can also couple PDT to other treatment modalities (conventional chemotherapy, PPT, radiotherapy) or imaging (MRI, fluorescence) propelling the application of PDT to the forefront of diagnosis and therapy of cancer.
Abbreviations
- 1O2 :
-
Singlet oxygen
- ABC:
-
Amphiphilic block copolymers
- AFPAA:
-
Amine-functionalized PAA
- AFPMMA:
-
Amine-functionalized polyacrylamide
- ALG:
-
Alginate
- AuNR:
-
Gold nanorods
- c(RGDfK):
-
Tumor targeting peptide
- Ce6:
-
Chlorin E6
- CHA2HB:
-
Cyclohexane-1,2-diamino hypocrellin B
- CpG-ODN:
-
5′-Purine–purine/T-CpG–pyrimidine–pyrimidine–3′–oligodeoxynucleotide
- CS:
-
Chitosan
- DOX:
-
Doxorubicin
- DR5:
-
Antibody targeting death receptor 5
- DTX:
-
Docetaxel
- EPR:
-
Enhanced permeability and retention
- GA:
-
Glutaraldehyde
- GC:
-
Glycol chitosan
- HA:
-
Hyaluronic acid/hyaluronan
- HB:
-
Hypocrellin B
- HMME:
-
Hematoporphyrin
- HpD:
-
Hematoporphyrin derivative
- HPPH:
-
2-(1-Hexyloxyethyl)-2-devinyl pyropheophorbide A
- HSA:
-
Human serum albumin
- ICG:
-
Indocyanine green
- MB:
-
Methylene blue
- MDR:
-
Multidrug resistance
- MRI:
-
Magnetic resonance imaging
- NIR:
-
Near infrared
- NPs:
-
Nanoparticles
- PAA:
-
Poly(acrylic acid)
- PAAm:
-
Poly(acrylamide)
- Pc4:
-
Silicon phthalocyanine
- PCL:
-
Poly(ε-caprolactone)
- PDEAEMA:
-
Poly(diethylaminoethyl methacrylate)
- PDLLA:
-
Poly(d,l-lactic acid)
- PDT:
-
Photodynamic therapy
- PEG:
-
Polyethylene glycol
- PEG-GEL:
-
Poly(ethylene glycol)-modified gelatin
- PEI:
-
Poly(ethylenimine)
- PheoA:
-
Pheophorbide A
- PLA:
-
Poly(lactic acid)
- PLGA:
-
Poly(lactic-co-glycolic acid)
- PLL:
-
Poly(l-lysine)
- PMA:
-
Poly(methacrylic acid)
- pNIPAM:
-
Poly(N-isopropylacrylamide)
- PpIX:
-
Protoporphyrin IX
- PS:
-
Photosensitizer
- PTT:
-
Photothermal therapy
- ROS:
-
Reactive oxygen species
- TCPP:
-
meso-tetra(Carboxyphenyl) porphyrin
- THPP:
-
tetra(Hydroxyphenyl)porphyrin
- TMP:
-
meso-tetra(N-Methyl-4-pyridyl) porphine tetratosylate
- TPPS4 :
-
tetrasodium-meso-tetra(4-sulfonatophenyl) porphyrine
- UDCA:
-
Ursodeoxycholic acid
- ZnPc:
-
Zinc phthalocyanine
References
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG (2013) Nat Rev Cancer 13:714–726
Wu Q, Yang Z, Nie Y, Shi Y, Fan DE (2014) Cancer Lett 347:159–166
Jain KK (2010) BMC Med 8:83
Jain RK, Stylianopoulos T (2010) Nat Rev Clin Oncol 7:653–664
Wang AZ, Langer R, Farokhzad OC (2012) Annu Rev Med 63:185–198
Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R, Jacks T, Anderson DG (2012) Nat Rev Cancer 12:39–50
Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY (2013) Adv Drug Deliv Rev 65:1866–1879
Palakurthi S, Yellepeddi VK, Vangara KK (2012) Expert Opin Drug Deliv 9:287–301
Iyer AK, Singh A, Ganta S, Amiji MM (2013) Adv Drug Deliv Rev 65:1784–1802
Webster DM, Sundaram P, Byrne ME (2013) Eur J Pharm Biopharm 84:1–20
Moghimi SM, Hunter AC, Andresen TL (2012) Annu Rev Pharmacol Toxicol 52:481–503
Bertrand N, Leroux JC (2012) J Control Release 161:152–163
Alexis F, Pridgen E, Molnar LK, Farokhzad OC (2008) Mol Pharm 5:505–515
Fang J, Nakamura H, Maeda H (2011) Adv Drug Deliv Rev 63:136–151
Maeda H (2012) J Control Release 164:138–144
Huynh NT, Roger E, Lautram N, Benoit JP, Passirani C (2010) Nanomedicine (Lond) 5:1415–1433
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC (2014) Adv Drug Deliv Rev 66:2–25
Kim CS, Duncan B, Creran B, Rotello VM (2013) Nano Today 8:439–447
Torchilin VP (2014) Nat Rev Drug Discov 13:813–827
Mura S, Nicolas J, Couvreur P (2013) Nat Mater 12:991–1003
Gao W, Chan JM, Farokhzad OC (2010) Mol Pharm 7:1913–1920
Tian L, Bae YH (2012) Colloids Surf B Biointerfaces 99:116–126
Du JZ, Mao CQ, Yuan YY, Yang XZ, Wang J (2013) Biotechnol Adv 32:789–803
Nowag S, Haag R (2014) Angew Chem Int Ed Engl 53:49–51
Abulateefeh SR, Spain SG, Aylott JW, Chan WC, Garnett MC, Alexander C (2011) Macromol Biosci 11:1722–1734
Dolmans DEJG, Fukumura D, Jain RK (2003) Nat Rev Cancer 3:380–387
Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J (2011) CA Cancer J Clin 61:250–281
Allison RR (2014) Future Oncol 10:123–142
Kawczyk-Krupka A, Bugaj AM, Latos W, Zaremba K, Wawrzyniec K, Sieron A (2015) Photodiagnosis Photodyn Ther 12:545–553
Huang Z (2005) Technol Cancer Res Treat 4:283–293
Avci P, Erdem SS, Hamblin MR (2014) J Biomed Nanotechnol 10:1937–1952
Lim CK, Heo J, Shin S, Jeong K, Seo YH, Jang WD, Park CR, Park SY, Kim S, Kwon IC (2013) Cancer Lett 334:176–187
Master A, Livingston M, Sen Gupta A (2013) J Control Release 168:88–102
Anand S, Ortel BJ, Pereira SP, Hasan T, Maytin EV (2012) Cancer Lett 326:8–16
Mohamed S, Parayath NN, Taurin S, Greish K (2014) Ther Deliv 5:1101–1121
Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC (2012) Chem Soc Rev 41:2971–3010
Kuimova MK, Yahioglu G, Ogilby PR (2009) J Am Chem Soc 131:332–340
Robertson CA, Evans DH, Abrahamse H (2009) J Photochem Photobiol B 96:1–8
Ormond AB, Freeman HS (2013) Materials 6:817–840
O’Connor AE, Gallagher WM, Byrne AT (2009) Photochem Photobiol 85:1053–1074
Allison RR, Bagnato VS, Sibata CH (2010) Future Oncol 6:929–940
Nokes B, Apel M, Jones C, Brown G, Lang JE (2013) J Surg Res 181:262–271
Fukuda H, Casas A, Batlle A (2006) J Environ Pathol Toxicol Oncol 25:127–143
Calzavara-Pinton PG, Venturini M, Sala R (2007) J Eur Acad Dermatol Venereol 21:293–302
Kreimer-Birnbaum M (1989) Semin Hematol 26:157–173
Garland MJ, Cassidy CM, Woolfson D, Donnelly RF (2009) Future Med Chem 1:667–691
Staneloudi C, Smith KA, Hudson R, Malatesti N, Savoie H, Boyle RW, Greenman J (2007) Immunology 120:512–517
Nowis D, Makowski M, Stoklosa T, Legat M, Issat T, Golab J (2005) Acta Biochim Pol 52:339–352
Brancaleon L, Moseley H (2002) Lasers Med Sci 17:173–186
Sandell JL, Zhu TC (2011) J Biophotonics 4:773–787
Lee YE, Kopelman R (2011) Methods Mol Biol 726:151–178
Barolet D (2008) Semin Cutan Med Surg 27:227–238
Vicente MG (2001) Curr Med Chem Anticancer Agents 1:175–194
Chowdhary RK, Sharif I, Chansarkar N, Dolphin D, Ratkay L, Delaney S, Meadows H (2003) J Pharm Pharm Sci 6:198–204
Jori G, Reddi E (1993) Int J Biochem 25:1369–1375
Woodburn KW, Vardaxis NJ, Hill JS, Kaye AH, Phillips DR (1991) Photochem Photobiol 54:725–732
Malik Z, Amit I, Rothmann C (1997) Photochem Photobiol 65:389–396
Reiners JJ Jr, Agostinis P, Berg K, Oleinick NL, Kessel D (2010) Autophagy 6:7–18
Castano AP, Mroz P, Hamblin MR (2006) Nat Rev Cancer 6:535–545
Krammer B (2001) Anticancer Res 21:4271–4277
Fingar VH, Wieman TJ, Wiehle SA, Cerrito PB (1992) Cancer Res 52:4914–4921
Fingar VH (1996) J Clin Laser Med Surg 14:323–328
Bhuvaneswari R, Gan YY, Soo KC, Olivo M (2009) Cell Mol Life Sci 66:2275–2283
Mitra S, Cassar SE, Niles DJ, Puskas JA, Frelinger JG, Foster TH (2006) Mol Cancer Ther 5:3268–3274
Pizova K, Tomankova K, Daskova A, Binder S, Bajgar R, Kolarova H (2012) Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 156:93–102
Gollnick SO, Owczarczak B, Maier P (2006) Lasers Surg Med 38:509–515
Patel G, Armstrong AW, Eisen DB (2014) JAMA Dermatol 150:1281–1288
Clark CM, Furniss M, Mackay-Wiggan JM (2014) Am J Clin Dermatol 15:197–216
Lansbury L, Bath-Hextall F, Perkins W, Stanton W, Leonardi-Bee J (2013) BMJ 347:f6153
Biel MA (2010) Methods Mol Biol 635:281–293
Kostron H (2010) Methods Mol Biol 635:261–280
Minnich DJ, Bryant AS, Dooley A, Cerfolio RJ (2010) Ann Thorac Surg 89:1744–1748
Simone CB, Friedberg JS, Glatstein E, Stevenson JP, Sterman DH, Hahn SM, Cengel KA (2012) J Thorac Dis 4:63–75
Wiedmann MW, Caca K (2004) Curr Pharm Biotechnol 5:397–408
Ortner MA (2011) Lasers Surg Med 43:776–780
Keane MG, Bramis K, Pereira SP, Fusai GK (2014) World J Gastroenterol 20:2267–2278
Bown SG, Rogowska AZ, Whitelaw DE, Lees WR, Lovat LB, Ripley P, Jones L, Wyld P, Gillams A, Hatfield AW (2002) Gut 50:549–557
Tejeda-Maldonado J, Garcia-Juarez I, Aguirre-Valadez J, Gonzalez-Aguirre A, Vilatoba-Chapa M, Armengol-Alonso A, Escobar-Penagos F, Torre A, Sanchez-Avila JF, Carrillo-Perez DL (2015) World J Hepatol 7:362–376
Marien A, Gill I, Ukimura O, Betrouni N, Villers A (2014) Urol Oncol 32:912–923
Cheung G, Sahai A, Billia M, Dasgupta P, Khan MS (2013) BMC Med 11:13. doi:10.1186/1741-7015-11-13
Estel R, Hackethal A, Kalder M, Munstedt K (2011) Arch Gynecol Obstet 284:1277–1282
Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S, Pogue BW, Hasan T (2010) Chem Rev 110:2795–2838
Zuluaga MF, Lange N (2008) Curr Med Chem 15:1655–1673
Lehar J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, Rickles RJ, Short GF III, Staunton JE, Jin X, Lee MS, Zimmermann GR, Borisy AA (2009) Nat Biotechnol 27:659–666
Kessel D, Erickson C (1992) Photochem Photobiol 55:397–399
Norum OJ, Selbo PK, Weyergang A, Giercksky KE, Berg K (2009) J Photochem Photobiol B 96:83–92
Selbo PK, Weyergang A, Hogset A, Norum OJ, Berstad MB, Vikdal M, Berg K (2010) J Control Release 148:2–12
Berg K, Berstad M, Prasmickaite L, Weyergang A, Selbo PK, Hedfors I, Hogset A (2010) Nucleic Acid Transfect 296:251–281
Rajendran L, Knolker HJ, Simons K (2010) Nat Rev Drug Discov 9:29–42
Allison RR, Mota HC, Sibata CH (2004) Photodiagnosis Photodyn Ther 1:263–277
Gelderblom H, Verweij J, Nooter K, Sparreboom A (2001) Eur J Cancer 37:1590–1598
Aggarwal LPF, Borissevitch IE (2006) Spectrochim Acta A Mol Biomol Spectrosc 63:227–233
Gabrielli D, Belisle E, Severino D, Kowaltowski AJ, Baptista MS (2004) Photochem Photobiol 79:227–232
Gao D, Agayan RR, Xu H, Philbert MA, Kopelman R (2006) Nano Lett 6:2383–2386
Chatterjee DK, Fong LS, Zhang Y (2008) Adv Drug Deliv Rev 60:1627–1637
Davis ME, Chen ZG, Shin DM (2008) Nat Rev Drug Discov 7:771–782
Parhi P, Mohanty C, Sahoo SK (2012) Drug Discov Today 17:1044–1052
Oh J, Yoon H, Park JH (2013) Biomed Eng Lett 3:67–73
Zhang D, Wu M, Zeng Y, Wu L, Wang Q, Han X, Liu X, Liu J (2015) ACS Appl Mater Interfaces 7:8176–8187
Mallidi S, Spring BQ, Chang S, Vakoc B, Hasan T (2015) Cancer J 21:194–205
Taratula O, Schumann C, Duong T, Taylor KL, Taratula O (2015) Nanoscale 7:3888–3902
Nichols JW, Bae YH (2014) J Control Release 190:451–464. doi:10.1016/j.jconrel.2014.03.057, Epub@2014 Apr 30
Monopoli MP, Aberg C, Salvati A, Dawson KA (2012) Nat Nanotechnol 7:779–786
Karmali PP, Simberg D (2011) Expert Opin Drug Deliv 8:343–357
Euliss LE, DuPont JA, Gratton S, DeSimone J (2006) Chem Soc Rev 35:1095–1104
Decuzzi P, Pasqualini R, Arap W, Ferrari M (2009) Pharm Res 26:235–243
Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE (2007) Nat Nanotechnol 2:249–255
Fujimori K, Covell DG, Fletcher JE, Weinstein JN (1989) Cancer Res 49:5656–5663
Ernsting MJ, Murakami M, Roy A, Li SD (2013) J Control Release 172:782–794
Owens DE III, Peppas NA (2006) Int J Pharm 307:93–102
Amoozgar Z, Yeo Y (2012) Wiley Interdiscip Rev Nanomed Nanobiotechnol 4:219–233
Gref R, Domb A, Quellec P, Blunk T, Muller RH, Verbavatz JM, Langer R (2012) Adv Drug Deliv Rev 64:316–326
Kettler K, Veltman K, van de Meent D, van Wezel A, Hendriks AJ (2014) Environ Toxicol Chem 33:481–492
Lee H, Fonge H, Hoang B, Reilly RM, Allen C (2010) Mol Pharm 7:1195–1208
Kim JA, Aberg C, Salvati A, Dawson KA (2012) Nat Nanotechnol 7:62–68
Mahon E, Salvati A, Baldelli Bombelli F, Lynch I, Dawson KA (2012) J Control Release 161:164–174
Mukherjee S, Ghosh RN, Maxfield FR (1997) Physiol Rev 77:759–803
Fleige E, Quadir MA, Haag R (2012) Adv Drug Deliv Rev 64:866–884
Bae YH, Huh KM, Kim Y, Park KH (2000) J Control Release 64:3–13
Xiong XB, Binkhathlan Z, Molavi O, Lavasanifar A (2012) Acta Biomater 8:2017–2033
Adams ML, Lavasanifar A, Kwon GS (2003) J Pharm Sci 92:1343–1355
Letchford K, Burt H (2007) Eur J Pharm Biopharm 65:259–269
Aliabadi HM, Lavasanifar A (2006) Expert Opin Drug Deliv 3:139–162
Grubbs RB, Sun Z (2013) Chem Soc Rev 42:7436–7445
Sutton D, Nasongkla N, Blanco E, Gao J (2007) Pharm Res 24:1029–1046
Hoffman AS (2013) Adv Drug Deliv Rev 65:10–16
Pasquier E, Kavallaris M, Andre N (2010) Nat Rev Clin Oncol 7:455–465
Li J, Wang Y, Zhu Y, Oupický D (2013) J Control Release 172:10
Tang W, Xu H, Kopelman R, Philbert MA (2005) Photochem Photobiol 81:242–249
Muthu MS, Wilson B (2012) Nanomedicine 7:307–309
Mahapatro A, Singh DK (2011) J Nanobiotechnol 9:55–55
Saraf S (2009) Expert Opin Drug Deliv 6:187–196
Anton N, Benoit JP, Saulnier P (2008) J Control Release 128:185–199
Quaglia F, Ostacolo L, De Rosa G, La Rotonda MI, Ammendola M, Nese G, Maglio G, Palumbo R, Vauthier C (2006) Int J Pharm 324:56–66
Venkataraman S, Hedrick JL, Ong ZY, Yang C, Ee PL, Hammond PT, Yang YY (2011) Adv Drug Deliv Rev 63:1228–1246
Valencia PM, Hanewich-Hollatz MH, Gao WW, Karim F, Langer R, Karnik R, Farokhzad OC (2011) Biomaterials 32:6226–6233
Holzer M, Vogel V, Mantele W, Schwartz D, Haase W, Langer K (2009) Eur J Pharm Biopharm 72:428–437
Wu L, Zhang J, Watanabe W (2011) Adv Drug Deliv Rev 63:456–469
Jayakumar R, Prabaharan M, Nair SV, Tamura H (2010) Biotechnol Adv 28:142–150
Hudson D, Margaritis A (2013) Crit Rev Biotechnol 34:161–179
Ren D, Yi H, Wang W, Ma X (2005) Carbohydr Res 340:2403–2410
Abdelghany SM, Schmid D, Deacon J, Jaworski J, Fay F, McLaughlin KM, Gormley JA, Burrows JF, Longley DB, Donnelly RF, Scott CJ (2013) Biomacromolecules 14:302–310
Philippova OE, Korchagina EV (2012) Polym Sci Ser A 54:552–572
Lee HM, Jeong YI, Kim dH, Kwak TW, Chung CW, Kim CH, Kang DH (2013) Int J Pharm 454:74–81
Lee SJ, Koo H, Jeong H, Huh MS, Choi Y, Jeong SY, Byun Y, Choi K, Kim K, Kwon IC (2011) J Control Release 152:21–29
Oh IH, Min HS, Li L, Tran TH, Lee YK, Kwon IC, Choi K, Kim K, Huh KM (2013) Biomaterials 34:6454–6463
Lim CK, Shin J, Kwon IC, Jeong SY, Kim S (2012) Bioconjug Chem 23:1022–1028
Chen R, Wang X, Yao X, Zheng X, Wang J, Jiang X (2013) Biomaterials 34:8314–8322
Khdair A, Gerard B, Handa H, Mao G, Shekhar MP, Panyam J (2008) Mol Pharm 5:795–807
Khdair A, Chen D, Patil Y, Ma LN, Dou QP, Shekhar MPV, Panyam J (2010) J Control Release 141:137–144
Usacheva M, Swaminathan SK, Kirtane AR, Panyam J (2014) Mol Pharm 11:3186–3195
Kratz F, Elsadek B (2012) J Control Release 161:429–445
Jiang C, Cheng H, Yuan A, Tang X, Wu J, Hu Y (2015) Acta Biomater 14:61–69
Fakirov S (2007) Handbook of engineering biopolymers. Hanser, München, pp 417–464
Jain SK, Gupta Y, Jain A, Saxena AR, Khare P, Jain A (2008) Nanomed Nanotechnol Biol Med 4:41–48
Babu A, Jeyasubramanian K, Gunasekaran P, Murugesan R (2012) J Biomed Nanotechnol 8:43–56
Babu A, Periasamy J, Gunasekaran A, Kumaresan G, Naicker S, Gunasekaran P, Murugesan R (2013) J Biomed Nanotechnol 9:177–192
Yan F, Zhang Y, Kim KS, Yuan HK, Vo-Dinh T (2010) Photochem Photobiol 86:662–666
Danhier F, Ansorena E, Silva JM, Coco R, Le BA, Preat V (2012) J Control Release 161:505–522
Sah H, Thoma LA, Desu HR, Sah E, Wood GC (2013) Int J Nanomedicine 8:747–765
Vargas A, Eid M, Fanchaouy M, Gurny R, Delie F (2008) Eur J Pharm Biopharm 69:43–53
Vargas A, Lange N, Arvinte T, Cerny R, Gurny R, Delie F (2009) J Drug Target 17:599–609
Hu Z, Pan Y, Wang J, Chen J, Li J, Ren L (2009) Biomed Pharmacother 63:155–164
da Volta SM, Oliveira MR, dos Santos EP, de Brito GL, Barbosa GM, Quaresma CH, Ricci-Junior E (2011) Int J Nanomedicine 6:227–238
Korbelik M, Madiyalakan R, Woo T, Haddadi A (2012) Photochem Photobiol 88:188–193
Fadel M, Kassab K, Fadeel DA (2010) Lasers Med Sci 25:283–292
Conte C, D’Angelo I, Miro A, Ungaro F, Quaglia F (2014) Curr Top Med Chem 14:1097–1114
Mikhail AS, Allen C (2009) J Control Release 138:214–223
Perry JL, Reuter KG, Kai MP, Herlihy KP, Jones SW, Luft JC, Napier M, Bear JE, DeSimone JM (2012) Nano Lett 12:5304–5310
Pridgen EM, Langer R, Farokhzad OC (2007) Nanomedicine 2:669–680
Dinarvand R, Sepehri N, Manoochehri S, Rouhani H, Atyabi F (2011) Int J Nanomedicine 6:877–895
Cohen EM, Ding H, Kessinger CW, Khemtong C, Gao J, Sumer BD (2010) Otolaryngol Head Neck Surg 143:109–115
Ding H, Mora R, Gao J, Sumer BD (2011) Otolaryngol Head Neck Surg 145:612–617
Master AM, Rodriguez ME, Kenney ME, Oleinick NL, Gupta AS (2010) J Pharm Sci 99:2386–2398
Knop K, Mingotaud AF, El-Akra N, Violleau F, Souchard JP (2009) Photochem Photobiol Sci 8:396–404
Rojnik M, Kocbek P, Moret F, Compagnin C, Celotti L, Bovis MJ, Woodhams JH, MacRobert AJ, Scheglmann D, Helfrich W, Verkaik MJ, Papini E, Reddi E, Kos J (2012) Nanomedicine 7:663–677
Ding H, Sumer BD, Kessinger CW, Dong Y, Huang G, Boothman DA, Gao J (2011) J Control Release 151:271–277
Peng CL, Lai PS, Lin FH, Yueh-Hsiu WS, Shieh MJ (2009) Biomaterials 30:3614–3625
Conte C, Ungaro F, Maglio G, Tirino P, Siracusano G, Sciortino MT, Leone N, Palma G, Barbieri A, Arra C, Mazzaglia A, Quaglia F (2013) J Control Release 167:40–52
Xiang GH, Hong GB, Wang Y, Cheng D, Zhou JX, Shuai XT (2013) Int J Nanomedicine 8:4613–4622
Lee DJ, Park GY, Oh KT, Oh NM, Kwag DS, Youn YS, Oh YT, Park JW, Lee ES (2012) Int J Pharm 434:257–263
Marrache S, Choi JH, Tundup S, Zaver D, Harn DA, Dhar S (2013) Integr Biol (Camb) 5:215–223
Zheng M, Yue C, Ma Y, Gong P, Zhao P, Zheng C, Sheng Z, Zhang P, Wang Z, Cai L (2013) ACS Nano 7:2056–2067
Master AM, Qi Y, Oleinick NL, Gupta AS (2012) Nanomedicine 8:655–664
Master A, Malamas A, Solanki R, Clausen DM, Eiseman JL, Sen GA (2013) Mol Pharm 10:1988–1997
Chen H, Tian J, He W, Guo Z (2015) J Am Chem Soc 137:1539–1547
Yan Y, Such GK, Johnston APR, Lomas H, Caruso F (2011) ACS Nano 5:4252–4257
Maiolino S, Moret F, Conte C, Fraix A, Tirino P, Ungaro F, Sortino S, Reddi E, Quaglia F (2015) Nanoscale 7:5643–5653
Maiolino S, Russo A, Pagliara V, Conte C, Ungaro F, Russo G, Quaglia F (2015) J Nanobiotechnol 13:29
Shieh MJ, Peng CL, Chiang WL, Wang CH, Hsu CY, Wang SJJ, Lai PS (2010) Mol Pharm 7:1244–1253
Koo H, Lee H, Lee S, Min KH, Kim MS, Lee DS, Choi Y, Kwon IC, Kim K, Jeong SY (2010) Chem Commun (Camb) 46:5668–5670
Qin M, Hah HJ, Kim G, Nie G, Lee YE, Kopelman R (2011) Photochem Photobiol Sci 10:832–841
Hah HJ, Kim G, Lee YE, Orringer DA, Sagher O, Philbert MA, Kopelman R (2011) Macromol Biosci 11:90–99
Wang S, Fan W, Kim G, Hah HJ, Lee YE, Kopelman R, Ethirajan M, Gupta A, Goswami LN, Pera P, Morgan J, Pandey RK (2011) Lasers Surg Med 43:686–695
Gupta A, Wang S, Pera P, Rao KV, Patel N, Ohulchanskyy TY, Missert J, Morgan J, Koo-Lee YE, Kopelman R, Pandey RK (2012) Nanomedicine 8:941–950
Fukumura D, Kashiwagi S, Jain RK (2006) Nat Rev Cancer 6:521–534
Fraix A, Manet I, Ballestri M, Guerrini A, Dambruoso P, Sotgiu G, Varchi G, Camerin M, Coppellotti O, Sortino S (2015) J Mater Chem B 3:3001–3010
Batrakova EV, Kabanov AV (2008) J Control Release 130:98–106
Jung YW, Lee H, Kim JY, Koo EJ, Oh KS, Yuk SH (2013) Curr Med Chem 20:3488–3499
Alakhova DY, Kabanov AV (2014) Mol Pharm 11:2566–2578
Chowdhary RK, Chansarkar N, Sharif I, Hioka N, Dolphin D (2003) Photochem Photobiol 77:299–303
Hioka N, Chowdhary RK, Chansarkar N, Delmarre D, Sternberg E, Dolphin D (2002) Can J Chem 80:1321–1326
Pellosi DS, Estevão BM, Semensato J, Severino D, Baptista MS, Politi MJ, Hioka N, Caetano W (2012) J Photochem Photobiol A Chem 247:8–15
Chu M, Li H, Wu Q, Wo F, Shi D (2014) Biomaterials 35:8357–8373
Sobczynski J, Kristensen S, Berg K (2014) Photochem Photobiol Sci 13:8–22
Vilsinski BH, Gerola AP, Enumo JA, Campanholi KSS, Pereira PCS, Braga G, Hioka N, Kimura E, Tessaro AL, Caetano W (2015) Photochem Photobiol 91:518–525
Lim CK, Shin J, Lee YD, Kim J, Park H, Kwon IC, Kim S (2011) Small 7:112–118
Chung CW, Kim CH, Choi KH, Yoo JJ, Kim DH, Chung KD, Jeong YI, Kang DH (2012) Eur J Pharm Biopharm 80:453–458
Lamch L, Bazylinska U, Kulbacka J, Pietkiewicz J, Biezunska-Kusiak K, Wilk KA (2014) Photodiagnosis Photodyn Ther 11:570–585
Park H, Na K (2013) Biomaterials 34:6992–7000
Park H, Park W, Na K (2014) Biomaterials 35:7963–7969
Lee YD, Cho HJ, Choi MH, Park H, Bang J, Lee S, Kwon IC, Kim S (2015) J Control Release 209:12–19
Sahu A, Choi WI, Lee JH, Tae G (2013) Biomaterials 34:6239–6248
Kim TH, Song C, Han YS, Jang JD, Choi MC (2014) Soft Matter 10:484–490
Ren Y, Wang R, Liu Y, Guo H, Zhou X, Yuan X, Liu C, Tian J, Yin H, Wang Y, Zhang N (2014) Biomaterials 35:2462–2470
Kim WL, Cho H, Li L, Kang HC, Huh KM (2014) Biomacromolecules 15:2224–2234
Gangopadhyay M, Singh T, Behara KK, Karwa S, Ghosh SK, Singh NDP (2015) Photochem Photobiol Sci 14:1329–1336
Jang WD, Nakagishi Y, Nishiyama N, Kawauchi S, Morimoto Y, Kikuchi M, Kataoka K (2006) J Control Release 113:73–79
Nishiyama N, Nakagishi Y, Morimoto Y, Lai PS, Miyazaki K, Urano K, Horie S, Kumagai M, Fukushima S, Cheng Y, Jang WD, Kikuchi M, Kataoka K (2009) J Control Release 133:245–251
Lu HL, Syu WJ, Nishiyama N, Kataoka K, Lai PS (2011) J Control Release 155:458–464
Fraix A, Kandoth N, Manet I, Cardile V, Graziano AC, Gref R, Sortino S (2013) Chem Commun (Camb) 49:4459–4461
Kandoth N, Vittorino E, Sciortino MT, Parisi T, Colao I, Mazzaglia A, Sortino S (2012) Chemistry 18:1684–1690
Sortino S (2010) Chem Soc Rev 39:2903–2913
Fraix A, Goncalves AR, Cardile V, Graziano AC, Theodossiou TA, Yannakopoulou K, Sortino S (2013) Chem Asian J 8:2634–2641
Swaminathan S, Garcia-Amoros J, Fraix A, Kandoth N, Sortino S, Raymo FM (2013) Chem Soc Rev 43:4167–4178
Kandoth N, Kirejev V, Monti S, Gref R, Ericson MB, Sortino S (2014) Biomacromolecules 15:1768–1776
Yoon HJ, Lim TG, Kim JH, Cho YM, Kim YS, Chung US, Kim JH, Choi BW, Koh WG, Jang WD (2014) Biomacromolecules 15:1382–1389
Thambi T, Park JH (2014) J Biomed Nanotechnol 10:1841–1862
Kaneda Y (2010) Expert Opin Drug Deliv 7:1079–1093
Huang Z, Yang Y, Jiang Y, Shao J, Sun X, Chen J, Dong L, Zhang J (2013) Biomaterials 34:746–755
Hammond P (2013) ACS Nano 7:3733–3735
Master AM, Livingston M, Oleinick NL, Sen GA (2012) Mol Pharm 9:2331–2338
Acknowledgments
The authors wish to thank the Italian Ministry of University and Research (PRIN 2010H834LS) and Italian Association for Cancer Research (IG2014 15764).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Conte, C., Maiolino, S., Pellosi, D.S., Miro, A., Ungaro, F., Quaglia, F. (2016). Polymeric Nanoparticles for Cancer Photodynamic Therapy. In: Sortino, S. (eds) Light-Responsive Nanostructured Systems for Applications in Nanomedicine. Topics in Current Chemistry, vol 370. Springer, Cham. https://doi.org/10.1007/978-3-319-22942-3_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-22942-3_3
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-22941-6
Online ISBN: 978-3-319-22942-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)