
Abstract
Cytochrome P450 enzymes (P450s or CYPs) catalyze an enormous variety of oxidative reactions in organisms from all major domains of life. Their monooxygenase activity relies on the reductive scission of molecular oxygen (O2) bound to P450 heme iron, and thus on the delivery of two electrons to the heme iron at discrete points in the catalytic cycle. Early studies suggested that P450 redox partner machinery fell into only two major classes: either the eukaryotic diflavin enzyme NADPH-cytochrome P450 oxidoreductase, or bacterial/mitochondrial NAD(P)H-ferredoxin reductase and ferredoxin partners. However, more recent studies, aided by genome sequence data, reveal a much more complex scenario. Several new types of P450 redox partner systems have now been characterized, including P450s naturally linked to their redox partners, or to a component protein of their P450 electron delivery system. Other P450s have evolved to bypass requirements for redox partners, and instead react directly with hydrogen peroxide or NAD(P)H to facilitate oxidative or reductive catalysis. Further P450s are fused to non-redox partner enzymes and can catalyse consecutive reactions in a common pathway. This chapter describes the biochemistry and the enormous natural diversity of P450 redox systems, including descriptions of novel P450s fused to non-redox partner proteins.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cytochrome P450
- Compound I
- P450 redox systems
- P450 oxidoreductase
- P450 BM3
- P450-flavodoxin fusion protein
- P450-ferredoxin fusion protein
11.1 Introduction
Cytochrome P450 enzymes (P450s or CYPs) are prodigious catalysts of oxidation reactions in Nature. P450s are heme b-containing enzymes that bind dioxygen (O2) to a ferrous heme iron, and then further reduce and protonate the iron-oxo species to form reactive intermediates capable of oxygen addition chemistry on a substrate bound close to the P450 heme iron [1]. The key species responsible for substrate oxidation reactions is the ferryl-oxo porphyrin radical cation species known as compound I. This transient species was definitively characterized for the first time in 2010 by Rittle and Green, who presented both the spectroscopic characteristics of compound I and a clear demonstration of its catalytic potency in substrate oxidation using a thermophilic P450 enzyme as a model system [2]. To achieve oxygen activation and substrate oxidation, the canonical P450 catalytic cycle (Fig. 11.1) involves two sequential single electron reduction reactions that first convert a ferric heme iron to the ferrous state (enabling the binding of O2), and then result in the further reduction of the ferrous oxo (formally ferric superoxo) form to the ferric peroxo state. To facilitate the formation of compound I, successive protonation reactions then occur to produce first the transient ferric hydroperoxo state (compound 0), followed by formation of compound I following the loss of a water molecule after the second protonation step. To achieve substrate hydroxylation (as shown in the model catalytic cycle in Fig. 11.1), compound I abstracts a hydrogen atom from the substrate bound close to the heme (forming a transient compound II or ferryl-hydroxo species), and then “rebounds” the hydroxyl to the substrate radical, according to the radical rebound model of P450 catalysis developed by Groves [3].

The cytochrome P450 catalytic cycle. Intermediate species in the canonical P450 catalytic cycle are shown. At the top of the cycle is the resting state species where the ferric P450 heme iron is proximally coordinated by cysteine sulfur (S) in its thiolate form. Binding of substrate (RH) displaces the distal water ligand and often shifts the heme iron equilibrium toward high spin. Electron delivery from a redox partner reduces the heme iron to the ferrous form, which can then bind to molecular oxygen (O2), and convert to the ferric-superoxo form. The delivery of a second electron from the redox partner forms the ferric-peroxo species, which becomes protonated to the reactive ferric-hydroperoxo (compound 0) form. A further protonation results in loss of a water molecule and production of the catalytically relevant compound I (ferryl-oxo porphyrin radical cation). Compound I abstracts a hydrogen atom from the substrate, and then “rebounds” a hydroxyl group to the substrate radical, forming a hydroxylated product (ROH), the dissociation of which enables rebinding of water in the distal position and restoration of the starting form
The requirement for electrons in P450 catalysis is almost invariably met by the cofactors NADH and/or NADPH, which deliver electrons by hydride transfer to a flavin cofactor in a P450 redox partner enzyme. Until relatively recently, the types of P450 redox systems used were thought to be quite limited (to two major types). However, studies in recent years have revealed a more complex arrangement in nature, with diverse systems used in different organisms and even cases in which protein-based redox systems are no longer used. These different P450 redox systems are described in more detail in the sections below.
11.2 Class I and II P450 Redox Systems
Several early studies on P450 systems focused on mammalian P450 enzymes, and led to the identification of the diflavin (FAD- and FMN-containing) enzyme cytochrome P450 reductase (also known as CPR, and more recently referred to as POR for P450 oxidoreductase) as a redox partner for the membrane-anchored P450 enzymes found in the endoplasmic reticulum in the liver and other tissues [4]. CPR, like its eukaryotic P450 partners, is attached to the membrane by an N-terminal “anchor” region consistent with a single membrane spanning alpha helical segment. Recent studies from Monk and coworkers provided the first structure of an intact P450 enzyme (the Saccharomyces cerevisiae lanosterol 14α-sterol demethylase, or CYP51), showing the conformation of the membrane-spanning segment and its influence on the orientation of the P450 itself, resulting in the positioning of P450 substrate access regions toward the membrane [5]. Structural studies on the rat CPR (using a soluble version from which the N-terminal transmembrane segment is removed) confirmed its predicted structural relationships with FAD/NAD(P)H-binding ferredoxin reductases (FDRs) and FMN-binding flavodoxins (FLDs) [6]. The CPR crystal structure revealed a modular arrangement of the CPR with a N-terminal FLD domain orientated toward the C-terminal FDR domain such that the two flavin cofactors face toward each other and are separated by only ~4 Å between the dimethyl groups at the ends of their respective FMN and FAD cofactors [7]. In this “closed” conformation, there is clearly scope for direct electron transfer between FAD and FMN cofactors. However, while this orientation is clearly effective for inter-flavin electron transfer, it is probably not conducive to electron transfer to the P450 partner. As shown in Fig. 11.2, a reorientation of the FMN domain to a more “open” conformation is necessary to position the reduced FMN cofactor close to the heme-binding site in the P450, with the docking of the FMN domain proposed to occur on the P450 protein surface at the proximal side of heme – i.e. on the opposite side of the heme from the substrate-binding cavity, but close to the cysteine thiolate bond to the P450 heme iron.

The structure of cytochrome P450 reductase. The structure of rat CPR (PDB 3ES9) is shown in an open conformation in which the FAD/NADP(H)-binding and FMN-binding domains are separated, such that the FMN domain may be able to interact with and pass an electron to a P450 partner protein. In a more “closed” conformation, the domains would be more compactly arranged with the isoalloxazine rings of the flavins placed adjacent to one another to enable direct inter-flavin electron transfer [7]. The FAD/NADP(H) domain is shown in red-to-green colour with the FAD-binding region in yellow/green and the NADP(H) binding portion in red/orange, and with the FAD cofactor in yellow spheres. The FMN domain is in blue with the FMN in orange spheres. The NADP+ cofactor is bound in the FAD/NADP(H) domain, close to the FAD cofactor. The NADP+ is also shown in spheres with a green carbon backbone [107]
CPR binds NADPH tightly and positions the redox active nicotinamide ring of the cofactor close to the FAD. Mutagenesis of several residues in CPR orthologues from different organisms has enabled identification of amino acids important for the binding of NADP(H) and for the progression of electron transfer to the FAD. The aromatic residues Trp677 (the indole ring thereof) and Tyr456 stack with the FAD isoalloxazine ring on opposite sides of the molecule in the rat CPR (a common arrangement in other CPRs) [7]. The displacement of the tryptophan side chain is an important catalytic step required for the reduced nicotinamide ring of NADPH to access the FAD and to transfer electrons (in the form of a hydride ion) to the flavin. The return of the conserved Trp side chain to its original position is likely also important to trigger the displacement of the oxidized NADP+ and to allow the enzyme to perform inter-flavin electron transfer as a prerequisite for reduction of the P450 partner [8, 9]. It is considered that the resting form of CPR in vivo is one in which the FAD cofactor is fully oxidized, while the more positive potential FMN cofactor is in a single electron reduced and relatively air-stable blue semiquinone form [10]. In this model, NADPH-dependent electron transfer results in a 3-electron reduced form in which the FAD is fully reduced (the hydroquinone or FADH2 form) and the FMN is a neutral semiquinone (FMNH). An internal electron transfer from FADH2 to the FMN produces the FMN hydroquinone, which is the electron donor to the P450 partner. The first electron is transferred from the FMN hydroquinone to the ferric heme iron, restoring the FMN semiquinone and forming ferrous heme iron, which is then competent to bind O2. A second electron is passed from the FAD to the FMN (restoring the resting, oxidized state of the FAD and forming the FMN hydroquinone again). The FMN hydroquinone then passes the second electron to the ferric-superoxo P450 species (forming the ferric peroxo state and enabling progression through the later protonation steps in the catalytic cycle) (Fig. 11.1). In this way, there is temporal control over electron transfer to the P450 (enabling dioxygen binding to heme iron to occur between the two individual electron transfer steps from CPR) and the CPR redox partner undergoes a 1-3-2-1 cycle, where the digits indicate the total number of electrons held on the CPR flavins at different stages in the process of P450 reduction.
The CPR-type redox partner system is widespread in eukaryotes and is often referred to as a class II (or E-class for eukaryotic-type) P450 redox partner. Its counterpart in early studies of P450 enzymes was the redox partner system supporting the activity of the cytochrome P450cam (CYP101A1) camphor 5-exo hydroxylase, an important member of the P450 enzyme superfamily and the first P450 for which a 3-dimensional structure was determined using X-ray crystallography [11]. P450cam is encoded by one of a suite of genes encoded on a transmissible plasmid in a strain of Pseudomonas putida, and the P450 plays a crucial role in initiating the breakdown of camphor as a source of energy for cell growth [12]. Also encoded on the same (CAM) plasmid are the redox partner proteins that supply electrons for P450cam catalysis. In this class I (or B-class for bacterial-type) system, electrons from NADH reduce the FAD-binding putidaredoxin reductase (PDR), which then transfers electrons one at a time to a ferredoxin (putidaredoxin or PD) [13]. PD binds a 2Fe-2S cluster which acts as a 1-electron carrier in this system. NADH reduces the PDR to its FADH2 form, which then passes single electrons in two successive reactions to the PD. The PD reduces the P450cam in two steps, essentially as described above for the class II system. Thus, PD is an electron “shuttling” protein that interacts in turn with reduced PDR and then P450cam to facilitate catalysis in this class I system. Figure 11.3 shows the crystal structure for a complex between the PDR/PD proteins [14, 15]. The flavodoxin domain in CPR enzymes plays a similar role to that of PD, but in this case is fused to its NADPH dehydrogenase (FDR-like) domain.

The crystal structure of the putidaredoxin reductase: putidaredoxin complex. The image shows the crystal structure for a covalently linked complex between the P450cam putidaredoxin reductase (PDR) and putidaredoxin (PD) (PDB 3LB8). The complex was formed by cross-linking the transiently formed PDR/PD complex using 1-ethyl 3-[3-(dimethylamino)propyl]carbodiimide (EDC). The PD is shown in cyan and magenta with the 2Fe-2S cluster in atom coloured spheres. The PD iron-sulfur cluster is docked close to the FAD cofactor in the PDR protein. The PDR is shown in multicolour with the FAD cofactor in yellow spheres [14]
Several bacterial P450 systems operate a class I redox system, including the Pseudomonas sp. terpineol oxidase P450terp (CYP108A1) and the Mycobacterium tuberculosis sterol demethylase CYP51B1 [16–18]. The ferredoxin component of the CYP51B1 redox system binds a 3Fe-4S iron-sulfur cluster [16, 17]. However, many such systems use 2Fe-2S cluster ferredoxins. These include the P450terp partner terpredoxin, with the most notable example being P450cam with its partner putidaredoxin [14, 18]. Figure 11.4 shows the crystal structure of a complex between P450cam and PD, illustrating how the ferredoxin docks on the proximal face of the P450 protein and places the iron-sulfur cluster close to the cysteine thiolate-heme iron bond and to the heme cofactor itself [19]. However, P450 redox systems using 4Fe-4S and even 7Fe cluster ferredoxins have been reported, with the Mycobacterium smegmatis FdxA protein shown to be a 7Fe ferredoxin (binding both a 3Fe-4S and a 4Fe-4S cluster) and suggested to support activity of one or more of the numerous P450s in this bacterium, and in the pathogen M. tuberculosis, which encodes 20 P450s [20–23]. The eukaryotic mitochondrial P450 systems also use a class I redox system, likely reflecting the prokaryotic origins of the mitochondrion. In these cases, the redox partners are the FAD-binding, NADPH-dependent adrenodoxin reductase and the 2Fe-2S ferredoxin adrenodoxin [24, 25]. However, it should also be noted that the FMN-binding flavodoxin proteins are also potential redox partners for prokaryotic P450s – as best demonstrated in the case of the Citrobacter braakii P450cin system (CYP176A1), where the flavodoxin cindoxin is the natural redox partner for the P450 and is itself reduced by a FAD-dependent reductase. P450cin can then oxidize the terpene cineole to enable the bacterium to use this molecule as a sole source of carbon for growth [26].

The crystal structure of the putidaredoxin:P450cam complex. The Pseudomonas putida P450cam/putidaredoxin complex (PDB 4JX1) is shown in cartoon representation with the P450cam alpha helices in green and beta sheets in blue. The I-helix is highlighted in yellow and the heme shown in purple spheres with the oxygen atoms in red. The hydroxylated product 5-exo-hydroxycamphor (also in spheres) is shown above the heme plane in the active site cavity with carbon atoms coloured in cyan. Putidaredoxin (in multicolour) docks on the proximal face of the P450cam and positions its 2Fe-2S cluster (atom coloured spheres) in close vicinity to the P450 heme and to the cysteine thiolate-heme iron bond. The complex was formed using the homobifunctional maleimide cross-linker 1,6-bismaleimidohexane, and using a D19C mutant of PD with a K344C mutant of P450cam to facilitate the cross-linking [19]
11.3 Flavocytochrome P450 BM3
The discovery of the Bacillus megaterium P450 BM3 enzyme by Armand Fulco and coworkers led to the characterization of the first “outlier” redox partner system from the class I/class II paradigm [27]. The 119 kDa P450 BM3 (CYP102A1) is a natural fusion of a soluble fatty acid hydroxylase P450 (at the N-terminus) to a soluble CPR module (at the C-terminus). The BM3 CPR domain lacks the membrane anchor region found in the eukaryotic CPRs, and is instead linked to the P450 domain by a short peptide linker region [28] (Fig. 11.5). BM3 is a high activity fatty acid hydroxylase, which hydroxylates a wide range of long chain fatty acids at the ω−1, ω−2 and ω−3 positions [28]. BM3 has the highest reported oxidase activity for a P450 (e.g. ~285 s−1 with arachidonic acid), facilitated by rapid electron transfer from the CPR module [29, 30]. Early studies on the BM3 enzyme demonstrated that the enzyme could oligomerize [31]. More recent work in this area has indicated that the dimeric form of the flavocytochrome is likely the catalytically relevant form, with electron transfer occurring between the CPR domain of one monomer and the P450 domain of the other [32–34]. A similar interdomain electron transfer process is thought to occur in the nitric oxide synthase (NOS) enzymes, which also use a CPR-like reductase domain to reduce a fused heme domain. In this case, the heme domain is another cysteine thiolate ligated hemoprotein that catalyzes successive oxidations of L-arginine to form first N-hydroxyl-L-arginine, and then L-citrulline and nitric oxide (NO) [35].

Biological diversity of P450 redox partners, redox partner fusions and non-redox partner P450 fusion enzymes. A schematic overview of a number of selected P450 redox systems where P450s interact with separate redox partners or other chemical entities to drive catalysis (a); or where P450s are parts of fusions in which the other protein is either a redox partner system or displays activity unrelated to P450 reduction (b and c, respectively). Section A shows “standard” class I and II redox systems (i and ii), where both CPR and P450 components of the class II system contain N-terminal membrane anchor domains. (iii) is the P450cin type system where a flavodoxin replaces a ferredoxin [26]. (iv) is the yeast/fungal system that requires only cytochrome b 5 reductase and b 5 partners [100]. (v) and (vi) show the NADH-dependent P450nor and H2O2-dependent peroxygenase-type P450s [76, 79, 80]. Section B shows a variety of P450-redox partner fusion enzymes. (vii) is the BM3-type CPR fusion and (viii) the CYP116B-type PDOR-P450 fusion enzyme [28, 29, 45, 46]. (ix) is a benzoate dioxygenase reductase-P450 fusion (CYP1049 family), (x) a XplA-type flavodoxin-P450 fusion [56, 60], (xi) the M. capsulatus MCCYP51FX fusion [63] and (xii) an example of a P450-HCP fusion, where HCP is a hybrid cluster protein family member and potential P450 redox partner [108]. Section C shows various examples of P450s fused to non-redox partner proteins. (xiii) is a CYP154 family P450 fused to a cinammyl alcohol dehydrogenase-type module, (xiv) and (xv) are P450 fusions to different types of peroxidases, with the animal-like peroxidase fusions typified by the Aspergillus nidulans Ppo proteins [67]. (xvi–xviii) show uncharacterized P450 fusions to IPPS (trans-isoprenyl diphosphate synthase family member), glycosyltranferase and lipoxygenase like modules
The binding of fatty acid substrates to P450 BM3 results in a substantial change in ferric heme iron spin-state from low-spin toward high-spin, and a concomitant large shift in P450 heme iron potential from −368 to −239 mV when bound to arachidonic acid [36]. This change favours electron transfer to the ferric heme iron and helps to ensure that electron transfer occurs only when substrate is available for oxidation. The catalytic cycle for the BM3 CPR is different to that for the eukaryotic CPRs, due mainly to alterations in the properties of the FMN-binding (flavodoxin) domain in BM3. The resting form of the BM3 CPR has both flavins (FAD and FMN) fully oxidized. Reduction with NADPH results in complete reduction of the FAD (to FADH2), and successive single electron transfers occur through the FMN, with the FMN semiquinone species passing electrons to the heme iron at the same reaction stages as described above (and as shown in Fig. 11.1). The BM3 reductase thus undergoes a 0-2-1-0 redox cycle during P450 catalysis [37]. Transient kinetic studies of the reduction of the isolated BM3 FMN domain with sodium dithionite showed formation of an anionic semiquinone, consistent with preceding EPR data from BM3 enzyme samples freeze-quenched during turnover, and pointing to the involvement of the BM3 FMN anionic semiquinone as the electron donor to the heme iron, as opposed to the FMN hydroquinone in the case of the eukaryotic CPRs [37, 38].
Several orthologues of P450 BM3 have been identified in other microbes, including two fatty acid hydroxylases (CYP102A2 and A3) in B. subtilis [39, 40]. Membrane-associated fatty acid hydroxylase activity was also found to be associated with CYP505A1 (P450foxy) from the fungus Fusarium oxysporum, a member of a distinct family of eukaryotic P450-CPR fusion enzymes [41] (Fig. 11.5). As with P450 BM3, P450foxy also catalyzes the ω−1 to ω−3 hydroxylation of fatty acids, and is also catalytically active with shorter chain fatty acids (C9 and C10) that are not efficient substrates for P450 BM3 [42].
11.4 The CYP116B Enzymes
Further complexity in the redox systems supporting P450 activities became clear with the identification of the CYP116B enzyme family, in which soluble bacterial P450s are fused to redox partner modules with structural similarity to the FMN and 2Fe-2S cluster binding phthalate dioxygenase reductase (PDOR) [43]. Analysis of microbial genomes provided the first evidence for these fusion enzymes, in which a redox partner distinct from the class I/II systems was recognized for the first time [44]. In the soluble bacterial CYP116B enzymes, the N-terminal P450 domain is fused to the C-terminal PDOR domain to form an ~86 kDa fusion enzyme [45] (Fig. 11.5). The CYP116B1 and CYP116B2 enzymes have been expressed and characterized, including studies to dissect the enzymes into their component P450 and PDOR domains, including the individual 2Fe-2S- and FMN-binding domains in the case of CYP116B2 from Rhodococcus sp. NCIMB 978 [45, 46]. Electron transfer in these enzymes proceeds from NAD(P)H through the FMN and 2Fe-2S clusters in the PDOR module, and then onto the P450 heme iron.
The related class I bacterial P450 CYP116A1 (from Rhodococcus erythropolis NI86/21) was previously shown to catalyze N-dealkylation reactions with the thiocarbamate herbicides vernolate (S-propyldipropylthiocarbamate) and EPTC (S-ethyldipropylthiocarbamate), as well as acting on the triazine herbicide atrazine (1-chloro-3-ethylamino-5-isopropylamino-2,4,6-triazine) [47, 48]. In view of the structural similarities between CYP116A1 and the heme domain of the Cupriavidus metallidurans CYP116B1, the thiocarbamate herbicides were tested for activity with CYP116B1, and both herbicides were shown to be hydroxylated on propyl chains, with the reaction progressing to N-dealkylation in the case of vernolate [45]. For the Rhodococcus sp. CYP116B2, activity in dealkylation of 7-ethoxycoumarin was demonstrated, along with hydroxylation and O-dealkylation of various alkyl aryl ethers [49], while the Rhodococcus ruber DSM 44319 CYP116B3 was also purified and shown to catalyze NADPH-dependent oxidation of a range of chemicals – including hydroxylation of naphthalene, fluorene, toluene and ethylbenzene [50]. A combination of rational and directed evolution approaches was also used to improve CYP116B3-dependent oxidative demethylation and deethylation reactions with 7-methoxycoumarin and 7-ethoxycoumarin, respectively [51]. Numerous protein engineering studies of P450 BM3 have demonstrated the catalytic versatility of mutants of this enzyme (e.g. [52, 53]), and studies on the CYP116B enzymes are now also beginning to point to their potential as biotechnologically important enzyme catalysts.
In other studies, the CYP116B PDOR modules have been used as surrogate electron donors for heterologous P450 enzymes, with potential applications in generating robust, catalytically self-sufficient P450-PDOR fusion enzymes [54]. Such fusions benefit from the relatively fast electron transfer kinetics of the PDOR modules, with NADPH-dependent reduction of the FMN/2Fe-2S centres in CYP116B1 occurring with a limiting rate constant of ~72 s−1 at 25 °C [45].
11.5 A Biotechnologically Important P450-Flavodoxin Fusion Protein
The discovery of an unusual cytochrome P450-flavodoxin fusion protein was made in Rhodococcus rhodochrous strain 11Y, isolated from soil contaminated with explosives. In this case, the flavodoxin forms the N-terminal domain of the enzyme. This P450 system (XplA or CYP177A1) was shown to catalyze the degradation of RDX (Royal Demolition eXplosive; hexahydro-1,3,5-trinitro-1,3,5-triazine) and to enable the bacterium to use this explosive as a sole source of nitrogen for growth. Electrons for the reaction are supplied by the adrenodoxin reductase-like XplB, encoded by the xplB gene in the same gene cluster as xplA. Thus, the electron transfer pathway in this system is from NADPH to the XplB FAD cofactor, and then on to the XplA FMN and finally to the P450 heme iron [55]. XplA degrades RDX under aerobic or aerobic conditions, with enzyme-catalyzed reductive denitration likely followed by hydration reactions to form products nitrite and formaldehyde, and either 4-nitro-2,4-diazabutanal (NDAB, under aerobic conditions) or methylenedinitramine (MEDINA, under anaerobic conditions) [56]. For applications in bioremediation, Arabidopsis thaliana was engineered to express the xplA gene, and plants grown in RDX-contaminated soil were shown to exhibit resistance to the explosive. Further studies with A. thaliana transformed with both xplA and xplB, along with the Enterobacter cloacae nitroreductase nsfl gene, showed that these plants could remove both RDX and the explosive 2,4,6-trinitrotoluene (TNT) from soil and allow plant growth at levels of these explosives that proved inhibitory to the growth of A. thaliana that expressed only xplA [57].
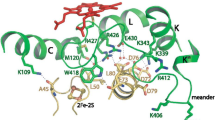
The structure of the XplA P450 domain was determined at 1.5 Å resolution, revealing a typical P450 fold and a compact active site with imidazole present as a ligand in the 6th (distal) position on the heme iron [58]. The structure highlighted the absence of the acid-alcohol pair (e.g. Asp251/Thr252 in the well-studied camphor hydroxylase P450cam) in XplA – which instead has Met394/Ala395 in the relevant positions in the P450 I helix [59]. The absence of amino acids with functions in stabilizing and protonating the P450 heme iron-oxo complexes is consistent with XplA having a predominantly reductive role (i.e. nitro group reduction). Other novel features of XplA relate to the unusually positive reduction potential of its flavodoxin FMN semiquinone/hydroquinone couple (−172 mV vs the normal hydrogen electrode, NHE) and the relatively weak binding of FMN to the protein (K d = 1.09 µM, compared to, for example, the Desulfovibrio vulgaris flavodoxin with a K d = 0.24 nM). These adaptations are likely made through evolution to facilitate a mainly reductive (rather than oxidative) role for this unusual P450 system [60, 61]. Approximately 30 different xplA genes have now been identified in different microbial genomes – with the majority of these being in Rhodococci [62].
11.6 A P450-Ferredoxin Fusion Protein
A further unusual P450 fusion protein was discovered in the methanotrophic bacterium Methylococcus capsulatus. In this case, the soluble P450 domain is at the N-terminal, with a ferredoxin linked at the C-terminal end via an alanine-rich linker [63]. The P450 (MCCYP51FX) has ~49 % identity between its heme domain and the CYP51B1 sterol demethylase enzyme from M. tuberculosis, while the ferredoxin domain has ~42 % identity to the 3Fe-4S cluster ferredoxin that is encoded by the Rv0763c gene located directly adjacent to the gene encoding CYP51B1 (Rv0764c) [17, 18]. Activity as a lanosterol demethylase was shown when MCCYP51FX was reconstituted with a heterologous (spinach) ferredoxin reductase, despite the observation that addition of lanosterol produced a type II P450 spectral change (Soret absorption shift to longer wavelength) that is more often associated with inhibitory interactions of a ligand with the P450 heme iron. Other studies indicated a propensity of the MCCYP51FX to self-aggregate, possibly indicating the formation of an oligomeric state in solution [63]. To date, this is the only characterized example of a P450-ferredoxin fusion enzyme, although other types of P450 fusion enzymes (to both redox and non-redox partners) have been identified, as described in the sections below.
11.7 Other Potential Catalytically Self-Sufficient P450s
Bioinformatics tools can be used to probe available genome sequences in order to identify P450s fused to other proteins. In a number of cases, these fusion partners are potential redox partners in view of their similarity to known redox enzymes. At the end of 2014, the CDART (Conserved Domain Architecture Retrieval Tool) program was used to identify potential P450 fusion enzymes and to assess whether these fused partners were likely to be electron donor systems for the P450, or else might play distinct roles in relation to the P450 [64]. Figure 11.5 shows selected results for the domain architecture of a range of P450 systems in which redox partners (either potential or proven) are found either as separate entities or are fused to the P450. Figure 11.5 also shows a number of P450 fusion enzymes in which the partner protein is not an electron transferase to the P450. In addition to the characterized proteins listed in the preceding sections, a number of uncharacterized potential P450-redox partner fusions are identified – including P450s fused to modules identified as (1) short chain dehydrogenases/reductases; (2) HCPs (hybrid cluster proteins) that contain a 4Fe-4S cluster; (3) Rieske-type 2Fe-2S iron cluster binding proteins; and (4) benzoate dioxygenase reductases – comprising 2Fe-2S ferredoxin, FAD-binding ferredoxin reductase and FCD (putative transcriptional regulator) genes. In the latter case, we have expressed and purified one such enzyme (CYP1049A1 from a Burkholderia sp.), confirming the predicted cofactor content of the reductase module and NAD(P)H-dependent reduction of the reductase portion of the enzyme (Luciakova et al. unpublished data). However, there are few other data available for the systems mentioned here, or for virtually all the rest of the other potentially new catalytically self-sufficient P450 systems identified from genome searches.
11.8 Non-redox Partner P450 Fusions
Of equal interest to the various likely P450-redox partner fusions identified in numerous prokaryotic and eukaryotic genomes are the several other P450 fusion enzymes in which the apparent fused partners are evidently not electron transfer modules. These protein modules may instead have catalytic functions that are associated with the activity of the P450 itself, e.g. providing a substrate for the linked P450, or using the P450 product as a substrate. Figure 11.5 shows examples for P450s fused (at either their N- or C-terminus) to cinammyl alcohol dehydrogenase (CAD), and to different types of peroxidase, glycosyltransferase, lipoxygenase and IPPS (isoprenyl diphosphate synthase) modules. Other types of P450 fusions from genome databases include those predicted to be fused to putative solute transporters (in ascomycete fungi), esterases/lipases, ubiquitin-like proteins, 2-oxoglutarate/Fe(II)-dependent oxygenase-type proteins and mannosyltransferases.
As is the case for the P450-redox partner fusion enzymes, very few of these non-redox partner fusion enzymes have been expressed and characterized. However, the fungal Ppo enzymes have been studied and are functional fusions of a N-terminal peroxidase/dioxygenase domain to a C-terminal P450. In Aspergillus nidulans, the Ppo’s are involved in synthesis of psi factors – molecules derived from oleic acid and linoleic acid, and which play key roles in regulation of the fungal life cycle, as well as being implicated in the synthesis of mycotoxins [65, 66]. The A. nidulans PpoA enzyme was demonstrated to catalyze the oxidation of linoleic acid to 8R-HPODE (8R-hydroperoxyoctadecadienoic acid) in the peroxidase/dioxygenase domain, with the product then isomerized to 5,8- dihydroxyoctadecadienoic acid by the P450 domain [67]. The A. nidulans PpoC enzyme was similarly shown to catalyze dioxygenation of linoleic acid, forming 10-HPODE. However, in this case the product is not further isomerized by the P450 domain, in which the heme coordinating cysteine thiolate is replaced by a glycine, leading to a heme-depleted P450 domain [68]. A further characterized example of a P450/non-redox partner fusion enzyme is the Penicillium brevicompactum P450-hydrolase fusion protein encoded by the mpaDE gene. The MpaDE enzyme catalyzes key steps in the production of mycophenolic acid (MPA), a molecule with important immunosuppressant, antimicrobial and antitumour activities [69]. Specifically, the P450 component (MpaD) is predicted to catalyze methyl hydroxylation of the precursor 5-methylorsellinic acid (5-MOA) to form 5,7-dihydroxy-4-methylphthalide, followed by a lactonization reaction on this product catalyzed by the Zn-dependent hydrolase domain (MpaE) to form 5,7-dihydroxy-4-methylphthalide (DHMP) as the next intermediate in the MPA synthesis pathway [69].
11.9 P450s that Bypass Redox Partners
Not all P450s are dependent on electron transfer from redox partner, and two particular classes of P450s are stand-alone enzymes that perform reductive or oxidative transformations of their substrates without the participation of any partner proteins. The peroxygenase P450s use hydrogen peroxide directly to form the reactive compound 0 state, which is then transformed via a further protonation and dehydration to generate the catalytically relevant compound I (Fig. 11.1). The first characterized examples of this enzyme class were P450 BSβ from Bacillus subtilis (CYP152A1) and P450 SPα from Sphingomonas paucimobilis (CYP152B1) [70, 71]. CYP152B1 was found to produce predominantly alpha-hydroxy fatty acid products from a range of fatty acid substrates, with C14 and C15 saturated fatty acids (myristic and pentadecanoic acids), and the polyunsaturated arachidonic acid being particularly good substrates, and with the S-enantiomeric hydroxy fatty acids being formed at levels of >98 % of the overall products formed [72]. CYP152A1 produces both α- and β-hydroxylated fatty acids, with the greater proportion being the β-hydroxy products [70, 73]. CYP152A1 and CYP152B1 have similar steady-state rate constants for fatty acid hydroxylation of around 1,000 min−1 with their preferred substrates [70, 73, 74]. A more recently studied member of the CYP152 family is the CYP152L1 enzyme from a Jeotgalicoccus species (OleT). Although strongly related to the CYP152A1/B1 enzymes, OleT catalyzes predominantly the oxidative decarboxylation of long chain fatty acids, producing terminal alkenes as the major products (with much smaller amounts of α- and β-hydroxylated fatty acids). CYP152A1 was also shown to produce 1-pentadecene from the saturated C16 fatty acid palmitic acid, but this product was not detected in parallel studies with CYP152B1. However, CYP152 enzymes from three other microbes (Corynebacterium efficiens, Kocuria rhizophila and Methylobacterium populi) were also shown to produce 1-pentadecene from palmitic acid [75]. The crystal structure of OleT was determined, confirming the strong structural similarity of this P450 to CYP152A1/B1. However, the factors determining preference for H2O2-dependent decarboxylation over hydroxylation in these enzymes remain uncertain. Nonetheless, the apparent rate constant for OleT-dependent oxidation of fatty acids is very fast at ~167 s−1 with 200 µM H2O2 [76]. Other studies used redox partner systems (the E. coli flavodoxin reductase [FLDR] and flavodoxin [FLD], and the P450 BM3 CPR domain) to drive NADPH-dependent catalysis in CYP152A1 and the Clostridium acetobutylicum CYP152A2 (P450CLA) enzymes. These studies showed that fatty acid hydroxylation could also be catalyzed by electron transfer using heterologous redox partner proteins [77]. NADPH-dependent fatty acid decarboxylation was also shown for OleT using either the fused PDOR domain of CYP116B1, or the E. coli FLDR/FLD redox partner combination [78].
The other major group of P450s that has evolved to function without the use of redox partners are the CYP55A P450 subfamily enzymes. The prototype enzyme of this class is CYP55A1 (P450nor) from the fungus Fusarium oxysporum (which also produces P450foxy), which catalyzes a purely reductive reaction in which two molecules of nitric oxide (NO) are converted to dinitrogen oxide (N2O) according to the reaction: 2NO + NADH + H+ → N2O + NAD+ + H2O. Electrons are delivered directly by NADH without the participation of any other proteins [79, 80]. This reaction is the final step in the respiratory conversion of nitrite/nitrate to N2O in the fungus [80]. Two distinct isoforms of CYP55A1 are produced that result from translation using (1) the first start codon (P450norA, which is then directed to the mitochondrion via an N-terminal targeting sequence), or (2) the subsequent start codon (P450norB that is cytoplasmically located) [81]. Orthologues of P450nor were also reported in other fungi – notably Trichosporum cutaneum (CYP55A4) and Cylindrocarpon tonkinense. As for the F. oxysporum P450nor, two different forms of the enzyme are found in C. tonkinense. However, in this case they originate from distinct genes – CYP55A2 (which encodes P450nor1) and CYP55A3 (which encodes P450nor2). Interestingly, P450nor1 is quite NADH-specific, whereas P450nor2 can use both NADH and NADPH, albeit with a higher affinity for NADPH [82].
Mechanistically, the P450nor reaction involves the binding of a molecule of NO to the heme iron of the P450 to form a ferric-NO complex that is then reduced to an intermediate with a red-shifted heme absorption maximum (at ~444 nm) that is considered to be a ferric-hydroxylamine radical species. The short lived intermediate then reacts with the second molecule of NO to generate the N2O product, with release of a molecule of water and NAD+ [83–85].
11.10 Cytochrome b 5 as a Redox Partner for P450 Enzymes
The cytochromes b 5 are small heme binding proteins found predominantly in eukaryotes, although b 5-like proteins and domains have now also been identified in bacteria, including in the purple bacterium Ectothiorhodospira vacuolata [86]. The b 5 proteins are small, membrane proteins (usually ~135 amino acids) with bis-His coordinated heme iron. Electron transfer to and from b 5 proteins occurs via an exposed edge of their heme cofactor, and the b 5 proteins shuttle between ferric and ferrous forms for single electron transfer reactions. Two distinct types of b 5 are found in mammalian tissues – one locating to the outer mitochondrial membrane, and the other to the endoplasmic reticulum membrane [87]. In the context of P450-dependent drug metabolism reactions, the b 5 isoform in the ER is of particular relevance, and is located on the cytoplasmic side of the ER membrane, attached via a C-terminal membrane anchor domain. The b 5 proteins are reduced by their partner cytochrome b 5 reductase, an NADH-dependent and FAD-binding protein that shares the same cellular location [88].
The relationships between b 5 and P450s are complex and remain incompletely understood. The b 5 heme iron has a relatively positive reduction potential (typically around 0 mV versus the normal hydrogen electrode, NHE) [89, 90]. This suggests that they should not be able to deliver the first of the two electrons required for P450 monooxygenation reactions, where the ferric P450 heme iron (in either low-spin or high-spin form) typically has a much more negative potential. However, delivery of the second electron from b 5 to a P450 ferric-superoxo heme species should be much more thermodynamically favourable. Early research on the roles of b 5 in P450 metabolism highlighted that addition of NADH could stimulate NADPH-dependent metabolism of drugs, and thus that a secondary system of electron transport involving cytochrome b 5 reductase and b 5 could provide the 2nd electron for P450 catalysis in reactions such as the activation of the anti-cancer drug ellipticine and the improved oxidation of nifedipine and testosterone by the major human drug metabolizing P450 CYP3A4 [91, 92]. In the case of human CYP2B4, the delivery of a 2nd electron to the P450 via b 5 was reported to occur much faster than from CPR, leading to more efficient catalysis in which NADPH oxidation was more tightly coupled to P450 product formation [93]. The b 5 protein also has a crucial role in the metabolism of steroid hormones. CYP17A1 in the adrenal cortex catalyzes the 17α-hydroxylation of both pregnenolone and progesterone, but 17-hydroxypregnenolone can be further oxidized by the same P450 to generate dehydroepiandrosterone in an acyl (17,20-lyase) bond cleavage reaction. 17-Hydroxypregnenolone can undergo a similar (but less efficient) lyase reaction to form androstenedione. The lyase reaction that forms androgens is enhanced by b 5, although there is still controversy relating to whether b 5’s role involves electron transfer or is achieved through imparting conformational change on the P450. In the latter case, studies suggest that b 5 may influence the binding mode of the substrate such that the iron-oxo species is orientated toward the C20 position (and away from the C17 position), favouring the lyase reaction [94–96]. Other recent in vivo studies using a conditional deletion of b 5 in mouse revealed diminished activities of P450s in the 3A and 2C families, as well as decreased CYP17A 17,20-lyase activity as a consequence of diminished production of b 5 [97, 98].
Studies in yeast and fungal systems have indicated that NADH-cytochrome b 5 reductase and b 5 could act as a functional redox partner system supporting (1) CYP51 (sterol demethylase)-dependent oxidative demethylation of 24-methylene-24,25-dihydrolanosterol in Candida albicans, and (2) hydroxylation of 4-propylbenzoic acid in the lignin degrading Phanaerochaete chrysosporium [99, 100]. However, given the thermodynamic considerations, the most likely mechanism would appear to be delivery of the first electron via the b 5 reductase, and the second (to the ferric-superoxo intermediate) from b 5 [101, 102].
11.11 Conclusions
The cytochromes P450 are an enormously divergent enzyme superfamily, members of which are found in all of the major domains of life. For most eukaryotes (and for several prokaryotes) there are large numbers of different CYP genes in the genome [103]. Early studies on P450 enzymes suggested a relatively simple set of redox partner systems from which these enzymes obtain the electrons required for activation of molecular oxygen and substrate monooxygenation. However, as genome sequence data continue to accumulate and novel types of P450s are characterized, it has become increasingly clear that there is considerable diversity in the types of enzyme machinery that can reduce different P450 enzymes, including various P450s that are fused to some or all of the accessory enzyme components required to drive P450 substrate oxidation [104]. These findings have inspired the creation of artificial fusions between P450s and heterologous redox partners to produce catalytically self-sufficient P450s for biotechnological applications [105, 106]. Other P450s have apparently evolved as fusions to non-redox partner proteins, and this may instead enable more catalytically efficient biochemical transformations if the two fused partners are component enzymes in the same pathway [65–67]. However, other P450s have also evolved to forego any requirement for redox partners – by using naturally the “peroxide shunt” mechanism for catalysis, or by deriving electrons directly from NAD(P)H [76, 84]. Undoubtedly, these recent discoveries on the complexity of redox processes in P450 enzymes are only the tip of the iceberg, and coming years will inevitably see identification of further diversity in the redox processes used in the P450 superfamily.
References
Guengerich FP, Munro AW (2013) Unusual cytochrome P450 enzymes and reactions. J Biol Chem 288:17065–17073
Rittle J, Green MT (2010) Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science 330:933–937
Groves JT (2006) High-valent iron in chemical and biological oxidations. J Inorg Biochem 100:434–437
Pandey AV, Flück CE (2013) NADPH P450 oxidoreductase: structure, function, and pathology of diseases. Pharmacol Ther 138:229–254
Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JD, O’Connell JD III, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, Stroud RM (2014) Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to the bilayer. Proc Natl Acad Sci U S A 111:3865–3870
Porter TD (1991) An unusual yet strongly conserved flavoprotein reductase in bacteria and mammals. Trends Biochem Sci 16:154–158
Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ (1997) Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci U S A 94:8411–8416
Gutierrez A, Doehr O, Paine M, Wolf CR, Scrutton NS, Roberts GC (2000) Trp-676 facilitates nicotinamide coenzyme exchange in the reductive half-reaction of human cytochrome P450 reductase: properties of the soluble W676H and W676A mutant reductases. Biochemistry 39:15990–15999
Gutierrez A, Munro AW, Grunau A, Wolf CR, Scrutton NS, Roberts GC (2003) Interflavin electron transfer in human cytochrome P450 reductase is enhanced by coenzyme binding. Relaxation kinetic studies with coenzyme analogues. Eur J Biochem 270:2612–2621
Iyanagi T, Xia C, Kim JJ (2012) NADPH-cytochrome P450 oxidoreductase: prototypic member of the diflavin reductase family. Arch Biochem Biophys 528:72–89
Poulos TL, Finzel BC, Howard AJ (1987) High-resolution crystal structure of cytochrome P450cam. J Mol Biol 195:687–700
Rheinwald JG, Chakrabarty AM, Gunsalus IC (1973) A transmissible plasmid controlling camphor oxidation in Pseudomonas putida. Proc Natl Acad Sci U S A 70:885–889
Sevrioukova IF, Garcia C, Li H, Bhaskar B, Poulos TL (2003) Crystal structure of putidaredoxin, the [2Fe-2S] component of the P450cam monooxygenase system from Pseudomonas putida. J Mol Biol 333:377–392
Sevrioukova IF, Poulos TL, Churbanova IY (2010) Crystal structure of the putidaredoxin reductase•putidaredoxin electron transfer complex. J Biol Chem 285:13616–13620
Churbanova IY, Poulos TL, Sevrioukova IF (2010) Production and characterization of a functional putidaredoxin reductase-putidaredoxin covalent complex. Biochemistry 49:58–67
Peterson JA, Lu JY, Geisselsoder J, Graham-Lorence S, Carmona C, Witney F, Lorence MC (1992) Cytochrome P-450terp. Isolation and purification of the protein and cloning and sequencing of its operon. J Biol Chem 267:14193–14203
Bellamine A, Mangla AT, Nes WD, Waterman MR (1999) Characterization and catalytic properties of the sterol 14α-demethylase from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 96:8937–8942
McLean KJ, Warman AJ, Seward HE, Marshall KR, Girvan HM, Cheesman MR, Waterman MR, Munro AW (2006) Biophysical characterization of the sterol demethylase P450 from Mycobacterium tuberculosis, its cognate ferredoxin, and their interactions. Biochemistry 45:8427–8443
Tripathi S, Li H, Poulos TL (2013) Structural basis for effector control and redox partner recognition in cytochrome P450. Science 340:1227–1230
Green AJ, Munro AW, Cheesman MR, Reid GA, von Wachenfeldt C, Chapman SK (2003) Expression, purification and characterisation of a Bacillus subtilis ferredoxin: a potential electron transfer donor to cytochrome P450 BioI. J Inorg Biochem 93:92–99
Puchkaev AV, Ortiz de Montellano PR (2005) The Sulfolobus solfataricus electron donor partners of thermophilic CYP119: an unusual non-NAD(P)H-dependent cytochrome P450 system. Arch Biochem Biophys 434:169–177
Ricagno S, de Rosa M, Aliverti A, Zanetti G, Bolognesi M (2007) The crystal structure of FdxA, a 7Fe ferredoxin from Mycobacterium smegmatis. Biochem Biophys Res Commun 360:97–102
McLean KJ, Munro AW (2008) Structural biology and biochemistry of cytochrome P450 systems in Mycobacterium tuberculosis. Drug Metab Rev 40:427–446
Ewen KM, Kleser M, Bernhardt R (2011) Adrenodoxin: the archetype of vertebrate-type [2Fe-2S] cluster ferredoxins. Biochim Biophys Acta 1814:111–125
Müller JJ, Lapko A, Bourenkov G, Ruckpaul K, Heinemann U (2001) Adrenodoxin reductase-complex structure suggests electron transfer path in steroid biosynthesis. J Biol Chem 276:2786–2789
Hawkes DB, Slessor KE, Bernhardt PV, De Voss JJ (2010) Cloning, expression and purification of cindoxin, an unusual FMN-containing cytochrome P450 redox partner. ChemBioChem 11:1107–1114
Narhi LO, Fulco AJ (1986) Characterization of a catalytically self-sufficient 119,000-dalton cytochrome P450 monooxygenase induced by barbiturates in Bacillus megaterium. J Biol Chem 261:7160–7169
Narhi LO, Fulco AJ (1987) Identification and characterization of two functional domains in cytochrome P-450 BM-3, a catalytically self-sufficient monooxygenase induced by barbiturates in Bacillus megaterium. J Biol Chem 262:6683–6690
Noble MA, Miles CS, Chapman SK, Lysek DA, Mackay AC, Reid GA, Hanzlik RP, Munro AW (1999) Roles of key active-site residues in flavocytochrome P450 BM3. Biochem J 339:371–379
Munro AW, Daff S, Coggins JR, Lindsay JG, Chapman SK (1996) Probing electron transfer in flavocytochrome P-450 BM3 and its component domains. Eur J Biochem 239:403–409
Black SD, Martin ST (1994) Evidence for conformational dynamics and molecular aggregation in cytochrome P450 102 (BM-3). Biochemistry 33:12056–12062
Neeli R, Girvan HM, Lawrence A, Warren MJ, Leys D, Scrutton NS, Munro AW (2005) The dimeric form of flavocytochrome P450 BM3 is catalytically functional as a fatty acid hydroxylase. FEBS Lett 579:5582–5588
Kitazume T, Haines DC, Estabrook RW, Chen B, Peterson JA (2007) Obligatory intermolecular electron-transfer from FAD to FMN in dimeric P450BM-3. Biochemistry 46:11892–11901
Girvan HM, Dunford AJ, Neeli R, Ekanem IS, Waltham TN, Joyce MG, Leys D, Curtis RA, Williams P, Fisher K, Voice MW, Munro AW (2011) Flavocytochrome P450 BM3 mutant W1046A is a NADH-dependent fatty acid hydroxylase: implications for the mechanism of electron transfer in the P450 BM3 dimer. Arch Biochem Biophys 507:75–85
Siddhanta U, Presta A, Fan B, Wolan D, Rouseau DL, Stuehr DJ (1998) Domain swapping in inducible nitric-oxide synthase. Electron transfer occurs between flavin and heme groups located on adjacent subunits in the dimer. J Biol Chem 273:18950–18958
Daff SN, Chapman SK, Turner KL, Holt RA, Govindaraj S, Poulos TL, Munro AW (1997) Redox control of the catalytic cycle of flavocytochrome P-450 BM3. Biochemistry 36:13816–13823
Murataliev MB, Klein M, Fulco AJ, Feyereisen R (1997) Functional interactions in cytochrome P450 BM3: flavin semiquinone intermediates, role of NADP(H), and mechanism of electron transfer by the flavoprotein domain. Biochemistry 36:8401–8412
Hanley SC, Ost TW, Daff S (2004) The unusual redox properties of flavocytochrome P450 BM3 flavodoxin domain. Biochem Biophys Res Commun 325:1418–1423
Gustafsson MC, Roitel O, Marshall KR, Noble MA, Chapman SK, Pessegueiro A, Fulco AJ, Cheesman MR, von Wachenfeldt C, Munro AW (2004) Expression, purification, and characterization of Bacillus subtilis cytochromes P450 CYP102A2 and CYP102A3: flavocytochrome homologues of P450 BM3 from Bacillus megaterium. Biochemistry 43:5474–5487
Chowdhary PK, Alemseghed M, Haines DC (2007) Cloning, expression and characterization of a fast self-sufficient P450: CYP102A5 from Bacillus cereus. Arch Biochem Biophys 468:32–43
Nakayama N, Takemae A, Shoun H (1996) Cytochrome P450foxy, a catalytically self-sufficient fatty acid hydroxylase of the fungus Fusarium oxysporum. J Biochem 119:435–440
Kitazume T, Tanaka A, Takaya N, Nakamura A, Matsuyama S, Suzuki T, Shoun H (2002) Kinetic analysis of hydroxylation of saturated fatty acids by recombinant P450foxy produced by an Escherichia coli expression system. Eur J Biochem 269:2075–2082
Correll CC, Batie CJ, Ballou DP, Ludwig ML (1992) Phthalate dioxygenase reductase: a modular structure for electron transfer from pyridine nucleotides to [2Fe-2S]. Science 258:1604–1610
De Mot R, Parrey AHA (2002) A novel class of self-sufficient cytochrome P450 monooxygenases in prokaryotes. Trends Microbiol 10:502–508
Warman AJ, Robinson JW, Luciakova D, Lawrence AD, Marshall KR, Warren MJ, Cheesman MR, Rigby SE, Munro AW, McLean KJ (2012) Characterization of Cupriavidus metallidurans CYP116B1 – a thiocarbamate herbicide oxygenating P450-phthalate dioxygenase reductase fusion protein. FEBS J 279:1675–1693
Hunter DJ, Roberts GA, Ost TW, White JH, Muller S, Turner NJ, Flitsch SL, Chapman SK (2005) Analysis of the domain properties of the novel cytochrome P450 RhF. FEBS Lett 579:2215–2220
Nagy I, Compernolle F, Ghys K, Vanderleyden J, De Mot R (1995) A single cytochrome-P-450 system is involved in degradation of the herbicides EPTC (S-ethyl dipropylthiocarbamate) and atrazine by Rhodococcus sp. strain ni86/21. Appl Environ Microbiol 61:2056–2060
Nagy I, Schoofs G, Compernolle F, Proost P, Vanderleyden J, De Mot R (1995) Degradation of the thiocarbamate herbicide EPTC (S-ethyl dipropylcarbamothioate) and biosafening by Rhodococcus sp. strain ni86/21 involves an inducible cytochrome P-450 system and aldehyde dehydrogenase. J Bacteriol 177:676–687
Celik A, Roberts GA, White JH, Chapman SK, Turner NJ, Flitsch SL (2006) Probing the substrate specificity of the catalytically self-sufficient cytochrome P450RhF from a Rhodococcus sp. Chem Commun 2006:4492–4494
Liu L, Schmid RD, Urlacher VB (2006) Cloning, expression, and characterization of a self-sufficient cytochrome P450 monooxygenase from Rhodococcus ruber DSM 44319. Appl Microbiol Biotechnol 72:876–882
Liu L, Schmid RD, Urlacher VB (2010) Engineering cytochrome P450 monooxygenase CYP116B3 for high dealkylation activity. Biotechnol Lett 32:841–845
Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM (2013) A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat Chem Biol 9:485–487
Butler CF, Peet C, Mason AE, Voice MW, Leys D, Munro AW (2013) Key mutations alter the cytochrome P450 BM3 conformational landscape and remove inherent substrate bias. J Biol Chem 288:25387–25399
Sabbadin F, Grogan G, Bruce NC (2013) LICRED: a versatile drop-in vector for rapid generation of redox-self-sufficient cytochromes P450. Methods Mol Biol 987:239–249
Seth-Smith HM, Rosser SJ, Basran A, Travis ER, Dabbs ER, Nicklin S, Bruce NC (2002) Cloning, sequencing, and characterization of the hexahydro-1,3,5-trinitro-1,3,5-triazine degradation gene cluster from Rhodococcus rhodochrous. Appl Environ Microbiol 68:4764–4771
Jackson RG, Rylott EL, Fournier D, Hawari J, Bruce NC (2007) Exploring the biochemical properties and remediation applications of the unusual explosive-degrading P450 system XplA/B. Proc Natl Acad Sci U S A 104:16822–16827
Rylott EL, Jackson RG, Sabbadin F, Seth-Smith HM, Edwards J, Chong CS, Strand SE, Grogan G, Bruce NC (2011) The explosive-degrading cytochrome P450 XplA: biochemistry, structural features and prospects for bioremediation. Biochim Biophys Acta 1814:230–236
Sabbadin F, Jackson R, Haider K, Tampi G, Turkenburg JP, Hart S, Bruce NC, Grogan G (2009) The 1.5-Å structure of XplA-heme, an unusual cytochrome P450 heme domain that catalyzes reductive biotransformation of royal demolition explosive. J Biol Chem 284:28467–28475
Raag R, Martinis SA, Sligar SG, Poulos TL (1991) Crystal structure of the cytochrome P-450CAM active site mutant Thr252Ala. Biochemistry 30:11420–11429
Bui SH, McLean KJ, Cheesman MR, Bradley JM, Rigby SE, Levy CW, Leys D, Munro AW (2012) Unusual spectroscopic and ligand binding properties of the cytochrome P450-flavodoxin fusion enzyme XplA. J Biol Chem 287:19699–19714
McCarthy AA, Walsh MA, Verma CS, O’Connell DP, Reinhold M, Yalloway GN, D’Arcy D, Higgins TM, Voordouw G, Mayhew SG (2002) Crystallographic investigation of the role of aspartate 95 in the modulation of the redox potentials of Desulfovibrio vulgaris. Biochemistry 41:10950–10962
Seth-Smith HM, Edwards J, Rosser SJ, Rathbone DA, Bruce NC (2008) The explosive-degrading cytochrome P450 system is highly conserved among strains of Rhodococcus spp. Appl Environ Microbiol 74:4550–4552
Jackson CJ, Lamb DC, Marczylo TH, Warrilow AG, Manning NJ, Lowe DJ, Kelly DE, Kelly SL (2002) A novel sterol 14α-demethylase/ferredoxin fusion protein from Methylococcus capsulatus represents a new class of the cytochrome P450 superfamily. J Biol Chem 277:46959–46965
Geer LY, Domrachev M, Lipman DJ, Bryant SH (2002) CDART: protein homology by domain architecture. Genome Res 12:1619–1623
Tsitsigiannis DI, Kowieski TM, Zarnowski R, Keller NP (2005) Three putative oxylipin biosynthetic genes integrate sexual and asexual development in Aspergillus nidulans. Microbiology 151:1809–1821
Tsitsigiannis DI, Keller NP (2006) Oxylipins act as determinants of natural product biosynthesis and seed colonization in Aspergillus nidulans. Mol Microbiol 59:882–892
Brodhun F, Gobel C, Hornung E, Feussner I (2009) Identification of PpoA from Aspergillus nidulans as a fusion protein of a fatty acid heme dioxygenase/peroxidase and a cytochrome P450. J Biol Chem 284:11792–11805
Brodhun F, Schneider S, Gobel C, Hornung E, Feussner I (2010) PpoC from Aspergillus nidulans is a fusion protein with only one active haem. Biochem J 425:553–565
Hansen BG, Mnich E, Nielsen KF, Nielsen JB, Nielsen MT, Mortensen UH, Larsen TO, Patil KR (2012) Involvement of a natural fusion of a cytochrome P450 and a hydrolase in mycophenolic acid biosynthesis. Appl Environ Microbiol 78:4908–4913
Lee DS, Yamada A, Sugimoto H, Matsunaga I, Ogura H, Ichihara K, Adachi S, Park SY, Shiro Y (2003) Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis − crystallographic, spectroscopic, and mutational studies. J Biol Chem 278:9761–9767
Fujishiro T, Shoji O, Nagano S, Sugimoto H, Shiro Y, Watanabe Y (2011) Crystal structure of H2O2-dependent cytochrome P450SPα with its bound fatty acid substrate: insight into the regioselective hydroxylation of fatty acids at the α position. J Biol Chem 286:29941–29950
Matsunaga I, Sumimoto T, Ueda A, Kusunose E, Ichihara K (2000) Fatty acid-specific, regiospecific, and stereospecific hydroxylation by cytochrome P450 (CYP152B1) from Sphingomonas paucimobilis: substrate structure required for α-hydroxylation. Lipids 35:365–371
Matsunaga I, Yokotani N, Gotoh O, Kusunose E, Yamada M, Ichihara K (1997) Molecular cloning and expression of fatty acid α-hydroxylase from Sphingomonas paucimobilis. J Biol Chem 272:23592–23596
Matsunaga I, Ueda A, Fujiwara N, Sumimoto T, Ichihara K (1999) Characterization of the ybdT gene product of Bacillus subtilis: novel fatty acid β-hydroxylating cytochrome P450. Lipids 34:841–846
Rude MA, Baron TS, Brubaker S, Alibhai M, Del Cardayre SB, Schirmer A (2011) Terminal olefin (1-alkene) biosynthesis by a novel P450 fatty acid decarboxylase from Jeotgalicoccus species. Appl Environ Microbiol 77:1718–1727
Belcher J, McLean KJ, Matthews S, Woodward LS, Fisher K, Rigby SE, Nelson DR, Potts D, Baynham MT, Parker DA, Leys D, Munro AW (2014) Structure and biochemical properties of the alkene producing cytochrome P450 OleT JE (CYP152L1) from the Jeotgalicoccus sp. 8456 bacterium. J Biol Chem 289:6535–6550
Girhard M, Schuster S, Dietrich M, Dürre P, Urlacher VB (2007) Cytochrome P450 monooxygenase from Clostridium acetobutylicum: a new α-fatty acid hydroxylase. Biochem Biophys Res Commun 362:114–119
Liu Y, Wang C, Yan J, Zhang W, Guan W, Lu X, Li S (2014) Hydrogen peroxide-independent production of α-alkenes by OleT JE P450 fatty acid decarboxylase. Biotechnol Biofuels 7:28
Daiber A, Shoun H, Ullrich V (2005) Nitric oxide reductase (P450nor) from Fusarium oxysporum. J Inorg Biochem 99:185–193
Shoun H, Tanimoto T (1991) Denitrification by the fungus Fusarium oxysporum and involvement of cytochrome P-450 in the respiratory nitrite reduction. J Biol Chem 266:11078–11082
Usuda K, Toritsuka N, Matsuo Y, Kim DH, Shoun H (1995) Denitrification by the fungus Cylindrocarpon tonkinense: anaerobic cell growth and two isozyme forms of cytochrome P-450nor. Appl Environ Microbiol 61:883–889
Shoun H, Fushinobu S, Jiang L, Kim SW, Wakagi T (2012) Fungal denitrification and nitric oxide reductase cytochrome P450nor. Phil Trans R Soc B Biol Sci 367:1186–1194
Shiro Y, Fujii M, Iizuka T, Adachi S, Tsukamoto K, Nakahara K, Shoun H (1995) Spectroscopic and kinetic-studies on reaction of cytochrome P450nor with nitric oxide − implication for its nitric oxide reduction mechanism. J Biol Chem 270:1617–1623
Lehnert N, Praneeth VKK, Paulat F (2006) Electronic structure of iron(II)-porphyrin nitroxyl complexes: molecular mechanism of fungal nitric oxide reductase (P450nor). J Comput Chem 27:1338–1351
Kostanjevecki V, Leys D, Van Driessche G, Meyer TE, Cusanovich MA, Fischer U, Guisez Y, Van Beeumen J (1999) Structure and characterization of Ectothiorhodospira vacuolata cytochrome b 558, a prokaryotic homologue of cytochrome b 5. J Biol Chem 274:35614–35620
Altuve A, Silchenko S, Lee KH, Kuczera K, Terzyan S, Zhang X, Benson DR, Rivera M (2001) Probing the differences between rat liver outer mitochondrial membrane cytochrome b 5 and microsomal cytochromes b 5. Biochemistry 40:9469–9483
Spatz L, Strittmatter P (1973) A form of reduced nicotinamide adenine dinucleotide cytochrome b 5 reductase containing both the catalytic site and an additional hydrophobic membrane-binding segment. J Biol Chem 248:793–799
Guzov VM, Houston HL, Murataliev MB, Walker FA, Feyereisen R (1996) Molecular cloning, overexpression in Escherichia coli, structural and functional characterization of house fly cytochrome b 5. J Biol Chem 271:26637–26645
Funk WD, Lo TP, Mauk MR, Brayer GD, MacGillivray RT, Mauk AG (1990) Mutagenic, electrochemical, and crystallographic investigation of the cytochrome b 5 oxidation-reduction equilibrium: involvement of asparagine-57, serine-64, and heme propionate-7. Biochemistry 29:5500–5508
Hildebrandt A, Estabrook RW (1971) Evidence for the participation of cytochrome b 5 in hepatic microsomal mixed-function oxidation reactions. Arch Biochem Biophys 143:66–79
Correia MA, Mannering GJ (1973) Reduced diphosphopyridine nucleotide synergism of the reduced triphosphopyridine nucleotide-dependent mixed-function oxidase system of hepatic microsomes. II. Role of the type I drug-binding site of cytochrome P-450. Mol Pharmacol 9:470–485
Sang-Choul I, Waskell L (2011) The interaction of microsomal cytochrome P450 2B4 with its redox partners, cytochrome P450 reductase and cytochrome b 5. Arch Biochem Biophys 507:144–153
Katagiri M, Kagawa N, Waterman MR (1995) The role of cytochrome b 5 in the biosynthesis of androgens by human P450C17. Arch Biochem Biophys 317:343–347
Akhtar M, Wright JN, Lee-Robichaud P (2011) A review of mechanistic studies on aromatase (CYP19) and 17α-hydroxylase-17,20-lyase (CYP17). J Steroid Biochem Mol Biol 125:2–12
Storbeck KH, Swart AC, Goosen P, Swart P (2013) Cytochrome b 5: novel roles in steroidogenesis. Mol Cell Endocrinol 371:87–99
Finn RD, McLaughlin LA, Ronseaux S, Rosewell I, Houston JB, Henderson CJ, Wolf CR (2008) Defining the in vivo role for cytochrome b 5 in cytochrome P450 function through the conditional hepatic deletion of microsomal cytochrome b 5. J Biol Chem 283:31385–31393
McLaughlin LA, Ronseaux S, Finn RD, Henderson CJ, Wolf CR (2010) Deletion of microsomal cytochrome b 5 profoundly affects hepatic and extrahepatic drug metabolism. Mol Pharmacol 78:269–278
Ichinose H, Wariishi H (2012) Heterologous expression and mechanistic investigation of a fungal cytochrome P450 (CYP5150A2): involvement of alternative redox partners. Arch Biochem Biophys 518:8–15
Syed K, Kattamuri C, Thompson TB, Yadav JS (2011) Cytochrome b 5 reductase-cytochrome b 5 as an active P450 redox enzyme system in Phanerochaete chrysosporium: atypical properties and in vivo evidence of electron transfer capability to CYP63A2. Arch Biochem Biophys 509:26–32
Henderson CJ, McLaughlin LA, Wolf CR (2013) Evidence that cytochrome b 5 and cytochrome b 5 reductase can act as sole electron donor to the hepatic cytochrome P450 system. Mol Pharmacol 83:1209–1217
Noble MA, Girvan HM, Smith SJ, Smith WE, Murataliev M, Guzov VM, Feyereisen R, Munro AW (2007) Analysis of the interactions of cytochrome b 5 with flavocytochrome P450 BM3 and its domains. Drug Metab Rev 39:599–617
Nelson DR, Goldstone JV, Stegeman JJ (2013) The cytochrome P450 genesis locus: the origin and evolution of animal cytochrome P450s. Philos Trans R Soc Lond B Biol Sci 368(1612):20120474
Munro AW, Girvan HM, McLean KJ (2007) Variations on a (t)heme − novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat Prod Rep 24:585–609
Sadeghi SJ, Gilardi G (2013) Chimeric P450 enzymes: activity of artificial redox fusions driven by different reductases for biotechnological applications. Biotechnol Appl Biochem 60:102–110
Munro AW, Girvan HM, McLean KJ (2007) Cytochrome P450 – redox partner fusion enzymes. Biochim Biophys Acta 1770:345–359
Hamdane D, Xia C, Im SC, Zhang H, Kim JJ, Waskell L (2009) Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J Biol Chem 284:11374–11378
Filenko N, Spiro S, Browning DF, Squire D, Overton TW, Cole J, Constantinidou C (2007) The NsrR regulon of Escherichia coli K-12 includes genes encoding the hybrid cluster protein and the periplasmic, respiratory nitrite reductase. J Bacteriol 189:4410–4417
Macedo S, Mitchell EP, RomÐo CV, Cooper SJ, Coelho R, Liu MY, Xavier AV, LeGall J, Bailey S, Garner DC, Hagen WR, Teixeira M, Carrondo MA, Lindley P (2002) Hybrid cluster proteins (HCPs) from Desulfovibrio desulfuricans ATCC 27774 and Desulfovibrio vulgaris (Hildenborough): X-ray structures at 1.25 Å resolution using synchrotron radiation. J Biol Inorg Chem 7:514–525
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
McLean, K.J., Luciakova, D., Belcher, J., Tee, K.L., Munro, A.W. (2015). Biological Diversity of Cytochrome P450 Redox Partner Systems. In: Hrycay, E., Bandiera, S. (eds) Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450. Advances in Experimental Medicine and Biology, vol 851. Springer, Cham. https://doi.org/10.1007/978-3-319-16009-2_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-16009-2_11
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-16008-5
Online ISBN: 978-3-319-16009-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)