
Abstract
The Notch pathway has been described as an oncogene in osteosarcoma, but the myriad functions of all the members of this complex signaling pathway, both in malignant cells and nonmalignant components of tumors, make it more difficult to define Notch as simply an oncogene or a tumor suppressor. The cell-autonomous behaviors caused by Notch pathway manipulation may vary between cell lines but can include changes in proliferation, migration, invasiveness, oxidative stress resistance, and expression of markers associated with stemness or tumor-initiating cells. Beyond these roles, Notch signaling also plays a vital role in regulating tumor angiogenesis and vasculogenesis, which are vital aspects of osteosarcoma growth and behavior in vivo. Further, osteosarcoma cells themselves express relatively low levels of Notch ligand, making it likely that nonmalignant cells, especially endothelial cells and pericytes, are the major source of Notch activation in osteosarcoma tumors in vivo and in patients. As a result, Notch pathway expression is not expected to be uniform across a tumor but likely to be highest in those areas immediately adjacent to blood vessels. Therapeutic targeting of the Notch pathway is likewise expected to be complicated. Most pharmacologic approaches thus far have focused on inhibition of gamma secretase, a protease of the presenilin complex. This enzyme, however, has numerous other target proteins that would be expected to affect osteosarcoma behavior, including CD44, the WNT/β-catenin pathway, and Her-4. In addition, Notch plays a vital role in tissue and organ homeostasis in numerous systems, and toxicities, especially GI intolerance, have limited the effectiveness of gamma secretase inhibitors. New approaches are in development, and the downstream targets of Notch pathway signaling also may turn out to be good targets for therapy. In summary, a full understanding of the complex functions of Notch in osteosarcoma is only now unfolding, and this deeper knowledge will help position the field to better utilize novel therapies as they are developed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction: The Notch Signaling Pathway
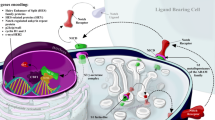
The Notch signaling pathway, a key component in normal bone development that is implicated as a key mediator in a number of various cancers, is initiated when a membrane-bound ligand belonging to the Delta–Serrate–Lag (DSL) family (jagged 1/Jag1, Jag2, delta-like-1/DLL1, DLL3, and DLL4) on the surface of a cell interacts with a membrane-bound Notch receptor (Notch1–4) on another cell. This interaction induces a two-step proteolytic cleavage of the receptor, first by ADAM10 (also known as Kuz) or ADAM17 (also known as TACE) and then by the γ-secretase complex which is made up of at least four individual proteins: presenilin, nicastrin, anterior pharynx-defective 1 (APH-1), and presenilin enhancer 2 (PEN-2). These cleavage events release the intracellular domain of Notch (ICN). Now activated, ICN enters the nucleus where it forms a transcriptional complex with Mastermind-like 1 (MAM1) to regulate transcriptional complexes containing the DNA-binding protein CBF1/RBPjk/Su(H)/Lag1 (CSL). This complex initiates the transcription of Hairy/Enhancer of Split-1 (Hes1), Hes5, Hes7, HES-related with YRPW motif (Hey1/HERP2), Hey2 (HERP1), and HeyL which encode basic helix loop helix (bHLH) transcription factors that perform a range of cellular activities that include promoting progenitor cell survival and suppressing differentiation [1, 2]. This pathway is displayed schematically in Fig. 1. The Notch signaling pathway, via cell–cell contacts, highly regulated feedback loops, and lateral inhibition/induction mechanisms, has been shown to influence multiple cellular processes including cell fate decisions, proliferation, apoptosis, migration, angiogenesis, and plasticity. In terms of bone homeostasis, skeletal cells express Notch1, Notch2, and low levels of Notch3, although Notch1 and 2 are considered responsible for the effects of Notch in the skeleton [3] (Notch signaling reviewed in [4, 5]).

Schematic diagram of Notch pathway signaling. Notch ligands, consisting of the jagged (Jag1 and Jag2) and delta-like (DLL1, DLL3, and DLL4) families, typically are presented on the surface of signal-sending cells, though these receptors can be shed by proteolytic cleavage in some circumstances. Prior to ligand binding, the Notch family receptors (Notch1, Notch2, Notch3, and Notch4) remain fixed at the plasma membrane, and the CSL transcription complex remains bound to corepressor elements, shutting off transcription of CSL target genes. Upon binding ligand, Notch1 is subject to a two-step proteolytic cleavage by ADAMS family protease and then γ-secretase. Cleavage by γ-secretase frees the cytoplasmic domain of the Notch1 from the plasma membrane; this fragment is termed intracellular Notch1 (ICN1). ICN1 binds to CSL, displacing corepressor elements and recruiting coactivator elements, including Mastermind-Like (MAML), turning on transcription of CSL target genes, including the Hes, Hey, Herp, NRARP, and Deltex families. Notch1 also mediates transcription of non-CSL target genes, which is termed the noncanonical Notch pathway. Regulation of Notch2, Notch3, and Notch4 is similar
Role of Notch Signaling in Normal Osteoblast Development
Mesenchymal stem cells (MSCs) can give rise to multiple lineages in response to environmental molecular cues: the presence of MyoD leads to the differentiation of MSCs into myocytes, PPARγ leads to the generation of adipocytes, the Sox family of genes drive chondrocytogenesis, and runt-related transcription factor-2 (RunX2) and osterix lead to osteoblastogenesis [6–8]. Normal osteoblast development and subsequent bone formation are meticulously regulated not only by RunX2 and osterix but also by a cascade of regulatory signaling that includes morphogens, signaling molecules, and transcriptional regulators [9–16]. A partial list of these factors includes the Wnt/β-catenin, TGFβ/bone morphogenic protein (BMP), FGF, Notch and Hedgehog signaling pathways, ATF4, TAZ, RANKL, and NFATc1 transcription factors [16–19]. Signaling molecules like RunX2, BMPs, and the Wnt/β-catenin canonical pathway are conducive to osteoblastogenesis, while others, such as the Notch signaling pathway, obstruct osteoblast differentiation [20–22]. In osteoblasts, RunX2 regulates the transcription of genes including osteocalcin, bone sialoprotein, osteopontin, type I collagen, fibronectin, galectin 3, MMP13, osteoprotegrin (OPG), Tram2, Lnx2 (an intracellular scaffolding protein that may play a role in Notch signaling), and Tnfrsf12a (a tumor necrosis factor receptor family member) by binding to sequences that resemble the 5′-ACACCA-3′ motif upstream from their transcription start sites [23–28]. Because of its importance in this process, RunX2 is labeled the “master regulator” of osteoblast differentiation; indeed homozygous RunX2 mutant mice have cartilaginous skeletons that fail to mineralize, owing to a complete arrest in osteoblast differentiation [24, 29, 30]. For further details of the role of RunX2 in osteoblast development and in osteosarcoma, please see the chapter on this book entitled “Developmental Pathways Hijacked by Osteosarcoma.”
The Notch signaling pathway plays an important and complex role in bone homeostasis [22, 31–34]. In bone marrow, Notch signaling normally acts to maintain a pool of mesenchymal progenitors by suppressing osteoblast differentiation by inhibiting RunX2 [3]. In osteoblasts, the Notch pathway has several mechanisms that inhibit osteoblastogenesis. Notch antagonizes Wnt signaling: ICN2 colocalizes with glycogen synthase kinase-3β (GSK3β) to mediate the degradation of β-catenin [22, 35]. It has been shown that NFATc1 and osterix form a complex that activates osterix-dependent transcription [36]; ICN and mastermind form a complex with Foxo1 which inhibits NFAT-mediated osteoblastogenesis, osteoblastic bone formation, as well as osteoclastogenesis and bone resorption [37, 38]. Furthermore, Engin et al. show that Notch both stimulates early osteoblastic proliferation by upregulating cyclin D, cyclin E, and osterix and represses osteoblast maturation through the binding of ICN to RunX2 [31]. Hilton et al. and others demonstrate an additional mechanism by which Notch signaling inhibits RunX2: RunX2 transcriptional activity is inhibited by its direct interactions with the HLH and Orange domains of Hey1 [3, 34]. The enzyme necessary for Notch receptor cleavage and activation, ADAM10, is expressed in cells of the osteoblast lineage and is localized at sites of active bone formation. Catalytically active ADAM10 was found to colocalize with Notch2 at these bone-forming sites [39]. This suggests that ADAM10 may play a role in controlling osteoblast differentiation; alternatively, it has been suggested that ADAM10 may work rather to cleave Notch receptor ligands to provide soluble activators of the receptor [39, 40].
Osteosarcoma and Differentiation. Osteosarcoma (OS) may be thought of as a disease of disrupted osteogenic differentiation [8, 10, 41–43]. With the prevention of the differentiation of MSCs into mature osteoblasts, there is an increased risk for malignant transformation as cells continue to proliferate uncontrollably [8, 44]. Osteosarcoma cells display similar characteristics to undifferentiated osteoblasts: early osteogenic markers like CTGF are high in OS cell lines, while markers of differentiation like RunX2, alkaline phosphatase, osteopontin, and osteocalcin are low [10, 41, 42, 45]. Interestingly, the aggressiveness of OS may depend on the stage of differentiation that was disrupted: more aggressive OS may develop from disruptions in the differentiation of early osteoblast progenitors, while benign tumors may arise from disruptions in late-stage osteoblasts [8, 41]. Considering the importance of Notch in normal osteoblast development, the Notch signaling pathway has become increasingly interesting to those studying the progression of osteosarcoma [46–52].
Notch and the Vasculature
Introduction. Blood vessels comprise an extensive tubular network that delivers oxygen and nutrients to all organs and tissues. Vital processes such as embryogenesis, wound healing, body temperature stabilization, and homeostatic balance maintenance all require highly adjustable blood supply and nutrient delivery. These demands are met through the meticulously regulated growth and expansion of the vascular network by angiogenesis. The process of sprouting angiogenesis is highly dynamic and requires a multitude of individual processes such as the proliferation of endothelial cells (ECs), selection of leading cells that develop filopodia and promote endothelial motility, elongation of the new sprout, formation of new cell–cell junctions, conversion into endothelial tubules, specification into arteries, veins, and capillaries, recruitment of mural cells (smooth muscle cells, SMCs, and pericytes), anastomosis with other vessels, remodeling and pruning, perfusion, and stabilization of the newly formed vessel.
The Notch signaling pathway is evolutionarily conserved and is an important mediator of cell–cell communication during the formation of new blood vessels [53]. Major components of the Notch pathway are expressed in the vasculature [54, 55], and genetic deletion of Notch pathway components, including Notch1 [56–58], Notch2 [59], Jag1 [60], DLL1 [61, 62], DLL4 [63, 64], Hey1/Hey2 [65], CSL [66], or presenilins which make up the γ-secretase complex [67, 68], as well as the ectopic activation of Notch1/Notch 4 [69, 70], results in embryonic lethality associated with defects in sprouting angiogenesis, arterial/venous specification, vascular remodeling, and vascular SMC organization (Table 1).
Role of Notch Signaling in Normal Vascular Development
Notch and Arterial/Venous Specification
One of the earliest roles for Notch is in the developing embryo; Notch functions in early vascular development to drive endothelial identity while suppressing venous identity [64, 71]. Later in development, arterial endothelial cells have been shown to require DLL1 to maintain their cellular identity [61]. A more detailed review of this subject has been published recently [72].
Notch and Sprouting Angiogenesis
Vascular endothelial growth factor (VEGF/VEGF-A) is the key regulator that promotes sprouting angiogenesis. In normal/physiologic angiogenesis, VEGF-A is secreted by astrocytes in the avascular region leading to the formation of a VEGF gradient [73, 74]. VEGF-A binds to the tyrosine kinase receptors VEGFR1 (Flt1) and VEGFR2 (KDR/Flk1/Flt2) expressed on the cell surface of nearby ECs. VEGFR2 is the primary receptor transmitting VEGF signals in ECs [75, 76], while VEGFR1, with weaker kinase activity, acts as a VEGF decoy [77, 78]. Newly sprouting blood vessels are made up of two important endothelial cell types: tip cells, which initiate new sprouting, and stalk cells which maintain connection with the parent vessel [79–83]. In response to VEGF-A/VEGFR2-mediated signaling, ECs at the leading front of angiogenic sprouts develop protruding filopodia and become tip cells that extend toward sources of pro-angiogenic growth factors. These tip cells respond to positive/negative guidance cues to allow for directional growth while preventing unorganized and random vessel development [84, 85]. Once such negative guidance cue involves VEGF-mediated induction of DLL4 as a negative feedback regulator, which acts to prevent uncontrolled angiogenic sprouting while promoting the timely formation of a well-differentiated vascular network [83, 86]. Expression of DLL4 stimulates Notch1 activation in adjacent ECs that trail tip cells and form the base of the protrusion and become stalk cells [87]. Whereas tip cells mainly express DLL4, stalk cells primarily express Jag1 which consequentially antagonizes DLL4 activity by competing for Notch receptors via DLL4/Notch1/Jag1-mediated lateral inhibition [82–84, 88–90]. Stalk cells are important in that they proliferate when stimulated with VEGF-A, form the vascular lumen, establish adherins and tight junctions to maintain integrity of the new sprout, and maintain connection with parental vessels so as to establish luminal/abluminal polarity which leads to basal lamina deposition and mural cell recruitment and attachment [84, 91, 92]. In stalk cells, Notch signaling potently inhibits VEGFR3 [93, 94]; VEGFC/VEGFR3 signaling activates PI3K and its downstream target FoxC2, which results in the downregulation of DLL4 in the stalk cell [95, 96]. High levels of activated Notch (ICN) lead to the production of soluble VEFGR1 which acts to enhance the steepness of the VEGF-A signaling gradient by sequestering VEGF-A and inhibiting its action with VEGFR2 in stalk cells [97]. Stalk cells express Hes1 and Hey1 which act to downregulate the levels of VEGFR2, VEGFR3, and DLL4, thereby transiently decreasing the responsiveness to VEGF-A and further enhancing the stalk cell phenotype [81, 82, 93]. This allows new tip cells to form along the front to form branching vessels [98, 99]. Vessel branching within the developing vascular network is also the consequence of another downstream Notch target, Notch-regulated ankyrin repeat protein (Nrarp), which counteracts Notch signaling and is expressed in stalk cells at the branch points [100, 101].
Considering that local changes in VEGF/Notch signaling can trigger the conversion of stalk cells into tip cells, and that the Notch pathway can act in a highly transient and oscillating manner [102], tip and stalk cell phenotypes are remarkably transitory and interchangeable as ECs dynamically shuffle position along the angiogenic sprout competing for the tip cell position [103]. This leads to highly regulated and organized vessel formation. In normal vascular development, these mechanisms work together to balance the numbers of tip cells and stalk cells required for effective sprouting and network formation [82, 104–107]. Tissue oxygenation eventually downregulates paracrine VEGF-A production and thus helps establish a quiescent state for the new vessels [108]. This process has been reviewed in detail [87, 109]. The role of Notch pathway signaling in regulating normal vascular development is shown schematically in Fig. 2.

Normal angiogenesis and the role of Notch pathway signaling. (a) Tip cell development through tubulogenesis. Upon exposure to VEGF-A, endothelial cells respond by taking on a tip cell signaling phenotype. The initial response is stochastic and cyclical, eventually allowing some cells to acquire the full tip cell phenotype, while adjacent cells are prevented from acquiring this phenotype through lateral inhibition, which is Notch mediated. Initial sprouting of tips is also a cyclic process, with individual tips extending and retracting back into the tip cell, leaving behind empty matrix sleeves that help to repattern the extracellular matrix needed in the sprouting blood vessel. Cells adjacent to tip cells become stalk cells, extending outwards toward the VEGF-A gradient, pushing the tip cell outward from the parent vessel. As the filopodia of nearby tip cells contact each other, macrophages are recruited to the site of anastomosis, facilitating fusion of tubes, with subsequent extension of these tubes. (b and c) Notch/VEGF signaling during activation, selection, and sprouting. VEGF-A binds to both VEGFR-1 and VEGFR-2 on adjacent endothelial cells, signaling through both receptors. Predominance of VEGFR-2 signaling favors a tip cell phenotype, which VEGFR-1 favors a stalk cell phenotype. VEGFR-2 signaling mediates upregulation of DLL4 which, in turn, activates Notch1 on the cells to either side of the endothelial cell. DLL4 reduces transcription of VEGFR-2 and promotes secretion of a soluble VEGFR-1 that serves as a ligand trap and reduces the ability of stalk cells to respond to VEGF-A. (d) Notch/VEGF signaling during anastomosis and the role of macrophages. Normal macrophages, without activation, express cell surface DLL4, Jag1, and Jag2 as well as Notch1, Notch2, and Notch4. Notch receptors, especially Notch2, allow macrophages to be recruited to the sites of tip cell anastomosis, where the high levels of DLL4 activate these macrophages. Through a process that is not fully understood, the activated macrophage then helps two tip cells to form a stable bridge that develops into a full vascular loop
Notch and Vascular Mural Functions
Notch signaling also plays an important role in vessel stability by regulating vascular mural cell function. Mural cells (SMCs and pericytes) are attached to the basal surface of certain vessels and help to stabilize the vessel wall, signal to ECs to inhibit their proliferation, promote survival, and regulate blood pressure [110, 111]. Mural cells express Notch1–3, Jag1, and DLL4 [112]. In vitro, it has been shown that endothelial Jag1 activates Notch3 on SMCs to induce Notch3 expression and regulate SMC differentiation [113]. Notch1 signaling is critical for mural cell recruitment to new vessels, whereas Notch3 plays a role in pericyte/SMC maturation once it arrives at its final destination. This process has been reviewed recently [72]. Notch pathway activity is essential for recruitment of bone marrow-derived pericytes to the blood vessels of Ewing sarcoma tumors, and inhibition of the Notch pathway with either shRNA or antibodies impeded Ewing sarcoma tumor growth in vivo and caused impaired vasculogenesis [114, 115]. Perivascular cells, in addition to the endothelium, also have been shown to play an important role in angiogenesis and are deregulated in pathological angiogenesis [110, 115].
Notch and Macrophage-Mediated Angiogenesis
Macrophages have been recognized as key angiogenic effector cells [116, 117]. Macrophages are closely associated with sprouting endothelial cells during retinal angiogenesis [118]. Importantly, tissue macrophages act as cellular chaperones during VEGF-mediated endothelial tip cell induction and anastomosis, allowing for the bridging of tip cells to form stable, perfused vessels [117, 119]. Inactive macrophages express Notch1, -2, and -4, DLL4, and Jag1-2; once activated, macrophages increase their expression of Notch1 and Jag1 [120–122]. Though it is known that VEGFR1 recruits macrophages to sites of inflammation and active angiogenesis [123], macrophage recruitment to sites of anastomosis remains an active area of research. It has been hypothesized that DLL4 expressed in tip cells attracts macrophages via Notch1–DLL4 signaling [117]. Mice with heterozygous mutations for Notch1 have decreased macrophage recruitment and, interestingly, also have decreased expression of VEGFR-1 [124]. Through these studies and others, it is clear that both VEGFR1 and Notch1 play an important role in macrophage recruitment to sites of angiogenesis. Recent publications are available with more complete reviews of the role of macrophages in angiogenesis [72, 125].
Role of Notch Signaling in Tumor Vascular
Notch Signaling at the Primary Tumor
Tumor angiogenesis relies on many of the same mechanisms involved in physiological angiogenesis. Tumors, restricted to 1–2 mm3 without an oxygen and nutrient source, release large amounts of VEGF in response to their hypoxic environment. Unlike normal angiogenesis, however, tumors continuously release pro-angiogenic factors despite the ever-growing expansion of blood vasculature into the well-oxygenated portions of the tumor. This vasculature not only feeds the tumor and allows for uncontrolled proliferation, but it also allows for the metastatic spread of the disease to distant loci, since osteosarcoma spreads almost exclusively via the hematogenous route.
VEGF-A has been shown to be over-expressed in many tumor types [126–128]. Although not much is known about the process of vasculogenesis in osteosarcoma, multiple studies have shown that VEGF overexpression in osteosarcoma unfavorably impacts the overall survival [129–131]. Similarly, the role of Notch has been well documented in other tumor types [115, 132, 133] but continues to be an active area of study in osteosarcoma. In multiple tumor types, it has been shown that either blockade [105, 106, 134–136] or forced activation of the Notch pathway [137–142] can inhibit angiogenesis. Genetic or pharmacologic inactivation/inhibition of either DLL4 or Notch1 signaling leads to an increase in the number of filopodia and sprouting tip cells at the angiogenic front which, together with EC proliferation, results in the formation of a hyperdense vascular network with immature, hyperplastic, and nonfunctional characteristics [81, 83, 86, 104, 107, 143]. Chronic blockade of the pathway, however, results in the formation of vascular neoplasms [144]. Conversely, activation of Notch signaling leads to a reduced number of tip cells and less dense vascular network [86, 107]. A schematic model of the role of Notch in tumor angiogenesis is shown in Fig. 3.

Tumor angiogenesis. (a) Heterogenic distribution of vasculature and O2 in a tumor. Because oxygen diffusion is limited in tissues to ~1 mm from capillaries, rapidly growing tumors will have regions of relative normoxia and other areas of profound hypoxia, with an oxygen gradient between these regions. The extremely high levels of VEGF-A secreted in the areas with the worst hypoxia override normal angiogenic controls, leading to large numbers of small, dysfunctional, and leaky blood vessels that can be observed on arteriograms (a, right hand panel) as a “vascular blush.” Other areas of the tumor do not appear to have any blood supply at all and often are necrotic when examined pathologically. (The right hand panel in (a) is taken from an osteosarcoma patient receiving an arteriogram prior to the delivery of intra-arterial chemotherapy. The method is exactly as described previously [205].) (b) Tumors hijack empty matrix sleeves for migration/invasion. As described above and in Fig. 2, normal angiogenesis involves cyclical extension and retraction of tips, repatterning the extracellular matrix, including spreading laminin away from the basement membrane toward the VEGF-A source. In tumors, these empty sleeves left behind by tip cell extension and retraction become pathways in which the extracellular matrix ceases to be a barrier to tumor cell migration, but rather a guide for tumor cells to “find” blood vessels. (c) Tumors promote uncontrolled angiogenesis. Growing tumors provide a sustained source of VEGF-A, either directly through their own secretion or by inducing hypoxia, thereby promoting VEGF-A secretion from nonmalignant cells within the tumor, such as tumor-associated fibroblasts. Unlike normal angiogenesis, in which VEGF-A levels eventually decline and new vessels are allowed to mature and stabilize, the sustained VEGF-A secretion in the tumor microenvironment causes uncontrolled, sustained angiogenesis, without the maturation and stabilization found in normal angiogenesis. (d) High expression of VEGF promotes an all tip cell phenotype. In areas with the highest VEGF-A secretion, the concentration of VEGF-A is sufficient to override the cellular processes that induce lateral inhibition and organized vessel formation. In this environment, endothelial cells may take on an “all tip cell” phenotype, leading to vascular leak and highly disorganized blood vessels that completely lack vessel wall components. Note that in any given tumor, aspects of abnormal blood vessel development shown in panels A–D may all be taking place, each in different regions of the tumor
Notch Signaling at the Metastatic Site
Judah Folkman first championed the concept that tumors require an “angiogenic switch” in the balance between pro- and anti-antigenic signals to establish a robust blood supply capable of supporting rapid tumor growth [145]. By extension, this model would suggest that, for dormant tumors, there is a balance between signals that increase angiogenesis and those that impede angiogenesis and that dormant micrometastases of osteosarcoma would be relatively poorly vascularized. Indraccolo and colleagues showed that expression of DLL4 on blood vessels in close proximity to colon cancer cells was necessary for these tumor cells to awaken from dormancy [146]. The same group had shown already that a short-term “spike” in angiogenesis was sufficient to awaken dormant tumors [147]. This awakening is associated with a transcriptional switch from expressing anti-angiogenic proteins to secreting pro-angiogenic ones [148]. While there is no direct experimental evidence in osteosarcoma models to support this role of the vasculature in osteosarcoma metastasis, it certainly seems plausible that a similar effect operates in these patients’ lungs.
There is a conception among some patients and families that major operations for osteosarcoma patients “spread the tumor.” While this has not been scientifically validated, it has been observed that pulmonary metastasis sometimes develops shortly after resection of the primary tumor or lung metastases. Since the healing of large wounds results in high levels of circulating growth factors and cytokines such as EGF and its related ERBB family ligands, these growth factors and angiogenic cytokines could stimulate the expansion of tumor vessels in micrometastases. The transient upregulation of Notch ligands on vessels near the dormant micrometastases may initiate the angiogenic response that facilitates growth. A more comprehensive review of the putative roles of the Notch pathway in regulating tumor escape from dormancy in the metastatic site has been published recently [149].
Notch Signaling in Osteosarcoma
The Notch pathway has been called “the stem cell master switch” [53, 150] because it influences multiple processes that drive morphogenesis, lineage specification, apoptosis, and proliferation, not only in normal tissues but also in some cancers [151]. Notch dysregulation serves as an oncogene for many cancers including T-cell leukemia [152] and solid tumors of pancreas, breast, prostate, melanoma, and colon [151, 153–157]. In these malignancies, it contributes to malignancy by promoting growth, survival, motility, neo-angiogenesis, drug resistance, invasion, and metastasis [158–163]. In other cancers, Notch functions as a tumor suppressor, impeding growth or causing apoptosis in B-lineage ALL [164], myeloid malignancies [165], squamous cell carcinomas [166], neuroblastoma [167], other neural crest-derived cancers [168], and the GI stromal tumor [169]. It was recently suggested that Notch1 signaling is activated in human OS and may play a role in tumor invasion and metastasis [47, 52, 170]. One possible reason for this association is the reported link between Notch pathway activity and behavior of tumor-initiating cells or the putative cancer stem cells [171–177].
Two popular models for tumorigenesis include the stochastic model and the cancer stem cell model. The traditional stochastic model presumes that cancer arises from a single cell which has become genetically unstable and initiates tumor growth. The cancer stem cell model proposes that tumor-initiating cells share important properties with normal stem cells, including self-renewal and resistance to stress [42]. Over the past 5 years or more, the theory of cancer stem cells in osteosarcoma has gained a great deal of acceptance, with numerous publications in recent years describing phenotype, behavior, and therapeutic potential [178–194]. Logically, cells with stem cell-like properties should be superior at tumorigenesis and metastasis.
This concept was studied recently using two murine cell lines, K7M2 and K12, which were derived from the same spontaneously occurring murine osteosarcoma. K7M2 metastasizes with high frequency to the lung in mouse models, whereas K12 is much less metastatic [195].
Several groups have published that K7M2 and K12 cells produce different quantities of cytokines and that inhibition of these cytokines alters OS cell behavior in vitro [195–198]. For example, we have demonstrated that highly metastatic K7M2 cells express and produce more bone morphogenetic protein-2 (BMP-2) and VEGF than less metastatic K12 cells. Additionally, we observed that the inhibition of these factors diminished the motility and viability of K7M2 cells [197, 198]. More recently, we have demonstrated important differences between K7M2 and K12 in terms of Notch1 expression and function [50].
To evaluate the role of Notch in regulating stemness behaviors, we first compared K7M2 and K12 cells with reverse transcription polymerase chain reaction (RT-PCR). We analyzed differences in the expression of Notch1, its downstream targets, and other important genes in OS biology. We observed a significant upregulation (nearly twofold) of Notch1, Notch2, and Notch4 expression [50]. We also observed the upregulation of the Notch1 target genes Hes1 and Stat3 in highly metastatic K7M2 cells compared with less metastatic K12 cells. Notch pathway inhibition using an inhibitor of γ-secretase (GSI) in K7M2 cells reduced expression of these genes down to levels similar to K12 and also reduced migration and invasiveness of K7M2. Activation of Notch in K12 using an exogenous ligand increased invasiveness and migration, confirming the vital role of the Notch pathway in regulating these processes in this model [50].
Aldehyde dehydrogenase (ALDH) is another putative cancer stem cell factor [199–202] that has been implicated in a variety of human cancers. ALDH is a tetrameric protein that oxidizes aldehydes to carboxylic acids and thus enables cells to withstand oxidative stress. Its activity has been associated with metastasis, drug resistance, and poor prognosis [199–203]. We have shown that K7M2 cells possess greater mean ALDH activity and a higher percentage of ALDH-positive cells than the less metastatic K12 cells [204]. GSI treatment of K7M2 cells reduced the expression of ALDH and rendered the cells less tolerant of oxidative stress (Fig. 4), while treatment of K12 cells with the Notch ligand jagged 1 increased ALDH expression and rendered cells more tolerant of oxidative stress (Fig. 5), confirming that Notch pathway signaling is upstream of ALDH expression [50].

K7M2 cells are resistant to H2O2 but become sensitive after treatment with DAPT. (a) K7M2 cells were treated with or without the γ-secretase inhibitor DAPT (10 μM) for 4 days and were then cultured with media containing H2O2 (0, 250, or 500 μM) for 6 h. Cell death was analyzed using PI exclusion assay. (b) The percentage of PI+ cells was determined for each group in (a). Asterisk indicates that the difference is significant comparing DAPT-treated or non-treated samples (p < 0.05). Figure taken from [50], used with permission

Notch1 activation with jagged 1 increases K12 cells’ resistance to oxidative stress. (a–d) K12 cells, which have low levels of Notch activation, were treated with soluble Jag1 to activate Notch pathway signaling. After Notch1 activation, K12 cells exhibited greater resistance to oxidative stress than untreated K12 cells. These images are presented as in Fig. 4. (e) K12 cell invasiveness increases with jagged 1 treatment. Jagged 1 treatment of K12 cells increased the in vitro invasion capacity of the cells through a semisolid matrigel matrix, suggesting that enhanced Notch expression contributes to metastatic potential in OS cells. Graphs depict the quantified mean invasion of tumor cells into matrigel over time. (f) Gene expression with Notch1 activation in less metastatic K12 cells. RT-PCR showed that the expression of Notch1, Hes1, and ALDH was upregulated in K12 cells with jagged 1 treatment, indicating a more aggressive phenotype. This figure is adapted from [50] and is used with permission
Conclusions
These studies, taken together, support the concept that Notch pathway signaling plays a key role in maintaining a stem cell-like phenotype for osteosarcoma and highlights the importance of Notch in osteosarcoma growth and metastasis. It is interesting to note, however, that the phenotype associated with Notch pathway expression could be induced by exposure to exogenous Notch ligand, calling into question the concept that tumor stem cells represent a discrete subpopulation in osteosarcoma. Given the importance of Notch signaling in tumor blood vessels and the high level of expression of Notch ligands in the vasculature, it is possible that the phenotype we associate with stemness in osteosarcoma really reflects proximity to tumor blood vessels and, therefore, exposure to Notch ligands. As therapies are developed to target Notch in cancer patients, the role of Notch in tumor vessel formation and expansion must also be considered.
References
Iso T, Kedes L, Hamamori Y (2003) HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol 194(3):237–255
Kageyama R, Ohtsuka T (1999) The Notch-Hes pathway in mammalian neural development. Cell Res 9(3):179–188
Hilton M, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, Kronenberg H, Teitelbaum S, Ross F, Kopan R, Long F (2008) Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med 14(3):306–314
Greenwald I, Kovall R (2013) Notch signaling: genetics and structure. WormBook : the online review of C elegans biology, pp 1–28
Kopan R (2012) Notch signaling. Cold Spring Harb Perspect Biol 4(10):a008904
Pittenger M, Mackay A, Beck S, Jaiswal R, Douglas R, Mosca J, Moorman M, Simonetti D, Craig S, Marshak D (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284(5411):143–147
Crigler L, Kazhanie A, Yoon T-J, Zakhari J, Anders J, Taylor B, Virador V (2007) Isolation of a mesenchymal cell population from murine dermis that contains progenitors of multiple cell lineages. FASEB J 21(9):2050–2063
Tang N, Song W-X, Luo J, Haydon R, He T-C (2008) Osteosarcoma development and stem cell differentiation. Clin Orthopaed Relat Res 466(9):2114–2130
Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, Amling M, Karsenty G (1999) A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Gen Dev 13(8):1025–1036
Haydon R, Luu H, He T-C (2007) Osteosarcoma and osteoblastic differentiation: a new perspective on oncogenesis. Clin Orthopaed Relat Res 454:237–246
Hong J-H, Hwang E, McManus M, Amsterdam A, Tian Y, Kalmukova R, Mueller E, Benjamin T, Spiegelman B, Sharp P, Hopkins N, Yaffe M (2005) TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309(5737):1074–1078
Lian J, Stein G, Javed A, van Wijnen A, Stein J, Montecino M, Hassan M, Gaur T, Lengner C, Young D (2006) Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endoc Metab Disord 7(1–2):1–16
Luu H, Song W-X, Luo X, Manning D, Luo J, Deng Z-L, Sharff K, Montag A, Haydon R, He T-C (2007) Distinct roles of bone morphogenetic proteins in osteogenic differentiation of mesenchymal stem cells. J Orthopaed Res 25(5):665–677
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng J, Behringer R, de Crombrugghe B (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108(1):17–29
Yamaguchi A, Komori T, Suda T (2000) Regulation of osteoblast differentiation mediated by bone morphogenetic proteins, hedgehogs, and Cbfa1. Endoc Rev 21(4):393–411
Deng Z-L, Sharff K, Tang N, Song W-X, Luo J, Luo X, Chen J, Bennett E, Reid R, Manning D, Xue A, Montag A, Luu H, Haydon R, He T-C (2008) Regulation of osteogenic differentiation during skeletal development. Front Biosci 13:2001–2021
Olsen B, Reginato A, Wang W (2000) Bone development. Annu Rev Cell Dev Biol 16:191–220
Harada S-i, Rodan G (2003) Control of osteoblast function and regulation of bone mass. Nature 423(6937):349–355
Ralston S, de Crombrugghe B (2006) Genetic regulation of bone mass and susceptibility to osteoporosis. Gen Dev 20(18):2492–2506
Gazzerro E, Canalis E (2006) Bone morphogenetic proteins and their antagonists. Rev Endoc Metab Disord 7(1–2):51–65
Krishnan V, Bryant H, Macdougald O (2006) Regulation of bone mass by Wnt signaling. J Clin Investig 116(5):1202–1209
Deregowski V, Gazzerro E, Priest L, Rydziel S, Canalis E (2006) Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. J Biol Chem 281(10):6203–6210
Banerjee C, McCabe L, Choi J, Hiebert S, Stein J, Stein G, Lian J (1997) Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone-specific complex. J Cell Biochem 66(1):1–8
Ducy P, Zhang R, Geoffroy V, Ridall A, Karsenty G (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89(5):747–754
Motohiko S, Natsuo Y, Takanobu N, Hirohisa K, Mizuo S, Seiichi H, Yukihiko K, Shintaro N, Takahiro O (1998) Expression of bone matrix proteins mRNA during distraction osteogenesis. J Bone Miner Res 13(8):1221–1231
Xiao Z, Hinson T, Quarles L (1999) Cbfa1 isoform overexpression upregulates osteocalcin gene expression in non-osteoblastic and pre-osteoblastic cells. J Cell Biochem 74(4):596–605
Prince M, Banerjee C, Javed A, Green J, Lian J, Stein G, Bodine P, Komm B (2001) Expression and regulation of Runx2/Cbfa1 and osteoblast phenotypic markers during the growth and differentiation of human osteoblasts. J Cell Biochem 80(3):424–440
Pregizer S, Barski A, Gersbach C, García A, Frenkel B (2007) Identification of novel Runx2 targets in osteoblasts: cell type-specific BMP-dependent regulation of Tram2. J Cell Biochem 102(6):1458–1471
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson R, Gao Y, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89(5):755–764
Otto F, Thornell A, Crompton T, Denzel A, Gilmour K, Rosewell I, Stamp G, Beddington R, Mundlos S, Olsen B, Selby P, Owen M (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89(5):765–771
Engin F, Yao Z, Yang T, Zhou G, Bertin T, Jiang M, Chen Y, Wang L, Zheng H, Sutton R, Boyce B, Lee B (2008) Dimorphic effects of Notch signaling in bone homeostasis. Nat Med 14(3):299–305
Sciaudone M, Gazzerro E, Priest L, Delany A, Canalis E (2003) Notch 1 impairs osteoblastic cell differentiation. Endocrinology 144(12):5631–5639
Tezuka K-I, Yasuda M, Watanabe N, Morimura N, Kuroda K, Miyatani S, Hozumi N (2002) Stimulation of osteoblastic cell differentiation by Notch. J Bone Miner Res 17(2):231–239
Zamurovic N, Cappellen D, Rohner D, Susa M (2004) Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J Biol Chem 279(36):37704–37715
Espinosa L, Inglés-Esteve J, Aguilera C, Bigas A (2003) Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J Biol Chem 278(34):32227–32235
Koga T, Matsui Y, Asagiri M, Kodama T, de Crombrugghe B, Nakashima K, Takayanagi H (2005) NFAT and Osterix cooperatively regulate bone formation. Nat Med 11(8):880–885
Winslow M, Pan M, Starbuck M, Gallo E, Deng L, Karsenty G, Crabtree G (2006) Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell 10(6):771–782
Ikeda F, Nishimura R, Matsubara T, Hata K, Reddy S, Yoneda T (2006) Activation of NFAT signal in vivo leads to osteopenia associated with increased osteoclastogenesis and bone-resorbing activity. J Immunol 177(4):2384–2390
Dallas D, Genever P, Patton A, Millichip M, McKie N, Skerry T (1999) Localization of ADAM10 and Notch receptors in bone. Bone 25(1):9–15
Qi H, Rand M, Wu X, Sestan N, Wang W, Rakic P, Xu T, Artavanis-Tsakonas S (1999) Processing of the notch ligand delta by the metalloprotease Kuzbanian. Science 283(5398):91–94
Luo X, Chen J, Song W-X, Tang N, Luo J, Deng Z-L, Sharff K, He G, Bi Y, He B-C, Bennett E, Huang J, Kang Q, Jiang W, Su Y, Zhu G-H, Yin H, He Y, Wang Y, Souris J, Chen L, Zuo G-W, Montag A, Reid R, Haydon R, Luu H, He T-C (2008) Osteogenic BMPs promote tumor growth of human osteosarcomas that harbor differentiation defects. Lab Investig 88(12):1264–1277
Reya T, Morrison S, Clarke M, Weissman I (2001) Stem cells, cancer, and cancer stem cells. Nature 414(6859):105–111
Thomas D, Kansara M (2006) Epigenetic modifications in osteogenic differentiation and transformation. Journal of cellular biochemistry 98(4):757–769
Wagner E, He B-C, Chen L, Zuo G-W, Zhang W, Shi Q, Luo Q, Luo X, Liu B, Luo J, Rastegar F, He C, Hu Y, Boody B, Luu H, He T-C, Deng Z-L, Haydon R (2010) Therapeutic Implications of PPARgamma in Human Osteosarcoma. PPAR Res 2010:956427
Thomas D, Johnson S, Sims N, Trivett M, Slavin J, Rubin B, Waring P, McArthur G, Walkley C, Holloway A, Diyagama D, Grim J, Clurman B, Bowtell D, Lee J-S, Gutierrez G, Piscopo D, Carty S, Hinds P (2004) Terminal osteoblast differentiation, mediated by runx2 and p27KIP1, is disrupted in osteosarcoma. J Cell Biol 167(5):925–934
Dailey D, Anfinsen K, Pfaff L, Ehrhart E, Charles J, Bønsdorff T, Thamm D, Powers B, Jonasdottir T, Duval D (2013) HES1, a target of Notch signaling, is elevated in canine osteosarcoma, but reduced in the most aggressive tumors. BMC Vet Res 9(1):130
Engin F, Bertin T, Ma O, Jiang MM, Wang L, Sutton RE, Donehower LA, Lee B (2009) Notch signaling contributes to the pathogenesis of human osteosarcomas. Hum Mol Genet 18(8):1464–1470
Hughes DPM (2009) How the NOTCH pathway contributes to the ability of osteosarcoma cells to metastasize. In: Jaffe N, Bruland OS, Bielack S (eds) Pediatric and adolescent osteosarcoma. Cancer treatment and research, 152nd edn. Springer, New York, NY, pp 479–496
Li Y, Zhang J, Ma D, Zhang L, Si M, Yin H, Li J (2012) Curcumin inhibits proliferation and invasion of osteosarcoma cells through inactivation of Notch-1 signaling. FEBS J 279(12):2247–2259
Mu X, Isaac C, Greco N, Huard J, Weiss K (2013) Notch signaling is associated with ALDH activity and an aggressive metastatic phenotype in murine osteosarcoma cells. Front Oncol 3:143
Tanaka M, Setoguchi T, Hirotsu M, Gao H, Sasaki H, Matsunoshita Y, Komiya S (2009) Inhibition of Notch pathway prevents osteosarcoma growth by cell cycle regulation. Br J Cancer 100(12):1957–1965
Zhang P, Yang Y, Nolo R, Zweidler-McKay PA, Hughes DP (2010) Regulation of NOTCH signaling by reciprocal inhibition of HES1 and Deltex 1 and its role in osteosarcoma invasiveness. Oncogene 29(20):2916–2926. doi:10.1038/onc.2010.62
Artavanis-Tsakonas S, Rand M, Lake R (1999) Notch signaling: cell fate control and signal integration in development. Science 284(5415):770–776
Shawber C, Kitajewski J (2004) Notch function in the vasculature: insights from zebrafish, mouse and man. Bioessays 26(3):225–234
Kume T (2009) Novel insights into the differential functions of Notch ligands in vascular formation. J Angiogen Res 1:8
Limbourg F, Takeshita K, Radtke F, Bronson R, Chin M, Liao J (2005) Essential role of endothelial Notch1 in angiogenesis. Circulation 111(14):1826–1832
Krebs L, Xue Y, Norton C, Shutter J, Maguire M, Sundberg J, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith G, Stark K, Gridley T (2000) Notch signaling is essential for vascular morphogenesis in mice. Gen Dev 14(11):1343–1352
Swiatek P, Lindsell C, del Amo F, Weinmaster G, Gridley T (1994) Notch1 is essential for postimplantation development in mice. Gen Dev 8(6):707–719
Hamada Y, Kadokawa Y, Okabe M, Ikawa M, Coleman J, Tsujimoto Y (1999) Mutation in ankyrin repeats of the mouse Notch2 gene induces early embryonic lethality. Development 126(15):3415–3424
Xue Y, Gao X, Lindsell C, Norton C, Chang B, Hicks C, Gendron-Maguire M, Rand E, Weinmaster G, Gridley T (1999) Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet 8(5):723–730
Sörensen I, Adams R, Gossler A (2009) DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 113(22):5680–5688
HrabÄ de Angelis M, McIntyre 2nd J, Gossler A (1997) Maintenance of somite borders in mice requires the Delta homologue DII1. Nature 386(6626):717–721
Gale N, Dominguez M, Noguera I, Pan L, Hughes V, Valenzuela D, Murphy A, Adams N, Lin H, Holash J, Thurston G, Yancopoulos G (2004) Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A 101(45):15949–15954
Duarte A, Hirashima M, Benedito R, Trindade A, Diniz P, Bekman E, Costa L, Henrique D, Rossant J (2004) Dosage-sensitive requirement for mouse DLL4 in artery development. Gen Dev 18(20):2474–2478
Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M (2004) The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Gen Dev 18(8):901–911
Krebs L, Shutter J, Tanigaki K, Honjo T, Stark K, Gridley T (2004) Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Gen Dev 18(20):2469–2473
Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, Serneels L, Umans L, Schrijvers V, Checler F, Vanderstichele H, Baekelandt V, Dressel R, Cupers P, Huylebroeck D, Zwijsen A, Van Leuven F, De Strooper B (1999) Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci U S A 96(21):11872–11877
Nakajima M, Yuasa S, Ueno M, Takakura N, Koseki H, Shirasawa T (2003) Abnormal blood vessel development in mice lacking presenilin-1. Mech Dev 120(6):657–667
Uyttendaele H, Ho J, Rossant J, Kitajewski J (2001) Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci U S A 98(10):5643–5648
Krebs L, Starling C, Chervonsky A, Gridley T (2010) Notch1 activation in mice causes arteriovenous malformations phenocopied by ephrinB2 and EphB4 mutants. Genesis 48(3):146–150
Shawber C, Das I, Francisco E, Kitajewski J (2003) Notch signaling in primary endothelial cells. Ann N Y Acad Sci 995:162–170
Kofler N, Shawber C, Kangsamaksin T, Reed H, Galatioto J, Kitajewski J (2011) Notch signaling in developmental and tumor angiogenesis. Gen Cancer 2(12):1106–1116
Koch S, Claesson-Welsh L (2012) Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med 2(7):a006502
Ferrara N, Gerber H-P, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9(6):669–676
Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck T, Pelletier N, Ferrara N (2001) Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem 276(5):3222–3230
Shalaby F, Rossant J, Yamaguchi T, Gertsenstein M, Wu X, Breitman M, Schuh A (1995) Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376(6535):62–66
Kappas N, Zeng G, Chappell J, Kearney J, Hazarika S, Kallianos K, Patterson C, Annex B, Bautch V (2008) The VEGF receptor Flt-1 spatially modulates Flk-1 signaling and blood vessel branching. J Cell Biol 181(5):847–858
Shibuya M (2006) Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol 39(5):469
Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea K, Powell-Braxton L, Hillan K, Moore M (1996) Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380(6573):439–442
Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380(6573):435–439
Suchting S, Freitas C, le Noble F, Benedito R, Bréant C, Duarte A, Eichmann A (2007) The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci U S A 104(9):3225–3230
Hellström M, Phng L-K, Hofmann J, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson A-K, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe M, Kalén M, Gerhardt H, Betsholtz C (2007) DLL4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445(7129):776–780
Lobov I, Renard R, Papadopoulos N, Gale N, Thurston G, Yancopoulos G, Wiegand S (2007) Delta-like ligand 4 (DLL4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci U S A 104(9):3219–3224
Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161(6):1163–1177
del Toro R, Prahst C, Mathivet T, Siegfried G, Kaminker J, Larrivee B, Breant C, Duarte A, Takakura N, Fukamizu A, Penninger J, Eichmann A (2010) Identification and functional analysis of endothelial tip cell-enriched genes. Blood 116(19):4025–4033
Mats H, Li-Kun P, Holger G (2007) VEGF and notch signaling: the yin and yang of angiogenic sprouting. Cell Adh Migr 1(3):133–136
Benedito R, Hellström M (2013) Notch as a hub for signaling in angiogenesis. Exp Cell Res 319(9):1281–1288
Eilken H, Adams R (2010) Turning on the angiogenic microswitch. Nat Med 16(8):853–854
Harrington L, Sainson R, Williams C, Taylor J, Shi W, Li J-L, Harris A (2008) Regulation of multiple angiogenic pathways by DLL4 and Notch in human umbilical vein endothelial cells. Microvasc Res 75(2):144–154
Eilken H, Adams R (2010) Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22(5):617–625
Iruela-Arispe M, Davis G (2009) Cellular and molecular mechanisms of vascular lumen formation. Dev Cell 16(2):222–231
Dejana E, Tournier-Lasserve E, Weinstein B (2009) The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell 16(2):209–221
Tammela T, Zarkada G, Wallgard E, Murtomäki A, Suchting S, Wirzenius M, Waltari M, Hellström M, Schomber T, Peltonen R, Freitas C, Duarte A, Isoniemi H, Laakkonen P, Christofori G, Ylä-Herttuala S, Shibuya M, Pytowski B, Eichmann A, Betsholtz C, Alitalo K (2008) Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454(7204):656–660
Benedito R, Rocha S, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams R (2012) Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 484(7392):110–114
Tammela T, Zarkada G, Nurmi H, Jakobsson L, Heinolainen K, Tvorogov D, Zheng W, Franco C, Murtomäki A, Aranda E, Miura N, Ylä-Herttuala S, Fruttiger M, Mäkinen T, Eichmann A, Pollard J, Gerhardt H, Alitalo K (2011) VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol 13(10):1202–1213
Hisaki H, Tsutomu K (2009) Foxc2 transcription factor as a regulator of angiogenesis via induction of integrin β3 expression. Cell Adh Migr 3(1):24–26
Chappell J, Taylor S, Ferrara N, Bautch V (2009) Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev Cell 17(3):377–386
Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M, Adams R (2009) The notch ligands DLL4 and Jagged1 have opposing effects on angiogenesis. Cell 137(6):1124–1135
Thomas J-L, Baker K, Han J, Calvo C, Nurmi H, Eichmann A, Alitalo K (2013) Interactions between VEGFR and Notch signaling pathways in endothelial and neural cells. Cell Mol Life Sci 70(10):1779–1792
Phng L-K, Potente M, Leslie J, Babbage J, Nyqvist D, Lobov I, Ondr J, Rao S, Lang R, Thurston G, Gerhardt H (2009) Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell 16(1):70–82
Sainson R, Aoto J, Nakatsu M, Holderfield M, Conn E, Koller E, Hughes C (2005) Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J 19(8):1027–1029
Kageyama R, Masamizu Y, Niwa Y (2007) Oscillator mechanism of Notch pathway in the segmentation clock. Dev Dyn 236(6):1403–1409
Jakobsson L, Franco C, Bentley K, Collins R, Ponsioen B, Aspalter I, Rosewell I, Busse M, Thurston G, Medvinsky A, Schulte-Merker S, Gerhardt H (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12(10):943–953
Leslie J, Ariza-McNaughton L, Bermange A, McAdow R, Johnson S, Lewis J (2007) Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 134(5):839–844
Noguera-Troise I, Daly C, Papadopoulos N, Coetzee S, Boland P, Gale N, Lin H, Yancopoulos G, Thurston G (2006) Blockade of DLL4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444(7122):1032–1037
Ridgway J, Zhang G, Wu Y, Stawicki S, Liang W-C, Chanthery Y, Kowalski J, Watts R, Callahan C, Kasman I, Singh M, Chien M, Tan C, Hongo J-AS, de Sauvage F, Plowman G, Yan M (2006) Inhibition of DLL4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444(7122):1083–1087
Siekmann A, Lawson N (2007) Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445(7129):781–784
Phng LK, Gerhardt H (2009) Angiogenesis: a team effort coordinated by notch. Dev Cell 16(2):196–208
Siekmann A, Affolter M, Belting H-G (2013) The tip cell concept 10 years after: new players tune in for a common theme. Exp Cell Res 319(9):1255–1263
Benjamin L, Hemo I, Keshet E (1998) A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 125(9):1591–1598
Armulik A, Abramsson A, Betsholtz C (2005) Endothelial/pericyte interactions. Circ Res 97(6):512–523
Villa N, Walker L, Lindsell C, Gasson J, Iruela-Arispe M, Weinmaster G (2001) Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev 108(1–2):161–164
Liu H, Kennard S, Lilly B (2009) NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ Res 104(4):466–475
Schadler KL, Zweidler-McKay PA, Guan H, Kleinerman ES (2010) Delta-like ligand 4 plays a critical role in pericyte/vascular smooth muscle cell formation during vasculogenesis and tumor vessel expansion in Ewing’s sarcoma. Clin Cancer Res 16(3):848–856
Stewart KS, Zhou Z, Zweidler-McKay P, Kleinerman ES (2011) Delta-like ligand 4-Notch signaling regulates bone marrow-derived pericyte/vascular smooth muscle cell formation. Blood 117(2):719–726
Crowther M, Brown N, Bishop E, Lewis C (2001) Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J Leukoc Biol 70(4):478–490
Schmidt T, Carmeliet P (2010) Blood-vessel formation: bridges that guide and unite. Nature 465(7299):697–699
Checchin D, Sennlaub F, Levavasseur E, Leduc M, Chemtob S (2006) Potential role of microglia in retinal blood vessel formation. Investig Ophthalmol Vis Sci 47(8):3595–3602
Fantin A, Vieira J, Gestri G, Denti L, Schwarz Q, Prykhozhij S, Peri F, Wilson S, Ruhrberg C (2010) Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 116(5):829–840
Bigas A, Martin D, Milner L (1998) Notch1 and Notch2 inhibit myeloid differentiation in response to different cytokines. Mol Cell Biol 18(4):2324–2333
Monsalve E, Pérez M, Rubio A, Ruiz-Hidalgo M, Baladrón V, García-Ramírez J, Gómez J, Laborda J, Díaz-Guerra M (2006) Notch-1 up-regulation and signaling following macrophage activation modulates gene expression patterns known to affect antigen-presenting capacity and cytotoxic activity. J Immunol 176(9):5362–5373
Singh N, Phillips R, Iscove N, Egan S (2000) Expression of notch receptors, notch ligands, and fringe genes in hematopoiesis. Exp Hematol 28(5):527–534
Murakami M, Zheng Y, Hirashima M, Suda T, Morita Y, Ooehara J, Ema H, Fong G-H, Shibuya M (2008) VEGFR1 tyrosine kinase signaling promotes lymphangiogenesis as well as angiogenesis indirectly via macrophage recruitment. Arterioscler Thromb Vasc Biol 28(4):658–664
Outtz H, Wu J, Wang X, Kitajewski J (2010) Notch1 deficiency results in decreased inflammation during wound healing and regulates vascular endothelial growth factor receptor-1 and inflammatory cytokine expression in macrophages. J Immunol 185(7):4363–4373
Outtz H, Tattersall I, Kofler N, Steinbach N, Kitajewski J (2011) Notch1 controls macrophage recruitment and Notch signaling is activated at sites of endothelial cell anastomosis during retinal angiogenesis in mice. Blood 118(12):3436–3439
Leung D, Cachianes G, Kuang W, Goeddel D, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246(4935):1306–1309
Hu P, Liu W, Wang L, Yang M, Du J (2013) High circulating VEGF level predicts poor overall survival in lung cancer. J Cancer Res Clin Oncol 139(7):1157–1167
Claesson-Welsh L, Welsh M (2013) VEGFA and tumour angiogenesis. J Intern Med 273(2):114–127
Yu X-W, Wu T-Y, Yi X, Ren W-P, Zhou Z-B, Sun Y-Q, Zhang C-Q (2013) Prognostic significance of VEGF expression in osteosarcoma: a meta-analysis. Tumour Biol 35(1):155–160
Chen D, Zhang Y-J, K-w Z, Wang W-C (2013) A systematic review of vascular endothelial growth factor expression as a biomarker of prognosis in patients with osteosarcoma. Tumour Biol 34(3):1895–1899
Lammli J, Fan M, Rosenthal H, Patni M, Rinehart E, Vergara G, Ablah E, Wooley P, Lucas G, Yang S-Y (2012) Expression of vascular endothelial growth factor correlates with the advance of clinical osteosarcoma. Int Orthopaed 36(11):2307–2313
Ranganathan P, Weaver K, Capobianco A (2011) Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer 11(5):338–351
Patel N, Li J-L, Generali D, Poulsom R, Cranston D, Harris A (2005) Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res 65(19):8690–8697
Rehman A, Wang C-Y (2006) Notch signaling in the regulation of tumor angiogenesis. Trend Cell Biol 16(6):293–300
Dufraine J, Funahashi Y, Kitajewski J (2008) Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene 27(38):5132–5137
Chi Sabins N, Taylor J, Fabian K, Appleman L, Maranchie J, Stolz D, Storkus W (2013) DLK1: a novel target for immunotherapeutic remodeling of the tumor blood vasculature. Mol Ther 21(10):1958–1968
Li J-L, Sainson R, Shi W, Leek R, Harrington L, Preusser M, Biswas S, Turley H, Heikamp E, Hainfellner J, Harris A (2007) Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res 67(23):11244–11253
Segarra M, Williams C, Sierra MDLL, Bernardo M, McCormick P, Maric D, Regino C, Choyke P, Tosato G (2008) DLL4 activation of Notch signaling reduces tumor vascularity and inhibits tumor growth. Blood 112(5):1904–1911
Trindade A, Kumar S, Scehnet J, Lopes-da-Costa L, Becker J, Jiang W, Liu R, Gill P, Duarte A (2008) Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 112(5):1720–1729
Williams C, Li J-L, Murga M, Harris A, Tosato G (2006) Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 107(3):931–939
Zhang J-P, Qin H-Y, Wang L, Liang L, Zhao X-C, Cai W-X, Wei Y-N, Wang C-M, Han H (2011) Overexpression of Notch ligand DLL1 in B16 melanoma cells leads to reduced tumor growth due to attenuated vascularization. Cancer Lett 309(2):220–227
Zhao X-C, Dou G-R, Wang L, Liang L, Tian D-M, Cao X-L, Qin H-Y, Wang C-M, Zhang P, Han H (2013) Inhibition of tumor angiogenesis and tumor growth by the DSL domain of human delta-like 1 targeted to vascular endothelial cells. Neoplasia 15(7):815–825
Scehnet J, Jiang W, Kumar S, Krasnoperov V, Trindade A, Benedito R, Djokovic D, Borges C, Ley E, Duarte A, Gill P (2007) Inhibition of DLL4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood 109(11):4753–4760
Yan M, Callahan C, Beyer J, Allamneni K, Zhang G, Ridgway J, Niessen K, Plowman G (2010) Chronic DLL4 blockade induces vascular neoplasms. Nature 463(7282):7
Folkman J (2002) Role of angiogenesis in tumor growth and metastasis. Semin Oncol 29(6, Supplement 16):15–18
Indraccolo S, Minuzzo S, Masiero M, Pusceddu I, Persano L, Moserle L, Reboldi A, Favaro E, Mecarozzi M, Di Mario G, Screpanti I, Ponzoni M, Doglioni C, Amadori A (2009) Cross-talk between tumor and endothelial cells involving the Notch3-DLL4 interaction marks escape from tumor dormancy. Cancer Res 69(4):1314–1323
Indraccolo S, Favaro E, Amadori A (2006) Dormant tumors awaken by a short-term angiogenic burst: the spike hypothesis. Cell Cycle 5(16):1751–1755
Almog N, Ma L, Raychowdhury R, Schwager C, Erber R, Short S, Hlatky L, Vajkoczy P, Huber PE, Folkman J, Abdollahi A (2009) Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res 69(3):836–844
Indraccolo S (2013) Insights into the regulation of tumor dormancy by angiogenesis in experimental tumors. Adv Exp Med Biol 734:37–52
Lai EC (2004) Notch signaling: control of cell communication and cell fate. Development 131(5):965–973
Bray SJ (2006) Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7(9):678–689
Zweidler-McKay P, Pear W (2004) Notch and T cell malignancy. Semin Cancer Biol 14(5):329–340
Hu YY, Zheng MH, Zhang R, Liang YM, Han H (2012) Notch signaling pathway and cancer metastasis. Adv Exp Med Biol 727:186–198
Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, Sriuranpong V, Iso T, Meszoely IM, Wolfe MS, Hruban RH, Ball DW, Schmid RM, Leach SD (2003) Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 3(6):565–576
van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, Clevers H (2005) Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435(7044):959–963
Wang Z, Li Y, Banerjee S, Kong D, Ahmad A, Nogueira V, Hay N, Sarkar FH (2010) Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-kappaB signaling pathways. J Cell Biochem 109(4):726–736
Weng AP, Ferrando AA, Lee W, Morris JPT, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306(5694):269–271
Bai F, Tagen M, Colotta C, Miller L, Fouladi M, Stewart CF (2010) Determination of the gamma-secretase inhibitor MK-0752 in human plasma by online extraction and electrospray tandem mass spectrometry (HTLC-ESI-MS/MS). J Chromatogr B Analyt Technol Biomed Life Sci 878(25):2348–2352
Cheng X, O’Neill HC (2009) Oncogenesis and cancer stem cells: current opinions and future directions. J Cell Mol Med 13(11–12):4377–4384
Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, Packer RJ, Goldman S, Gururangan S, Gajjar A, Demuth T, Kun LE, Boyett JM, Gilbertson RJ (2011) Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol 29(26):3529–3534
Mazumdar J, Dondeti V, Simon MC (2009) Hypoxia-inducible factors in stem cells and cancer. J Cell Mol Med 13(11–12):4319–4328
Shih Ie M, Wang TL (2007) Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res 67(5):1879–1882
Wang Z, Li Y, Banerjee S, Sarkar FH (2009) Emerging role of Notch in stem cells and cancer. Cancer Lett 279(1):8–12
Zweidler-McKay PA, He Y, Xu L, Rodriguez CG, Karnell FG, Carpenter AC, Aster JC, Allman D, Pear WS (2005) Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood 106(12):3898–3906
Kannan S, Sutphin RM, Hall MG, Golfman LS, Fang W, Nolo RM, Akers LJ, Hammitt RA, McMurray JS, Kornblau SM, Melnick AM, Figueroa ME, Zweidler-McKay PA (2013) Notch activation inhibits AML growth and survival: a potential therapeutic approach. J Exp Med 210(2):321–337
Proweller A, Tu L, Lepore JJ, Cheng L, Lu MM, Seykora J, Millar SE, Pear WS, Parmacek MS (2006) Impaired Notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res 66(15):7438–7444
Zage P, Nolo R, Fang W, Stewart J, Garcia-Manero G, Zweidler-McKay P (2012) Notch pathway activation induces neuroblastoma tumor cell growth arrest. Pediatr Blood Cancer 58(5):682–689
Kunnimalaiyaan M, Chen H (2007) Tumor suppressor role of Notch-1 signaling in neuroendocrine tumors. Oncologist 12(5):535–542
Dumont AG, Yang Y, Reynoso D, Katz D, Trent JC, Hughes DP (2012) Anti-tumor effects of the Notch pathway in gastrointestinal stromal tumors. Carcinogenesis 33(9):1674–1683
Hughes DP (2009) How the NOTCH pathway contributes to the ability of osteosarcoma cells to metastasize. Cancer Treat Res 152:479–496
Capaccione KM, Pine SR (2013) The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 34(7):1420–1430
Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li Y-M, Eberhart CG (2006) Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res 66(15):7445–7452
Farnie G, Clarke R (2007) Mammary stem cells and breast cancer – role of Notch signalling. Stem Cell Rev 3(2):169–175
Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP, Wicha MS (2013) Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res 19(8):1972–1980
Korkaya H, Wicha MS (2009) HER-2, Notch, and breast cancer stem cells: targeting an axis of evil. Clin Cancer Res 15(6):1845–1847
Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, Schuster K, Shao C, Larsen JE, Sullivan LA, Honorio S, Xie Y, Scaglioni PP, DiMaio JM, Gazdar AF, Shay JW, Wistuba II, Minna JD (2010) Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res 70(23):9937–9948
Wang J, Sullenger B, Rich J (2012) Notch signaling in cancer stem cells. Adv Exp Med Biol 727:174–185
Adhikari AS, Agarwal N, Wood BM, Porretta C, Ruiz B, Pochampally RR, Iwakuma T (2010) CD117 and Stro-1 identify osteosarcoma tumor-initiating cells associated with metastasis and drug resistance. Cancer Res 70(11):4602–4612
Williams S, Maecker H, French D, Liu J, Gregg A, Silverstein L, Cao T, Carano R, Dixit V (2011) USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell 146(6):918–930
Wang L, Park P, Lin C (2009) Characterization of stem cell attributes in human osteosarcoma cell lines. Cancer Biol Ther 8(6):543–552
Siclari V, Qin L (2010) Targeting the osteosarcoma cancer stem cell. J Orthop Surg Res 5:78
Gibbs C, Levings P, Ghivizzani S (2011) Evidence for the osteosarcoma stem cell. Curr Orthop Pract 22(4):322–326
Di Fiore R, Fanale D, Drago-Ferrante R, Chiaradonna F, Giuliano M, De Blasio A, Amodeo V, Corsini L, Bazan V, Tesoriere G, Vento R, Russo A (2012) Genetic and molecular characterization of the human osteosarcoma 3AB-OS cancer stem cell line: a possible model for studying osteosarcoma origin and stemness. J Cell Physiol 228(6):1189–1201
Di Fiore R, Santulli A, Ferrante R, Giuliano M, De Blasio A, Messina C, Pirozzi G, Tirino V, Tesoriere G, Vento R (2009) Identification and expansion of human osteosarcoma-cancer-stem cells by long-term 3-aminobenzamide treatment. J Cell Physiol 219(2):301–313
Tang Q, Zhao Z, Li J, Liang Y, Yin J, Zou C, Xie X, Zeng Y, Shen J, Kang T, Wang J (2011) Salinomycin inhibits osteosarcoma by targeting its tumor stem cells. Cancer Lett 311(1):113–121
Wilson H, Huelsmeyer M, Chun R, Young K, Friedrichs K, Argyle D (2008) Isolation and characterisation of cancer stem cells from canine osteosarcoma. Vet J 175(1):69–75
Martins-Neves S, Lopes Á, do Carmo A, Paiva A, Simoes P, Abrunhosa A, Gomes C (2012) Therapeutic implications of an enriched cancer stem-like cell population in a human osteosarcoma cell line. BMC Cancer 12:139
Liu B, Ma W, Jha R, Gurung K (2011) Cancer stem cells in osteosarcoma: recent progress and perspective. Acta Oncol 50(8):1142–1150
Basu-Roy U, Basilico C, Mansukhani A (2012) Perspectives on cancer stem cells in osteosarcoma. Cancer Lett 338(1):158–167
Wang L, Park P, Zhang H, La Marca F, Lin C (2011) Prospective identification of tumorigenic osteosarcoma cancer stem cells in OS99-1 cells based on high aldehyde dehydrogenase activity. Int J Cancer 128(2):294–303
Wang L, Park P, Zhang H, La Marca F, Claeson A, Valdivia J, Lin C (2011) BMP-2 inhibits the tumorigenicity of cancer stem cells in human osteosarcoma OS99-1 cell line. Cancer Biol Ther 11(5):457–463
Rainusso N, Man T, Lau C, Hicks J, Shen J, Yu A, Wang L, Rosen J (2011) Identification and gene expression profiling of tumor-initiating cells isolated from human osteosarcoma cell lines in an orthotopic mouse model. Cancer Biol Ther 12(4):278–287
Yang M, Yan M, Zhang R, Li J, Luo Z (2011) Side population cells isolated from human osteosarcoma are enriched with tumor-initiating cells. Cancer Sci 102(10):1774–1781
Huang Y, Dai H, Guo Q (2012) TSSC3 overexpression reduces stemness and induces apoptosis of osteosarcoma tumor-initiating cells. Apoptosis 17(8):749–761
Khanna C, Prehn J, Yeung C, Caylor J, Tsokos M, Helman L (2000) An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clin Exp Meta 18(3):261–271
Khanna C, Khan J, Nguyen P, Prehn J, Caylor J, Yeung C, Trepel J, Meltzer P, Helman L (2001) Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res 61(9):3750–3759
Weiss KR (2010) Inhibition of osteosarcoma metastatic potential with noggin and s-Flt. In: 2010 Meeting of the Musculoskeletal Tumor Society, Philadelphia, PA, 2010
Weiss KR, Cooper GM, Jadlowiec JA, McGough RL 3rd, Huard J (2006) VEGF and BMP expression in mouse osteosarcoma cells. Clin Orthop Relat Res 450:111–117
Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, Houvenaeghel G, Extra JM, Bertucci F, Jacquemier J, Xerri L, Dontu G, Stassi G, Xiao Y, Barsky SH, Birnbaum D, Viens P, Wicha MS (2010) Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res 16(1):45–55
Cheung AM, Wan TS, Leung JC, Chan LY, Huang H, Kwong YL, Liang R, Leung AY (2007) Aldehyde dehydrogenase activity in leukemic blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia 21(7):1423–1430
Cho HJ, Lee TS, Park JB, Park KK, Choe JY, Sin DI, Park YY, Moon YS, Lee KG, Yeo JH, Han SM, Cho YS, Choi MR, Park NG, Lee YS, Chang YC (2007) Disulfiram suppresses invasive ability of osteosarcoma cells via the inhibition of MMP-2 and MMP-9 expression. J Biochem Mol Biol 40(6):1069–1076
Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, Fields JZ, Wicha MS, Boman BM (2009) Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res 69(8):3382–3389
Honoki K, Fujii H, Kubo A, Kido A, Mori T, Tanaka Y, Tsujiuchi T (2010) Possible involvement of stem-like populations with elevated ALDH1 in sarcomas for chemotherapeutic drug resistance. Oncol Rep 24(2):501–505
Mu X, Isaac C, Schott T, Huard J, Weiss K (2013) Rapamycin Inhibits ALDH Activity, Resistance to Oxidative Stress, and Metastatic Potential in Murine Osteosarcoma Cells. Sarcoma 2013:11
Hughes DP (2009) Strategies for the targeted delivery of therapeutics for osteosarcoma. Expert Opin Drug Deliv 6(12):1311–1321
Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE, The Mouse Genome Database Group (2014) The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res 42(D1):D810–D817
Jiang R, Lan Y, Chapman HD, Shawber C, Norton CR, Serreze DV, Weinmaster G, Gridley T (1998) Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev 12(7):1046–1057
Sorensen I, Adams RH, Gossler A (2009) DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 113(22):5680–5688. doi:10.1182/blood-2008-08-174508
Dunwoodie SL, Clements M, Sparrow DB, Sa X, Conlon RA, Beddington RS (2002) Axial skeletal defects caused by mutation in the spondylocostal dysplasia/pudgy gene Dll3 are associated with disruption of the segmentation clock within the presomitic mesoderm. Development 129(7):1795–1806
Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A (2004) Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev 18(22):2730–2735
Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S (1997) Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 89(4):629–639
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
McManus, M.M., Weiss, K.R., Hughes, D.P.M. (2014). Understanding the Role of Notch in Osteosarcoma. In: Kleinerman, M.D., E. (eds) Current Advances in Osteosarcoma. Advances in Experimental Medicine and Biology, vol 804. Springer, Cham. https://doi.org/10.1007/978-3-319-04843-7_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-04843-7_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-04842-0
Online ISBN: 978-3-319-04843-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)