
Abstract
Fungi are organisms, which can serve as models for investigation of physiological processes and additionally, they have a high potential for industrial applications. In order to leverage their full potential, their genomes are modified for analysis of gene functions, elucidation of metabolic processes and also for their adjustment to biotechnological processes for efficient production of enzymes and metabolites. One of the most prominent methods for genome manipulation by transformation is protoplast transformation. In this review, we give an overview on prerequisites for protoplasting and transformation as well as methods and conditions for every single step as applied in diverse fungi. Thereby, we aim to provide a basis for troubleshooting with established methods and for development of new protocols to be applied to species for which transformation has not yet been achieved. Thereafter, we briefly summarize tools for enhanced efficiency of transformation, which have been applied in combination with protoplast transformation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Genome modification
- Protoplast transformation
- Protoplasting
- Cell wall degradation
- Gene deletion
- Complementation
- Functional genomics
1 Introduction
Fungi have gained attention in recent years for their importance in health and economy. Acting in symbiotic relationships with plants as biofungicides, producing numerous drugs as antibiotics and hydrolytic enzymes, for example, for second generation biofuels, and causing plant and animal diseases, the importance of fungi is remarkable nowadays (Borkovich and Ebbole 2010). Furthermore, the increase of available sequenced genomes of different fungi allows functional genomic analyses to elucidate mechanisms of gene regulation for enzyme production, substrate sensing, signaling cascades, etc. as well as their interconnections and conserved metabolic pathways in different fungi. Available molecular genetic tools allow the transfer of DNA to a targeted locus in the genome (Olmedo-Monfil et al. 2004). Combinations of these tools with different transformation systems are being used for efficient genetic modifications of fungi for research purposes and industrial strain improvement. High throughput approaches have been developed to facilitate construction of whole genome knockout libraries, for example, for Neurospora crassa (Collopy et al. 2010) or Trichoderma reesei (Schuster et al. 2012).
Protoplasting is one of the methods commonly used to prepare cells in order to genetically modify fungi. More than six decades have passed since the first reports appeared on protoplast isolation from yeasts (Bachmann and Bonner 1959; Eddy and Williamson 1959) and filamentous fungi (Bachmann and Bonner 1959; Fawcett et al. 1973). Since then, the interest in the improvement of this technique has enabled scientists to achieve higher transformation rates and more efficient targeting to the appropriate genomic locus. Protoplast preparation requires removal of the fungal cell wall, nowadays predominantly by using enzymes. Mechanical and other nonenzymatic methods have been reported, but they have not been used extensively due to their inconvenience. Nevertheless, some of these disadvantages could be due to the specific procedure for each particular organism and the physiological state that might be induced in the protoplasts as a consequence of the treatments used (Sun et al. 1992; Von Klercker 1982). Alternative methods to protoplast transformation include electroporation, biolistics, and Agrobacterium transformation (see other chapters of this book), which were established to improve the transformation efficiency for some species that are not suitable for protoplast transformation. The simplicity in technical operations and materials required for this method make the protoplast transformation the most commonly selected method to perform transformations in filamentous fungi. Therefore, enzymatic methods for protoplasting have early on become the choice for most labs (Peberdy 1979). However, with the recent high throughput projects for gene manipulation, electroporation is increasingly applied (Park et al. 2011; Schuster et al. 2012).
In this chapter we provide an overview about different steps of genetic transformation of fungi and a review of different protocols for protoplast transformation in fungi.
2 The Fungal Cell Wall: A Barrier to be Removed
The first obstacle found when intending to transfer exogenous DNA into the fungal cell is the cell wall. Most protoplasting procedures remove this cell wall by using enzymatic digestion, mainly using fungal enzymes (Gruber and Seidl-Seiboth 2012). Therefore, the structure of fungi cell wall determines the enzymes needed to digest it (Adams 2004).
2.1 Composition of the Fungal Cell Wall
The main components of the fungal cell wall are chitin, 1,3-β- and 1,6-β-glucans, proteins, mannans, and other polymers, which are cross-linked forming this complex structure. The crystalline polysaccharides, chitin and β-glucans, constitute the skeletal portion of the wall, whereas the amorphous polysaccharides and protein–polysaccharide complexes are components of the wall matrix. The shape, integrity, and mechanical strength of the fungus is determined by the chemical composition and the cell wall, which is responsible for the interaction of the fungus with its environment as well as biological activity (Adams 2004; Gooday 1995; Lesage and Bussey 2006).
For most of fungi, the central core of the cell wall is a branched β-1,3/1,6-glucan that is linked to chitin via a β-1,4 linkage. β-1,6 glucosidic linkages between chains account for 3 % and 4 % of the total glucan linkages, respectively, in Saccharomyces cerevisiae and A. fumigatus (Adams 2004; Fleet 1991; Fontaine et al. 2000; Gooday 1995; Kollar et al. 1995; Lesage and Bussey 2006; Perez and Ribas 2004). The cell wall structures of these fungi were investigated in detail. This structural core, which determines the mechanical strength, is suggested to be fibrillar and embedded in an amorphous cement (usually removed by alkali treatment) (Latgé 2007). This structure varies markedly between species of fungi, although there is a close correlation between taxonomic classification and cell wall composition among fungi determined by their evolution (Bartnicki-Garcia 1968; Borkovich and Ebbole 2010). The basic composition of all fungal cell walls, including those of the distant chytrids, consists of a branched polysaccharide of β-1,3/1,6 glucan that is linked to chitin via a β-1,4 linkage. This ancient ancestral fungal cell wall has been further modified and decorated in the various fungal orders (Borkovich and Ebbole 2010). As an example, α-l,3 glucan is present in cell walls of Ascomycetes and Basidiomycetes. Following their bifurcation, β-1,6 glucan was added to Ascomycetes and xylose to Basidiomycetes (Kollar et al. 1997; Ruiz-Herrera et al. 1996; Vaishnav et al. 1998). Within the Ascomycetes, more differences have been developed between the yeasts and filamentous fungi. The yeast S. cerevisiae contains β-1,6 glucan, whereas filamentous fungi such as A. fumigatus contain linear α-1,3/1,4 glucan and galactomannan with galactofuran side chains (Fontaine et al. 2000; Kollar et al. 1997). Regarding to chitin content, cell walls of filamentous fungi contain higher levels than those of yeasts (approximately 15 % vs. 2–3 %, respectively). The reason may be that the cell wall of filamentous fungi is cylindrical and under high turgor pressure, therefore it needs to increase its rigidity (Borkovich and Ebbole 2010). The attachment to embedded proteins and mannans also differs between filamentous fungal cell walls from those of the yeasts. Mannan chains in S. cerevisiae cell wall are only found attached to cell wall proteins (CWPs), whereas in A. fumigatus, mannan chains are also found directly linked to glucans (Borkovich and Ebbole 2010; Latgé 2007; Lesage and Bussey 2006). These differences in the cell wall composition between yeast and filamentous fungi may have appeared because of different evolutionary pressures. The adaptation of the filamentous fungal cell wall to extremely rapid deposition and growth at the hyphal tip and its ability to penetrate hard surfaces (Collinge and Trinci 1974) differ from the isotropic growth usually confined to surfaces in yeast (Borkovich and Ebbole 2010). This could be the reason why mutations in secretory and transport-related genes in yeast usually result in less severe phenotypic consequences than in filamentous fungi (Borkovich and Ebbole 2010). The morphological complexity of filamentous fungi makes slight disturbances in homeostasis more obvious than they are in yeast (Seiler and Plamann 2003; Whittaker et al. 1999).
2.2 Functions and Biological Activity of the Fungal Cell Wall
Concerning the biological activity of fungal cell wall, it provides protective and aggressive functions. It acts as an initial barrier in contact with hostile environments. Its absence can cause the death of the fungus unless there is an osmotic stabilizer. Regarding to the aggressive function, it harbors hydrolytic and toxic molecules, required for a fungus to invade its ecological niche. Furthermore, its rigid structure is useful as a force for the penetration of insoluble substrates that it colonizes or invades (Latgé 2007).
Polysaccharides, which represent 80–90 % of the dry matter of fungal cell walls, include amino sugars, hexoses, hexuronic acids, methylpentoses, and pentoses (Bartnicki-Garcia 1970). Glucose and N-acetyl-d-glucosamine (GlcNAc) usually represent the chemical elements of skeletal wall polysaccharides, such as chitin, non-cellulosic β-glucans, and a-glucans.
Each component of cell wall performs specific functions, owing to distinctive physicochemical properties. Chitin and β-glucans are responsible for the mechanical strength of the wall, while the amorphous homo- and heteropolysaccharides, often in association with proteins, act as cementing substances and constitute the carbohydrate moieties of extracellular enzymes and cell wall antigens (Ballou and Raschke 1974; Gander 1974; Lampen 1968). The location within of cell wall components the wall structure also plays a role in the functional specialization. The outer surface of the cell wall is composed of amorphous material (often glycoprotein in nature), leading to a smooth or slightly granular texture. The interfibrillar spaces of the inner wall layer are filled with amorphous material, similar to that of the outer wall layer. In contrast, the layer of the wall adjacent to the plasmalemma is mainly composed by skeletal microcrystalline components. Due to plasticity of fungal cell walls, the structure varies during different growth phases. Newly synthesized portions of the walls are thin and smooth, whereas older portions have the primary wall covered with secondary layers composed of amorphous matrix material (Hunsley 1973; Hunsley and Gooday 1974; Hunsley and Kay 1976; Trinci 1978; Trinci and Coolinge 1975). Consequently, young mycelium is more vulnerable to enzymatic degradation than aged hyphae.
2.2.1 Chitin
Chitin, an α,β-(1,4) polymer of N-acetyl-glucosamine (GlcNAc), is the second main cell wall fiber (Borkovich and Ebbole 2010). It forms microfibrils that are stabilized by hydrogen bonds. Chitin provides tensile strength to the cell wall and composes approximately 2 % of the total cell wall dry weight in yeast, and 10–15 % in filamentous fungi (Borkovich and Ebbole 2010; Klis et al. 2002; Roncero 2002), even though previous literature reports that chitin represents between 0.3 and 40 % of the filamentous fungi cell wall dry weight (Mol and Wessels 1990; Molano et al. 1980; Sietsma and Wessels 1990). The absence of chitin and most glucans from plant and mammalian species makes these components of the fungal cell wall potential and actual targets for antifungal drugs (Beauvais and Latgé 2001; Gooday 1977; Latgé 2007; Nimrichter et al. 2005; Selitrennikoff and Nakata 2003). Synthesis of chitin is well studied in yeast and occurs mainly in the plasmalemma (Braun and Calderone 1978; Jan 1974), specifically, at sites of polarized growth (Lenardon et al. 2010). Depending on the growth phase, chitin synthesis can occur at the bud tip (early bud growth) (Sheu et al. 2000) or over the entire bud surface (isotropic growth) in S. cerevisiae. After nuclear division, chitin is directed towards the mother-bud neck to prepare for cytokinesis (Lenardon et al. 2010).
Chitin is folded to form antiparallel chains, attached through intra-chain hydrogen bonds that lead to formation of strong fibrous microfibrils (Lenardon et al. 2010). In the zygomycetes the polymer chitosan is produced when part of the chitin is deacetylated immediately after synthesis but before chains crystallize by one or more chitin deacetylases (Ariko and Ito 1975; Davis and Bartnicki-Garcia 1984). However, in C. albicans less than 5 % of chitin is deacetylated to chitosan, while zygomycetes and the basidiomycete Cryptococcus neoformans have more than two-thirds deacetylated to chitosan (Baker et al. 2007; Bartnicki-Garcia 1968). Chitosan could be more elastic and resistant to the action of hostile chitinases than chitin (Lenardon et al. 2010). Nevertheless, partially deacetylated glucosaminoglycans are also observed (Datema et al. 1977b). Through ionic interactions, these insoluble polycationic polymers bind to essentially soluble polyanionic glycuronans (containing glucuronic acid, fucose, mannose, and galactose), which are maintained insoluble in the wall (Datema et al. 1977a, b). In contrast, the chitin (glucosaminoglycan) of the walls of ascomycetes and basidiomycetes is fully acetylated and associated with (1-3)-β-/(1-6)-β-glucan in an alkali-insoluble complex (Wessels 1994).
2.2.2 Glucans
Glucans, d-glucose polymers, represent the main fibrous component of the cell wall (Borkovich and Ebbole 2010). Glucans differ both in type and in relative proportions of individual glycosidic bonds. β-glucans are most abundant in fungal cell walls, present usually as constituents of the skeletal microfibrillar portions of the walls, although some fungi also contain glucose polymers linked by α-glycosidic bonds, such as S. pombe (Hochstenbach et al. 1998). Mainly, glucans can be found in long β-1,3 or short β-1,6-linked chains forming the main bulk (30–60 %, dry weight) of the cell wall (Borkovich and Ebbole 2010). β-1,3 glucans have a coiled spring-like structure that confers elasticity and tensile strength to the cell wall (Borkovich and Ebbole 2010). β-1,6 glucan acts as a flexible glue by forming covalent cross-links to β-1,3 glucan and chitin and to cell wall mannoproteins (Kapteyn et al. 2000; Kollar et al. 1997; Lowman et al. 2003; Shahinian and Bussey 2000; Sugawara et al. 2004). α-1,3 glucan harbors an amorphous structure and forms an alkali-soluble cement within the β glucan and chitin fibrils (Beauvais et al. 2005; Grün et al. 2005).
Synthesis of glucans almost exclusively occurs in the cell wall or at the outer surface of the plasmalemma, due to their insolubility and high degree of crystallinity (Farkas 1979). Furthermore, the fungal cell wall contains abundant branched 1,3-β- and 1,6-β-cross-links between proteins and homo- and heteropolysaccharides, chitin, glucan, and other wall components (Cabib et al. 2001; Klis et al. 2006). Nevertheless, the linkages between the individual macromolecular components of the wall do not involve ligase-type enzymes. There is a self-assembly of subunits in the formation of wall fabric through physicochemical interactions, apart from the non-catalytic formation of disulfide bridges between the protein moieties of wall glycoproteins. The number of chemical and physicochemical links increases with the cell age. Hence, older portions of the walls are more resistant to attacks by endogenous (Polacheck and Rosenberger 1975) as well as exogenous (Brown 1971; Necas 1971; Villanueva 1966) polysaccharide hydrolases. Therefore, protocols to remove fungal cell walls require young cells—generally between 16 and 20 h after germination—for protoplast preparation. Changes during germination vary depending on the fungus. For example, P. notatum spores increase the cell wall content of glucosamine, galactosamine, and glucose during the transition from resting spores to swollen spores, to germlings, and to grown mycelium, while galactose content is decreased when spores reached the swollen stage (Martin et al. 1973). However, Colletotrichum lagenarium conidia show a decrease in the content of mannose in the cell wall, and xylose and rhamnose disappear from the mycelial wall (Auriol 1974). Surprisingly, it was reported that in T. viride, the cell wall from conidia does not contain chitin, in contrast to grown mycelium (Benitez et al. 1976). The fungus Puccinia graminis var. tritici was shown to undergo less changes during germination, such as decreasing the amounts of neutral sugars in the cell walls and increasing the content of chitin and protein in the mycelial wall (Ellis and Griffiths 1974).
2.2.3 Non-cellulosic β-Glucans
(1,3;1,4)-β-d-Glucans consist of unbranched and unsubstituted chains of (1,3)- and (1,4)-β-glucosyl residues. The physicochemical properties of the polysaccharides and the functional properties in cell walls depend on the ratio of (1,4)-β-d-glucosyl residues to (1,3)-β-d-glucosyl residues (Burton and Fincher 2009). These polysaccharides are not extensively found in fungi, only in certain species as the pathogenic fungi Rhynchosporium secalis (Pettolino et al. 2009). The regular distribution of polysaccharides in the cell wall due to the molecular links formed by (1,3)-β-glucosyl residues results in less soluble and less suitable gel formation in the matrix phase of the wall. The gel-like material offers some structural support for the wall, combined with flexibility and porosity. Moreover, these interactions can be influenced by associations with other polysaccharides or proteins in the cell wall (Burton and Fincher 2009).
2.2.4 Mannans
Mannans are polymers of mannose and can be found as α-1,6/α-1,2 or α-1.3/β-1,2 mannan chains either attached directly to glucans or covalently attached to proteins via asparagine (N-linked) or serine/threonine (O-linked) amino acid residues (Cutler 2001; Shibata et al. 2007). They comprise 10–20 % of the dry weight of the cell wall (Borkovich and Ebbole 2010; Lesage and Bussey 2006).
2.2.5 Proteins
Polysaccharide–protein complexes represent a principal cell wall constituent, especially in yeasts (Bartnicki-Garcia 1968; Calderone and Braun 1991; Phaff 1971). The amount of glycoproteins in the cell wall is variable, between 10 and 40 %, whereas the actual polypeptide content is about 4 % (de Groot et al. 2007; Klis et al. 2002). The main role of proteins in the wall is to modify and crosslink the wall polymers, instead of being involved in structural maintenance. Moreover, proteins exposed to the outer surface can also participate in determining antigenic and adhesive properties (Calderone and Braun 1991; Hazen 1990).
2.3 Enzymes, Biosynthesis, and Degradation of the Fungal Cell Wall
The structure of the fungal cell wall is highly dynamic and changes constantly during cell expansion and division in yeast, growth, morphogenesis, spore germination, hyphal branching, and septum formation in filamentous fungi. Hydrolytic enzymes, which are closely associated with the cell wall, are responsible for the maintenance of wall plasticity and functions during mycoparasitism (Gruber and Seidl-Seiboth 2012). Among hydrolases identified to date, chitinases, glucanases, and transglycosylases are found to be responsible for breaking and reforming of bonds within and between polymers, leading to re-modeling of the cell wall during growth and morphogenesis (Adams 2004; Lesage and Bussey 2006). The presence of these polysaccharide hydrolases in fungi indicates an autolytic activity in the fungal cell wall (Barras 1972; Fevre 1977; Fleet and Phaff 1974). This autolytic function involves not only cell wall weakening during growth and other morphogenetic processes (Adams 2004; Lesage and Bussey 2006). Therefore, the cell wall of fungi is a dynamic structure whose functions may vary with environmental conditions and in the course of cell and life cycles (Farkas 1979).
Composition of the cell wall determines the enzymes required to digest this structure. Therefore, removal of the fungal cell wall composed by glucans or cellulose, chitin and proteases involves the presence of chitinases, proteases, cellulases, β-glucanases, etc. in commercial preparations. Examples are the lysing enzymes form Trichoderma harzianum (provided by Sigma-Aldrich; L1412), which is frequently used for protoplast preparation from filamentous fungi, but also for yeast spheroplast transformation by hydrolyzing poly (1-3)-glucose of the yeast cell wall glucan (Kelly and Nurse 2011). This preparation contains β-glucanase, cellulase, protease, and chitinase activities. Lysing enzymes from Rhizoctonia solani, apart from β-(1-3)-glucanases, also contain protease, pectinase, and amylase activities (Liu et al. 2010). Also Driselase from Basidiomycetes sp. has been used for digestion of arabinoxylans by fungal glycanases (Liu et al. 2010). This commercial preparation contains laminarinase, xylanase, and cellulase (Product from Sigma-Aldrich). Bovine serum albumin (100 mg) (Sigma, St. Louis, MO, USA) and β-d-glucanase (1 g) (InterSpex Products, San Mateo, CA, USA) can be also used for cell wall digestion (Pratt and Aramayo 2002).
3 Getting Foreign DNA into the Fungal Cell
Filamentous fungi have gained an increased importance in recent years for production of organic chemicals, enzymes, and antibiotics. Most of strains used in industry for those purposes have been developed by screening and/or mutagenesis. Moreover, the genome of different species has been published, and genome sequences available for numerous fungi provide fast access to sequence data of individual genes to be modified. Versatility and convenience of molecular genetic tools nowadays available are constantly increasing, also as a consequence of high throughput functional genomics studies such as the Neurospora crassa whole genome knockout project (Colot et al. 2006). Those tools allow for transferring exogenous DNA to the genome. One of the most common techniques to transform different filamentous fungi is protoplast transformation. This procedure has a general application for different host strains.
However, alternative methods to protoplast transformation have been developed for fungal transformation, such as electroporation, biolistic transformation (Ruiz-Díez 2001), and Agrobacterium-mediated transformation (AMT) (Michielse et al. 2005). A. tumefaciens transfers a part of its DNA (transferred DNA (T-DNA)) to a high number of fungal species and its application with fungi is still increasing. AMT is an alternative for those fungi that are difficult to transform with traditional methods or for which the traditional protocols failed to yield stable DNA integration. The simplicity and efficiency of AMT allows for generation of a large number of stable transformants. Furthermore, T-DNA is integrated randomly and mainly as a single copy, which makes this method suitable for insertional mutagenesis in fungi, obtaining high homologous recombination frequencies (Michielse et al. 2005). Biolistic transformation is a powerful method when protoplasts are difficult to obtain and/or the organisms are difficult to culture. This is particularly applicable to arbuscular mycorrhizal (AM) fungi (Harrier and Millam 2001).
Nevertheless, different integration events are observed using different approaches for fungal transformation. Hence, single-copy integration events were detected when AMT was used for transformation (de Groot et al. 1998; Malonek and Meinhardt 2001; Meyer 2008; Meyer et al. 2003; Mullins and Kang 2001). Protoplast transformation preferentially leads to multicopy integration events (Meyer 2008; Meyer et al. 2003; Mullins and Kang 2001) and lower homologous integration than other methods (Grallert et al. 1993). The influence of the transformation technique on the outcome of the DNA integration event can determine the design of the genetic modification approach. For instance, for targeted integration or gene deletion, AMT would be the method of choice, since it has been shown to favor homologous recombination (HR). Nevertheless, protoplast transformation is still the standard method in most labs (besides electroporation), because laborious vector construction and preparation largely alleviate the advantages of AMT. Moreover, protoplasting transformation would be choice if multiple copies of a gene of interest should integrate at random sites in the genome (heterologous recombination). If AMT does not result in a good efficiency of transformation, protoplast transformation using a NHEJ-deficient recipient strain could be used instead (Meyer 2008).
The first protoplast transformation was made in Saccharomyces cerevisiae. Hutchison and Hartwell (1967) had designed a protocol for protoplast preparation by dissolving the cell walls with a commercial glucanase preparation (Glusulase) and stabilizing the resulting protoplasts with sorbitol. The goal of this first fungal transformation was to use the protoplasts for studies on macromolecular synthesis. The next approach using protoplasts for transformation was made on a leu2 mutant to revert the auxotrophy to prototrophy by introducing wild type DNA in the presence of calcium chloride (Hinnen et al. 1978). Later on, the use of protoplasts for transformation was extended to the filamentous members of the class ascomycetes, N. crassa (Case et al. 1979) and Aspergillus nidulans (Tilburn et al. 1983), and to several other species over the next years till nowadays. Although the original protocols have been improved, the main steps were not fundamentally changed: first, the fungal cell wall needs to be removed by enzymatic degradation. During and after this step, osmotic stabilization of the generated protoplasts is required for survival. Thereafter, several chemicals are used to render the remaining cell membrane permeable for foreign DNA. Finally, DNA is added to the protoplasts and shuttled into the cells. In the following we give an overview on protocols used for these steps, which can serve as a basis for trouble shooting of existing protocols and for development of new protocols for fungi, for which transformation has not yet been attempted or achieved. A summary of conditions used in various organisms is provided in Table 2.1.
3.1 Removing the Cell Wall: Protoplast Preparation
3.1.1 Cell Type and Growth Phase
For protoplast preparation, different cell types have been used in different species. Germinating micro- and macroconidia can be the choice for protoplast preparation (Olmedo-Monfil et al. 2004). Generally, as protoplasting involves removing the cell wall, the growth phase should be chosen in a way that the fungal cell wall is vulnerable to the attack by hydrolytic enzymes, which is more likely in an early stage after germination.
The time necessary to grow the mycelium plays an important role in protoplast preparation, and is related to the growth phase (Naseema et al. 2008). The components of the cell wall vary during different stages of the growth phase, leading to different degrees of composition/digestion of the cell wall. Moreover, components and their relative ratios in the fungal cell wall also differ between different species. Usually, the mycelium is more sensitive to lytic enzymes in the log phase (Naseema et al. 2008). In Neurospora sp. usually young mycelium (after 4–6 h of growth at 25–30 °C) is used to release protoplasts from hyphae after enzymatic treatment to break the cell wall (Buxton and Radford 1984; Vollmer and Yanofsky 1986). A more recent protocol uses conidial spheroplasts for transformation, growing N. crassa for 3 days at 34 °C followed by 2 days at room temperature (Pratt and Aramayo 2002). However, depending on the needed protoplast preparation efficiency, different alternatives can be considered. For Aspergillus and Penicillium species both germinating conidia and mycelium were used (Fincham 1989). A. niger was grown for 20 h (Azizi et al. 2013), while A. nidulans can be grown for 10 h (Mania et al. 2010), both at 30 °C. A pre-culture of Penicillum chrysogenum is grown for 24 h at 30 °C (Flanagan et al. 1990; Hamlyn et al. 1981; Sukumar et al. 2010). Mycelium of Podospora anserina (Brygoo and Debuchy 1985) and Ascobolus immersus (Goyon and Faugeron 1989) is used for protoplast preparation since those species do not produce conidia. Nevertheless, basidiospores (used for Schizophyllum commune (Munoz-Rivas et al. 1986)), dikaryotic mycelium, or, in some cases, the very small vegetatively produced oidia can be used (Agaricales and other Basidiomycetes). Coprinopsis cinerea (Coprinus cinereus) was grown for 48 h at 37 °C (Binninger et al. 1987) while other Basidiomycetes as Ustilago maydis were grown for 18 h at 30 °C (Waard 1976) or for 24 h at 24 °C (Liu et al. 2010). For protoplasts from Fusarium pallidoroseum (mycelium is obtained by centrifugation for 10 min at 10,000g from a liquid culture), the optimal incubation time is 18 h at 25 °C (Naseema et al. 2008), while for F. graminearum incubation takes 12–16 h at room temperature (Goswami 2012). The optimal age of cultures reported for T. harzianum was 16–24 h at 25–29 °C (Balasubramanian and Lalithakumari 2008). Mycelium of T. atroviride was used for protoplast preparation, being 13–14 h at 28 °C the conditions for pre-growth (Cardoza et al. 2006). For T. reesei the optimal pre-growth is 16 h at 28 °C. Paracoccidioides brasiliensis was grown for 72 h (De Borba et al. 1994). For Stagonospora nodorum, Cochliobolus sativus, Pyrenophora teres, and Cercospora beticola the protocol indicates the growth of cells for 4 days at 27 °C (Liu and Friesen 2012). The choice of cell type in any fungus is a matter of experience, and the efficiency of protoplasting can hardly be predicted in advance.
However, from the examples given above, a good starting point for developing a protoplasting protocol for a new species is young mycelium from conidia germinated and grown for 12–24 h at temperatures of 25–30 °C.
3.1.2 Growth Conditions
In addition to the growth phase, the growth conditions applied to reach this growth phase are crucial. Besides the suitable temperature of 25–30 °C, the medium composition of the culture is important. Again, depending on the organism, different media compositions proved successful. N. crassa is grown in complete medium with sucrose (Case et al. 1979; Mautino et al. 1996; Vollmer and Yanofsky 1986) or Vogel’s minimal medium with sucrose (Akins and Lambowitz 1985; Schweizer et al. 1981). Acremonium chrysogenum can be grown on a modified medium suggested by Demain and colleagues, which contains meat extract, fish meal, corn steep solids, ammonium acetate, sucrose, and glucose (Demain et al. 1963; Demain and Newkirk 1962; Hamlyn et al. 1981). A. niger can be grown in transformation medium (Kusters-van Someren et al. 1991) or in SAB-UU broth (sabouraud agar, pH 6.5, supplemented with uridine and uracil) (Azizi et al. 2013). Other Aspergillus strains were grown in minimal medium supplemented with yeast extract, casamino acids, glucose, and uridine (Hamlyn et al. 1981; Pontecorvo et al. 1953). T. harzianum and T. atroviride are grown in PDB (Potato Dextrose Broth) (Balasubramanian and Lalithakumari 2008), while T. reesei is grown on malt extract plates (Gruber et al. 1990). P. chrysogenum was grown on the medium containing mineral salts, corn steep liquor, yeast nucleic acid hydrolysates, methionine, phenylacetylethanolamine, sucrose, and agar (Macdonald et al. 1963) supplemented with yeast extract and casamino acids (Hamlyn et al. 1981) and also in PDB (Flanagan et al. 1990; Sukumar et al. 2010). A similar medium was used for F. pallidoroseum (Naseema et al. 2008). Nevertheless, for F. graminearum YPD medium (yeast extract, bactopeptone, and dextrose) is the medium of choice (Goswami 2012), as well as for S. cerevisiae (Ezeronye and Okerentugba 2001). Protoplasts from C. cinerea were prepared using oidia grown in YMG (yeast extract, malt extract, and glucose) media (Binninger et al. 1987). The medium used for U. maydis pre-growth is composed of yeast extract, bactopeptone, and sucrose (Eppendorf 2002; Waard 1976). PDA (Potato Dextrose Agar) was the medium used for Lentinus lepideus protoplast preparation (Kim et al. 2000). Rhizoctonia solani was grown in V8 juice agar (Liu et al. 2010), P. brasiliensis in PYG (peptone, yeast extract, and glucose) medium (De Borba et al. 1994), and Pseudozyma flocculosa in YMPD (yeast extract, malt extract, peptone, and dextrose) (Cheng and Belanger 2000) for protoplast preparation. S. nodorum, C. sativus, P. teres, and C. beticola are grown in Fries medium (Liu and Friesen 2012). Germination of spores and initial cultivation can be done on solid media (ideally covered with cellophane to enable easy removal of mycelia) or in liquid culture.
3.1.3 Optimal Enzyme Combination for Lysing the Cell Wall
After reaching the optimal growth phase for protoplasting, the fungal cell wall as major obstacle for uptake of DNA has to be removed (Olmedo-Monfil et al. 2004). The structure of the cell wall is different among different fungi, requiring different enzyme combinations together with specific conditions to sufficiently degrade the cell wall and release protoplasts. Furthermore, combination of enzymes was found to be more efficient than using single enzymes for this purpose (Gallmetzer et al. 1999), which is in accordance with the presence of diverse compounds in the fungal cell wall. Therefore, the selection of enzymes is a key factor in protoplast preparation. The effectiveness of cell wall degradation depends on the combination of choice for different batches of lytic enzymes.
Traditionally, commercially available enzymes have been used for protoplast isolation from yeasts. The digestive juice from Helix pomatia was the first lytic enzyme preparation used, which contains muramidases and β-glucuronidases (Eddy and Williamson 1959). Later, the same enzyme mixture has been also used extensively by many other groups with C. neoformans and S. cerevisiae (Deutch and Parry 1974; Foury and Golfeau 1973; Partridge and Drew 1974; Peterson et al. 1976; Shahin 1972; Whittaker and Andrews 1969) and it was commercially available as “helicase”, “sulfatase,” and “glusulase” (Peberdy 1979). A second enzyme, zymolyase, commercially known as Zymolase 100T and derived from Arthrobacter luteus, has also been used since then for the isolation of protoplasts from different fungi, as S. cerevisiae (Dziengel et al. 1977; Ezeronye and Okerentugba 2001; Tomo et al. 2013) and P. brasiliensis (Zymolase 20T) (De Borba et al. 1994).
For N. crassa, Glusulase and Novozym 234, the commercial name of an enzyme mixture from T. viride, which was most frequently used, were the most common enzyme preparations for protoplasting (Case et al. 1979; Vollmer and Yanofsky 1986). Production of Novozym 234 (Novo Nordisk) was however discontinued and the product was replaced by “Lysing enzymes from Trichoderma harzianum” (Sigma-Aldrich), also known as Glucanex, in many research groups (used for P. chrysogenum Hamlyn et al. 1981; Flanagan et al. 1990; Sukumar et al. 2010), P. flocculosa (Cheng and Belanger 2000), T. atroviride (Cardoza et al. 2006), and F. pallidoroseum (Naseema et al. 2008). More recently, bovine serum albumin (Sigma) in combination with β-d-glucanase (InterSpex Products, San Mateo, CA, USA) was used for cell wall digestion of N. crassa (Pratt and Aramayo 2002). A. niger and A. nidulans protoplasts were prepared using Glucanex, which contains cellulase, protease, and chitinase activities (Azizi et al. 2013; Mania et al. 2010). Likewise, Glucanex was also used for protoplast preparation of Sclerotium rolfsii (Fariña et al. 1998). For Trichoderma the new mixture seems to work equally well as the Novozyme product (Steiger 2013). Both enzyme mixtures contain mainly 1,3-glucanases and chitinases, among other hydrolytic enzymes such as cellulases and proteases. For U. maydis cellulases and the commercial product Driselase (enzyme mixture containing cellulases, pectinase, laminarinase, xylanase, and amylase) were used for cell wall digestion (Waard 1976). Likewise, for cell wall digestion of R. solani (Liu et al. 2010) and F. graminearum (Goswami 2012), a combination of lysing enzymes and Driselase were used. For S. nodorum, C. sativus, P. teres, and C. beticola a combination of β-1,3 glucanase and Driselase (from Sigma-Aldrich) is used for cell wall digestion (Liu and Friesen 2012). Some researchers have preferred to use defined enzyme mixtures of cellulases and chitinases for preparing protoplasts of C. cinerea (Binninger et al. 1987). For the taxol-producing ascomycete Ozonium, lywallzyme was the most efficient enzyme to produce protoplasts as tested with several combinations of enzymes (Zhou et al. 2008).
The concentration of enzyme mixture necessary for digestion of cell wall has to be tested for each enzyme combination and each fungus. For F. pallidoroseum 20 mg of lytic enzyme/mL was reported as optimal concentration for achieving the maximum yield of protoplasts (Naseema et al. 2008). For T. harzianum and T. viride it was shown that 5 mg/mL of the lytic enzyme Novozym 234 in osmotic stabilizer (0.6 M KCl) was the optimal concentration for protoplast preparation (Balasubramanian and Lalithakumari 2008). For T. reesei, 5 mg/mL of Novozym 234 were used (Gruber et al. 1990) and the same concentration of Glucanex is currently used. Hundred milligrams of bovine serum albumin (Sigma, St. Louis, MO, USA) in combination with 1 g of β-d-glucanase (InterSpex Products, San Mateo, CA, USA) were used for cell wall digestion of N. crassa (Pratt and Aramayo 2002).
An alternative to improve the protoplast preparation efficiency is to make a pretreatment to modify or influence the structure of cell wall. As a result, the cell wall can be more flexible or sensitive during the treatment with enzymes. Addition of 2-mercaptoethanol was found to improve the release of protoplasts (Ezeronye and Okerentugba 2001; Peberdy 1979). Likewise, pretreatment with 2-mercaptoethanol containing 0.1 M Tris and 0.1 M EDTA improved the protoplast preparation efficiency (Zhou et al. 2008). Mercapto-compounds are assumed to accelerate cell wall degradation due to the rupture of disulphide bonds of cell wall proteins (Okerentugba 1984).
Also for S. cerevisiae transformation, the use of lithium ions to make cell walls permeable to DNA without forming protoplasts was adopted as the alternative by some groups (Das et al. 1984; Dhawale et al. 1984; Ito et al. 1983; Limura et al. 1983).
3.1.4 Stabilizing Protoplasts During and After Removal of the Cell Wall
During digestion of the cell wall, fungi need to keep the osmotic balance to avoid rupture of cells. Rigid cellular walls are necessary for fungal cells to survive in hypotonic environments. When the cell wall is removed, a hypertonic environment is necessary to keep the cell stable and avoid lysis. Therefore, all solutions used for protoplast preparation have to contain an osmotic stabilizer to prevent lysis of protoplasts. At the same time, contact with any agents that could possibly damage the cell membrane once the cell wall is removed (such as traces of soap on glassware) has to be avoided. Sorbitol, at concentrations between 0.8 and 1.2 M, has been most commonly used and seems to be satisfactory for many species including N. crassa (Case et al. 1979; Vollmer and Yanofsky 1986; Mautino et al. 1996), Aspergillus sp. (Azizi et al. 2013; De Bekker et al. 2009; Mania et al. 2010), Trichoderma sp. (Gruber et al. 1990), P. brasiliensis (De Borba et al. 1994), F. graminearum (Goswami 2012), and U. maydis (Waard 1976; Eppendorf 2002). For Aspergillus and Penicillium sp., potassium chloride at concentrations between 0.6 and 0.7 M is used as standard osmotic stabilizer (Ballance and Turner 1985; Díez et al. 1987; Picard et al. 1987). A similar solution is applied for Ozonium sp. (Zhou et al. 2008). Magnesium sulfate at 1.2 M was also used for A. niger, A. nidulans, and S. rolfsii protoplast preparation (Fariña et al. 1998; Tilburn et al. 1983). Other alternatives are 0.5 M mannitol which was applied for protoplasting of C. cinerea (Binninger et al. 1987) and sucrose for Podospora sp. (Brygoo and Debuchy 1985), L. lepideus (Kim et al. 2000), P. flocculosa (Cheng and Belanger 2000), and R. solani (Liu et al. 2010) as osmotic stabilizers. For T. virens, sorbitol is the osmotic stabilizer of choice for protoplast preparation (Catalano et al. 2011). Nevertheless, for T. harzianum and T. viride, a test for the optimal concentration of the optimal osmotic stabilizer revealed 0.6 KCl as ideal (Balasubramanian and Lalithakumari 2008), which also works for Trichothecium roseum (Balasubramanian et al. 2003) and F. pallidoroseum (Naseema et al. 2008). Likewise, KCl as osmotic stabilizer increased the regeneration efficiency for Talaromyces flavus when added to regeneration medium (Santos and De Melo 1991). A similar effect was observed for S. nodorum, C. sativus, P. teres, and C. beticola (Liu and Friesen 2012). In general, it is assumed that inorganic salts are more effective for fungi while sugar and sugar alcohols are considered more advisable with yeasts and higher plants (Lalithakumari 1996). However, as the protocols reviewed above show, protoplasting is possible with both agents in concentrations of 0.5–1 M and has to be optimized for every fungus individually. The concentration of osmotic stabilizer is critical to keep the protoplasts alive, and low concentrations of sorbitol or sucrose around 0.2 M in single layer cultures or 1 M in overlay medium can increase the fast growth of colonies, while higher concentrations could inhibit them (Ruiz-Díez 2001).
3.1.5 Incubation Time and Temperature for Lysing the Cell Wall
Efficient uptake of foreign DNA and subsequent integration into the genome is crucially dependent on complete removal of the fungal cell wall, but without destabilizing the vulnerable protoplasts. It is important to determine the temperature range in which a maximal rate of lysing cell walls is achieved with different enzymes. The enzymolysis temperature for most filamentous fungi was found to be between 24 and 35 °C (Sun et al. 2001). In addition to osmotic shock, protoplasts can also be damaged by temperature shocks, which have to be avoided. The incubation time necessary to break the cell wall varies from 2 to 3 h in most protocols for fungi, with increasing protoplast production of mycelia gradually until 3 h. Longer exposure time (also in dependence of enzyme combination) was reported to lead to rupture of protoplasts due to damage of the cell membrane (Zhou et al. 2008). In contrast, shorter exposure times to lytic enzymes resulted in higher capacity to regenerate protoplasts (Zhou et al. 2008). However, at the same time efficiency of DNA uptake and genomic integration is likely to decrease if the cell wall is not completely removed. Therefore the optimum time and temperature for a given species, cell type, and enzyme mixture used has to be found.
For Ozonium sp., 3 h at 30 °C were found to be the optimal conditions for maximum protoplast release (Zhou et al. 2008), as well as for S. nodorum, C. sativus, P. teres, and C. beticola (Liu and Friesen 2012). For F. pallidoroseum, 3 h at 30 °C with constant stirring at 30 rpm were the conditions used for protoplast preparation, while after 4 h the mycelium was completely lysed (Naseema et al. 2008). Here it is important to note that also shearing forces due to stirring, but also subsequently due to rapid pipetting can cause the newly formed protoplasts to rupture. The maximum release of protoplasts from T. harzianum and T. viride was reported after 3 h of incubation at 100 rpm, 28 °C and pH 5.5 (Balasubramanian and Lalithakumari 2008). T. reesei and T. virens protoplast preparation requires an incubation time of 2 h at 30 °C with only gentle agitation, as well as for A. niger and A. nidulans protoplasts (Azizi et al. 2013; Mania et al. 2010) and C. cinerea (Binninger et al. 1987). However, for N. crassa, 60–90 min at 30 °C was enough for protoplast release (Pratt and Aramayo 2002; Vollmer and Yanofsky 1986). For P. chrysogenum, 2 h at room temperature are sufficient for removal of the cell wall (Hamlyn et al. 1981; Flanagan et al. 1990; Sukumar et al. 2010). Digestion of the L. lepideus cell wall requires an incubation time of 6 h of the mycelium for the release of protoplasts (Kim et al. 2000). The time and temperature used for R. solani protoplast release consist on a first incubation at 37 °C for 15 min followed by 34 °C during 105 min incubation (Liu et al. 2010). In case of S. rolfsii cell wall digestion requires 1 h of incubation at 45 °C (Fariña et al. 1998). For U. maydis the exact time for digestion of cell wall is determined by checking the number of protoplast released under the microscope, using around 30 °C for incubation (Waard 1976; Eppendorf 2002), likewise for F. graminearum (Goswami 2012).
3.1.6 Evaluation and Storage of Protoplasts
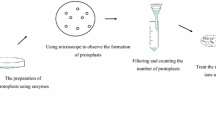
In order to achieve high transformation efficiency, protoplasts should be checked under the microscope in order to confirm their integrity. Usually this is done by adding distilled water to the microscope slide. Due to the altered osmotic pressure, protoplasts will lyse, while cells with insufficiently degraded cell wall will remain intact and prevent efficient uptake of DNA, which will decrease overall transformation efficiency. If this is the case, incubation time should be increased (Becker and Lundblad 2001).
Concentration of protoplasts can be checked directly under the microscope or in a hemocytometer. The ratio of protoplasts to DNA can strongly influence transformation efficiency. While a high number of protoplasts decreases the amount of DNA per cell and hence likelihood of integration, a low number of protoplasts and/or high DNA concentration will increase the total number of transformants obtained although their number per μg of DNA is lower (Forsburg 2003). However, increased amounts of DNA per protoplast can also result in multicopy integration.
After incubation for digestion, protoplasts are normally purified by filtration through glass wool, glass filters, or nylon filters such as Miracloth (Calbiochem), followed by gentle centrifugation to remove the supernatant and resuspension of the protoplasts in a solution containing osmotic stabilizers. The speed and time of centrifugation depend on how sensitive protoplasts are. Longer centrifugation times normally result in a higher number of protoplasts to be obtained, but at the risk of damaging them. For T. reesei 10 min at 600g is appropriate, while for F. pallidoroseum 6 min at only 100g is used (Naseema et al. 2008). In general, it is important to avoid shearing forces by using swing out rotors instead of fixed angle rotors, as the sliding along the wall of the tube might kill protoplasts. Similarly, vigorous vortexing or pipetting has to be avoided.
Once protoplasts are stabilized with sorbitol, they can be snap frozen and stored at −70 to −80 °C for many fungi. With S. cerevisiae and S. pombe this is commonly done with tolerable loss in transformation efficiency (Altherr et al. 1983; Gietz and Schiestl 2007; Jimenez 1991).
Neurospora protoplasts have been found to remain viable indefinitely at −70 °C, and so a single batch can be used for several successive transformation experiments (Pratt and Aramayo 2002; Vollmer and Yanofsky 1986). Protoplasts of Aspergillus spp. are reported to be similarly robust (De Bekker et al. 2009). However, this is not the case for all protoplast preparations. For example, for transformation of T. reesei freshly prepared protoplasts are required, because storage of protoplasts reduces drastically their efficiency (Penttilä et al. 1987).
3.1.7 Uptake of DNA/Transformation
The common protocol for transformation of protoplasts starts with mixing the purified DNA (either linear or circular double-stranded), with the protoplast suspension and a solution containing polyethylene glycol (PEG). In contrast to transformation protocols for yeast, using carrier DNA is not common during transformation of filamentous fungi. The incubation time necessary is between 15 and 30 min on ice for most of fungi in order to allow for DNA attachment to protoplasts (Ruiz-Díez 2001). Although double-stranded DNA is normally used, single-stranded DNA was successfully transformed into Saccharomyces sp. (Singh et al. 1982) and A. immersus (Goyon and Faugeron 1989).
The stabilization buffer, in which the protoplasts are suspended, is usually composed of calcium ions and an osmotic stabilizer. Calcium ions are assumed to be responsible for opening channels or pores in the cell membrane to facilitate DNA uptake (Olmedo-Monfil et al. 2004). The osmotic stabilizer is present to keep the osmotic balance, which is usually maintained by the cell wall (see above). PEG is considered responsible for clumping and fusion of protoplasts, precipitation of DNA and consequent induction of interaction between DNA and the cell surface (Bird 1996; Fincham 1989). Although a more efficient alternative to deliver DNA to the cells was proposed, the role of PEG in DNA uptake remains largely unknown (Radford et al. 1981). Nevertheless, PEG is important for efficient transformation and if poor frequencies are obtained, different lots of PEG should be tested (Becker and Lundblad 2001). Generally, lower molecular weight PEG (such as 3,350) works better than high molecular weight PEG (such as 8,000), but also here optimization for an individual species may improve results. For yeast transformations, it is recommended to remove PEG prior to plating the transformation mixture in order to increase efficiency (Becker and Lundblad 1997). This may also be beneficial for transformation of filamentous fungi.
In an alternative protocol, DNA can be encapsulated in liposomes (artificially constructed lipid vesicles), which can fuse with protoplasts. Nonetheless, this is a time-consuming procedure compared to the routine protocols using free DNA (Fincham 1989).
Additional components can be added to the transformation mixture, such as 1 % (w/v) dimethyl sulfoxide (DMSO), to stimulate electrofusion of protoplasts (Case et al. 1979; Nea and Bates 1987), or 0.05–0.1 mg of heparin per mL (Kinsey and Rambosek 1984) in successful protocols only for Neurospora. Although several functions of heparin are known, such as assisting serine protease inhibitors, it is unknown how this can influence transformation efficiency (Bird 1996). Nevertheless, there is no evidence to confirm that these additional components could improve the yields of transformants in other fungi.
The DNA concentration needed for transformation usually varies between 3 and 10 μg, with 10 μg being the ideal concentration, for example, for T. reesei, although lower amounts also may yield satisfactory results. Regarding protoplast concentration, variations according to laboratory preferences are common. Usual amounts range from 5 × 107–5 × 108 (Kubicek and Harman 1998) to 1 × 108–1 × 109 mL−1 (Gruber et al. 1990).
Also the final concentration of PEG is critical for the efficiency of DNA uptake. Therefore, PEG added should be adjusted to final volume of the solution including protoplasts and DNA (Gietz and Schiestl 2007). The concentration of calcium chloride can vary from 10 mM, as most commonly used for S. cerevisiae, Aspergillus sp., and other filamentous fungi, to 50 mM for Neurospora species (Fincham 1989). PEG solution can contain a final concentration of 25 % (w/v) PEG 6000 (Gruber et al. 1990), although up to 10 volumes of 40 % (w/v) PEG 4000 can be used for majority of fungal species (Fincham 1989). Nevertheless, a protocol for S. commune specifies only a little over 1 volume of 44 % (w/v) PEG 4000 (Munoz-Rivas et al. 1986). pH seems to play a role to keep protoplasts stable and a range of 6–8 is used for different species of fungi (Ruiz-Díez 2001). Some protocols include the addition of lipofectin, a liposome preparation formulated from cationic lipids (Bethesda Research Laboratories, Gaithersburg, MD), increasing the transformation efficiency, as reported for N. crassa (Selitrennikof and Sachs 1991).
After 15–30 min of incubation with exogenous DNA, protoplasts, and PEG, some protocols include the addition of 1–2 mL of PEG, followed by an incubation time of 5–30 min (depending on the species) at room temperature (Gruber et al. 1990). However, normally protoplasts are quite stable and rounds for washing can be increased for convenience. Finally, stabilization buffer is added to the solution and in case of using an agar-overlay, mixed with the overlay medium or alternatively spread out on selective medium (Gruber et al. 1990). Interestingly, also the brand of agar used for plate preparation can influence transformation efficiency (likely due to impurities) and should be considered when optimizing protocols.
3.1.8 Incubation Time and Medium for Protoplast Regeneration
Regeneration of protoplasts can be improved depending on the media prior to application to selective medium. Therefore, protoplasts are usually transferred to regeneration media without selective pressure. However, in many cases, transformed protoplasts are transferred directly to media complementing an auxotrophic marker or containing a drug depending on the selectable marker gene used on the introduced DNA construct, without prior cultivation on media lacking the selection reagent. In these cases, recovery of the fungus from the protoplast state is often called regeneration. The media used for regeneration have to be osmotically buffered to allow the recovery of protoplasts.
N. crassa transformed protoplasts can be regenerated overnight at 25 °C in regeneration solution (Vogel’s salts + Sucrose + MgSO4 + any necessary metabolic supplements) (Pratt and Aramayo 2002). Regeneration of A. niger protoplasts is done on a nonselective medium (minimal medium pH 6.0, sucrose and agar) (De Bekker et al. 2009). T. reesei protoplasts after transformation were recovered on malt extract medium containing sorbitol, hygromycin B, and uridine (Guangtao et al. 2009). Later on, once protoplasts are regenerated, only those ones which harbor the transforming DNA can grow in the medium including a selection reagent. The maintenance of osmotic balance is critical for recovering protoplasts. Additionally, for protoplast recovery in Ozonium sp., 0.1 % (w/v) Triton X-100 and 0.04 ‰ (w/v) sodium deoxycholate were used as colony restrictors. The use of these detergents or others is important to avoid spore clumping, in order to isolate single transformant colonies (Jensen et al. 2013). However, such colony restrictors are most often only added for single spore isolation steps after initial transformant isolation. Thereby decreased recovery of protoplasts due to a destabilizing effects of, for example, the detergent Triton X-100 can be avoided.
4 Concluding Remarks
Protoplasting is well established as a method for fungal transformation nowadays. Therefore, to develop a transformation system for a fungus, protoplasting is a good choice. Nevertheless, for many species there is considerable room for improvement with efficiency of protoplast transformation and we provided some strategies to optimize existing protocols. Developing and optimizing protoplast transformation with fungi still remains of process of trial and error to some extent and due to the varying cell wall composition and maybe also some defense mechanisms, protocols cannot be generalized.
References
Adams DJ (2004) Fungal cell wall chitinases and glucanases. Microbiology 150:2029–2035
Akins RA, Lambowitz AM (1985) General method for cloning Neurospora crassa nuclear genes by complementation of mutants. Mol Cell Biol 5:2272–2278
Altherr MR, Quinn LA, Kado CI, Rodroguez RL (1983) Transformation and storage of competent yeast cells. In: Lurquin P, Kleinhofs A (eds) Genetic engineering in eukaryotes, vol 61, 4th edn. Springer, New York, pp 33–36
Ariko Y, Ito E (1975) A pathway of chitosan formation in Mucor rouxii. Enzymatic deacetylation of chitin. Eur J Biochem 55:71–78
Auriol P (1974) On the polyosides of the cell walls of conidia and mycelia of Colletotrichum lagenarium Pass Ell and Halst. C R Seances Acad Sci 279(25):1867–1869
Azizi M, Yakhchali B, Ghamarian A, Enayati S, Khodabandeh M, Khalaj V (2013) Cloning and expression of gumboro VP2 antigen in Aspergillus niger. Avicenna J Med Biotechnol 5:35–41
Bachmann BJ, Bonner DM (1959) Protoplasts from Neurospora crassa. J Bacteriol 78:550–556
Baker LG, Specht CA, Donlin MJ, Lodge JK (2007) Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot Cell 6:855–867
Balasubramanian N, Lalithakumari D (2008) Characteristics of protoplast inter, intra-fusant and regeneration of antagonistic fungi Trichoderma harzianum and Trichoderma viride. Afr J Biotechnol 7:18
Balasubramanian N, Juliet GA, Srikalaivani P, Lalithakumari D (2003) Release and regeneration of protoplasts from the fungus Trichothecium roseum. Can J Microbiol 49:263–268
Ballance DJ, Turner G (1985) Development of a high-frequency transforming vector for Aspergillus nidulans. Gene 36:321–331
Ballou CE, Raschke WC (1974) Polymorphism of the somatic antigen of yeast. Science 184:127–134
Barras DR (1972) A b-glucan endohydrolase from Schizosaccharomyces pombe and its role in cell wall growth. Antonie van Leeuwenhoek 38:65–80
Bartnicki-Garcia S (1968) Cell wall chemistry, morphogenesis, and taxonomy of fungi. Annu Rev Microbiol 22:87–108
Bartnicki-Garcia S (1970) Cell wall composition and other biochemical markers in fungal phylogeny. Academic, New York
Beauvais A, Latgé J-P (2001) Membrane and cell wall targets in Aspergillus fumigatus. Drug Resist Updat 4:38–49
Beauvais A, Maubon D, Park S, Morelle W, Tanguy M, Huerre M, Perlin DS, Latge JP (2005) Two alpha(1-3) glucan synthases with different functions in Aspergillus fumigatus. Appl Environ Microbiol 71:1531–1538
Becker DM, Lundblad V (1997) Introduction of DNA into yeast cells. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (eds) Current protocols, vol 2. Wiley, New York
Becker DM, Lundblad V (2001) Introduction of DNA into yeast cells. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA et al (eds) Current protocols in molecular biology. Wiley, New York
Benitez T, Villa TG, Garcia Acha I (1976) Some chemical and structural features of the conidial wall of Trichoderma viride. Can J Microbiol 22:318–321
Binninger DM, Skrzynia C, Pukkila PJ, Casselton LA (1987) DNA-mediated transformation of the basidiomycete Coprinus cinereus. EMBO J 6:835–840
Bird DM (1996) Transformation and gene targeting in Aspergillus nidulans. Microbiology and genetics. Massey University, Palmerston North
Borkovich K, Ebbole DJ (eds) (2010) Cellular and molecular biology of filamentous fungi. APS, Washington
Braun PC, Calderone RA (1978) Chitin synthesis in Candida albicans. Comparison of yeast and hyphal forms. J Bacteriol 133:1472–1477
Brown JP (1971) Susceptibility of the cell walls of some yeasts to lysis by enzymes of Helix pomatia. Can J Microbiol 17:205–208
Brygoo Y, Debuchy R (1985) Transformation by integration in Podospora anserina. 1. Methodology and phenomenology. Mol Gen Genet 200:128–131
Burton RA, Fincher GB (2009) (1,3;1,4)-β-D-glucans in cell walls of the poaceae, lower plants, and fungi: a tale of two linkages. Mol Plant 2:873–882
Buxton FP, Radford A (1984) The transformation of mycelial spheroplasts of Neurospora crassa and the attempted isolation of an autonomous replicator. Mol Gen Genet 196:337–344
Cabib E, Roh D-H, Schmidt M, Crotti LB, Varma A (2001) The yeast cell wall and septum as paradigms of cell growth and morphogenesis. J Biol Chem 276:19679–19682
Calderone RA, Braun PC (1991) Adherence and receptor relationships of Candida albicans. Microbiol Rev 55:1–20
Cardoza RE, Vizcaino JA, Hermosa MR, Monte E, Gutierrez S (2006) A comparison of the phenotypic and genetic stability of recombinant Trichoderma spp. generated by protoplast- and Agrobacterium-mediated transformation. J Microbiol 44:383–395
Case ME, Schweizer M, Kushner SR, Giles NH (1979) Efficient transformation of Neurospora crassa by utilizing hybrid plasmid DNA. Proc Natl Acad Sci U S A 76:5259–5263
Catalano V, Vergara M, Hauzenberger JR, Seiboth B, Sarrocco S, Vannacci G, Kubicek CP, Seidl-Seiboth V (2011) Use of a non-homologous end-joining-deficient strain (delta-ku70) of the biocontrol fungus Trichoderma virens to investigate the function of the laccase gene lcc1 in sclerotia degradation. Curr Genet 57:13–23
Cheng Y, Belanger RR (2000) Protoplast preparation and regeneration from spores of the biocontrol fungus Pseudozyma flocculosa. FEMS Microbiol Lett 190:287–291
Collinge AJ, Trinci APJ (1974) Hyphal tips of wild-type and spreading colonial mutants of Neurospora crassa. Arch Microbiol 99:353–368
Collopy PD, Colot HV, Park G, Ringelberg C, Crew CM, Borkovich KA, Dunlap JC (2010) High-throughput construction of gene deletion cassettes for generation of Neurospora crassa knockout strains. Methods Mol Biol 638:33–40
Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, Weiss RL, Borkovich KA, Dunlap JC (2006) A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A 103:10352–10357
Cutler JE (2001) N-glycosylation of yeast, with emphasis on Candida albicans. Med Mycol 1:75–86
Das S, Kellermann E, Hollenberg CP (1984) Transformation of Kluyveromyces fragilis. J Bacteriol 158:1165–1167
Datema R, Van den Ende H, Wessels JH (1977a) The hyphal wall of Mucor mucedo 1. Polyanionic polymers. Eur J Biochem 80:611–619
Datema R, Wessels JH, van den Ende H (1977b) The hyphal wall of Mucor mucedo 2. Hexosamine containing polymers. Eur J Biochem 80:621–626
Davis LL, Bartnicki-Garcia S (1984) Chitosan synthesis by the tandem action of chitin synthetase and chitin deacetylase from Mucor rouxii. Biochem J 23:1065–1068
De Bekker C, Wiebenga A, Aguilar G, Wosten HA (2009) An enzyme cocktail for efficient protoplast formation in Aspergillus niger. J Microbiol Methods 76:305–306
De Borba CM, Meirelles MN, Da Silva AM, De Oliveira PC (1994) Paracoccidioides brasiliensis protoplast production by enzymatic treatment. Mycoses 37:317–323
de Groot MJ, Bundock P, Hooykaas PJ, Beijersbergen AG (1998) Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat Biotechnol 16:839–842
de Groot PW, Yin QY, Weig M, Sosinska GJ, Klis FM, de Koster CG (2007) Mass spectrometric identification of covalently bound cell wall proteins from the fission yeast Schizosaccharomyces pombe. Yeast 24:267–278
Demain AL, Newkirk JF (1962) Biosynthesis of cephalosporin C. Appl Microbiol 10:321–325
Demain AL, Newkirk JF, Hendlin D (1963) Effect of methionine, norleucine and lysine derivatives on cephalosporin C formation in chemically defined media. J Bacteriol 85:339–344
Deutch CE, Parry JM (1974) Sphaeroplast formation in yeast during the transition from exponential phase to stationary phase. J Gen Microbiol 80:259–268
Dhawale S, Paietta J, Marzluf G (1984) A new, rapid and efficient transformation procedure for Neurospora. Curr Genet 8:77–79
Díez B, Alvarez E, Cantoral JM, Barredo JL, Martín JF (1987) Selection and characterization of pyrG mutants of Penicillium chrysogenum lacking orotidine-5′-phosphate decarboxylase and complementation by the pyr4 gene of Neurospora crassa. Curr Genet 12:277–282
Dziengel A, Held W, Schlanderer G, Dellweg H (1977) Eur J Appl Microbiol 4:21–27
Eddy AA, Williamson DH (1959) Formation of aberrant cell walls and of spores by the growing yeast protoplast. Nature 183:1101–1104
Ellis DH, Griffiths DA (1974) The location and analysis of melanins in the cell walls of some soil fungi. Can J Microbiol 20:1379–1386
Eppendorf (2002) Protocol Ustilago maydis No. 4308 91. Eppendorf AG, Hamburg
Ezeronye OU, Okerentugba PO (2001) Optimum conditions for yeast protoplast release and regeneration in Saccharomyces cerevisiae and Candida tropicalis using gut enzymes of the giant African snail Achatina achatina. Lett Appl Microbiol 32:190–193
Fariña JI, Siñeriz F, Molina OE, Perotti NI (1998) High scleroglucan production by Sclerotium rolfsii: influence of medium composition. Biotechnol Lett 20:825–831
Farkas V (1979) Biosynthesis of cell walls of fungi. Microbiol Rev 43:117–144
Fawcett PA, Loder PB, Duncan MJ, Beesley TJ, Abraham EP (1973) Formation and properties of protoplasts from antibiotic-producing strains of Penicillium chrysogenum and Cephalosporium acremonium. J Gen Microbiol 79:293–309
Fevre M (1977) Subcellular localization of glucanase and cellulase in Saprolegnia monoica Pringsheim. J Gen Microbiol 103:287–295
Fincham JR (1989) Transformation in fungi. Microbiol Rev 53:148–170
Flanagan WP, Klei HE, Sundstrom DW, Lawton CW (1990) Optimization of a pellicular biocatalyst for penicillin G production by Penicillium chrysogenum. Biotechnol Bioeng 36:608–616
Fleet GH (1991) Cell walls. In: Rose AH, Harrisson JD (eds) The yeast, vol 4. Academic, New York, pp 199–277
Fleet GH, Phaff HJ (1974) Glucanases in Schizosaccharomyces. Isolation and properties of the cell wall associated b-(1-3)-glucanases. J Biol Chem 249:1717–1928
Fontaine T, Simenel C, Dubreucq G, Adam O, Delepierre M, Lemoine J, Vorgias CE, Diaquin M, Latge JP (2000) Molecular organization of the alkali-insoluble fraction of Aspergillus fumigatus cell wall. J Biol Chem 275:27594–27607
Forsburg SL (2003) Introduction of DNA into S. pombe Cells. In Current protocols in molecular biology. Wiley. http://dx.doi.org/10.1002/0471142727.mb1317s64
Foury F, Golfeau A (1973) J Gen Microbial 75:227–229
Gallmetzer M, Burgstaller W, Schinner F (1999) An optimized method for the isolation of protoplasts from Penicillium simplicissimum to produce sealed plasma membrane vesicles. Mycologia 91:206–212
Gander JE (1974) Fungal cell wall glycoproteins and peptido-polysaccharides. Annu Rev Microbiol 28:103–119
Gietz RD, Schiestl RH (2007) Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:35–37
Gooday GW (1977) Biosynthesis of the fungal cell wall-mechanism and implications. J Gen Microbiol 99:1–11
Gooday GW (1995) Cell walls (3). In: Gow NR, Gadd G (eds) The growing fungus. Springer, Netherlands, pp 43–62
Goswami RS (2012) Targeted gene replacement in fungi using a split-marker approach. In: Bolton MD, Thomma BPHJ (eds) Plant fungal pathogens, vol 835, 16th edn. Humana, New York, pp 255–269
Goyon C, Faugeron G (1989) Targeted transformation of Ascobolus immersus and de novo methylation of the resulting duplicated DNA sequences. Mol Cell Biol 9:2818–2827
Grallert B, Nurse P, Patterson TE (1993) A study of integrative transformation in Schizosaccharomyces pombe. Mol Gen Genet 238:26–32
Gruber S, Seidl-Seiboth V (2012) Self versus non-self: fungal cell wall degradation in Trichoderma. Microbiology 158:26–34
Gruber F, Visser J, Kubicek CP, Graaff LH (1990) The development of a heterologous transformation system for the cellulolytic fungus Trichoderma reesei based on a pyrG-negative mutant strain. Curr Genet 18:71–76
Grün CH, Hochstenbach F, Humbel BM, Verkleij AJ, Sietsma JH, Klis FM, Kamerling JP, Vliegenthart JF (2005) The structure of cell wall alpha-glucan from fission yeast. Glycobiology 15:245–257
Guangtao Z, Hartl L, Schuster A, Polak S, Schmoll M, Wang T, Seidl V, Seiboth B (2009) Gene targeting in a nonhomologous end joining deficient Hypocrea jecorina. J Biotechnol 139:146–151
Hamlyn PF, Bradshaw RE, Mellon FM, Santiago CM, Wilson JM, Peberdy JF (1981) Efficient protoplast isolation from fungi using commercial enzymes. Enzyme Microb Technol 3:321–325
Harrier LA, Millam S (2001) Biolistic transformation of arbuscular mycorrhizal fungi. Mol Biotechnol 18:25–33
Hazen KC (1990) Cell surface hydrophobicity of medically important fungi, especially Candida species. In: Doyle RJ, Rosenberg M (eds) Microbial cell surface hydrophobicity. American Society for Microbiology, Washington, pp 249–295
Hinnen A, Hicks JB, Fink GR (1978) Transformation of yeast chimaeric ColEl plasmid carrying LEU2. Proc Natl Acad Sci U S A 75:1929–1933
Hochstenbach F, Klis FM, Van den Ende H, Van Donselaar E, Peters PJ, Klausner RD (1998) Identification of a putative alpha-glucan synthase essential for cell wall construction and morphogenesis in fission yeast. Proc Natl Acad Sci U S A 95:9161–9166
Hunsley D (1973) Apical wall structure in hyphae of Phytophthora parasitica. New Phytol 72:985–990
Hunsley D, Gooday GW (1974) The structure and development of septa in Neurospora crassa. Protoplasma 82:125–146
Hunsley D, Kay D (1976) Wall structure of Neurospora hyphal apex-immunofluorescent localization of wall surface antigens. J Gen Microbiol 95:233–248
Hutchison HT, Hartwell LH (1967) Macromolecule synthesis in yeast spheroplasts. J Bacteriol 94:1697–1705
Ito H, Fukuda Y, Murata K, Kimura A (1983) Transformation of intact yeast cells treated with alkali cations. J Bacteriol 153:163–168
Jan YN (1974) Properties and cellular localization of chitin synthetase in Phycomyces blakesleanus. J Biol Chem 249:1973–1979
Jensen AB, Aronstein K, Flores JM, Vojvodic S, Palacio MA, Spivak M (2013) Standard methods for fungal brood disease research. J Apic Res 52:13
Jimenez J (1991) Cryopreservation of competent Schizosaccharomyces pombe protoplasts. Trends Genet 7:40
Kapteyn JC, Hoyer LL, Hecht JE, Muller WH, Andel A, Verkleij AJ, Makarow M, Van Den Ende H, Klis FM (2000) The cell wall architecture of Candida albicans wild-type cells and cell wall-defective mutants. Mol Microbiol 35:601–611
Kelly FD, Nurse P (2011) De Novo growth zone formation from fission yeast spheroplasts. PLoS One 6:e27977
Kim BK, Kang JH, Jin M, Kim HW, Shim MJ, Choi EC (2000) Mycelial protoplast isolation and regeneration of Lentinus lepideus. Life Sci 66:1359–1367
Kinsey JA, Rambosek JA (1984) Transformation of Neurospora crassa with the cloned am (glutamate dehydrogenase) gene. Mol Cell Biol 4:117–122
Klis FM, Mol P, Hellingwerf K, Brul S (2002) Dynamics of cell wall structure in Saccharomyces cerevisiae. FEMS Microbiol Rev 26:239–256
Klis FM, Boorsma A, De Groot PWJ (2006) Cell wall construction in Saccharomyces cerevisiae. Yeast 23:185–202
Kollar R, Petrakova E, Ashwell G, Robbins PW, Cabib E (1995) Architecture of the yeast cell wall. The linkage between chitin and b(1,3)-glucan. J Biol Chem 270:1170–1178
Kollar R, Reinhold BB, Petrakova E, Yeh HJ, Ashwell G, Drgonova J, Kapteyn JC, Klis FM, Cabib E (1997) Architecture of the yeast cell wall. b(1-6)-glucan interconnects mannoprotein, b(1-)3-glucan, and chitin. J Biol Chem 272:17762–17775
Kubicek CP, Harman GE (1998) Trichoderma and gliocadium, basic biology, taxonomy and genetics, vol 1. CRC, London
Kusters-van Someren MA, Harmsen JA, Kester HC, Visser J (1991) Structure of the Aspergillus niger pelA gene and its expression in Aspergillus niger and Aspergillus nidulans. Curr Genet 20:293–299
Lalithakumari D (1996) Protoplasts—a biotechnological tool for plant pathological studies. Ind Phytopathol 49:199–212
Lampen J (1968) External enzymes of yeast: their nature and formation. Antonie van Leeuwenhoek 34:1–18
Latgé J-P (2007) The cell wall: a carbohydrate armour for the fungal cell. Mol Microbiol 66:279–290
Lenardon MD, Munro CA, Gow NAR (2010) Chitin synthesis and fungal pathogenesis. Curr Opin Microbiol 13:416–423
Lesage G, Bussey H (2006) Cell wall assembly in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 70:317–343
Limura Y, Gotoh K, Ouchi K, Nishima T (1983) Transformation of yeast without the spheroplasting process. Agric Biol Chem 47:897–901
Liu Z, Friesen TL (2012) Polyethylene glycol (PEG)-mediated transformation in filamentous fungal pathogens. Methods Mol Biol 835:365–375
Liu TH, Lin MJ, Ko WH (2010) Factors affecting protoplast formation by Rhizoctonia solani. N Biotechnol 27:64–69
Lowman DW, Ferguson DA, Williams DL (2003) Structural characterization of (1-3)-β-d-glucans isolated from blastospore and hyphal forms of Candida albicans. Carbohydr Res 338:1491–1496
Macdonald KD, Hutchinson JM, Gillett WA (1963) Isolation of auxotrophs of Penicillium chrysogenum and their penicillin yields. J Gen Microbiol 33:365–374
Malonek S, Meinhardt F (2001) Agrobacterium tumefaciens-mediated genetic transformation of the phytopathogenic ascomycete Calonectria morganii. Curr Genet 40:152–155
Mania D, Hilpert K, Ruden S, Fischer R, Takeshita N (2010) Screening for antifungal peptides and their modes of action in Aspergillus nidulans. Appl Environ Microbiol 76:7102–7108
Martin JF, Nicolas G, Villanueva JR (1973) Chemical changes in the cell walls of conidia of Penicillium notatum during germination. Can J Microbiol 19:789–796
Mautino MR, Barra JL, Rosa AL (1996) eth-1, the Neurospora crassa locus encoding S-adenosylmethionine synthetase: molecular cloning, sequence analysis and in vivo overexpression. Genetics 142:789–800
Meyer V (2008) Genetic engineering of filamentous fungi: progress, obstacles and future trends. Biotechnol Adv 26:177–185
Meyer V, Mueller D, Strowig T, Stahl U (2003) Comparison of different transformation methods for Aspergillus giganteus. Curr Genet 43:371–377
Michielse CB, Hooykaas PJ, Van den Hondel CA, Ram AF (2005) Agrobacterium-mediated transformation as a tool for functional genomics in fungi. Curr Genet 48:1–17
Mol PC, Wessels JGH (1990) Differences in wall structure between substrate hyphae and hyphae of fruit-body stipes in Agaricus bisporus. Mycol Res 94:472–479
Molano J, Bowers B, Cabib E (1980) Distribution of chitin in the yeast cell wall. An ultrastructural and chemical study. Cell BioI 261:15147–15152
Mullins ED, Kang S (2001) Transformation: a tool for studying fungal pathogens of plants. Cell Mol Life Sci 58:2043–2052
Munoz-Rivas A, Specht CA, Drummond BJ, Froeliger E, Novotny CP, Ullrich RC (1986) Transformation of the basidiomycete Schizophyllum commune. Mol Gen Genet 205:103–106
Naseema A, Dhanya B, AnjanadeviI P, Sheena KG, Alex S (2008) Isolation and regeneration of protoplasts from the mycelium of Fusarium pallidoroseum—a potential biocontrol agent of water hyacinth [Eichhornia crassipes (Mart.) Solms]. J Trop Agric 46(1–2):67–69
Nea LJ, Bates GW (1987) Factors affecting protoplast electrofusion efficiency. Plant Cell Rep 6:337–340
Necas O (1971) Cell wall synthesis in yeast protoplasts. Bacteriol Rev 35:149–170
Nimrichter L, Rodrigues ML, Rudrigues EG, Travassos LR (2005) The multitude targets for the inmune system and drug therapy in the fungal cell wall. Microbes Infect 7:789–798
Okerentugba PO (1984) Production and genetic analysis of hybrid strains of Saccharomyces cerevisiae by spheroplast fusion. PhD thesis, University of Strathclyde, Glasgow
Olmedo-Monfil V, Cortés-Penagos C, Herrera-Estrella A (2004) Three decades of fungal transformation. In: Balbás P, Lorence A (eds) Recombinant gene expression, vol 267, 21st edn. Humana, New York, pp 297–313
Park G, Servin JA, Turner GE, Altamirano L, Colot HV, Collopy P, Litvinkova L, Li L, Jones CA, Diala F-G, Dunlap JC, Borkovich KA (2011) Global analysis of serine-threonine protein kinase genes in Neurospora crassa. Eukaryot Cell 10:1553–1564
Partridge BM, Drew JA (1974) Candida protoplasts and their ultrastructure. Sabouraudia 12:166–178
Peberdy JF (1979) Fungal protoplasts: isolation, reversion, and fusion. Annu Rev Microbiol 33:21–39
Penttilä M, Nevalainen H, Rättö M, Salminen E, Knowles J (1987) A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei. Gene 61:155–164
Perez P, Ribas JC (2004) Cell wall analysis. Methods 33:245–251
Peterson EM, Hawley RJ, Calderone RA (1976) An ultrastructural analysis of protoplast-spheroplast induction in Cryptococcus neoformans. Can J Microbiol 22:1518–1521
Pettolino F, Sasaki I, Turbic A, Wilson SM, Bacic A, Hrmova M, Fincher GB (2009) Hyphal cell walls from the plant pathogen Rhynchosporium secalis contain β(1,3/1,6)-D-glucans, galacto- and rhamnomannans, β(1,3/1,4)-D-glucans and chitin. FEBS J 276:3698–3709
Phaff HJ (1971) Structure and biosynthesis of the yeast cell envelope, vol 2. Academic, New York
Picard M, Debuchy R, Julien J, Brygoo Y (1987) Transformation by integration in Podospora anserina. Mol Gen Genet 210:129–134
Polacheck Y, Rosenberger RF (1975) Autolytic enzymes in hyphae of Aspergillus nidulans: their action on old and newly formed walls. J Bacteriol 121:332–337
Pontecorvo G, Roper JA, Hemmons LM, MacDonald KD, Bufton AWJ (1953) The genetics of Aspergillus nidulans. Adv Genet 5:141–238
Pratt RJ, Aramayo R (2002) Improving the efficiency of gene replacements in Neurospora crassa: a first step towards a large-scale functional genomics project. Fungal Genet Biol 37:56–71
Radford A, Pope S, Sazci A, Fraser MJ, Parish JH (1981) Liposome-mediated genetic transformation of Neurospora crassa. Mol Gen Genet 184:567–569
Roncero C (2002) The genetic complexity of chitin synthesis in fungi. Curr Genet 41:367–378
Ruiz-Díez B (2001) Strategies for the transformation of filamentous fungi. J Appl Microbiol 92:189–195
Ruiz-Herrera J, Leon CG, Carabez-Trejo A, Reyes-Salinas E (1996) Structure and chemical composition of the cell walls from the haploid yeast and mycelial forms of Ustilago maydis. Fungal Genet Biol 20:133–142
Santos TMC, De Melo IS (1991) Preparation and regeneration of protoplasts of Talaromyces flavus. Rev Bras Genet 14:335–340
Savitha S, Sadhasivam S, Swaminathan K (2010) Regeneration and molecular characterization of an intergeneric hybrid between Graphium putredinis and Trichoderma harzianum by protoplasmic fusion. Biotech Adv 28:285–292
Schuster A, Bruno KS, Collett JR, Baker SE, Seiboth B, Kubicek CP, Schmoll M (2012) A versatile toolkit for high throughput functional genomics with Trichoderma reesei. Biotechnol Biofuels 5:1754–6834
Schweizer M, Case ME, Dykstra CC, Giles NH, Kushner SR (1981) Identification and characterization of recombinant plasmids carrying the complete qa gene cluster from Neurospora crassa including the qa-1þ regulatory gene. Proc Natl Acad Sci U S A 78:5086–5090
Seiler S, Plamann M (2003) The genetic basis of cellular morphogenesis in the filamentous fungus Neurospora crassa. Mol Biol Cell 14:4352–4364
Selitrennikof CP, Sachs MS (1991) Lipofectin increases the efficiency of DNA-mediated transformation of Neurospora crassa. Fungal Genet Newsl 38:90–91
Selitrennikoff CP, Nakata M (2003) New cell wall targets for antifungal drugs. Curr Opin Investig Drugs 4:200–205
Shahin MM (1972) Relationship between yield of protoplasts and growth phase in Saccharomyces. J Bacteriol 110:769–771
Shahinian S, Bussey H (2000) b(1,6)-Glucan synthesis in Saccharomyces cerevisiae. Mol Microbiol 35:477–489
Sheu YJ, Barral Y, Snyder M (2000) Polarized growth controls cell shape and bipolar bud site selection in Saccharomyces cerevisiae. Mol Cell Biol 20:5235–5247
Shibata N, Suzuki A, Kobayashi H, Okawa Y (2007) Chemical structure of the cell-wall mannan of Candida albicans serotype A and its difference in yeast and hyphal forms. Biochem J 404:365–372
Sietsma JH, Wessels JGH (1990) The occurrence of glucosaminoglycan in the wall of Schizosaccharomyces pombe. Gen Microbiol 136:2261–2265
Singh H, Bieker JJ, Dumas LB (1982) Genetic transformation of Saccharomyces cerevisiae with single-stranded circular DNA vectors. Gene 20:441–449
Steiger MG (2013) Molecular tools in Trichoderma genetic studies. In: Mukherjee PK, Horwitz BA, Singh US, Mukherjee M, Schmoll M (eds) Trichoderma: biology and applications. CABI, Walllingford
Sugawara T, Takahashi S, Osumi M, Ohno N (2004) Refinement of the structures of cell-wall glucans of Schizosaccharomyces pombe by chemical modification and NMR spectroscopy. Carbohydr Res 339:2255–2265
Sukumar M, Sundar M, Sivarajan M (2010) Penicillin production from transformed protoplast of Penicillium chrysogenum by fermentation. Ferment J Pharmacogenom Pharmacoproteomics 1:102
Sun S, Furtula V, Nothnagel EA (1992) Mechanical release and lectin labeling of maize root protoplasts. Protoplasma 169:49–56
Sun JQ, Yu HY, Zhang P, Zhang YF, Ling HB, Xie YH, Zhou DP, Ping WX (2001) The formation and regeneration of Nodulisporium sylviforme protoplast. Chin J Appl Environ Biol 7:375–381
Tilburn J, Scazzocchio C, Taylor GG, Zabicky-Zissman JH, Lockington RA, Davies RW (1983) Transformation by integration in Aspergillus nidulans. Gene 26:205–221
Tomo N, Goto T, Morikawa Y (2013) Trans-packaging of human immunodeficiency virus type 1 genome into Gag virus-like particles in Saccharomyces cerevisiae. Microb Cell Fact 12:28
Trinci APJ (1978) Wall and hyphal growth. Sci Prog London 65:75–99
Trinci APJ, Coolinge AJ (1975) Hyphal wall growth in Neurospora crassa and Geotrichium candidum. J Gen Microbiol 91:355–361
Vaishnav VV, Bacon BE, O’Neill M, Cherniak R (1998) Structural characterization of the galactoxylomannan of Cryptococcus neoformans Cap67. Carbohydr Res 306:315–330
Villanueva JR (1966) Protoplasts of fungi. In: Ainsworth GC, Sussman AE (eds) The fungi, vol 2. Academic, New York, pp 3–62
Vollmer SJ, Yanofsky C (1986) Efficient cloning of genes of Neurospora crassa. Proc Natl Acad Sci U S A 83:4869–4873
Von Klercker J (1982) Eine Methode zur isolier lebender protoplasten. Ofvers Vetensk Akad Forhandl 49:463–474
Waard MA (1976) Formation of protoplasts from Ustilago maydis. Antonie Van Leeuwenhoek 42:211–216
Wessels JGH (1994) Developmental regulation of fungal cell wall formation. Annu Rev Phytopathol 32:413–437
Whittaker PA, Andrews KR (1969) Protoplast production in haploid Saccharomyces cerevisiae and petite mutants of this yeast. Microbios 1(1B):99–104
Whittaker SL, Lunness P, Milward KJ, Doonan JH, Assinder SJ (1999) sodVIC is an alpha-COP-related gene which is essential for establishing and maintaining polarized growth in Aspergillus nidulans. Fungal Genet Biol 26:236–252
Zhou X, Wei Y, Zhu H, Wang Z, Lin J, Liu L, Tang K (2008) Protoplast formation, regeneration and transformation form the taxol-producing fungus Ozonium sp. Afr J Biotechnol 7:2017–2024
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Rodriguez-Iglesias, A., Schmoll, M. (2015). Protoplast Transformation for Genome Manipulation in Fungi. In: van den Berg, M., Maruthachalam, K. (eds) Genetic Transformation Systems in Fungi, Volume 1. Fungal Biology. Springer, Cham. https://doi.org/10.1007/978-3-319-10142-2_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-10142-2_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-10141-5
Online ISBN: 978-3-319-10142-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)