
Abstract
Interleukin (IL)-18, a member of the IL-1 family of cytokines, has emerged as a key regulator of mucosal homeostasis within the gastrointestinal tract. Like other members of this family, IL-18 is secreted as an inactive protein and is processed into its active form by caspase-1, although other contributors to precursor processing are emerging.
Numerous studies have evaluated the role of IL-18 within the gastrointestinal tract using genetic or complementary pharmacological tools and have revealed multiple roles in tumorigenesis. Most striking among these are the divergent roles for IL-18 in colon and gastric cancers. Here, we review our current understanding of IL-18 biology and how this applies to colorectal and gastric cancers.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cancer
- Colon
- Caspase-1
- Cytokines
- Gastric
- Inflammation
- IL-1
- IL-12
- IL-18
- IL-18BP
- IFNγ, Gastrointestinal
- Microenvironment
- NfkB
- Therapeutics
5.1 Introduction
The development of cancer is a multistep process whereby normal cells acquire characteristics that promote malignant transformation. The “hallmarks of cancer” describe a series of processes that enable a cancer cell to resist death, evade growth suppressors, induce angiogenesis, continually proliferate, avoid immune destruction, deregulate cell metabolism, and invade and metastasize to other organs [1]. Tumor-promoting inflammation is also an important cancer hallmark, with innate immune cells including macrophages, dendritic cells (DC), and natural killer (NK) cells, as well as adaptive immune cells including T and B cells present in the tumor microenvironment. These tumor-infiltrating immune cells are the primary source of cytokines that can further perpetuate tumor development [2]. In this review, we discuss the biology of IL-18, its receptor, and our current appreciation of its immunoregulatory functions in cancer. We focus on the expression and role of IL-18 in two gastrointestinal (GI) cancers, colorectal cancer (CRC) and gastric cancer (GC).
5.2 The Biology of IL-18
IL-18 is a pro-inflammatory and immunoregulatory cytokine that belongs to the IL-1 superfamily, which also includes IL-1α and β; IL-33; IL-36α, β, and γ; IL-1Rα; IL-36Rα; and IL-38 [3]. IL-18 was discovered in 1989 as “IFN-γ-inducing factor,” following the observation that it induced production of IFN-γ in mouse spleen cells following injection with bacterial lipopolysaccharide (LPS) [4]. Subsequently, murine IL-18 was purified and cloned from liver cells of mice treated with Propionibacterium acnes (P. acnes) in 1995 [5].
5.2.1 IL-18 Is a Beta-Trefoil Structured Protein
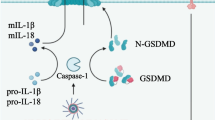
Cloned murine and human IL-18 cDNA encodes proteins consisting of 192 and 193 amino acids, respectively, and shares approximately 65% sequence homology with each other [5,6,7]. Similar to IL-1β, IL-18 lacks the usual leader sequence necessary for secretion from the cell membrane at the N-terminus, so it is synthesized as a 24-kDa inactive precursor protein (pro-IL-18), which becomes active following cleavage by the IL-1β-converting enzyme, also known as caspase-1 [8]. The crystal structure of IL-18 highlights that it folds into a β-trefoil sheet, similar to IL-1β, although the surface residues are dissimilar [6].
5.2.2 Multiple Mechanisms of IL-18 Secretion
Using in vivo murine models, it was shown that the intracellular cysteine protease caspase-1 cleaves pro-IL-18 at an Asp35-Asn36 cleavage site, creating a bioactive protein that is secreted from the cell [8]. Subsequent experiments demonstrated that caspase-1 was essential for secretion of IL-18, as serum IL-18 was not increased in P. acnes-primed caspase-1-deficient mice (caspase-1−/−) following LPS challenge when compared to wild-type control mice (caspase-1+/+) [8]. In addition, transgenic mice overexpressing human caspase-1 showed elevated serum levels of mature IL-18, further suggesting an important role for caspase-1 in mature IL-18 production [9].
More recently, caspase-1-independent IL-18 production has been documented [10]. This is facilitated by proteinase-3 (PR3), a 29-kDa serine proteinase, that is able to induce the production of mature IL-18 after IFN-γ priming in epithelial cells [10]. In combination with a proteinase inhibitor, IFN-γ-primed cells treated with PR3 did not produce IL-18, suggesting that the extracellular enzymatic activity of PR3 contributes to pro-IL-18 cleavage [10]. Most importantly, using a caspase-1-specific inhibitor revealed that PR3-induced mature IL-18 production was independent of caspase-1 [10].
In addition, Fas signaling which triggers apoptosis and induces the production of many pro-inflammatory cytokines has been shown to lead to an increase in IL-18 [11]. P. acnes-primed macrophages can secrete mature IL-18 upon stimulation with Fas ligand independent of caspase-1 [11]. It has also been shown that Fas signaling activates capase-8 in macrophages and dendritic cells and leads to the maturation of IL-18 independent of inflammasome-induced capase-1 pathway [12].
Finally, upon stimulation with vascular endothelial growth factor (VEGF)-D, IL-18 is secreted in GC cell lines and is linked to a metalloprotease 33 (ADAM33) which has a metalloprotease activity [13]. VEGF-D-induced IL-18 secretion is inhibited by knocking down ADAM33 expression, suggesting ADAM33 could be a novel regulator for mature IL-18 secretion [13].
5.2.3 IL-18-Producing Cells in the Gastrointestinal Tract
Pro-IL-18 is constitutively expressed by numerous cell types within the GI tract, including endothelial cells, keratinocytes, and intestinal epithelial cells [14]. Recent studies have shown that IL-22 can induce the expression of pro-IL-18 in intestinal epithelial cells and in turn provides protection against gut bacterial infections [15]. However, both hemopoietic and epithelial cell-derived mature IL-18 have been shown to mediate intestinal inflammation in a murine DSS-induced experimental colitis model [16]. Interestingly, elevated human IL-18 mRNA transcripts were found in both epithelial cells and other mucosal cells from patients with Crohn’s disease (CD) and ulcerative colitis (UC) [17]. In addition, immunohistochemical staining of IL-18 was increased mainly in epithelial cells from severely inflamed UC patients compared to that of healthy control patients [17]. On the other hand, intense staining of IL-18 was found in both the colonic epithelium and inflammatory cells, including macrophages and dendritic cells, in the lamina propria of CD patients [17]. These results suggest that IL-18 may play a different role in UC and CD.
5.3 IL-18 Signaling
The receptor for IL-18, IL-18R, was first purified and characterized from a Hodgkin’s disease cell line L428 in 1997 [18]. IL-18R forms a heterodimer at the cell surface, which contains an α chain (IL-18R1) responsible for extracellular binding of IL-18 and a nonbinding signal-transducing β chain (IL-18RAP) [19, 20]. As the binding of IL-18 and IL-18R1 is a low-affinity interaction, the expression of a co-receptor, IL-18RAP, is required for functional IL-18 signaling [18]. Murine IL-18R1 is 65% homologous to human IL-18R1 in overall amino acid sequence [18]. It was previously reported that the binary complex of human IL-18/IL18-R1 is identical to that of IL-1β and its receptor; however, X-ray crystallography of the human tertiary complex IL-18/IL18-R1/IL-18RAP revealed substantial differences from IL-1β and its receptor [6, 21]. For example, the second domain (D2) of the two IL-18Rs lacks one β-strand, which is conserved among other IL-1-related receptors and was shown to contribute to the inter-receptor interaction [21]. This could explain the binding specificity of IL-18 and its receptor.
5.3.1 Expression of IL-18R in the Gastrointestinal Tract
In the GI tract, IL-18R is expressed by multiple cell types, including neutrophils, T cells, NK cells, macrophages, and endothelial cells [22]. IL-18R is also expressed by colonic epithelial cells, with genetic ablation of IL-18R protecting mice from DSS-induced experimental colitis [16]. IL-18R is also expressed on the surface of T cells isolated from both UC and CD patients [23].
5.3.2 IL-18 Activates Multiple Signaling Pathways
Upon engagement of the IL-18 signaling complex (Fig. 5.1), IL-1R-associated kinase (IRAK) is recruited via the adaptor protein, myeloid differentiation primary response 88 (MyD88). IRAK then dissociates from the receptor complex and interacts with TNF-receptor-associated factor (TRAF6), which then relays the signal via NF-κB-inducing kinase (NIK) to two I-κB kinases (IKK-1 and IKK-2) leading to NF-κB activation [22]. The requirement of each component for the signaling pathway has been demonstrated using mice deficient in each individual molecule. For example, IL-18-induced IFN-γ production was abolished in MyD88-deficient mice, while IL-18-induced NF-κB and JNK activation was blocked in MyD88-deficient Th1 cells [24]. These findings suggest that MyD88 is essential for IL-18-mediated signaling and function. Most importantly, co-immunoprecipitation revealed an interaction between MyD88 and IL-18R1 and that this interaction was required to induce NF-κB and AP-1 activation [24]. Similarly, IL-18-induced IFN-γ production was impaired in IRAK-deficient mice in response to P. acnes and LPS stimulation [25]. Moreover, IRAK-deficient Th1 and NK cells also exhibit defects in IFN-γ production, which is induced by IL-18 [25]. Additionally, IL-18-stimulated mouse lymphoma cells, EL-4, showed recruitment of IRAK to IL-18R1 and the formation of complex between IRAK and TRAF6 [26].

Schematic of IL-18 signaling. IL-18 forms a signaling complex by binding to the IL-18R1 which then recruits the co-receptor, IL-18RAP, to form a high-affinity complex [14]. Upon ligand binding and the formation of the heterodimer, the Toll–IL-1-receptor (TIR) domains of the receptor trigger the binding of a signaling molecule, MyD88 [22]. The death domain of MyD88 interacts and phosphorylates IRAK4, which then associates with TRAF-6 [22]. This results in the activation of IKK and finally NFκB, which translocates to the nucleus and regulates gene expression. Alternatively, TRAF-6 can activate ERK and JUN which will translocate to the nucleus and regulate gene expression through binding to AP1. The IL-18BP is a negative regulator, present in the extracellular compartment where it binds to mature IL-18 and prevents binding to the IL-18 receptor
Increasing evidence suggests that IL-18 can activate other signaling pathways including mitogen-activated protein kinase (MAPK) p38. Stimulation of IL-18 in epithelial [27] and hippocampal cell lines [28] showed enhanced phosphorylation of MAPKp38 and STAT3. Interestingly, IL-18-induced IFN-γ production is mainly mediated through MAPK and STAT3 pathway in NK cells [29].
5.3.3 Negative Regulation: IL-18 Binding Protein
IL-18 binding protein (BP) was discovered from human urine as a soluble receptor for IL-18 [30]. It was then isolated, characterized, and cloned in 1999 [30]. IL-18BP is a constitutively expressed and secreted protein that is found at high levels in the serum of healthy humans [31]. It was found that IL-18BP binds IL-18 with an affinity significantly higher than that of IL-18R1 [30]. Most importantly, IL-18BP blocks IL-18-induced IFN-γ production in vitro and in vivo, suggesting that IL-18BP is a natural negative regulator of IL-18-mediated immune responses [30]. Indeed, stimulation of different cell lines with IFN-γ induces the expression of IL-18BP via IFN regulatory factor 1 activation [32, 33]. These results suggest that the activity of IL-18 is modulated by a negative feedback loop which is mediated by IFN-γ-induced IL-18BP.
5.4 Immunomodulatory Role of IL-18 in the Gastrointestinal Tract
The emergence of a role for IL-18 as an immunoregulatory cytokine originated from the observation that it modulates IFN-γ production from Th1 and NK cells [34, 35]. Subsequently, other functions of IL-18 in these immune cell types, as well as other T cell subsets, have been explored.
5.4.1 Synergistic Action of IL-18 and IL-12 in IFN-γ-Producing Th1 Cells
IL-18 itself is not able to induce Th1 differentiation since naïve CD4+ T cells do not express IL-18R; however, IL-18 synergizes with IL-12 to promote IFN-γ production in murine T cells in vitro [35]. This was then confirmed in IL-18-deficient (Il18−/−) mice which exhibited reduced IFN-γ production by Th1 cells [34]. Furthermore, IL-18- and IL-12-double-deficient (Il12−/−;Il18−/−) mice showed a severely defective Th1 response when compared to the single cytokine-deficient mice [34]. This synergistic effect is mediated by the induction of IL-18R on the surface of Th1, but not Th2, cells by IL-12 [36, 37]. Moreover, it is believed that IFN-γ production by IL-18 and IL-12 is mediated at the transcriptional level [38]. IL-12-dependent IFN-γ activation requires both AP-1 and STAT4 binding sites in the IFN-γ promoter, while IL-18 activates the IFN-γ promoter through AP-1 and NFκB binding sites [22, 38].
Similarly, human T cells also require both IL-18 and IL-12 for optimal production of IFN-γ [39]. Additionally, IL-12 can induce IL-18R expression in human CD4+ T cells and triggers dose-dependent production of IFN-γ following IL-18 stimulation [39].
5.4.2 IL-18 Upregulates NK Cell Cytotoxic Activity
IL-18 enhances NK cell cytotoxic activity in vitro, and this activity is further augmented when co-stimulated with IL-12 [39]. Although Il18−/− mice have similar numbers of NK cells compared to wild-type mice, Il18−/− mice showed reduced NK cell activity, and Il12−/−;Il18−/− mice showed a marked reduction in NK cell activity [39]. This observation suggested that the cooperative action of both IL-12 and IL-18 is essential for cytolytic function of NK cells. However, unlike the IFN-γ production activity, IL-18 can facilitate NK cell activity in the absence of IL-12, demonstrated by the lack of change in NK cell activity following IL-18 stimulation in IL-12-deficient mice [40]. This can be explained by the expression of both IL-18R and IL-12R on NK cells [40]. Consistent with murine IL-18, human IL-18 can also induce the cytotoxic activity of NK cells [41]. Instead of IL-12, IL-18 synergizes with IL-2 to enhance the IFN-γ-production, cytotoxic activity, and expansion of NK cells in human blood [41]. Interestingly, IL-2 receptor was not expressed on NK cells, but its expression was substantially upregulated when isolated human NK cells were cultured with both IL-18 and IL-2 [42].
5.4.3 Effect of IL-18 on Other Immune Cell Populations
The role for IL-18 in the behavior of CD8+ T cells is controversial. Several studies have shown that IL-18 and IL-12 mediated cytotoxic activity, including IFN-γ and granzyme production, of CD8+ T cells using murine spleen cell cultures [43]. Moreover, proliferation of CD8+ T cells was impaired in Il18−/− mice, and blocking IL-18 signaling in wild-type CD8+ T cells showed reduced live cells, suggesting that IL-18 promotes survival of CD8+ T cells [44]. Flow cytometry analysis demonstrated the upregulation of IL-18RAP on anti-CD3-activated CD8+ T cells when compared to naïve CD8+ T cells [44]. However, in vivo studies showed that CD8+ T cell numbers were not affected in IL-18R-deficient (Il18r−/−) mice following infection with the parasite Trypanosoma cruzi [45].
It has also been reported that there is an increase in frequency of Th17 cells in the lamina propria of Il18−/− and Il18r−/− mice, suggesting that IL-18 limits Rorγt+ Th17 cell differentiation [46]. Moreover, IL-18 is not required for Foxp3+ Treg cell differentiation but promotes optimal Treg cell function by enhancing the expression of key effector molecules, including Furin, Bcl11b, Tnfrsf4, and Stat3 in the colonic lamina propria [46]. These molecules have been shown to play a crucial role in suppressing T cell-mediated colitis by Treg cells [46]. However, a recent study suggested that NLRP1/IL-18 signaling regulates the gut microbiome rather than immune response in a mouse colitis model [47].
5.5 The Role of IL18 in the Gastrointestinal Microenvironment: From Homeostasis to Tumorigenesis
5.5.1 Microbial Colonization
In order to maintain a homeostatic state in the gut, several mechanisms are required to limit the interaction with microbiota in the lumen. These mechanisms include a physical barrier provided by epithelial cells, the production of antimicrobial proteins, and a mucus layer produced by goblet cells. The gut microbiota has been shown to induce activation of the inflammasome, as germ-free mice showed abolished caspase-1 cleavage and a subsequent reduction in IL-18 production [48]. Interestingly, at steady state, the colonic tissue from Il18−/− mice showed reduced expression of antimicrobial peptides (AMPs), in particular angiogenin-4 (Ang4), when compared to WT mice [48]. These results suggest that IL-18 helps maintain gut homeostasis through induction of AMPs. Moreover, it has also been shown that IL-18-mediated AMP production is downstream of the NLRP6 inflammasome, rather than the NLRP3 inflammasome [48]. Most importantly, specific metabolites from the microbiome activate an NLRP6-IL-18-AMP axis which, in turn, optimize commensal colonization and persistence [49]. Nlrp6-deficient mice had concomitant dysbiosis, characterized by an expansion of bacterial phyla Bacteroidetes and TM7 [48]. Interestingly, nlrp1-deficient mice (nlrp1−/−) are less susceptible to a murine model of acute colitis, although these mice have no difference in immune cell infiltration, in particular Th1 and Th17, in the colon when compared to their control [47]. Instead, nlrp1−/−-deficient mice have increased butyrate-producing Clostridiales in the gut, suggesting that the NLRP1/IL-18 axis promotes IBD through limiting butyrate-producing microbiome [47]. Taken together, these results suggest that IL-18 is a crucial regulator of mucosal homeostasis.
5.5.2 Inflammatory Bowel Disease
Colitis is one of the predisposing factors for tumorigenesis. IL-18 is highly expressed in both the colonic epithelium and immune cells within the lamina propria of inflammatory bowel disease (IBD) patients [17], suggesting that IL-18 could play a role in contributing to the pathogenesis of these diseases. CD is an IBD associated with a Th1 cytokine profile and accumulation of IL-12 [50]. One animal model of CD is induced by 2,4,6-trinitrobenzene sulfonic acid (TNBS), which disrupts the mucosal epithelial barrier and modifies cell surface proteins [51]. In this model, administration of a neutralizing antibody against IL-18 resulted in less weight-loss and decreased histological scores and inflammation compared to control mice [51]. Similarly, TNBS immunization in Il18−/− mice showed a reduction in colonic thickening and immune cell infiltration in the colon when compared to Il18+/+ mice [51].
In contrast to CD, UC is an IBD associated with a Th2 cytokine profile. A commonly used murine UC model is induced by dextran sulfate sodium (DSS), which damages the epithelial barrier defenses against luminal bacterial products. In this model, administration of DSS to wild-type mice together with a neutralizing antibody against IL-18 [52], and the negative regulator, IL-18BP [53], showed reduced disease severity including decreased weight-loss, clinical scores, rectal bleeding, and inflammation when compared to controls [52, 53]. These results suggest that IL-18 causes detrimental effects in this model .
As mentioned previously, mature IL-18 production requires caspase-1; therefore, studies have also investigated in the role of caspase-1 in IBD using caspase-1-deficient (caspase1−/−) mice. In one study, it was shown that caspase1−/− mice are protected from DSS-induced colitis, which is consistent with DSS mice treated with an antibody blocking IL-18 signaling [52, 54]. However, conflicting results have been reported whereby caspase1−/− mice had increased disease in the DSS-induced colitis model [55]. The disparity in these results may be attributed to differences in the intestinal microflora between animal facilities. In caspase1−/− mice, IL-18 was barely detected in serum, suggesting that the caspase-1 protective effect is due to IL-18 production [55]. To confirm this, recombinant IL-18 was administered to caspase1−/− mice, which reversed the phenotype, suggesting that the caspase-1/IL-18 axis is protective [55]. Furthermore, inflammasomes such as NLRP3 [56] and NLRP6 [57], which are required for mature IL-18 production, also showed worsening of disease following DSS treatment. Similarly, Il18−/− and Il18r−/− mice developed severe colitis associated with high lethality and increased histopathological abnormalities compared with control mice [58], suggesting that IL-18 is required for gut barrier protection.
In order to resolve the conflicting role of IL-18 in DSS-induced colitis, IL-18- and IL-18R-deficient tissue-specific mice were developed to delineate the involvement of IL-18 in epithelial and hematopoietic cells [16]. Mice with deletion of IL-18R in epithelial cells showed protection against DSS-induced colitis, suggesting that IL-18 signaling may disrupt the epithelial cell barrier [16]. Most importantly, mice deficient for IL-18BP showed severe colitis and progressive loss of mature goblet cells, which could be reversed by deleting the epithelial IL-18R in these mice [16]. It was also shown that IL-18 could disrupt the maturation of goblet cells through transcriptional regulation of goblet cell differentiation factors [16]. These results suggest that IL-18 is the key regulator for goblet cell dysfunction, which may be a key pathological event in human UC [59].
5.5.3 Anti-tumorigenic Role for IL-18 in Colon Cancer
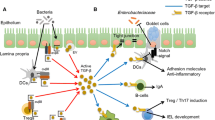
Humans heterozygous for the IL-18 A607C polymorphism exhibit increased risk of CRC development [60]. Since IL-18 has a strong immunoregulatory role associated with the production of IFN-γ by Th1 cells and enhances the cytolytic activity of CD8+ cells and NK cells, it has been suggested that IL-18 may play an important role in anti-tumor immunity (Fig. 5.2).

Schematic of IL-18 signaling in GI tract tumorigenesis. In both colon and gastric cancers, IL-18 is mainly produced by epithelial cells. (Left) In colon cancer, IL-18 has an anti-tumorigenic role through cooperation with IL-12 resulting in IFN-γ production by Th1 cells and NK cells. Moreover, IL-18 acts on intestinal epithelial cells to trigger the release of antimicrobial peptides (AMPs) regulating the microbiota and maintaining gut barrier homeostasis [48]. (Right) In contrast, IL-18 has a pro-tumorigenic role in gastric cancer, which includes activation of the inflammasome [69]. However, the role of microbes/IL-18/inflammasome in gastric cancer has not yet been explored
The role of IL-18 in CRC has mainly been explored using the azoxymethane (AOM) DSS model, in which cyclic administration of DSS in the drinking water following the administration of AOM results in the formation of colonic tumors [61]. DSS-induced damage of epithelial cells leads to the activation of inflammasome-induced inflammation through NLRP3, NLRP6, caspase-1, and IL-18 [56]. In this model, Il18−/− mice showed increased tumor number and burden, which was associated with severe colitis histopathology compared with wild-type mice [56]. The increase in tumorigenesis was attributed to the lack of IFN-γ production in this model. In addition, mice deficient in Nlrp3, Nlrp6, and caspase-1 also developed more tumors and had defective IL-18 production, suggesting that the inflammasome provides protection against tumor formation via IL-18 [56]. Similarly, myeloid cell-restricted Card9-deficient mice have been used to demonstrate the crucial role of inflammation-induced IL-18 production by myeloid cells in colon cancer development [62]. In this model, the commensal gut fungi recognition signaling pathway, the caspase recruitment domain 9/spleen tyrosine kinase (Card9-Syk) axis, was abrogated.
Approximately 40% of CRC patients progress to fatal metastasis (80% being confined to the liver); therefore, the role of the inflammasome and IL-18 in CRC metastasis has also recently been explored [63]. In order to mimic liver metastasis in CRC, a mouse model of intrasplenic injection of transplantable tumor cells derived from a murine primary colon carcinoma (MC38) is often used. In this model, mice deficient in Nlrp3 showed reduced IL-18 production and increased liver metastasis [63]. Moreover, Il18−/− and Il18r−/− mice also developed an increase in liver metastasis. This was attributed to IL-18-mediated immune surveillance being independent to T and B cells but dependent on hepatic NK cell maturation and priming Fas-mediated cytotoxicity [63]. In line with this, treatment of NK cells with recombinant IL-18 enhances NK cell-mediated death and eliminates tumor cells in a co-culture system [64]. More recently, IL-18 has been shown to promote the anti-tumor properties of eosinophils against colon cell lines by enhancing cell-cell contact between the two cell types [65].
5.5.4 Pro-tumorigenic Role for IL-18 in Gastric Cancer
The expression of IL-18 is elevated in the sera of patients with GC and CRC [66, 67]. IL-18 is also expressed in both GC and CRC biopsies, but in CRC its expression does not correlate with survival [68]. However, in contrast to CRC, a number of studies examining IL-18 in GC have suggested a pro-tumorigenic role for this cytokine [69, 70] (Fig. 5.2). Initial studies demonstrated that administration of recombinant IL-18 increased cell proliferation in four GC cell lines (MKN28, MKN45, NUGC3, and KATOIII) through the activation of NF-κB [70]. This was consistent with observations made in xenograft experiments, where transplantation of GC cell lines into immunocompromised mice followed by administration of IL-18 resulted in a reduction in survival compared to control mice [70].
More recently, in addition to IL-18, components of the inflammasome upstream have also been demonstrated to have a pro-tumorigenic role in murine models of GC. Using the Gp130F/F model of GC, where mice develop spontaneous gastric adenomas from 4 weeks of age, it has been shown that mice deficient in IL-18 (Gp130F/F:Il18−/−) had reduced tumor burden, which was also observed in Asc-deficient mice (Gp130F/F:Asc−/−), signifying an important role of inflammasome activation in tumor formation in this model [69]. The reduction in tumor burden was found to not be due to a change in cell proliferation, as previous studies have suggested, but rather was a result of increased apoptosis of tumor cells due to increased expression of caspase 8, as well as a reduction in NF-κB activation [69]. However, unlike previous findings in vitro that demonstrated a role for immune cell responses in the tumor, no significant differences were found in immune subsets between Gp130F/F and Gp130F/F:Asc−/− mice, suggesting a dispensable role for inflammation in this model [69]. While these studies demonstrate an apparent pro-tumorigenic role for IL-18 in GC, it remains to be seen whether the same inflammasomes that are activated in CRC, such as NLRP3 and NLRP6, also have a role in GC.
IL-18 has also been implicated in metastasis in GC. In vitro and xenograft experiments have shown that IL-18 induces the expression of CD44, COX2, and VEGF, which were involved in metastasis of GC cell xenografts and angiogenesis [67]. Additionally, neutralization of IL-18 in these models through siRNA knockdown or administration of IL-18BP was found to significantly reduce tumor burden while promoting expression of CD70, which has been suggested to increase susceptibility of cancer cells to NK cell-mediated anti-tumor responses [67].
5.6 Therapeutic Activation or Inhibit IL-18 Signaling
Several studies have suggested that recombinant IL-18 may be a useful therapeutic modality for cancer. This stems from the observation that recombinant IL-18 administration to mice before or after inoculation of CL8-1, a mouse melanoma cell line, showed decreased tumor development associated with elevated serum IFN-γ levels [71]. A similar observation was obtained in mice inoculated with the Meth A sarcoma cell line, where all mice administered recombinant IL-18 survived 3 weeks longer than controls [72]. The anti-tumor effect in this model was abrogated when treating mice with a neutralizing antibody against NK cells, suggesting that IL-18 induced NK cell anti-tumor activity [72]. Other more recent studies have shown that IL-18 promoted the expansion of NK cells from lung cancer patients and enhanced the anti-tumor phenotype of NK cells [41]. In line with our understanding of the role of capase-1 in CRC, caspase-1 mice that received recombinant IL-18 in the AOM/DSS model showed mild disease with significantly less weight loss and less inflammation, ulceration, and hyperplasia on histological colon sections [56]. The combined use of IL-18 with a low dose of IL-2 showed a synergistic anti-tumor effect in mice subcutaneously inoculated with a sarcoma cell line, MCA205 [73]. This effect was also largely dependent on NK cell-mediated IFN-γ production [73]. Moreover, combined treatment of IL-18 and IL-12 also demonstrated anti-tumor efficacy in MCA205 tumor-bearing mice [71]. However, it was found that administration of both IL-18 and IL-12 resulted in a dose-dependent systemic inflammatory response in mice, with elevated serum levels of IFN-γ and other cytokines resulting in acute multiorgan pathology, particularly in the liver [74]. More recently, the combination of recombinant IL-18 and immune checkpoint inhibitors resulted in synergistic inhibition tumor growth without adverse events in animal models of peritoneal dissemination of CT-26 colon carcinoma, which was associated with the accumulation of NK cells [75].
5.6.1 Therapeutics in Clinical Development
Currently, there are a number of therapeutic agents in clinical trials that target the IL-18 signaling pathway. These include recombinant IL-18 (SB-485232) and IL-18BP (Tadekinig alfa) and a neutralizing IL-18 monoclonal antibody (GSK1070806).
SB-485232 is a recombinant IL-18 protein developed by GlaxoSmithKline (GSK), which has been trailed in patients with metastatic melanoma and non-Hodgkin’s lymphoma [76, 77]. In a separate phase II trial, stage IV metastatic melanoma patients were administered single agent SB-485232 at doses from 0.01 to 1.0 mg/kg each day for five consecutive days, followed by a 23-day rest period [77]. While the treatment was generally well tolerated, complete response was not observed in any of the patients, while only two patients achieved unconfirmed partial responses, suggesting a limited role for single-agent recombinant IL-18 in this disease. Combinatorial therapies involving IL-18 may yield more promising results, such as in a phase I clinical trial combining SB-485232 with the monoclonal antibody rituximab, a standard-of-care treatment, in patients with non-Hodgkin’s lymphoma [76]. In this context, responses were observed in approximately 25% of patients, with two patients achieving a complete response; however, further clinical trials would be required to determine proper efficacy of this treatment regime [76]. It is important to note that administration of recombinant IL-18 has been associated with adverse events. These included chills, fever, fatigue, and nausea, while more serious adverse events included pleural effusion, increased lipase, deep vein thrombosis, and pulmonary embolism [77]. Other reported complications include deep vein thrombosis and abnormal EKG; however, this was not attributed to the study drug.
The pharmaceutical company AB2 Bio has developed a human recombinant IL-18BP, called Tadekinig alfa, and has demonstrated that it was safe and well tolerated in phase I/Ib clinical trials and a phase II trial, with injection site reactions, upper airway infections, and arthralgia the most common adverse events reported [78]. And, recently, they have conducted a phase III clinical trial in patients with single-point mutations in the NLRC4 gene. These patients have gain-of-function mutations and give rise to severe, life-threatening systemic inflammation associated with extremely high levels of IL-18 (NCT03113760). However, trials in patients with IBD or cancer have not been investigated.
Recently, a phase II trial of a neutralizing IL-18 monoclonal antibody, GSK1070806, was registered for patients with moderate to severe CD after a phase I trial demonstrated safety in healthy and obese subjects [79]; however, results have yet to be reported. Additionally, this therapeutic will also be investigated in a phase II trial in Behcet’s disease, an inflammatory disease affecting the blood vessels, highlighting the potential for targeting the IL-18 pathway in other inflammatory conditions (NCT03522662).
5.6.2 Emerging Opportunities to Alter IL18 Signaling
As an alternative to systemic administration of recombinant protein, gene transfer technology has also been developed. Gene therapy works by inserting the gene of interest into a cell aiming to increase the amount of a specific gene in a targeted cell/tissue rather than systemically [80]. Interestingly, intravenous injection of plasmid encoding both IL-18 and IL-12 showed anti-tumor efficacy with diminished toxicity in mice [81]. Moreover, a cancer vaccine that delivered IL-18 also showed promising results in eradicating tumor cells in mice inoculated with murine colon cancer cell lines. This was associated with increased in CD8 and CD4 T cell infiltration in the tumor [82].
5.7 Concluding Remarks
IL-18 is clearly associated with a number of physiological functions throughout the GI tract. As further studies elucidate the cell-, disease-, and context-specific role of IL-18 signaling, the evolutionary significance of the divergent role of IL-18 in CRC and GC will become more apparent.
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
Nguyen PM, Putoczki TL (2018) Could the inhibition of IL-17 or IL-18 be a potential therapeutic opportunity for gastric cancer? Cytokine 118:8. https://doi.org/10.1016/j.cyto.2018.01.008
Garlanda C, Dinarello CA, Mantovani A (2013) The interleukin-1 family: back to the future. Immunity 39:1003–1018. https://doi.org/10.1016/j.immuni.2013.11.010
Nakamura K, Okamura H, Wada M, Nagata K, Tamura T (1989) Endotoxin-induced serum factor that stimulates gamma interferon production. Infect Immun 57:590–595
Okamura H et al (1995) Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 378:88–91. https://doi.org/10.1038/378088a0
Kato Z et al (2003) The structure and binding mode of interleukin-18. Nat Struct Biol 10:966–971. https://doi.org/10.1038/nsb993
Ushio S et al (1996) Cloning of the cDNA for human IFN-gamma-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. J Immunol 156:4274–4279
Ghayur T et al (1997) Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386:619–623. https://doi.org/10.1038/386619a0
Yamanaka K et al (2000) Skin-specific caspase-1-transgenic mice show cutaneous apoptosis and pre-endotoxin shock condition with a high serum level of IL-18. J Immunol 165:997–1003. https://doi.org/10.4049/jimmunol.165.2.997
Sugawara S et al (2001) Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol 167:6568–6575. https://doi.org/10.4049/jimmunol.167.11.6568
Tsutsui H et al (1999) Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity 11:359–367
Bossaller L et al (2012) Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol 189:5508–5512. https://doi.org/10.4049/jimmunol.1202121
Kim KE et al (2009) Expression of ADAM33 is a novel regulatory mechanism in IL-18-secreted process in gastric cancer. J Immunol 182:3548–3555. https://doi.org/10.4049/jimmunol.0801695
Dinarello CA, Novick D, Kim S, Kaplanski G (2013) Interleukin-18 and IL-18 binding protein. Front Immunol 4:289. https://doi.org/10.3389/fimmu.2013.00289
Munoz M et al (2015) Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity 42:321–331. https://doi.org/10.1016/j.immuni.2015.01.011
Nowarski R et al (2015) Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 163:1444–1456. https://doi.org/10.1016/j.cell.2015.10.072
Pizarro TT et al (1999) IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol 162:6829–6835
Torigoe K et al (1997) Purification and characterization of the human interleukin-18 receptor. J Biol Chem 272:25737–25742. https://doi.org/10.1074/jbc.272.41.25737
Born TL, Thomassen E, Bird TA, Sims JE (1998) Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. J Biol Chem 273:29445–29450. https://doi.org/10.1074/jbc.273.45.29445
Hoshino K et al (1999) Cutting edge: generation of IL-18 receptor-deficient mice: evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J Immunol 162:5041–5044
Tsutsumi N et al (2014) The structural basis for receptor recognition of human interleukin-18. Nat Commun 5:5340. https://doi.org/10.1038/ncomms6340
Akira S (2000) The role of IL-18 in innate immunity. Curr Opin Immunol 12:59–63
Kanai T et al (2000) Interleukin 18 is a potent proliferative factor for intestinal mucosal lymphocytes in Crohn’s disease. Gastroenterology 119:1514–1523
Adachi O et al (1998) Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143–150
Kanakaraj P et al (1999) Defective interleukin (IL)-18-mediated natural killer and T helper cell type 1 responses in IL-1 receptor-associated kinase (IRAK)-deficient mice. J Exp Med 189:1129–1138. https://doi.org/10.1084/jem.189.7.1129
Kojima H et al (1998) Interleukin-18 activates the IRAK-TRAF6 pathway in mouse EL-4 cells. Biochem Biophys Res Commun 244:183–186. https://doi.org/10.1006/bbrc.1998.8236
Lee JK et al (2004) Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A 101:8815–8820. https://doi.org/10.1073/pnas.0402800101
Alboni S et al (2014) Interleukin 18 activates MAPKs and STAT3 but not NF-kappaB in hippocampal HT-22 cells. Brain Behav Immun 40:85–94. https://doi.org/10.1016/j.bbi.2014.02.015
Kalina U et al (2000) IL-18 activates STAT3 in the natural killer cell line 92, augments cytotoxic activity, and mediates IFN-gamma production by the stress kinase p38 and by the extracellular regulated kinases p44erk-1 and p42erk-21. J Immunol 165:1307–1313. https://doi.org/10.4049/jimmunol.165.3.1307
Novick D et al (1999) Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity 10:127–136
Kim SH et al (2000) Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc Natl Acad Sci U S A 97:1190–1195. https://doi.org/10.1073/pnas.97.3.1190
Hurgin V, Novick D, Rubinstein M (2002) The promoter of IL-18 binding protein: activation by an IFN-gamma -induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein beta. Proc Natl Acad Sci U S A 99:16957–16962. https://doi.org/10.1073/pnas.262663399
Muhl H et al (2000) Interferon-gamma mediates gene expression of IL-18 binding protein in nonleukocytic cells. Biochem Biophys Res Commun 267:960–963. https://doi.org/10.1006/bbrc.1999.2064
Takeda K et al (1998) Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8:383–390
Robinson D et al (1997) IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity 7:571–581
Xu D et al (1998) Selective expression and functions of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells. J Exp Med 188:1485–1492. https://doi.org/10.1084/jem.188.8.1485
Yoshimoto T et al (1998) IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol 161:3400–3407
Barbulescu K et al (1998) IL-12 and IL-18 differentially regulate the transcriptional activity of the human IFN-gamma promoter in primary CD4+ T lymphocytes. J Immunol 160:3642–3647
Tominaga K et al (2000) IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int Immunol 12:151–160. https://doi.org/10.1093/intimm/12.2.151
Hyodo Y et al (1999) IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J Immunol 162:1662–1668
Senju H et al (2018) Effect of IL-18 on the expansion and phenotype of human natural killer cells: application to cancer immunotherapy. Int J Biol Sci 14:331–340. https://doi.org/10.7150/ijbs.22809
Son YI et al (2001) Interleukin-18 (IL-18) synergizes with IL-2 to enhance cytotoxicity, interferon-gamma production, and expansion of natural killer cells. Cancer Res 61:884–888
Okamoto I, Kohno K, Tanimoto T, Ikegami H, Kurimoto M (1999) Development of CD8+ effector T cells is differentially regulated by IL-18 and IL-12. J Immunol 162:3202–3211
Li W, Kashiwamura S, Ueda H, Sekiyama A, Okamura H (2007) Protection of CD8+ T cells from activation-induced cell death by IL-18. J Leukoc Biol 82:142–151. https://doi.org/10.1189/jlb.0706431
Oliveira AC et al (2017) Crucial role for T cell-intrinsic IL-18R-MyD88 signaling in cognate immune response to intracellular parasite infection. Elife 6. https://doi.org/10.7554/eLife.30883
Harrison OJ et al (2015) Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) Treg cell function in the intestine. Mucosal Immunol 8:1226–1236. https://doi.org/10.1038/mi.2015.13
Tye H et al (2018) NLRP1 restricts butyrate producing commensals to exacerbate inflammatory bowel disease. Nat Commun 9:3728. https://doi.org/10.1038/s41467-018-06125-0
Levy M et al (2015) Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163:1428–1443. https://doi.org/10.1016/j.cell.2015.10.048
Hand TW (2015) Interleukin-18: the bouncer at the mucosal bar. Cell 163:1310–1312. https://doi.org/10.1016/j.cell.2015.11.041
Peluso I, Pallone F, Monteleone G (2006) Interleukin-12 and Th1 immune response in Crohn’s disease: pathogenetic relevance and therapeutic implication. World J Gastroenterol 12:5606–5610. https://doi.org/10.3748/wjg.v12.i35.5606
Kanai T et al (2001) Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn’s disease. Gastroenterology 121:875–888. https://doi.org/10.1053/gast.2001.28021
Siegmund B et al (2001) Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol 281:R1264–R1273. https://doi.org/10.1152/ajpregu.2001.281.4.R1264
Sivakumar PV et al (2002) Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut 50:812–820. https://doi.org/10.1136/gut.50.6.812
Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA (2001) IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A 98:13249–13254. https://doi.org/10.1073/pnas.231473998
Dupaul-Chicoine J et al (2010) Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32:367–378. https://doi.org/10.1016/j.immuni.2010.02.012
Zaki MH et al (2010) The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32:379–391. https://doi.org/10.1016/j.immuni.2010.03.003
Elinav E et al (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145:745–757. https://doi.org/10.1016/j.cell.2011.04.022
Takagi H et al (2003) Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand J Gastroenterol 38:837–844
Gersemann M et al (2009) Differences in goblet cell differentiation between Crohn’s disease and ulcerative colitis. Differentiation 77:84–94. https://doi.org/10.1016/j.diff.2008.09.008
Nikiteas N, Yannopoulos A, Chatzitheofylaktou A, Tsigris C (2007) Heterozygosity for interleukin-18 -607 A/C polymorphism is associated with risk for colorectal cancer. Anticancer Res 27:3849–3853
Fung KY, Putoczki T (2018) In vivo models of inflammatory bowel disease and colitis-associated cancer. Methods Mol Biol 1725:3–13. https://doi.org/10.1007/978-1-4939-7568-6_1
Malik A et al (2018) SYK-CARD9 signaling axis promotes gut fungi-mediated inflammasome activation to restrict colitis and colon cancer. Immunity 49:515–530 e515. https://doi.org/10.1016/j.immuni.2018.08.024
Dupaul-Chicoine J et al (2015) The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 43:751–763. https://doi.org/10.1016/j.immuni.2015.08.013
Tanaka F et al (2000) Rapid generation of potent and tumor-specific cytotoxic T lymphocytes by interleukin 18 using dendritic cells and natural killer cells. Cancer Res 60:4838–4844
Gatault S et al (2015) IL-18 is involved in eosinophil-mediated tumoricidal activity against a colon carcinoma cell line by upregulating LFA-1 and ICAM-1. J Immunol 195:2483–2492. https://doi.org/10.4049/jimmunol.1402914
Haghshenas MR et al (2009) IL-18 serum level and IL-18 promoter gene polymorphism in Iranian patients with gastrointestinal cancers. J Gastroenterol Hepatol 24:1119–1122. https://doi.org/10.1111/j.1440-1746.2009.05791.x
Kang JS et al (2009) Interleukin-18 increases metastasis and immune escape of stomach cancer via the downregulation of CD70 and maintenance of CD44. Carcinogenesis 30:1987–1996. https://doi.org/10.1093/carcin/bgp158
Uhlen M et al (2015) Proteomics. Tissue-based map of the human proteome. Science 347:1260419. https://doi.org/10.1126/science.1260419
Deswaerte V et al (2018) Inflammasome adaptor ASC suppresses apoptosis of gastric cancer cells by an IL18-mediated inflammation-independent mechanism. Cancer Res 78:1293–1307. https://doi.org/10.1158/0008-5472.Can-17-1887
Majima T et al (2006) Exploitation of interleukin-18 by gastric cancers for their growth and evasion of host immunity. Int J Cancer 118:388–395. https://doi.org/10.1002/ijc.21334
Osaki T et al (1998) IFN-gamma-inducing factor/IL-18 administration mediates IFN-gamma- and IL-12-independent antitumor effects. J Immunol 160:1742–1749
Micallef MJ et al (1997) In vivo antitumor effects of murine interferon-gamma-inducing factor/interleukin-18 in mice bearing syngeneic Meth A sarcoma malignant ascites. Cancer Immunol Immunother 43:361–367
Son YI, Dallal RM, Lotze MT (2003) Combined treatment with interleukin-18 and low-dose interleukin-2 induced regression of a murine sarcoma and memory response. J Immunother 26:234–240
Carson WE et al (2000) Coadministration of interleukin-18 and interleukin-12 induces a fatal inflammatory response in mice: critical role of natural killer cell interferon-gamma production and STAT-mediated signal transduction. Blood 96:1465–1473
Ma Z et al (2016) Augmentation of immune checkpoint cancer immunotherapy with IL18. Clin Cancer Res 22:2969–2980. https://doi.org/10.1158/1078-0432.CCR-15-1655
Robertson MJ et al (2013) A dose-escalation study of recombinant human interleukin-18 in combination with rituximab in patients with non-Hodgkin lymphoma. J Immunother 36:331–341. https://doi.org/10.1097/CJI.0b013e31829d7e2e
Tarhini AA et al (2009) A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer 115:859–868. https://doi.org/10.1002/cncr.24100
Gabay C et al (2018) Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis 77:840–847. https://doi.org/10.1136/annrheumdis-2017-212608
Mistry P et al (2014) Safety, tolerability, pharmacokinetics, and pharmacodynamics of single-dose antiinterleukin- 18 mAb GSK1070806 in healthy and obese subjects. Int J Clin Pharmacol Ther 52:867–879. https://doi.org/10.5414/CP202087
O’Reilly M et al (2014) Gene therapy: charting a future course--summary of a National Institutes of Health Workshop, April 12, 2013. Hum Gene Ther 25:488–497. https://doi.org/10.1089/hum.2014.045
Rodriguez-Galan MC et al (2009) Coexpression of IL-18 strongly attenuates IL-12-induced systemic toxicity through a rapid induction of IL-10 without affecting its antitumor capacity. J Immunol 183:740–748. https://doi.org/10.4049/jimmunol.0804166
Higashi K et al (2014) A novel cancer vaccine strategy with combined IL-18 and HSV-TK gene therapy driven by the hTERT promoter in a murine colorectal cancer model. Int J Oncol 45:1412–1420. https://doi.org/10.3892/ijo.2014.2557
Acknowledgments
The Walter and Eliza Hall Institute receives funding from the Victorian State Government Operational Infrastructure Support Program. TLP receives funding from the National Health and Medical Research Council (NHMRC) of Australia Project Grants (1080498, 1098643). TLP is a Victorian Cancer Agency Fellow and WEHI Dyson Bequest Centenary Fellow.
Conflict of Interest
The authors declare no conflicts.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Fung, K.Y., Nguyen, P.M., Putoczki, T.L. (2020). Emerging Roles for Interleukin-18 in the Gastrointestinal Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1240. Springer, Cham. https://doi.org/10.1007/978-3-030-38315-2_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-38315-2_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-38314-5
Online ISBN: 978-3-030-38315-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)