
Abstract
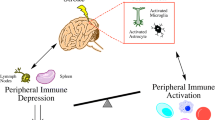
Stroke activates an inflammatory response that results in the infiltration of peripheral immune cells into the ischemic area, contributing to exacerbation of tissue damage. However, evidence indicates that inflammatory cell infiltration can also promote neuroprotection through regulatory immune cells that mitigate injury. These immune regulatory cells may also be important mediators of neuroprotection associated with preconditioning, a phenomenon whereby small exposure to a potential harmful stimulus is able to induce protection against a subsequent ischemic event. The elucidation of mechanisms that allow these immune cells to confer neuroprotection is critical to developing new therapeutic strategies against acute stroke. In the present review, we discuss the dual role of peripheral immune cells in stroke-related brain injury and neuroprotection. Furthermore, we report new data from our laboratory that supports the important role of peripheral cells and their interaction with the brain endothelium for the establishment of the protective phenotype in preconditioning.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
The Good and the Bad of Immune Cell Activation in Stroke
Brain injury caused by ischemic stroke is characterized by complex spatial and temporal events evolving over several days in which inflammation plays a key role. The inflammatory response is a complex regulatory process that begins early and lasts for days and even weeks after ischemia, involving many different cell types, inflammatory mediators, and extracellular receptors. The sudden loss of oxygen and glucose causes cells in the ischemic area to be rapidly killed by lipolysis and proteolysis resulting from total bioenergetic failure. As cells die, they release intracellular components that activate neighboring cells to produce pro-inflammatory mediators such as cytokines and chemokines, and promote transmigration of inflammatory cells from the periphery to the ischemic brain area, resulting in exacerbation of damage [1]. The critical role of inflammatory cells in stroke-induced brain injury has been demonstrated by the attenuation of ischemic damage in mice that are unable to mount an inflammatory response [2, 3]. However, certain aspects of the inflammatory response are critical to remove dead cells and promote regeneration after ischemic injury [4]. Thus, the inflammatory cascade can be beneficial or damaging, depending on the stage of tissue injury, the magnitude of the response, and whether the inflammatory response also activates neuroprotective pathways [4].
Recent studies indicate that brain injury activates populations of inflammatory-related cells that can limit ischemic damage. For example, subpopulations of regulatory lymphocytes are induced after stroke that suppress inflammation, thus promoting protection against injury [5, 6]. In addition, peripheral inflammatory cells have been implicated in the induction of protection associated with preconditioning [7]. Preconditioning is a phenomenon whereby exposure to a small but potentially harmful stimulus is able to induce protection against a subsequent ischemic event. These studies point to a role for inflammatory cells prior to an ischemic event in mediating the evolution of injury.
This review discusses the role of circulating immune cell activation in the pathology of stroke injury and neuroprotection, and reports data from our laboratory supporting the role of circulating cells in mediating preconditioning.
Peripheral Immune Cells Contribute to Ischemic Injury Following Stroke
Following ischemia, neutrophils, macrophages, and lymphocytes have been shown to infiltrate the brain [8]. Neutrophils are the most abundant cell population present at the site of injury, with a peak influx between 1 and 3 days after ischemia [8, 9]. They have been widely considered key contributors to inflammatory brain injury because they are a main source of free oxygen radicals that can directly cause neuronal death. In addition neutrophils cause endothelial cell dysfunction and blood-brain barrier disruption through release of matrix metalloproteinase 9 [10]. Multiple studies using both neutrophil-depleted animals and antagonists or blocking antibodies support neutrophils as key players in inflammatory-induced ischemic injury [11–16]. However, other studies report no effect on infarct size when neutrophils are depleted or blocked [2, 17]. Some of these differences may be attributed to the particular experimental model system employed, with those models involving reperfusion more likely to demonstrate a role for neutrophils in the exacerbation of injury. Clinical trials reflect these differences with slight improvement seen with an antagonist for CD11b, an integrin highly expressed on neutrophils, used in combination with tissue plasminogen activator, an inducer of reperfusion [18]. However, other clinical trials have shown no effect on stroke outcome when neutrophil infiltration is blocked [19, 20]. These results suggest that neutrophils may not be the only contributors to ischemic injury, but instead a complex interaction of multiple cell populations may mediate inflammatory damage.
Resident microglia and infiltrating macrophages also represent a significant immune population identified in ischemic tissue following stroke. These cells appear as early as 12 h after ischemia and peak between 3 and 7 days [8, 9]. Macrophages and microglia produce reactive oxygen species (ROS), nitric oxide, and other cytokines that can have direct toxic effects on brain tissue following stroke. In addition, macrophages and microglia amplify the inflammatory response through recruitment of other immune cells such as lymphocytes. Involvement of macrophages and microglia in the development of infarct damage after stroke was demonstrated by Tang et al. [21]. In this study, attenuation of proliferation and infiltration of microglia and macrophages was obtained by knocking down the chemokine receptor 1 (CX3CR1), which is highly expressed on resident brain microglia and peripheral macrophages. Knockout animals demonstrated a significant decrease in the ischemic lesion, number of apoptotic cells, and post-ischemic brain inflammatory responses (including ROS and pro-inflammatory cytokine production), indicating that suppression of macrophage and microglia activation reduces ischemia-induced inflammation and neurotoxicity.
The importance of lymphocytes (B and T cells) to the inflammatory response following ischemia has become increasingly evident. Although lymphocyte infiltration in the brain after ischemia peaks around day 3 [8, 9], several studies have found both B and T lymphocytes in the brain as early as 4–6 h after ischemia, suggesting a role in the development of inflammation in the ischemic area [2, 9]. Severe combined immunodeficient (SCID) mice lacking both T and B cells showed significant reduction of ischemic volume and suppression of post-ischemic induction of inflammatory mediators in the brain, demonstrating the important role of T and B cells in development of the ischemic damage [3].
Protective Role of Peripheral Immune Cells Responding to Stroke
In addition to the damaging effects of peripheral immune cells, studies have shown that inflammatory cells also play a role in protecting the brain from ischemic injury. Downes et al. [22] showed that hematopoietic cells have a critical role in mitigating the extent of injury following stroke. In particular, they showed that hematopoietic cells exhibit a neuroprotective function after stroke that is mediated by myeloid differentiation factor-88 (MyD88), the adaptor protein for Toll-like receptors (TLRs), and IL-1 signaling cascades. They demonstrated that mice lacking MyD88 signaling on hematopoietic cells had exacerbated ischemic volume, indicating that particular peripheral cell populations can promote protection against brain injury.
Microglia and macrophages have also been implicated in neuroprotection after stroke. These cells phagocytize debris and dead cells, and microglia can produce neurotrophic factors that can promote neuronal growth and survival, both mechanisms that are important in resolution of inflammation and tissue regeneration after injury. Selective ablation of proliferating microglia has been shown to exacerbate ischemic injury [23], indicating an important role of microglia in modulating protection against ischemia. In addition, exogenous microglial cells administered peripherally 24 h before or after global ischemia have been shown to reach the ischemic area and protect against neuronal injury by releasing neurotrophic factors [24].
B cells may represent another hematopoietic population with potential to confer protection against ischemic injury. Two conflicting reports using B cell-deficient mice have defined a neutral or protective role for B cells in ischemic injury. Yilmaz et al. [2] reported no difference in infarct size in mice deficient in B cells, whereas Ren et al. [25] found exacerbation of injury in B cell-deficient mice, supporting a protective role for B cells. Ren et al. were also able to reduce the infarct size in their model by adoptively transferring B cells into the deficient mice, further supporting a protective role for B cells in ischemic injury. Further work in this model has defined IL10 secreting B cells (B regulatory cells; Bregs) as the subset of B cells mediating protection [6, 26].
Regulatory lymphocytes represent a small percentage of infiltrating B and T lymphocytes—referred to as Bregs and Tregs, respectively. They are characterized by their ability to control immune responses, regulate the function of effector T cells, and regulate the activity of antigen-presenting cells. The anti-inflammatory cytokine, IL10, is the primary effector of regulatory lymphocytes. A growing amount of evidence points to the importance of these regulatory cells in modulating the inflammatory response after stroke, leading to suppressed inflammation and added protection against ischemic injury. Similar to the protective role of Bregs, Liesz et al. [5] found that depletion of Tregs significantly increased brain infarct volume and worsened functional outcome, indicating a protective effect of Tregs. Protection was mediated through Treg induction of IL10 and the subsequent suppression of inflammatory cytokines and modulation of the invasion and activation of lymphocytes and microglia in the ischemic brain [5].
It is not clear whether regulatory cells need to infiltrate the brain to induce the protective phenotype. Tregs, for example, have been detected in the brain 3 days after stroke, while their effect on modulating cytokine levels in the brain is already evident 6 h after ischemia [5]. Therefore, it is more likely that Tregs are able to monitor and regulate the inflammatory response from the periphery. Understanding the mechanisms by which peripheral cells communicate with the central nervous system in the context of injury as well as how they may mediate protection may be critical to the development of new therapeutic strategies against stroke.
Involvement of Peripheral Immune Cells in Preconditioning Induced Neuroprotection
Peripheral immune cells have been implicated as contributors to the induction of neuroprotection associated with preconditioning. Preconditioning involves mild treatment with an otherwise harmful stimulus to reprogram the cellular response to injury, thereby leading to a reduction of damage. A particularly effective means of inducing systemic preconditioning against cerebral ischemia occurs through activation of the innate immune response with ligands for TLR. We have shown that prior systemic administration using one of several TLR agonists (e.g., lipopolysaccharide, CpG oligonucleotides) induces robust neuroprotection against subsequent cerebral ischemia in a mouse stroke model [27–30]. We have also shown significant protection with the TLR9 agonist, CpG, in a nonhuman primate model of cerebral ischemia [31]. TLR9 is expressed on multiple cells both in the periphery (i.e., leukocytes) and in the brain (i.e., neurons, astrocytes, microglia, endothelium). We have published that effective preconditioning and neuroprotection with CpG stimulation requires TLR9 expression on at least two distinct cell populations, one of hematopoietic origin and one of non-hematopoietic origin [32]. We generated bone marrow chimeric mice by irradiating WT mice and repopulating their leukocytes with cells from TLR9-deficient mice. Thus, these animals lacked TLR9 expression on leukocytes, while still expressing TLR9 on parenchymal cells such as endothelial cells, astrocytes, microglia, and neurons. We found that mice lacking TLR9 expression on leukocytes were not protected by CpG preconditioning, demonstrating that TLR9-mediated leukocyte responses are required to confer CpG-induced neuroprotection. In addition, irradiated TLR9KO mice repopulated with leukocytes from WT mice also were not protected by CpG preconditioning, demonstrating that TLR9 expression on leukocytes was not sufficient for CpG-induced neuroprotection. These data indicate a strict requirement for TLR9-mediated responses on both leukocytes and parenchymal cells for protection induced by systemic CpG preconditioning and suggest a need for cross-talk between these compartments to achieve neuroprotection [32].
Systemic administration of CpG allows direct contact with brain microvascular endothelial cells belonging to the blood-brain barrier (BBB). The BBB is the interface between the blood and the brain and strictly regulates the passage of molecules and cells between the periphery and the CNS compartment [33]. Stroke dramatically impairs BBB integrity, leading to increased permeability and expression of endothelial adhesion molecules, resulting in increased leukocyte adhesion and transmigration [1]. We have found, using an in vitro BBB model consisting of a co-culture of primary brain microvascular endothelial cells (BMECs) and mixed glial cells (Fig. 1a), that CpG preconditioning can signal and protect the endothelium from ischemic injury. Cell cultures were preconditioned with CpG 24 h before modeled ischemia consisting of 5 h of oxygen-glucose deprivation (OGD). CpG significantly attenuated both the drop in trans-endothelial-electrical resistance (TEER) (Fig. 1b) and the increase of BBB permeability to Na-fluorescein induced by OGD (Fig.1c). These results indicate that CpG preconditioning stabilizes the BBB, possibly by affecting endothelial interactions with circulating immune cells that could alter the inflammatory response to ischemia. In support of this, we have found, using in vivo 2-photon microscopy, that CpG preconditioning induces leukocyte rolling and adhesion to brain microvascular endothelium before an ischemic event that may contribute to neuroprotection (Fig. 2).

CpG protects the BBB in an in vitro model of ischemic injury. (a) Schematic diagram of in vitro BBB model consisting of brain microvascular endothelial cells (BMECs) co-cultured with glial cells. Pretreatment with CpG attenuates OGD-induced decrease of trans-endothelial-electrical resistance (TEER) (b) and increase of permeability for Na Fluorescein (c) in an in vitro BBB model of ischemic injury. Values are group means ± SEM; *p < 0.001 versus control (CTR) and °p < 0.01 versus OGD

CpG-induces leukocyte-endothelial interactions. (a) Representative images of cortical vessels in mice acquired by in vivo two-photon microscopy 24 h after treatment with saline or CpG (0.8 mg/kg). Leukocytes stained in red. (b) Quantification of leukocytes per mm of vessel from five separate video clips +/− SEM of an individual mouse for each treatment
It remains to be elucidated which leukocyte populations are required for CpG induction of neuroprotection. Bregs offer potential as they are known to be immunosuppressive, and CpG has been reported to drive the differentiation of B cells into Bregs [34]. In line with a protective role for Bregs prior to stroke, Bodhankar et al. [26] showed that adoptive transfer of Bregs 24 h before cerebral ischemia reduces infarct size. Mice receiving IL-10-secreting B cells showed an increase of regulatory cell populations in the periphery and reduced infiltration of T cells and proinflammatory cytokines levels in the ischemic hemisphere. These results show that the increased presence of Bregs before stroke can modulate the inflammatory response, potentially contributing to protection against subsequent ischemia. In further support, Monson et al. [7] found that repetitive hypoxic preconditioning (RHP) induces an immunosuppressive phenotype in resident B cells before stroke and results in decreased infiltration of leukocytes in the brain following stroke. It appeared that RHP reprograms B cells to downregulate genes involved in T cell differentiation and B-T cell interactions [7].
Conclusions
These studies clearly demonstrate the complex nature of the inflammatory response associated with stroke. The interplay between damaging and protective effects is a delicate balancing act that determines the extent of injury. Understanding this interplay and developing ways to modulate the immune cell effectors could greatly advance therapeutic treatment of ischemic brain injury, including stroke. Recent data suggest that endogenous mechanisms engaged by preconditioning include modulation of immune cells, highlighting the effectiveness of targeting these responses in mitigating cerebral ischemic injury.
References
Huang J, Upadhyay UM, Tamargo RJ (2006) Inflammation in stroke and focal cerebral ischemia. Surg Neurol 66(3):232–245
Yilmaz G et al (2006) Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 113(17):2105–2112
Hurn PD et al (2007) T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab 27(11):1798–1805
Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17(7):796–808
Liesz A et al (2009) Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 15(2):192–199
Bodhankar S et al (2013) IL-10-producing B-cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis 28(3):375–386
Monson NL et al (2014) Repetitive hypoxic preconditioning induces an immunosuppressed B cell phenotype during endogenous protection from stroke. J Neuroinflammation 11:22
Gelderblom M et al (2009) Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40(5):1849–1857
Gronberg NV et al (2013) Leukocyte infiltration in experimental stroke. J Neuroinflammation 10:115
Rosell A et al (2008) MMP-9-positive neutrophil infiltration is associated to blood–brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 39(4):1121–1126
Matsuo Y et al (1994) Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke 25(7):1469–1475
Matsuo Y et al (1994) Role of cell adhesion molecules in brain injury after transient middle cerebral artery occlusion in the rat. Brain Res 656(2):344–352
Hudome S et al (1997) The role of neutrophils in the production of hypoxic-ischemic brain injury in the neonatal rat. Pediatr Res 41(5):607–616
Mori E et al (1992) Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke 23(5):712–718
Zhang Y et al (1996) Propentofylline inhibits polymorphonuclear leukocyte recruitment in vivo by a mechanism involving adenosine A2A receptors. Eur J Pharmacol 313(3):237–242
McCarter JF et al (2001) FK 506 protects brain tissue in animal models of stroke. Transplant Proc 33(3):2390–2392
Harris AK et al (2005) Effect of neutrophil depletion on gelatinase expression, edema formation and hemorrhagic transformation after focal ischemic stroke. BMC Neurosci 6:49
Krams M et al (2003) Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose–response study of UK-279,276 in acute ischemic stroke. Stroke 34(11):2543–2548
Becker KJ (2002) Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr Med Res Opin 18(Suppl 2):s18–s22
Enlimomab Acute Stroke Trial Investigators (2001) Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology 57(8):1428–1434
Tang Z et al (2014) CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation 11:26
Downes CE et al (2013) MyD88 is a critical regulator of hematopoietic cell-mediated neuroprotection seen after stroke. PLoS One 8(3), e57948
Lalancette-Hebert M et al (2007) Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 27(10):2596–2605
Imai F et al (2007) Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab 27(3):488–500
Ren X et al (2011) Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci 31(23):8556–8563
Bodhankar S et al (2014) Treatment of experimental stroke with IL-10-producing B-cells reduces infarct size and peripheral and CNS inflammation in wild-type B-cell-sufficient mice. Metab Brain Dis 29(1):59–73
Rosenzweig HL et al (2004) Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 35(11):2576–2581
Rosenzweig HL et al (2007) Endotoxin preconditioning protects against the cytotoxic effects of TNFalpha after stroke: a novel role for TNFalpha in LPS-ischemic tolerance. J Cereb Blood Flow Metab 27(10):1663–1674
Stevens SL et al (2008) Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab 28(5):1040–1047
Marsh B et al (2009) Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci 29(31):9839–9849
Bahjat FR et al (2011) Proof of concept: pharmacological preconditioning with a Toll-like receptor agonist protects against cerebrovascular injury in a primate model of stroke. J Cereb Blood Flow Metab 31(5):1229–1242
Packard AE et al (2012) TLR9 bone marrow chimeric mice define a role for cerebral TNF in neuroprotection induced by CpG preconditioning. J Cereb Blood Flow Metab 32(12):2193–2200
Abbott NJ et al (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37(1):13–25
Miyazaki D et al (2009) Regulatory function of CpG-activated B cells in late-phase experimental allergic conjunctivitis. Invest Ophthalmol Vis Sci 50(4):1626–1635
Conflict of Interest Statement
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gesuete, R., Stevens, S.L., Stenzel-Poore, M.P. (2016). Role of Circulating Immune Cells in Stroke and Preconditioning-Induced Protection. In: Applegate, R., Chen, G., Feng, H., Zhang, J. (eds) Brain Edema XVI. Acta Neurochirurgica Supplement, vol 121. Springer, Cham. https://doi.org/10.1007/978-3-319-18497-5_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-18497-5_7
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-18496-8
Online ISBN: 978-3-319-18497-5
eBook Packages: MedicineMedicine (R0)