
Abstract
The interactions between tumor cells and the non-malignant stromal and immune cells that make up the tumor microenvironment (TME) are critical to the pathophysiology of cancer. Mesenchymal stem cells (MSCs) are multipotent stromal stem cells found within most cancers and play a critical role influencing the formation and function of the TME. MSCs have been reported to support tumor growth through a variety of mechanisms including (i) differentiation into other pro-tumorigenic stromal components, (ii) suppression of the immune response, (iii) promotion of angiogenesis, (iv) enhancement of an epithelial-mesenchymal transition (EMT), (v) enrichment of cancer stem-like cells (CSC), (vi) increase in tumor cell survival, and (vii) promotion of tumor metastasis. In contrast, MSCs have also been reported to have antitumorigenic functions including (i) enhancement of the immune response, (ii) inhibition of angiogenesis, (iii) regulation of cellular signaling, and (iv) induction of tumor cell apoptosis. Although literature supporting both arguments exists, most studies point to MSCs acting in a cancer supporting role within the confines of the TME. Tumor-suppressive effects are observed when MSCs are used in higher ratios to tumor cells. Additionally, MSC function appears to be tissue type dependent and may rely on cancer education to reprogram a naïve MSC with antitumor effects into a cancer-educated or cancer-associated MSC (CA-MSC) which develops pro-tumorigenic function. Further work is required to delineate the complex crosstalk between MSCs and other components of the TME to accurately assess the impact of MSCs on cancer initiation, growth, and spread.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Mesenchymal stem cells
- Tumor microenvironment
- Cancer
- Immune response
- Angiogenesis
- Cancer stem cell
- Stroma
- Epithelial-mesenchymal transition
3.1 Introduction
In the course of neoplasia, tumor cells (TCs) extensively interact with adjacent cell populations in the “tumor microenvironment (TME).” The TME is a complex network of non-malignant stromal and immune cells which surround the cancerous tissue. Interactions with microenvironment cells cause TCs to undergo genetic and functional changes that increase metastasis, enhance proliferation, and induce chemotherapeutic resistance [1, 2]. In addition, the TME also contains a non-cellular component consisting of the extracellular matrix (ECM) and soluble factors . Studies have shown that ECM and soluble factors in the TME play an important role in supporting tumor progression, and these factors are strongly associated with tumorigenesis [3].
Mesenchymal stem cells (MSCs) (also known as multipotent mesenchymal stromal cells) are non-hematopoietic multipotent stromal stem cells that can be found in a variety of tissues, such as ovary, brain, spleen, liver, kidney, lung, muscle, thymus, pancreas, adipose, and bone marrow. MSCs are distinct from other stromal cells, such as fibroblasts , and MSCs have a unique expression profile that is positive for stromal cell markers (CD73, 105, 44, 29, and 90) but negative for endothelial (CD34, 31, and vWF) and hematopoietic (CD45 and 14) markers [4]. MSCs are progenitor cells to multiple stromal components, possessing the ability to differentiate into osteocytes (bone), adipocytes (adipose), chondrocytes (cartilage), and fibroblasts [5]. Given the lack of one specific marker and the fact that they are closely related to more terminally differentiated stromal cells, the identification of MSCs can be challenging. The international society for cellular therapy published minimal criteria for defining multipotent mesenchymal stroma cells which state MSCs (1) must be plastic adherent; (2) must express CD105, CD73, and CD90 and lack expression of CD45, CD34, CD14 or CD11b, CD79a, or CD19 and HLA-DR surface molecules; and (3) must differentiate to osteoblasts, adipocytes, and chondroblasts in vitro [4] (Fig. 3.1).

The definition of mesenchymal stem cell (MSC). MSCs express CD105, CD73, and CD90 and lack the expression of CD45, CD34, CD14, CD11b, CD79a, or CD19. MSCs also differentiate to fibroblasts, osteoblasts, adipocytes, and chondrocytes in vitro
Scholarly literature presents divergent evidence on the role of MSCs in the TME and cancer progression. Both pro-tumorigenic and antitumorigenic functions have been ascribed to MSCs; this dichotomous relationship can be attributed to the heterogeneity in MSC definition, source of MSC derivation, and methods of study. Thus, this chapter will present evidence of the pro- and antitumorigenic functions of MSCs and will discuss potential reasons for the existence of this apparent contradiction.
3.2 Pro-tumorigenic Function of MSCs in the TME
Within the confines of the tumor microenvironment, tumor-secreted factors and direct TC-MSC interactions induce a pro-tumorigenic phenotype in the MSC population, creating carcinoma-associated mesenchymal stem cells (CA-MSCs) [6, 7]. CA-MSCs retain their differentiation capacity and stromal surface markers, but they contribute to tumor progression via several mechanisms: (i) differentiation into other pro-tumorigenic components of the TME, (ii) suppression of immune response, (iii) promotion of angiogenesis, (iv) enhancement of an epithelial-mesenchymal transition (EMT), (v) enrichment of cancer stem-like cells (CSC), (vi) increase in tumor cell survival, and (vii) promotion of tumor metastasis [6, 8,9,10,11,12,13].
3.2.1 Differentiation into Pro-tumorigenic Components of the TME
A defining characteristic of MSCs is their ability to differentiate into multiple cell lineages, such as fibroblasts, adipocytes, osteocytes, and chondrocytes. These multipotent properties suggest that MSCs may play a key role in the generation of most stromal components of the TME. Multiple reports have demonstrated that CA-MSCs differentiate into tumor supporting carcinoma-associated fibroblasts (CAFs) and adipocytes (CAAs) in the presence of tumor cells.
3.2.1.1 Carcinoma-Associated Fibroblasts
The traditional role of fibroblasts is to facilitate wound healing by regulating extracellular matrix remodeling [14]. Within the confines of the TME, CAFs constitute the majority of the local stroma and contribute significantly to tumorigenesis [15]. While CAFs can be derived from local stromal fibroblasts, both resident and distally recruited MSCs have been shown to acquire a CAF-like phenotype within the TME niche [16]. Interestingly, CA-MSCs demonstrate an even greater ability to differentiate into CAFs versus normal MSCs within the TME [17]. While the exact mechanism underlying CA-MSC to CAF differentiation has not yet been elucidated, there is growing evidence that tumor-secreted factors induce the TGF-β/Smad signaling pathway in MSCs drive differentiation into a CAF phenotype [18, 19]. Additionally, the CAF phenotype is stable and persists in in vitro cell culture sans tumor stimulation [20].
Pro-tumorigenic functions of CAFs include increased tumor cell invasion, enhanced EMT through Hedgehog signaling, ECM remodeling resulting in increased desmoplasia, promotion of tumor initiation in pre-malignant cells, increased CSC profile, promotion of migration and metastasis, and increased chemotherapeutic resistance [21,22,23,24,25,26,27].
3.2.1.2 Carcinoma-Associated Adipocytes
Adipocytes are a major component of adipose tissue, and they function in both lipid storage and signaling regulation. Adipocytes generate a variety of growth factors, hormones, cytokines, and adipokines. Specifically, CAAs have a unique secretome that aids in extracellular matrix remodeling, invasion, therapeutic resistance, and EMT [28]. Increased insulin-like growth factor binding protein-2 (IGFBP-2) expression and secretion in CAAs was shown to enhance migration and invasion in in vitro and in vivo breast cancer models [29]. Additionally, co-culture of ovarian cancer cells and CAAs exhibited enhanced migration and invasion of the cancer cells through increased production and secretion of IL-8/fatty acid binding protein-4 [30].
3.2.2 Suppression of Immune Response
Canonically , MSCs play a role in healing damaged tissues, engaging in direct and paracrine crosstalk with immune cells [31]. MSCs demonstrate chemotaxis towards inflammatory chemokines released by damaged tissues, migrating to the wound and suppressing both innate and adaptive immunes responses [32]. Dendritic cell (DC) differentiation is suppressed when MSCs downregulate interferon-γ (IFN-γ) and TNF-α expression [33]. Direct cell-to-cell interactions between MSCs and natural killer (NK) cells alter the phenotype of NK cells, suppressing proliferation and cytokine secretion [34]. Macrophages co-cultured with MSCs favor M2 polarization, leading to an increase in phagocytic activity and decreased expression of inflammatory cytokines IFNγ, TNF-α, IL-1β, and IL-12 [35, 36]. Additionally, soluble factors secreted by MSCs have been shown to repress T- and B-cell proliferation while increasing apoptosis in activated T cells [37,38,39].
In the context of the TME , CA-MSCs use similar mechanisms to support tumor growth. Mounting evidence suggests that CA-MSCs can regulate the proliferation and maturation of DCs, NK cells, T cells, and B cells [34, 40,41,42]. Additionally, CA-MSCs promote immunosuppression by secreting the cytokines IL-10, TGFβ, nitric acid, indoleamine 2,3-dioxygenase, and prostaglandin E2 [43, 44]. In vivo studies using murine melanoma tumor models have shown that IFN-γ and TNF-α promote the immunosuppressive role of CA-MSCs, enabling increased tumor growth [11, 45]. A mouse model of pancreatic cancer likewise demonstrated CA-MSCs promote cancer growth through M2 macrophage polarization [46]. Another study using a prostate cancer model demonstrated that MSCs significantly increase tumor initiation and growth through suppression of the immune response [47].
3.2.3 Promotion of Angiogenesis
The induction of angiogenesis is a hallmark of cancer and is considered one of the early steps in the development of invasive cancers [48]. Angiogenesis is the development of new blood vessels from existing vasculature and is necessary to sustain expanding tumor growth. An increasing amount of evidence suggests that angiogenesis is governed by MSCs within the TME. Work in syngeneic mouse models has shown that co-injection of MSCs supports the formation of tumor neo-vasculature by localizing close to the vascular walls and by expressing CD31 [10]. There is also evidence that MSCs secrete a number of soluble pro-angiogenic factors, such as LIF, M-CSF, MIP-2, VEGF, IFN-γ, and TNFα. Moreover, MSCs can enhance angiogenesis through induction of the ERK1/2 and p38 MAPK pathways, which enhance the expression of VEFG and CXCR4 in tumor cells [49]. CA-MSCs, via a paracrine signaling loop involving BMP4 and Hedgehog, also induce angiogenesis in ovarian cancer models [13]. Collectively, this research suggests that CA-MSCs appear to play a role in tumorigenesis via promotion of neovascularization .
3.2.4 Enhancement of the Epithelial-Mesenchymal Transition (EMT)
The detachment of cancer cells from the primary tumor, otherwise known as dissemination, is the initial step in metastatic spread. Dissemination is found to be tightly associated with the epithelial-mesenchymal transition (EMT), a process in which epithelial cells undergo multiple changes to gain mesenchymal properties. EMT is typically an embryonic process. However, increasing evidence shows that the TME stimulates EMT in cancer cells through the activation of the same pathways stimulated during embryogenesis. Both embryonic and cancerous EMT are characterized by loss of E-cadherin, which often results from change-of-function mutations in the CDH1 gene or from decreased E-cadherin expression. This altered expression affects downstream steps, such as the activation of transcriptional factors Snail, Slug, Twist, and FOXC2 [50]. In addition, the disruption of E-cadherin is associated with expression of N-cadherin, or mesenchymal cadherin, which facilitates motility and migration of cancer cells within the surrounding stroma [51]. MSCs can stimulate EMT in cancer cells through CCL5 production. CCL5 promotes the secretion of matrix metalloproteinase (MMPs) which act by breaking down the extracellular matrix (ECM), thereby increasing the motility of cancer cells and enhancing their metastatic ability [52]. In a pancreatic cancer model, MSCs stimulated EMT through a Notch-dependent mechanism [53].
3.2.5 Enrichment of Cancer Cell Stemness
Cancer stem-like cells (CSCs) , also known as tumor initiating cells, are a subpopulation of cancer cells with the ability to recapitulate the entire tumor population and are the cells thought to be responsible for cancer initiation, chemotherapy resistance, and metastasis. A growing body of work demonstrates that MSCs enhance CSC proliferation and invasiveness via multiple pathways and in a variety of cancer types. Secretion of IL-6 by MSCs increases JAK2/STAT3 pathway activation in cancer cells, which has been shown to enhance sphere formation and tumor initiation in lung cancer [54]. Following MSC co-culture, breast cancer cells exhibit upregulated CXCL7 and IL-6 pathways and demonstrate enhanced mammosphere formation and increased self-renewal capacity [55]. In another breast cancer study, MSCs were linked to the promotion of stem cell proliferation via P2X-mediated purinergic signaling [56]. Furthermore, activation of the WNT and TGF-β signaling pathways in gastric cancer resulted in an increase of the CSC population [57]. A Hedgehog/BMP4 signaling loop between CA-MSCS and ovarian cancer cells likewise increases ovarian CSCs [13]. Taken together, these data suggest that MSCs play a significant role in enriching the CSC population and driving disease initiation, resistance, and progression .
3.2.6 Increasing Tumor Cell Survival
MSCs contribute to tumor cell survival in several ways. Within the TME, tumor progression is accompanied by hypoxia and energy starvation. Within these otherwise treacherous conditions, it has been reported that MSCs increase their cellular proliferation and stemness through the expression of Rex-1 and Oct-4 [58]. MSCs have also been shown to release many soluble factors that promote tumor survival and proliferation including VEGF, FGF-2, PDGF, HGF, brain-derived neurotropic factor (BDNF), SDF-1α, IGF-1, IGF-2, TGF-β, and IGFBP-2 [59,60,61]. Many of these molecules, namely, VEGF and FGF-2, mediate the expression of anti-apoptotic factor Bcl-2 in order to promote tumor cell survival [62, 63]. A study by Burger et al. demonstrated that SDF-1α expressed by MSCs can prevent drug-induced apoptosis of chronic lymphocytic leukemia (CLL) cells [64]. Another study showed that direct cell-to-cell contact with MSCs significantly enhances the viability and proliferation of glioblastoma [65]. Thus, MSCs appear to make a noteworthy contribution to the survival of tumor cells .
3.2.7 Promotion of Tumor Metastasis
During metastasis , cancer cells escape the primary tumor and eventually lead to the formation of secondary tumors in distant parts of the body. In order for primary tumors to form secondary tumors, cancer cells need to go through the sequential events of invasion, intravasation, extravasation, and colonization [66]. The process of invasion starts once cancer cells break away from the primary tumor mass. The detached cancer cells invade the basement membrane and migrate through the surrounding stroma to reach nearby blood vessels. Cancer cells then intravasate as they penetrate the lymphatic or vascular wall and travel through the circulatory system. The traveling cancer cells extravasate from the vasculature by exiting through the vascular wall and implanting into distant organs. Ultimately, the cancer cells proliferate and form tumors in their new location via a process known as colonization. The successful completion of the metastatic process is determined by the ability of cancer cells to colonize distant organs [48, 67].
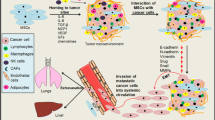
Studies have shown that MSCs play a crucial role in promoting metastasis through multiple mechanisms. It has been reported that MSCs secrete TGF-β which increases cancer cells’ invasive and migratory potential [65]. In the breast cancer cell line MCF7, cancer cells exhibited an enhanced migratory capacity after MSC-exosome treatment, specifically through induction of the WNT pathway. Exosome treatment led to an increase in the expression of WNT target genes Axin2 and Dkk1, as well as β-catenin [68]. A different investigation identified that MCF-7 breast cancer cells have increased migration potential when co-cultured with MSCs in vitro which is mediated through ER-SDF-1/CXCR4 crosstalk [69]. It has also been reported that bone marrow-derived MSCs enhance the migratory capacity of breast cancer cell lines through the CXCR2 receptor [12]. Finally, as discussed above, MSCs promote cancer cell metastasis through inducing EMT and enrichment of CSCs [53] (Fig. 3.2).

The role of MSCs in supporting tumor progression. MSCs (1) differentiate to form cancer-associated fibroblasts (CAFs), (2) dampen the anti-tumor immune response, and (3) induce cancer cell EMT, cancer cell stemness, angiogenesis, cancer cell survival, and metastasis
3.3 Antitumorigenic Function of MSCs in the TME
As previously mentioned, significant controversy exists regarding the role of MSCs in cancer. In addition to the pro-tumorigenic effects described above, other studies have shown that MSCs act in an ant-tumorigenic manner to suppress disease progression. Studies both in vivo and in vitro have shown that MSCs can inhibit tumor growth and metastasis through several mechanisms such as (i) modulation of immune responses, (ii) inhibition of angiogenesis, (iii) regulation of cellular signaling, and (iv) induction of apoptosis.
3.3.1 Modulation of Immune Responses
Although MSCs have been mainly shown to suppress immune responses, there are reports of MSCs inducing an antitumorigenic immune response. In a rat colon cancer model, MSCs inhibited cancer growth by increasing monocyte and granulocyte infiltration in the TME [70]. Further, Toll-like receptor 3 (TLR3)-activated MSCs enhance neutrophil function, and MSCs have been reported to stimulate resting T cells and act as antigen-presenting cells; however it is unclear if this happens within the TME [71, 72]. MSCs may also play a role in recruitment of different immune populations into the TME altering the ratio of Treg and myeloid-derived suppressor cells to CD8+ T cells shifting the balance towards an antitumorigenic state [73]. Interestingly, this change in immune infiltration was associated only with MSCs injected distant from the tumor rather than co-injected with tumor cells indicating naïve or non-tumor-associated MSCs may have divergent functions compared to MSCs in direct association with tumor cells .
3.3.2 Inhibition of Angiogenesis
While the pro-angiogenic functions of MSCs have been well described, there is evidence that MSCs can inhibit angiogenesis under certain circumstances. Direct injection of MSCs into an in vitro Matrigel angiogenesis assay led to the induction of apoptosis in endothelial cells. This assay showed that endothelial apoptosis was accompanied by increase in reactive oxygen species, which ultimately led to capillary degeneration. Further, direct in vivo injection of MSCs into mouse melanomas exhibited tumor devascularization via a reduction in endothelial markers PECAM1 and VE-cadherin [74].
Additional research has demonstrated the anti-angiogenic effects of MSCs in gliomas. Bone marrow-derived MSCs suppress the growth of both patient-derived primary glioma cells in vitro and human glioma cell lines in vivo. Co-injection of human-derived MSCs and glioma cell lines resulted in a significant reduction of microvessel density, as demonstrated with CD31 staining. Further proteomic analysis of these samples showed downregulation of the pro-angiogenic factors PDFG-BB, IGF-1, FGF-2, and IL-1β. In vivo glioma-MSC co-cultures also demonstrated a decrease in PDGF-BB and IL-1β expression and a reduction in tumor volume compared to glioma-only tumors [75].
Given the data presented in mouse melanomas and human gliomas, MSCs may play a role in both the enhancement and inhibition of angiogenesis .
3.3.3 Regulation of Cellular Signaling
Within the tumor microenvironment , various cellular signals regulate tumor cell survival, proliferation, migration, and metabolism. Increasing evidence shows that MSCs influence the cellular signaling of tumor cells. In addition to pro-tumorigenic regulation, MSCs regulate signaling pathways that inhibit tumor progression. The phosphoinositide 3-kinase/AKT and WNT/β-catenin signaling pathways are associated with the development of carcinomas of the breast, liver, colon, skin, stomach, and ovary. Studies report that MSCs inhibit tumor proliferation through inhibition of the PI3K/AKT pathway and suppression of the WNT/β-catenin pathway. MSCs specifically induced expression of DKK1 in human carcinoma cell lines (hepatocellular, H7402 and HepG2; breast, MCF-7; hematopoietic, K562 and HL60) via WNT signaling, which inhibited cell proliferation [76,77,78].
3.3.4 Induction of Apoptosis
MSCs have also been reported to induce tumor cell apoptosis and cell cycle arrest [79]. MSCs had an inhibitory effect on mouse hepatoma, lymphoma, and insulinoma cells through induction of p21 and the caspase 3 pathway [80]. Moreover, MSCs cultured at a high density expressed type I IFN, leading to the cell death of breast cancer cells, MCF-7, and MDR-MB-231 cells. Furthermore, MSCs primed with IFN-γ can induce tumor cell-specific apoptosis [81, 82] (Fig. 3.3).

The role of MSCs in suppressing tumor progression through increasing monocyte and granulocyte infiltration, inhibiting angiogenesis and tumor cell proliferation, and inducing tumor cell apoptosis
3.4 Conclusions
While examples of MSCs functioning in an antitumorigenic manner exist, the majority of evidence points to MSCs acting in a cancer supporting role within the confines of the TME. These antitumorigenic findings cannot be merely discarded however, but rather contextualized. Tumor-suppressing effects are observed in higher ratios of MSCs to tumor cell (~2:1 and greater) which are significantly greater than the TME MSC population [74, 83, 84]. These findings support the development and use of ex vivo MSCs in a therapeutic role but lack the physiological relevancy representative of the natural TME (Fig. 3.4).

MSCs source and number affect the role of MSCs within tumor microenvironment into pro-tumorigenic versus anti-tumorigenic
MSC/CA-MSC function also appears to develop in a tissue- and disease-dependent manner. Bone marrow-derived MSCs (BM-MSCs) developed a cancer supporting phenotype in a breast cancer TME model but not an ovarian cancer TME model. However, omental-derived MSCs were able to promote growth in the ovarian cancer TME model, while BM-MSCs inhibited tumor growth in the ovarian cancer TME model [85]. As breast cancer typically metastasizes to bone while ovarian cancer rarely does and prefers to metastasize to omentum, these findings suggest the importance in MSC source in the development of tumor supporting/suppressing phenotypes and may explain some of the divergent findings regarding MSC function. Further, most of the reports demonstrating antitumorigenic roles for MSCs are from experiments using MSCs without prior exposure to cancer cells or without direct association with cancer cells. This speaks to an important difference in the function of cancer-naïve MSCs vs cancer-educated MSCs.
Despite the divergence in evidence describing the role of MSCs in tumor promotion or suppression, it is apparent that MSCs play a dynamic role within the TME. Further work is required to unravel the complex crosstalk between MSCs and tumor, immune, and other stromal cells. Given the heterogeneity of MSCs, additional work is required to identify and adequately describe various subpopulations that may have differing functions dependent on cancer type. This will be essential to understanding how MSCs contribute to cancer development and progression and may lead to the identification of new therapeutic targets or biomarkers as well as the use of MSCs as therapeutic agents.
References
Ansell SM, Vonderheide RH (2013) Cellular composition of the tumor microenvironment. Am Soc Clin Oncol Educ Book 33:e91
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19(11):1423–1437
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y et al (2017) Role of tumor microenvironment in tumorigenesis. J Cancer 8(5):761–773
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8(4):315–317
Tropel P, Noel D, Platet N, Legrand P, Benabid AL, Berger F (2004) Isolation and characterisation of mesenchymal stem cells from adult mouse bone marrow. Exp Cell Res 295(2):395–406
Coffman LG, Pearson AT, Frisbie LG, Freeman Z, Christie E, Bowtell DD, Buckanovich RJ (2019) Ovarian carcinoma-associated mesenchymal stem cells arise from tissue-specific normal stroma. Stem Cell 37(2):257–269
McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S et al (2011) Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J Clin Invest 121(8):3206–3219
Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B et al (2009) Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One 4(4):e4992
Guan J, Chen J (2013) Mesenchymal stem cells in the tumor microenvironment. Biomed Rep 1(4):517–521
Suzuki K, Sun R, Origuchi M, Kanehira M, Takahata T, Itoh J et al (2011) Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol Med 17(7–8):579–587
Han Z, Tian Z, Lv G, Zhang L, Jiang G, Sun K et al (2011) Immunosuppressive effect of bone marrow-derived mesenchymal stem cells in inflammatory microenvironment favours the growth of B16 melanoma cells. J Cell Mol Med 15(11):2343–2352
Halpern JL, Kilbarger A, Lynch CC (2011) Mesenchymal stem cells promote mammary cancer cell migration in vitro via the CXCR2 receptor. Cancer Lett 308(1):91–99
Coffman LG, Choi YJ, McLean K, Allen BL, di Magliano MP, Buckanovich RJ (2016) Human carcinoma-associated mesenchymal stem cells promote ovarian cancer chemotherapy resistance via a BMP4/HH signaling loop. Oncotarget 7(6):6916–6932
Bainbridge P (2013) Wound healing and the role of fibroblasts. J Wound Care 22(8):407–408. 10-12
Ohlund D, Elyada E, Tuveson D (2014) Fibroblast heterogeneity in the cancer wound. J Exp Med 211(8):1503–1523
Paunescu V, Bojin FM, Tatu CA, Gavriliuc OI, Rosca A, Gruia AT et al (2011) Tumour-associated fibroblasts and mesenchymal stem cells: more similarities than differences. J Cell Mol Med 15(3):635–646
Arena S, Salati M, Sorgentoni G, Barbisan F, Orciani M (2018) Characterization of tumor-derived mesenchymal stem cells potentially differentiating into cancer-associated fibroblasts in lung cancer. Clin Transl Oncol 20(12):1582–1591
Barcellos-de-Souza P, Comito G, Pons-Segura C, Taddei ML, Gori V, Becherucci V et al (2016) Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-derived TGF-beta1. Stem Cells 34(10):2536–2547
Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z et al (2012) Inhibition of TGF-beta/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells 30(12):2810–2819
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R et al (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121(3):335–348
Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W et al (2012) SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett 322(2):169–176
Cukierman E, Bassi DE (2010) Physico-mechanical aspects of extracellular matrix influences on tumorigenic behaviors. Semin Cancer Biol 20(3):139–145
Shimoda M, Mellody KT, Orimo A (2010) Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin Cell Dev Biol 21(1):19–25
Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L et al (2010) Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res 70(17):6945–6956
Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV et al (2012) Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 22(5):571–584
Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V et al (2009) A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med 15(1):68–74
Chandler C, Liu T, Buckanovich R, Coffman LG (2019) The double edge sword of fibrosis in cancer. Transl Res 209:55–67
Mazo EB, Kan Ia D (1974) [Angiotensin test in the diagnosis of vasorenal hypertension]. Ter Arkh 46(7):110–113
Wang C, Gao C, Meng K, Qiao H, Wang Y (2015) Human adipocytes stimulate invasion of breast cancer MCF-7 cells by secreting IGFBP-2. PLoS One 10(3):e0119348
Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR et al (2011) Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17(11):1498–1503
Le Blanc K (2006) Mesenchymal stromal cells: tissue repair and immune modulation. Cytotherapy 8(6):559–561
Wang M, Yuan Q, Xie L (2018) Mesenchymal stem cell-based immunomodulation: properties and clinical application. Stem Cells Int 2018:3057624
Gao WX, Sun YQ, Shi J, Li CL, Fang SB, Wang D et al (2017) Effects of mesenchymal stem cells from human induced pluripotent stem cells on differentiation, maturation, and function of dendritic cells. Stem Cell Res Ther 8(1):48
Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M (2006) Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 24(1):74–85
Zhang QZ, Su WR, Shi SH, Wilder-Smith P, Xiang AP, Wong A et al (2010) Human gingiva-derived mesenchymal stem cells elicit polarization of m2 macrophages and enhance cutaneous wound healing. Stem Cells 28(10):1856–1868
Selleri S, Bifsha P, Civini S, Pacelli C, Dieng MM, Lemieux W et al (2016) Human mesenchymal stromal cell-secreted lactate induces M2-macrophage differentiation by metabolic reprogramming. Oncotarget 7(21):30193–30210
Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P et al (2002) Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 99(10):3838–3843
Akiyama K, Chen C, Wang D, Xu X, Qu C, Yamaza T et al (2012) Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell 10(5):544–555
O'Connor BP, Vogel LA, Zhang W, Loo W, Shnider D, Lind EF et al (2006) Imprinting the fate of antigen-reactive B cells through the affinity of the B cell receptor. J Immunol 177(11):7723–7732
Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E et al (2003) Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 101(9):3722–3729
Tabera S, Perez-Simon JA, Diez-Campelo M, Sanchez-Abarca LI, Blanco B, Lopez A et al (2008) The effect of mesenchymal stem cells on the viability, proliferation and differentiation of B-lymphocytes. Haematologica 93(9):1301–1309
Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, Yu XD et al (2005) Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood 105(10):4120–4126
Batten P, Sarathchandra P, Antoniw JW, Tay SS, Lowdell MW, Taylor PM et al (2006) Human mesenchymal stem cells induce T cell anergy and downregulate T cell allo-responses via the TH2 pathway: relevance to tissue engineering human heart valves. Tissue Eng 12(8):2263–2273
Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T et al (2007) Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood 109(1):228–234
Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J et al (2003) Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood 102(10):3837–3844
Mathew E, Brannon AL, Del Vecchio A, Garcia PE, Penny MK, Kane KT et al (2016) Mesenchymal stem cells promote pancreatic tumor growth by inducing alternative polarization of macrophages. Neoplasia 18(3):142–151
Cheng J, Li L, Liu Y, Wang Z, Zhu X, Bai X (2012) Interleukin-1alpha induces immunosuppression by mesenchymal stem cells promoting the growth of prostate cancer cells. Mol Med Rep 6(5):955–960
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Zhu W, Huang L, Li Y, Zhang X, Gu J, Yan Y et al (2012) Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett 315(1):28–37
El-Haibi CP, Bell GW, Zhang J, Collmann AY, Wood D, Scherber CM et al (2012) Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci U S A 109(43):17460–17465
Bates RC, Mercurio AM (2003) Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell 14(5):1790–1800
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW et al (2007) Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449(7162):557–563
Kabashima-Niibe A, Higuchi H, Takaishi H, Masugi Y, Matsuzaki Y, Mabuchi Y et al (2013) Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci 104(2):157–164
Hsu HS, Lin JH, Hsu TW, Su K, Wang CW, Yang KY et al (2012) Mesenchymal stem cells enhance lung cancer initiation through activation of IL-6/JAK2/STAT3 pathway. Lung Cancer 75(2):167–177
Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F et al (2011) Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res 71(2):614–624
Maffey A, Storini C, Diceglie C, Martelli C, Sironi L, Calzarossa C et al (2017) Mesenchymal stem cells from tumor microenvironment favour breast cancer stem cell proliferation, cancerogenic and metastatic potential, via ionotropic purinergic signalling. Sci Rep 7(1):13162
Nishimura K, Semba S, Aoyagi K, Sasaki H, Yokozaki H (2012) Mesenchymal stem cells provide an advantageous tumor microenvironment for the restoration of cancer stem cells. Pathobiology 79(6):290–306
Berniakovich I, Giorgio M (2013) Low oxygen tension maintains multipotency, whereas normoxia increases differentiation of mouse bone marrow stromal cells. Int J Mol Sci 14(1):2119–2134
Efimenko A, Starostina E, Kalinina N, Stolzing A (2011) Angiogenic properties of aged adipose derived mesenchymal stem cells after hypoxic conditioning. J Transl Med 9:10
Hung SC, Pochampally RR, Chen SC, Hsu SC, Prockop DJ (2007) Angiogenic effects of human multipotent stromal cell conditioned medium activate the PI3K-Akt pathway in hypoxic endothelial cells to inhibit apoptosis, increase survival, and stimulate angiogenesis. Stem Cells 25(9):2363–2370
Crisostomo PR, Wang Y, Markel TA, Wang M, Lahm T, Meldrum DR (2008) Human mesenchymal stem cells stimulated by TNF-alpha, LPS, or hypoxia produce growth factors by an NF kappa B- but not JNK-dependent mechanism. Am J Physiol Cell Physiol 294(3):C675–C682
Konig A, Menzel T, Lynen S, Wrazel L, Rosen A, Al-Katib A et al (1997) Basic fibroblast growth factor (bFGF) upregulates the expression of bcl-2 in B cell chronic lymphocytic leukemia cell lines resulting in delaying apoptosis. Leukemia 11(2):258–265
Dias S, Shmelkov SV, Lam G, Rafii S (2002) VEGF(165) promotes survival of leukemic cells by Hsp90-mediated induction of Bcl-2 expression and apoptosis inhibition. Blood 99(7):2532–2540
Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell'Aquila M, Kipps TJ (2000) Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 96(8):2655–2663
Rodini CO, Goncalves da Silva PB, Assoni AF, Carvalho VM, Okamoto OK (2018) Mesenchymal stem cells enhance tumorigenic properties of human glioblastoma through independent cell-cell communication mechanisms. Oncotarget 9(37):24766–24777
Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331(6024):1559–1564
Massague J, Obenauf AC (2016) Metastatic colonization by circulating tumour cells. Nature 529(7586):298–306
Lin R, Wang S, Zhao RC (2013) Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol Cell Biochem 383(1–2):13–20
Rhodes LV, Antoon JW, Muir SE, Elliott S, Beckman BS, Burow ME (2010) Effects of human mesenchymal stem cells on ER-positive human breast carcinoma cells mediated through ER-SDF-1/CXCR4 crosstalk. Mol Cancer 9:295
Ohlsson LB, Varas L, Kjellman C, Edvardsen K, Lindvall M (2003) Mesenchymal progenitor cell-mediated inhibition of tumor growth in vivo and in vitro in gelatin matrix. Exp Mol Pathol 75(3):248–255
Cassatella MA, Mosna F, Micheletti A, Lisi V, Tamassia N, Cont C et al (2011) Toll-like receptor-3-activated human mesenchymal stromal cells significantly prolong the survival and function of neutrophils. Stem Cells 29(6):1001–1011
Stagg J, Pommey S, Eliopoulos N, Galipeau J (2006) Interferon-gamma-stimulated marrow stromal cells: a new type of nonhematopoietic antigen-presenting cell. Blood 107(6):2570–2577
Zheng H, Zou W, Shen J, Xu L, Wang S, Fu YX et al (2016) Opposite effects of coinjection and distant injection of mesenchymal stem cells on breast tumor cell growth. Stem Cells Transl Med 5(9):1216–1228
Otsu K, Das S, Houser SD, Quadri SK, Bhattacharya S, Bhattacharya J (2009) Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood 113(18):4197–4205
Ho IA, Toh HC, Ng WH, Teo YL, Guo CM, Hui KM et al (2013) Human bone marrow-derived mesenchymal stem cells suppress human glioma growth through inhibition of angiogenesis. Stem Cells 31(1):146–155
Qiao L, Xu Z, Zhao T, Zhao Z, Shi M, Zhao RC et al (2008) Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res 18(4):500–507
Zhu Y, Sun Z, Han Q, Liao L, Wang J, Bian C et al (2009) Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia 23(5):925–933
Qiao L, Xu ZL, Zhao TJ, Ye LH, Zhang XD (2008) Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett 269(1):67–77
Ji X, Zhang Z, Han Y, Song J, Xu X, Jin J et al (2016) Mesenchymal stem cells derived from normal gingival tissue inhibit the proliferation of oral cancer cells in vitro and in vivo. Int J Oncol 49(5):2011–2022
Lu YR, Yuan Y, Wang XJ, Wei LL, Chen YN, Cong C et al (2008) The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biol Ther 7(2):245–251
Dasari VR, Kaur K, Velpula KK, Gujrati M, Fassett D, Klopfenstein JD et al (2010) Upregulation of PTEN in glioma cells by cord blood mesenchymal stem cells inhibits migration via downregulation of the PI3K/Akt pathway. PLoS One 5(4):e10350
Ryu H, Oh JE, Rhee KJ, Baik SK, Kim J, Kang SJ et al (2014) Adipose tissue-derived mesenchymal stem cells cultured at high density express IFN-beta and suppress the growth of MCF-7 human breast cancer cells. Cancer Lett 352(2):220–227
Brennen WN, Chen S, Denmeade SR, Isaacs JT (2013) Quantification of Mesenchymal Stem Cells (MSCs) at sites of human prostate cancer. Oncotarget 4(1):106–117
Poggi A, Varesano S, Zocchi MR (2018) How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive. Front Immunol 9:262
Coffman LG, Pearson AT, Frisbie LG, Freeman Z, Christie E, Bowtell DD et al (2019) Ovarian carcinoma-associated mesenchymal stem cells arise from tissue-specific normal stroma. Stem Cells 37(2):257–269
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Atiya, H., Frisbie, L., Pressimone, C., Coffman, L. (2020). Mesenchymal Stem Cells in the Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1234. Springer, Cham. https://doi.org/10.1007/978-3-030-37184-5_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-37184-5_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-37183-8
Online ISBN: 978-3-030-37184-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)