
Abstract
To interact with the external environments, bacteria often display long proteinaceous appendages on their cell surface, called pili or fimbriae. These non-flagellar thread-like structures are polymers composed of covalently or non-covalently interacting repeated pilin subunits. Distinct pilus classes can be identified on basis of their assembly pathways, including chaperone-usher pili, type V pili, type IV pili, curli and fap fibers, conjugative and type IV secretion pili, as well as sortase-mediated pili. Pili play versatile roles in bacterial physiology, and can be involved in adhesion and host cell invasion, DNA and protein secretion and uptake, biofilm formation, cell motility and more. Recent advances in structure determination of components involved in the various pilus systems has enabled a better molecular understanding of their mechanisms of assembly and function. In this chapter we describe the diversity in structure, biogenesis and function of the different pilus systems found in Gram-positive and Gram-negative bacteria, and review their potential as anti-microbial targets.
Magdalena Lukaszczyk, Brajabandhu Pradhan—Authors contributed equally.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Bacterial cells are frequently decorated with non-flagellar proteinaceous cell surface appendages, referred to as pili or fimbriae. The appendages usually have low nanometer scale width, but can be multiple microns in length, often exceeding the diameter of the producing bacterium. The structures are made up of many hundreds or thousands of pilus subunits, which are covalently or non-covalently associated depending on the pilus system. The function of bacterial pili is varied. The vast majority is implicated in adherence and/or multicellular behaviour. Pili frequently mediate adherence and/or invasion (in)to eukaryotic host cells, but can also be implication in biofilm formation through pilus self-association, binding of neighbouring cells or giving shape to the extracellular matrix. In other systems, though, the pili serve a role as a hollow conduit or a scaffolding structure for the secretion or uptake of proteins and nucleic acids, or in rare instances extracellular electron transport. In this review we summarize the current molecular and structural understanding of the function, build-up and assembly of the major pilus systems found in Gram-positive and Gram-negative bacteria. For each system, we provide a review of the main architecture of the pili and its constituent components, as well as the prevailing mechanistic understanding of the assembly pathways. Since pili are frequently first line virulence factors, a great interest has been gathered for their chemical inhibition as a means towards the development of non-antibiotic, virulence targeted antibacterials (Steadman et al. 2014; Ruer et al. 2015). For some systems, like the type 1 fimbriae implicated in urinary tract infections (UTIs), significant progress has been made, to the point that anti-adhesive compounds are undergoing clinical trials and may reach the market in coming years. For many more systems, the molecular understanding is now such that selective inhibitory compounds can be sought and are being identified. Coming years will show if more of these pathways can be targeted in a clinical setting.
The Chaperone-Usher Pilus System
Arguably the most abundant pilus assembly system in Gram-negative bacteria is the chaperone-usher (CU) pathway. It is a conserved protein secretion-assembly system found in the Gram-Negative classes α-, β-, γ- and δ-proteobacteria, where it is primarily associated with human and animal pathogenic genera including Escherichia, Shigella, Proteus, Klebsiella, Salmonella, Pseudomonas, Yersinia and many others (Sauer et al. 2004). CU fimbriae are linear non-covalent multi-subunit polymers that require two accessory proteins for their assembly and translocation to the cell surface: a periplasmic chaperone and an outer membrane usher. The chaperone stabilizes fimbrial subunits in the periplasm and targets them to the usher, a pilus assembly platform in the outer membrane (OM) that facilitates subunit polymerization and transport to the cell surface (Thanassi et al. 1998).
Pili assembled by the chaperone-usher pathway are prime virulence factors of proteobacteria. They contribute to the establishment and persistence of the infection by mediating host- and tissue-specific adherence, and can play a role in the evasion of host defence mechanisms by contributing to biofilm formation or inducing host cell invasion (Sauer et al. 2004). Chaperone-usher pili are associated with a wide range of diseases, such as urinary and gastrointestinal tract infections, meningitis and sepsis (Proft and Baker 2009). Amongst the earliest and most comprehensively studied chaperone-usher systems are type 1 and P fimbriae produced by uropathogenic E. coli (UPEC) (Hultgren et al. 1993). The type 1 fimbrial adhesin FimH binds to the D-mannosylated receptors of the human and animal bladder, whereas the P pilus adhesin PapG binds galabiose-containing glycosphingolipids in the kidney epithelium. These two systems formed the basis of extensive structural and molecular studies, which together with those on the Yersinia pestis capsular antigen Caf, have revealed the canonical principles of CU biogenesis (Sauer et al. 2004).
Morphology and Structure of CU Pili
CU pili are encoded in gene clusters comprising a cognate chaperone and usher, and up to seven different pilus subunits of approximately 10–30 kDa (Sauer et al. 2004; Nuccio and Bäumler 2007). The assembled pili are homo- or heteropolymers of hundreds to thousands of pilus subunits organized into linear single-start filaments. These filaments can undergo additional quaternary condensation to form rigid, helically wound rods or are found as long flexible filaments that often collapse into a dense capsular mass on the bacterial cell surface. Archetypal examples of rod-forming pili are type 1 and P pili of E. coli. These are monoadhesive structures, capped by a single copy of an adhesive subunit. In this review, the general description of pilus assembly by chaperone-usher pathway will be based primarily on the type 1 pilus, encoded by the fim operon. The type 1 pilus consists of a long, rigid and helical pilus rod composed of several thousand copies of the major pilus subunit FimA (Fig. 12.1) and is terminated with a short flexible tip fibrillum built of the two adaptor subunits FimG and FimF and the adhesin FimH, which is located in a single copy at the distal tip of the pilus. Consecutive subunits interact through non-covalent contacts. Each pilin subunit is characterized by an incomplete immunoglobulin-like fold, lacking its C-terminal β-strand, and by the presence of an unstructured N-terminal extension (Nte) of 10–20 amino acids (Fig. 12.1). In the mature pilus, the Nte of one subunit complements the incomplete Ig-fold of the consecutive subunit in a mechanism called donor strand exchange (DSE) (Choudhury et al. 1999; Sauer et al. 1999) (Fig. 12.1d). Although non-covalent, these fold complementation interactions between pilus subunits and their complementary Nte peptides have extremely high activation barriers for dissociation, displaying extrapolated dissociation half-lives of 109–1011 years (Puorger et al. 2008, 2011). The long-lived fold complementation interactions effectively protect the pili from loss-of-function by breakage at one of the several hundred DSE contacts. In addition, the pilus rods are dynamic structures characterized by remarkable spring-like properties (Fällman et al. 2005). Force spectroscopy and EM imaging have shown reversible uncoiling of the helical structure in the pilus rod. The recent atomic cryoEM models of both type 1 and P pilus provide the details of the extensive subunit-subunit interaction network within the rod (Hospenthal et al. 2016, 2017; Spaulding et al. 2018). In their coiled state, both pili are right-handed superhelical structures, where the Nte of one pilus subunit complements the hydrophobic groove of the adjacent subunit. The type 1 pilus rod has a diameter of ~70 Å with a ~14 Å wide central hollow lumen, whereas the P pilus has a diameter of ~81 Å with a ~21 Å lumen. In both structures the pilin subunits form a continuous ascending path, with the N-terminal portion of Ntes facing towards the pilus exterior and the C-terminal part towards the lumen. Both pili have a similar helical pitch of ~25 Å, though the type 1 structure comprises 3.13 FimA pilin subunits per turn, compared to 3.28 PapA subunits in the more tightly packed P pilus. Subsequently, the axial rise of type 1 pilus is slightly higher (8 Å for type 1 and 7.7 Å for P pilus). In the helical rod, each PapA subunit interacts with five preceding and five succeeding subunits (Hospenthal et al. 2016), whereas each FimA subunit interacts with four preceding and four succeeding subunits (Spaulding et al. 2018); (Hospenthal et al. 2017). The overall interaction network responsible for maintaining the helical quaternary structure involves mostly weak hydrophilic contacts (Hospenthal et al. 2016). The combination of both strong DSE interaction and weak hydrophilic forces explains the ability of pilus rod to uncoil elastically when the shear force is applied, without breaking apart. At the distal tip, FimF and FimG provide a short flexible linker between the rigid FimA pilus rod and the FimH adhesin. FimH lacks an N-terminal extension. Instead, it has a two-domain structure with a C-terminal pilin domain and a full N-terminal β-sandwich domain that carries the adhesive function (Choudhury et al. 1999). The pilin domain interacts with the penultimate pilus subunit (FimG) by DSE, whereas the lectin domain mediates binding to the host receptors (Fig. 12.1). In the P pilus, the PapA pilus rod is separated from the PapG tip adhesin by a longer flexible fibrillum made up of the linker subunits PapK, PapE and PapF (Rose et al. 2008). Most chaperone-usher pili are monoadhesive, displaying rigid pili with tip-localized two-domain adhesins similar to the type 1 pilus (Nuccio and Bäumler 2007). The adhesins’ lectin domains share an elongated β-jelly-roll topology. However, the structures of their ligand binding pocket may differ remarkably (Moonens and Remaut 2017). For example, FimH adhesin has a deep, negatively charged binding pocket which is located at the tip of the lectin domain (Hung et al. 2002) whereas the PapG adhesin has a shallow cavity localized on the side of the lectin domain (Dodson et al. 2001).

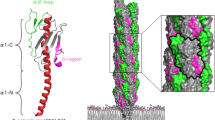
The pilus architecture and schematic assembly pathways for chaperone-usher pilus (left) and type V pilus systems (right). Fimbrial subunits are shown in blue, tip adhesins/pilins in pink, transmembrane pores in green, chaperones in yellow. At the bottom representative operons coding for both systems are shown. Left: Type 1 pilus biogenesis as model chaperone-usher assembly pathway. The chaperone FimC (yellow) binds the fimbrial subunit FimA (light blue) (PDB entry: 4DWH) in the periplasm. The pilus tip fibrillum complex composed of FimC:FimF:FimG:FimH traverses the outer membrane usher FimD (PDB entry: 4j3O). Type 1 pilus is composed of a pilus rod (EMDB entry: EMD-7342, PDB entry: 6c53) built of over 1000 FimA subunits and a tip fibrillum (FimF:FimG:FimH) (PDB entry: 3JWN). a Cryo-EM map of type 1 pilus rod (EMDB entry: EMD-7342, PDB entry: 6c53) with the neighbouring subunits n−1, n0, n+1. b Structure of FimH binding pocket interacting with α-D-mannose (PDB entry: 1KLF). The D-mannose (yellow) and the mannose-interacting residues (Phe1, Asn46, Asp47, Asp54, Gln133, Asn135, Asp140 and Phe142) (red) are shown in ball and stick model. c Donor strand complementation (DSC) of the FimA (blue) pilus subunit by the G1 strand (in orange) of the chaperone FimC (yellow) (PDB entry: 4DWH). G1 strand of the FimC is located parallel to the strand F of FimA during DSC. d A topological diagram of donor strand exchange (DSE) of adjacent subunits of type 1 pilus. The incomplete Ig-fold of one subunit (light blue) is complemented by an Nte of the previous subunit (dark blue). Right: The proposed pathway scheme of type V pilus biogenesis. e A topological diagram of proposed strand-exchange mechanism of adjacent subunits of type V pilus. The hydrophobic groove exposed along both NTD and CTD of one subunit (light blue) is complemented by the A1′ and A2′ strands of the consecutive subunit. f Left: “open” conformation of anchor pilin BovFim4b (PDB entry: 5CAG). Right: “closed” conformation of FimA4 (PDB entry: 4Q98). The conserved C-terminal appendage composed of A1′ and additional disordered region is shown in dark blue. In the “open” conformation this appendage is extended, while in the “closed” conformation it folds back to the C-terminal domain
In polyadhesive CU pili, subunits polymerize through DSE-mediated subunit contacts, but lack the coil-forming quaternary interaction seen in the pilus rods of monoadhesive pili. They form flexible filaments of one or more types of a major structural subunit, which each contain one to two independent receptor binding sites (Zav’yalov 2013). In such a way, the entire pilus structure is directly involved in the adhesion. Examples include E. coli F4 (Fae) and Afa/Dr fimbriae (Moonens et al. 2015; Keller et al. 2002; Anderson et al. 2004), or the Y. pestis F1 (Caf) or pH6 (Psa) antigens (Zavialov et al. 2003; Bao et al. 2013). Although the individual subunits have low micromolar to millimolar affinity for their glycan or protein ligands, their high valency results in high binding avidity (Moonens and Remaut 2017).
Biogenesis of CU Pili
Chaperone-usher pili are encoded in gene clusters that hold both the structural subunits as well as the assembly machinery, consisting of a pilus-specific chaperone and usher protein. The individual pilin subunits are translocated across the inner membrane via the SecYEG translocon, after which chaperone and usher are sufficient for the folding and stabilization of pilus subunits, their ordered assembly into pili, and translocation to the cell surface (Sauer et al. 2004). The secretion-assembly pathway is here described based on the fim operon. CU pilus subunits do not stably fold in the periplasm as a result of an incomplete immunoglobulin (Ig)-like fold that lacks seventh, C-terminal β-strand. In the periplasm, initial subunit folding is catalysed by the chaperone (FimC) (Vetsch et al. 2004; Bann et al. 2004). Prior to incorporation into the pilus, the missing C-terminal β-strand in the incomplete Ig-like fold of pilus subunits is complemented by an extended β strand (G1) of chaperone (Fig. 12.1c), an interaction referred to as “donor strand complementation” (DSC) (Choudhury et al. 1999; Sauer et al. 1999). However, the G1 strand of the chaperone lies parallel to the subunit strand F during DSC, rather than the antiparallel pairing in a canonical Ig-fold and seen for the Nte during the DSE interaction. As a result, the chaperone traps pilus subunits into a more loosely packed, higher energy folding intermediate compared to the subunit–Nte interaction (Sauer et al. 2002; Zavialov et al. 2005). The chaperone:subunit complexes are recruited to the outer membrane usher (FimD), which acts as a platform for pilus polymerization and OM translocation. The usher is a five domain protein composed of a 24-stranded β-barrel that forms the pilus translocation channel, a plug domain that occludes the pore in the resting state, two C-terminal (CTD1 and CTD2) and an N-terminal domain (NTD) that reside in the periplasm and forms the chaperone:subunit binding sites (Remaut et al. 2008; Phan et al. 2011). The usher NTD domain recruits the chaperone:adhesin complex and this interaction is mostly mediated by the chaperone (Nishiyama et al. 2005). The subunits are sequentially incorporated at the base of the growing pilus, starting from the tip adhesin. In type 1 and P pilus assembly, the affinity of the usher (FimD/PapC) NTD is highest for the chaperone:adhesin complex (FimC:FimH/PapD:PapG) and decreases for later subunits (Dodson et al. 1993). Together with the differential affinities of the usher for different subunit:chaperone complexes, favourable DSE kinetics of cognate versus non-cognate subunit–Nte interactions direct the order of pilus assembly (Nishiyama et al. 2003; Rose et al. 2008). Binding of the chaperone:adhesin complex primes the usher for pilus assembly (Nishiyama et al. 2008). The pre-initiation step was recently captured for in a crystal structure of the P pilus usher PapC bound to the PapD:PapG chaperone:adhesin complex (Omattage et al. 2018). In this pre-initiation complex, the chaperone:subunit is bound to the usher NTD, whilst the usher is still in a locked conformation with the plug domain residing inside the lumen of the β-barrel. A previous crystal structure of the FimD usher bound to the FimC:FimH chaperone:adhesin complex capture the usher in its primed conformation and gave exquisite insight into the mechanism of pilus assembly at the usher (Phan et al. 2011) (Fig. 12.1). In this structure, the lectin domain of FimH displaces the usher plug domain and traverses the lumen of the usher β-barrel, whilst its C-terminal pilin domain remains bound to the FimC chaperone, itself bound to the usher by the CTD1 and CTD2 domains. This primed usher configuration leaves the NTD, the primary chaperone:subunit recruitment platform of the usher, accessible to bind the next chaperone:subunit (FimC:FimG). When the new chaperone:subunit complex binds the NTD, the subunit is oriented such that its N-terminal extension is ideally positioned to undergo DSE with the subunit at the base of the growing fiber and bound at the CTDs. During this DSE reaction, the Nte of the incoming subunit competes out the donor β-strand of the chaperone in the chaperone:subunit at the CTDs (Remaut et al. 2006; Phan et al. 2011). DSE is a concerted zip-in-zip-out process that initiates at the so-called P5 pocket, a hydrophobic pocket accepting the incoming Nte and not occupied by the chaperone donor strand (Remaut et al. 2006). At the end of DSE process the chaperone dissociates from the now penultimate subunit and the usher CTDs, after which the newly added chaperone:subunit complex shifts from the NTD to the now liberated binding site at the CTD. This makes the NTD accessible for a new recruitment round and translocates the pilus outward with one subunit step. Consecutive iterations of the recruitment, DSE and translocation steps result in the stepwise build-up of the pili. At all times, the pilus is tethered to the usher via the last incorporated chaperone:subunit complex. In type 1 and P pili, the length of individual pili is rather uniform and depends on the incorporation of a dedicated terminator subunit (FimI and PapH, respectively) (Verger et al. 2006; Bečárová 2015). These terminator subunits show occluded P5 pockets, so that they cannot undergo DSE and remain anchored in the usher. In many operons, however, there is no indication for a terminator subunit and pilus growth is likely controlled by protein expression and the availability of free subunits.
CU Pili as Antibacterial Targets
Because of their prime importance in bacterial virulence, chaperone-usher pili have raised considerable interest as novel target for vaccine or antibacterial drug development (Steadman et al. 2014). Let us consider the example of uropathogenic E. coli, where type 1 pili are strongly associated with symptomatic bladder infections (cystitis) by mediating strong attachment and invasion of the superficial umbrella cells in the urothelium, resulting in tissue damage and inflammation (Mulvey et al. 1998). S and P pili are associated with ascending UTIs by the binding of sialosyl oligosaccharides and globoside receptors in the kidney epithelium, respectively, leading to pyelonephritis (Roberts et al. 1994; Korhonen et al. 1986). More recently F9 fimbriae were shown important for inflammation-associated adherence in ongoing and chronic bladder infections (Conover et al. 2016), and the type 1 and F17-like pili were associated with the establishment of a gut reservoir of uropathogenic E. coli (Spaulding et al. 2017). Strains lacking these pili are severely compromised in the initiation, persistence and recurrence of UTIs. Indeed, a promising approach has been proposed to use anti-adhesive agents that interfere with the bacterial adherence to the host tissue (Ruer et al. 2015). As such drugs would target bacterial virulence factor rather than being bactericidal, the spread of antibiotic resistance mechanisms is thought less likely to occur. Competitive inhibitors of FimH adhesin in the form of α-D-mannose derivatives called mannosides, are highly efficacious in murine models of UTI (Cusumano et al. 2011; Spaulding et al. 2017). Mannosides reach low nanomolar affinities and can be formulated as orally available anti-adhesive compounds that successfully treat UPEC UTI. Anti-adhesive receptor-analogues have also been reported for the P and F9 pili (Ohlsson et al. 2002; Kalas et al. 2018). An alternative to competitive inhibition of the adhesin is the chemical attenuation of pilus biogenesis. This has been achieved for type 1, P and S pili by inhibiting chaperone docking to the usher using a family of bicyclic 2-pyridones, called pilicides (Pinkner et al. 2006), or by competitive inhibition of the DSE reaction with organic compounds (AL1) binding the subunit P5 pocket of FimH (Lo et al. 2014). Chaperone-usher pili also make promising vaccine candidates. The immunization of mice and primates with type 1 pilus adhesin FimH reduced the in vivo colonization of bladder as well as recurrent UTIs (Langermann et al. 1997, 2000). In animal husbandry, immunization of sows with cocktails including F4, F5, F6 and F41 fimbrial subunits from enterotoxigenic E. coli protects the litter from neonatal diarrhea (Matias et al. 2017), and passive immunization by addition of neutralizing antibodies of the F4 adhesin FaeG to pig feed protects young animals from post-weaning diarrhea (Virdi et al. 2013).
Type V Pilus
In 2016 Xu et al. characterized a novel type of pili with a biogenesis mechanism distinct from all known pilus systems and described as proteinase-mediated donor-strand exchange (Xu et al. 2016). These unique pilus system, termed the Type V or Bacteroidia pilus, is found exclusively in the Bacteroidia class. They were first coined in Porphyromonas gingivalis, a human oral pathogen associated with severe adult periodontitis and gingivitis (Xu et al. 2016). Bacteroidia pili play a role in bacterial adhesion, co-aggregation and biofilm formation. Two morphologically different pili of P. gingivalis have been described: major or long (0.3–1.6 µm), with a major pilin subunit FimA (not to be confused with FimA major subunit of CU type 1 pilus), and minor or short (80–120 nm), with a major pilus subunit Mfa1 (Hamada et al. 1996; Yoshimura et al. 1984). Both major and minor pili are encoded by similar operons, containing the genes of structural pilins forming the pilus stalk (FimA or Mfa1, respectively), anchoring pilins (FimB and Mfa2, respectively), tip pilins (Mfa4), as well as other accessory subunits and regulatory elements.
Type V Pilus Structure and Biogenesis
Type V pilins are composed of two domains: an N-terminal domain (NTD) and a slightly larger C-terminal domain (CTD). Both domains have a transthyretin-like fold that is composed of seven core β-strands organized into two β-sheets. The NTD has an archetypal fold with 7 β-strands (A1–G1), whereas the CTD (except for Mfa4) possess an C-terminal extension of two highly conserved amphipathic β-strands (A1′ and A2′). Crystal structures of the P. gingivalis FimA superfamily stalk subunits showed that this appendage may be present in two conformations: “open”—exposed along the CTD, and “closed”—folded back to the CTD, with a flexible loop between A1′ and A2′ strands (Xu et al. 2016) (Fig. 12.1f). The structural characterization of type V tip pilins show that these subunits terminate the pilus structure, as they either lack the A1′–A2′ appendage (e.g. Mfa4), or the appendage interacts with a fused C-terminal lectin domain (e.g. BovFim1C) (Kloppsteck et al. 2016; Xu et al. 2016).
The biogenesis of type V pilus superficially resembles the donor strand-mediated fold complementation mechanism seen in CU pilus, however, it additionally requires the lipoprotein precursors of pilin subunits and outer membrane proteinase. It is thought that the mechanism of type V pilus assembly, both in P. gingivalis and B. fragilis, is based on the lipoprotein sorting pathway, although this has not been unambiguously demonstrated (Shoji et al. 2004). Type V prepilins are produced as lipoprotein precursors. Importantly, prepilins possess exceptionally long signal peptides, as compared to other bacterial pilins. The C-terminal part of the signal peptide is called a lipobox and contains the lipidated cysteine residue. Pilin subunits are transported by SecYEG machinery to the periplasmic side of inner membrane, where they are folded, and their signal peptide is cleaved off by a type II signal proteinase. Subsequently, the C-terminal cysteine residue is lipidated and the modified pilin subunits are secreted to the extracellular environment. How the periplasmic transport and outer membrane translocation step occur is presently unknown. Whether this requires the help of a periplasmic chaperone and outer membrane usher-like protein remain to be identified. The folded pilins occur in the periplasm in the “closed” state, therefore they are predicted to be stable and the presumed chaperone most likely binds the lipid moiety present on the pilin N-terminus. The mechanism of type V pilus assembly at the outer membrane has not been described yet, but it is hypothesized to be based on the lipoprotein-sorting machinery. The first pilin subunit bound to the outer membrane is an anchor pilin, which is docked in the membrane via the N-terminal lipid moiety. Next pilin subunits, stalk and tip pilins, undergo the cleavage of the N-terminal short peptide, by trypsin-like arginine and lysine specific proteinase located in the outer membrane: gingipain R or gingipain K (Rgp or Kgp). The proteolytic step yields the mature, assembly-prone form of pilin (Nakayama et al. 1996). During proteolytic maturation, the A1 β-strand is removed, and an extended hydrophobic groove is generated in the NTD. This groove can now be occupied by the β-strand from the neighbouring subunit, similarly as in the chaperone-usher pilus. Although both N-terminal and C-terminal regions of the adjacent pilin have been suggested to act as donors of complementing strand (Kloppsteck et al. 2016; Xu et al. 2016), the involvement of the flexible C-terminal appendage of A1′ and A2′ was validated by the cross-linking experiments (Xu et al. 2016). Accordingly, the hydrophobic groove of NTD can be occupied by the extended C-terminal appendage of the neighbouring subunit, with the A1′ strand filling the NTD groove to restore seven β-stranded fold, and the A2′ strand complementing the CTD groove, parallel to G2 strand, which in “closed” conformation was occupied by its own A1′ and A2′ strands (Xu et al. 2016) (Fig. 12.1e). A recent study confirmed the crucial role of the conserved sequences in both N- and C-termini of the pilus subunit Mfa1 in subunit polymerization of Mfa fimbriae. Treatment of P. gingivalis with peptides analogous to these sequences inhibits the Mfa fimbriae assembly and impedes biofilm formation (Alaei et al. 2019).
Type IV Pili
Type IV pili (T4P) are several micrometres long, flexible surface appendages that are bacterial virulence factors, widely distributed in many Gram-negative bacteria, including Pseudomonas aeruginosa, Neisseria gonorrhoeae, N. meningitidis, Myxococcus xanthus. Some of the bacterial species, like Vibrio cholerae and enteropathogenic E. coli (EPEC), produce bundle-forming pili, required for adherence to epithelial cells and auto-aggregation (Ramboarina et al. 2005). Apart from Gram-negative bacteria, type IV-related pili have also been identified in Gram-positive genera Clostridia and Ruminococcus, in Cyanobacteria and in archaea, where they form the archellum, suggesting an early evolutionary origin of this system (Imam et al. 2011; Proft and Baker 2009; Szabó et al. 2007). Type IV pili are multifunctional organelles with a distinguishing ability to extend and retract by reversible polymerization and depolymerization. They play a role in adhesion to host cells and solid substrates, in biofilm formation, DNA and phage uptake, cell motility, cellular invasion as well as microcolony formation. Apart from their function in virulence, type IV pili machinery powers a flagella-independent type of bacterial movement known as twitching motility (Mattick 2002). This is possible thanks to the repeated cycles of extension, adhesion and retraction powered by cytoplasmic adenosine triphosphatases (ATPases). Although T4P are very thin (6–9 nm) structures, they are remarkably strong molecular machines that can endure extensive forces of over 100 pN (Maier et al. 2002).
Type IV pilus subunits share a distinguishing N-methylated N-terminus, and a conserved hydrophobic N-terminal 25-residue α-helical domain and a C-terminal disulphide bonded β-domain (Craig et al. 2004). Based on the sequence similarity and length, type IV pilins are divided into two subclasses: type IVa and type IVb. The type IVa pilins are shorter (average length of mature protein: 150 amino acids) than the type IVb pilins (190 amino acids), and hold a shorter signal peptide (5–6 amino acids for type IVa and 15–30 amino acids for type IVb) (Craig and Li 2008). The N-methylated N-terminal residue of type IVa pilins is always phenylalanine, whereas for type IVb it may be methionine, leucine or valine. Comparison of the available pilin structures shows notably different protein topologies in the β-domain of type IVa and type IVb pilins. The conserved N-terminal hydrophobic α-helix of Type IV pilus subunits acts both as a transmembrane and coiled-coil protein interaction domain in the structural core of the pili (Giltner et al. 2012) (Fig. 12.2). Although the general build-up is equivalent, Type IVa and type IVb pilins form pili that differ in diameter and helical structure. The occurrence of these two pilus types is also different: type IVa are found in many Gram-negative bacteria, including pathogens like Neisseria spp. or P. aeruginosa, whereas type IVb pili have only been found in human enteric bacteria, like V. cholerae, Salmonella enterica serovar Typhi, enteropathogenic E. coli (EPEC) and enterotoxigenic E. coli (ETEC). The host range of type IVa pili expressing pathogens is much broader and includes humans and other mammals, plants and possibly other bacteria (Craig et al. 2004). Interestingly, the type IVa pili assembly requires a complex machinery built of many components, which genes are scattered around the whole bacterial genome. In contrast, type IVb pili assembly systems are composed of smaller number of proteins usually encoded by gene clusters, sometimes present on the plasmids (Pelicic 2008).

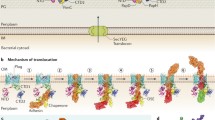
The architecture of type IV pilus systems. Left: Schematic representation of the archetypal type IV pilus system. The structures presented in the figure come from different organisms. The pilus rod is composed of thousands of helically arranged pilin subunits (represented by PilE from N. meningitidis, EMDB entry: EMD-8287, PDB entry: 5KUA). The pilus traverses the outer membrane pore (PilQ from N. meningitidis, EMDB entry: EMD-2105, PDB entry: 4AV2). The pilus elongation and retraction is catalysed by cytoplasmic ATPases (PilF from T. thermophilus, PDB entry: 5OIU and PilT from A. aeolicus, PDB entry: 2GSZ). a Cryo-EM map of type IV pilus rod of N. meningitidis (EMDB entry: EMD-8287, PDB entry: 5KUA) with the neighbouring PilE subunits shown. b Structures of type IVa pilins: pilin GC from N. gonorrhoeae (PDB entry: 2PIL) and PAK pilin from P. aeruginosa (PDB entry: 1OQW) and type IVb pilin: truncated TcpA pilin from Vibrio cholerae (PDB entry: 1OQV). The αβ-loops are shown in green and the D-regions are shown in magenta. Right: The architectural model of the type IVa pilus machinery from M. xanthus in a non-piliated and piliated state (PDB entries: 3JC9 and 3JC8, respectively). PilA pilus subunits are shown in light blue. The components of secretin subcomplex, N-terminal motif of TsaP, LysM (turquoise), together with PilQ AMIN domains (green) anchor the entire basal body to the peptidoglycan layer. The alignment subcomplex components, PilN (grey) and PilO (pink) form coiled coils that is a link between the secretin and motor complexes. Lipoprotein PilP (khaki) is anchored in the inner membrane and PilM (beige) is located in the cytoplasm. The motor complex components, hexameric ATPase PilB (red) and inner membrane PilC (orange) are present at the base of the system. Strikingly, the distance between inner and outer membrane has been found to be longer in the piliated state of the system
Overall, the architecture of both type IVa and type IVb pilins is similar. They are small (15–20 kDa) proteins having a conserved lollipop-like fold with an extended hydrophobic N-terminal α-helical spine (α1) and a globular C-terminal head domain, typically composed of four to seven-stranded antiparallel β-sheet. The hydrophobic N-terminal part of α1 helix, α1-N, protrudes from the protein core, while the amphipathic C-terminal part, α1-C, is embedded in the globular domain and packs against it. The head domains hold two regions involved in T4P interactions: a D-region, containing the conserved cysteines, and exposed αβ-loop that can undergo post-translational modifications (Fig. 12.2b). For example, the N. gonorrhoeae gonococcal (GC) pilin D-region forms a ridge that displays hypervariable surface regions, and the second ridge is formed by αβ-loop containing two post-translational modifications (Craig and Li 2008). The structural diversity of type IV pilins results from the loop sequence differences as well as the topology of the secondary structure elements in the globular domain (Craig et al. 2004; Giltner et al. 2012). Type IVa pilins are characterized by four contiguous β-strands as in the N. gonorrhoeae GC pilin (Parge et al. 1995), whereas the type IVb pilins display more variable β-sheet topology, with five to seven β strands, as in the case of EPEC BfpA pilin or V. cholerae TcpA pilin (Craig and Li 2008; Craig et al. 2003).
Type IV Pilus Assembly Components and Biogenesis
Type IV pilus (T4P) machinery is a multimeric protein assembly that spans both inner and outer membrane in Gram-negative bacteria. It is homologous to the type 2 secretion system (T2SS), which is involved in the transport of folded proteins from the periplasm to the extracellular environment across the cell membrane (Berry and Pelicic 2015). T2SS assembles a periplasmic pseudopilus that is implicated in the secretion of toxins and hydrolytic enzymes, whereas type IV pilus machinery is responsible for the assembly and disassembly of pilin subunits in the core of the pilus. Moreover, in contrast to CU pili and curli, the biogenesis of type IV pili requires ATP as an energy source (Turner et al. 1993). The same holds true for pilus disassembly (Merz et al. 2000). The nomenclature of T4P components varies drastically amongst different organisms, in this chapter the P. aeruginosa nomenclature will be used.
The biogenesis of type IV pilus requires the action of a specialized machinery, including an outer membrane secretin subcomplex (PilQ, PilF, TsaP), an inner membrane motor subcomplex (PilC, PilB, PilT) and an alignment subcomplex (PilM, PilN, PilO, PilP) (Leighton et al. 2015a). The outer membrane secretin subcomplex is composed of two elements: PilQ, a large gated multimeric outer membrane pore and a pilotin PilF, which function is to ensure correct localization, assembly and outer membrane insertion of the secretin subcomplex. In N. gonorrhoeae and M. xanthus there is another component of secretin subcomplex present, the T4P secretin-associated protein, TsaP. This component was suggested to anchor the outer membrane secretin subcomplex to the peptidoglycan cell wall by its LysM motif (Siewering et al. 2014), but its role requires further investigation. The motor subcomplex is made up of the inner membrane protein PilC and the cytoplasmic ATPases PilB and PilT, which drive the processes of pilus elongation and retraction, respectively (Whitchurch et al. 1991; Chiang et al. 2005). The alignment subcomplex, composed of highly conserved PilM, PilN, PilO and PilP proteins, functions as a platform connecting the secretin and the motor complexes (Thanassi et al. 2012; Takhar et al. 2013). The function of particular subunits has not been described, however recent study show that PilNO complex is a dynamic link between the secretin and motor complexes, involved in both elongation and retraction of type IV pilus (Leighton et al. 2015b). PilP subunit is bound to the PilQ pore, which stabilizes its assembly during pilus secretion as the passage of the pilus through the pore requires the disassembly of membrane-spanning domains of PilQ (Berry et al. 2012). PilN and PilO subunits form a dimer via their periplasmic domains. This dimer serves as an anchor of the whole alignment subcomplex in the inner membrane and binds the periplasmic lipoprotein PilP, which interacts with the PilQ pore. Finally, PilN binds the actin-like protein PilM, so that all the cellular compartments are connected via the PilMNOPQ protein interaction network. This trans-envelope complex interacts also with the PilA major pilus subunit and facilitates the passage of the pilus through the periplasm (Tammam et al. 2013). The final pilus rod is made of major subunit PilA (PilE in N. meningitidis) and minor pilus subunits, FimU, PilV, PilW, PilX and PilE, which together form a pilus rod, organized in a helical manner.
The pilus formation starts from the insertion of the prepilin subunits into the inner membrane by the SecYEG translocon. Prepilin maturation requires the cleavage of the signal peptide and methylation of the mature pilin N-terminus by the action of prepilin peptidase PilD (Zhang et al. 1994; Lory and Strom 1997). Prior to incorporation into the pilus, the conserved hydrophobic N-terminal α-helix in the mature pilins anchors them in the inner membrane, with the globular domain exposed to the periplasm. Mature pilin subunits are then extracted from the inner membrane and incorporated into the base of a growing pilus by the assembly machinery, in a process that involves coiled-coil interactions between the N-terminal α-helices in the central hydrophobic core of the pili (Craig et al. 2006). The assembled T4P core passes through the outer membrane secretin PilQ, which is a multimeric gated channel, homologs of which are also present in type II and type III secretion systems (Bitter et al. 1998). The assembly-disassembly of the pilus core requires ATP hydrolysis, by the action of the cytoplasmic ATPases, PilB (PilF in T. thermophilus) and PilT, respectively (Misic et al. 2010; Collins et al. 2018; Turner et al. 1993). Finally, the T4P assembly requires an integral inner membrane protein PilG, as well as several minor pilins (PilH-K) (Tonjum et al. 1995; Winther-Larsen et al. 2005).
Structure of Type IV Pili and Their Assembly Machinery
Since the T4P machinery is a large multiprotein assembly, the reconstruction of its overall structure in an intact state is a challenging task, despite the many structures of individual T4P components being available (Misic et al. 2010; Karuppiah and Derrick 2011; Kim et al. 2006; Berry et al. 2012; Craig et al. 2003). Cryo-electron tomography (cryo-ET) enabled reconstruction of type IV pilus machinery from M. xanthus and T. thermophilus intact cells, at resolutions 30–40 Å and 32–45 Å, respectively (Chang et al. 2016; Gold et al. 2015). The structures from both organisms showed the T4P machineries to span the entire cell envelope and to be present in a closed state in the absence of pilus and an open state when the pilus is present. Models with the available structure of individual components docked into the cryoET volumes indicate that in general the same core components form the overall T4P machinery in both organisms (Fig. 12.2). The differences include longer PilQ secretin in T. thermophilus, which is characterized by larger distance between inner and outer membrane. Moreover, Thermus lacks the PilP homologue, which in M. xanthus is a part of mid-periplasmic ring. The T4P secretion apparatus is made of four interconnected rings, three in the periplasm and one in the cytoplasm, a cytoplasmic disc and dome, a periplasmic stem and an outer membrane pore (Fig. 12.2). In the resting, non-piliated state the PilA major pilus subunits form the short stem, whereas in the piliated state this structure transverse through the periplasmic rings and the PilQ pore to the external environment. The pilus arises at the cytoplasmic dome formed by cytoplasmic domains of IM PilC, surrounded by PilM cytoplasmic ring. The coiled coils across the inner membrane connect the cytoplasmic ring with the lower periplasmic ring, which is composed of the globular domains of PilO and PilN. Periplasmic domains of PilQ with PilP form the mid-periplasmic ring and the upper periplasmic ring is composed of TsaP around PilQ. The structure docking suggests a 1:1 stoichiometry of ring components PilP, PilN, PilO, PilM and TsaP (Fig. 12.2). Although the modelling suggests 12 copies of each subunit, the exact number of particular subunits is still in question (Leighton et al. 2015a; Tammam et al. 2013). The hexameric ATPases PilB and PilT bind interchangeably to the base of the T4P machinery and occur as a cytoplasmic disc in the piliated structure during elongation and retraction, respectively. The other differences between piliated and non-piliated state include the opening of the PilQ gate (PilQ of T. thermophilus has 2 gates) and the pilus traversing the periplasm and PilQ pore into the extracellular environment. Interestingly, the distance between inner and outer membrane is longer in the piliated state, possibly due to the presence of the pilus. The model of pilus assembly was proposed, where ATP hydrolysis by PilB induces PilC rotation and relocation to facilitate incorporation of PilA major pilus subunits, one at a time, from the IM to the base of nascent pilus. In contrast, PilT bound to the basal body induces pilus retraction, by rotating PilC into location that induces expulsion of PilA subunits (Chang et al. 2016). Furthermore, PilNO heterodimer was found to participate in both pilus retraction and elongation (Leighton et al. 2015a). The structure of type IV pilus rod from N. meningitidis was modelled by fitting the 1.44 Å crystal structure of major pilin PilE subunit into the ~6 Å cryo-EM density map (Fig. 12.2). The fully assembled T4P core is composed of the N-terminal α-helices packed together in a coiled arrangement in the centre of the structure, with the globular domains facing to the outside. Notably, the α-helical order is lost in the central part of α1 in the intact pilus (between residues Gly14 and Pro22), making this region more flexible. The extension of this region may be responsible for the spring-like properties of type IV pilus under shear forces (Kolappan et al. 2016). Importantly, the glutamate residue Glu5 of each subunit was confirmed to be essential for the assembly of T4P, as it forms a salt bridge with the positively charged N-terminal amine of the adjacent subunit. This interaction is believed to be involved in the incorporation of subsequent pilins from the inner membrane into the nascent pilus (Craig et al. 2006; Kolappan et al. 2016).
Mycobacterial Pili
For a long time, there was no evidence of pili produced by mycobacteria. However, in 2005 Alteri observed by negative staining and transmission electron microscopy (TEM) that mycobacteria produce two morphologically distinct types of pili: type IV- and curli-like pili (MTP) (Alteri 2005). Mycobacterium type IV pili are flexible appendages forming rope-like bundles. The analysis of M. tuberculosis genome showed that this organism contains the type IVb pili gene cluster, encoding small type IVb prepilins of the Flp pili family (fimbrial low-molecular weight protein). The Flp protein expression and secretion by M. tuberculosis was confirmed by gene expression analysis and immunofluorescent microscopy. M. tuberculosis type IVb pili are encoded by a 5-kb genomic island, which contains seven genes, including type IVb flp prepilin, a transmembrane protein and secreted proteins (Danelishvili et al. 2010). In addition, two Flp family prepilin peptidases were found distant from the flp locus. These two proteins encoded by Rv0990c and Rv2551c ORFs are thought to be involved in the secretion or cleavage of the Flp prepilin substrate. The flp locus of M. tuberculosis is homologous to flp-tad locus of Aggregatibacter actinomycetemcomitans and was presumably acquired by horizontal gene transfer. In many pathogens Flp/Tad pili serve as colonization factors and promote biofilm formation (Ramsugit and Pillay 2015). As M. tuberculosis is a non-motile organisms, its type IV pilus potentially functions as adherent factor, which was also initially proved by Alteri (2005). Its function, however, needs to be further elucidated.
Bacterial Amyloid Fibers
Curli and Fap are functional bacterial amyloid fibers secreted by many Gram-negative bacteria as part of an extracellular matrix that binds cells together to form bacterial communities known as biofilms. Biofilms are a commonly found sedimentary life-form for bacteria. The extracellular matrix helps adherence to the substrate and provides a protective shield that helps cells cope with various environmental stress such as oxidative damage, desiccation, antibiotics as well as host immune responses (Branda et al. 2005; Depas et al. 2015; Hall-Stoodley and Stoodley 2009). Biofilms can be found on both biotic and abiotic surfaces in almost all kinds of environments (Chai et al. 2013; Jeter and Matthysse 2005; Otter et al. 2015; Ryu and Beuchat 2005; Uhlich et al. 2006). The secreted extracellular matrix is composed of polysaccharides, nucleic acids and protein fibers. Functional amyloids can constitute a major fraction (up to 85%) of the biofilm matrix (Larsen et al. 2008; Reichhardt and Cegelski 2014). Historically, amyloids are best known for their association with neurodegenerative diseases such as Alzheimer’s, Parkinson’s and Huntington’s disease and are considered off-pathway protein folding products that are associated with cytotoxicity (Chiti and Dobson 2006). However, over the last two decades an increasing number of examples emerged of so-called functional amyloids, where the amyloid state comprises the native state of the protein and fulfils an adapted biological function (Blanco et al. 2012). In bacteria, several such functional amyloids are involved in biofilm formation, adherence, persistence and pathogenesis (Fowler et al. 2007; Van Gerven et al. 2018). Curli is one of the first and most extensively studied bacterial amyloids. Curli were first identified in Salmonella (Grund and Weber 1988) and E. coli (Olsén et al. 1989) biofilms and later found to be broadly distributed amongst Proteobacteria and Bacteroidetes (Dueholm et al. 2012). A recently discovered bacterial amyloid system is Fap, found in Pseudomonas (Dueholm et al. 2010). Curli and Fap both form surface-localized linear fibers implicated in biofilm formation. Although non-homologous, both systems share a number of common characteristics, such as an amyloid-like cross β architecture of the fibers, the presence of a minor and major subunit that act as a specific nucleator and the main polymerizing subunit, respectively, as well as diffusion-driven secretion channels in the outer membrane. This section will discuss in detail, the structure and function of components of Curli and Fap systems.
Curli
The curli pathway is one of the most extensively studied functional amyloid system. Curli subunits are secreted through the type VIII secretion system, also known as the nucleation-precipitation pathway (Chapman et al. 2002; Olsén et al. 1989). In E. coli, curli subunits and assembly machinery are encoded in two csg (curli structural gene) operons: csgBAC and csgDEFG (Chapman et al. 2002; Hammar et al. 1995). CsgD is the master regulator of the curli biogenesis as it activates the transcription of csgBAC operon through a coordinated complex signalling network (Brombacher et al. 2003; Chirwa and Herrington 2003; Gerstel and Römling 2003). CsgA is the major structural component of the curli fibers while CsgB is the nucleator of amyloid self-assembly (Olsén et al. 1989; Chapman et al. 2002). CsgC is a periplasmic protein responsible for inhibition of premature polymerization of CsgA and therefore possibly helps the bacteria in keeping a check on the cytotoxic aspects of amyloids (Evans et al. 2015). CsgG is an outer membrane protein forming a pore responsible for transport of the curli subunits from the periplasm to the extracellular space. It forms a pore complex by interacting with CsgE in the periplasmic side and CsgF in the extracellular side of the outer membrane. CsgE helps in fine-tuning the regulation of transport of CsgA and CsgB through the pore complex (Goyal et al. 2014; Nenninger et al. 2011) while CsgF interacts with CsgG and CsgB to possibly act as the interface between the assembled fibers and the outer membrane (Nenninger et al. 2009; Schubeis et al. 2018).
Architecture of Curli Fibers
Curli fibers are composed of two components, the major structural component CsgA, and minor nucleator component CsgB. In E. coli, both subunits are approximately 13 kDa polypeptides. A 20 residue long N-terminal signal peptide targets the subunits for export to the periplasm by the SecYEG translocon, after which it is cleaved off (Chapman et al. 2002; Olsén et al. 1989). The first 22 residues of the mature subunits (after proteolytic removal of the signal peptide), termed N22, are believed to form a pathway-specific targeting signal. N22 is sufficient to direct native and non-native proteins to the outer surface through the curli secretion machinery (Robinson et al. 2006). N22 is followed by the amyloidogenic core domain of the subunits, formed by five pseudo repeat sequences of ~22 amino acids, termed R1 to R5. In other species, CsgA can be considerably longer, encompassing over twenty pseudo-repeats. The pseudo-repeats hold an average 30 percent pairwise sequence identity. Isolated pseudo-repeat peptides are in themselves amyloidogenic, though can differ strongly in their assembly propensity. In E. coli CsgA for instance R1, R3 and R5 will readily self-assemble, whilst R2 and R4 only poorly do so (Hammer et al. 2007; Wang et al. 2007, #128). Although in vitro CsgA will self-assemble into curli-like fibers, the minor subunit CsgB acts as a potent nucleator for CsgA fibrillation. In vivo, the formation of cell surface associated curli requires the nucleator CsgB, in a process that is dependent on the curli accessory protein CsgF (Nenninger et al. 2009). R4 and R5 of CsgB (with its markedly different primary sequence containing four positively charged residues Lys133, Arg140, Arg147, and Arg151) has been implicated in interaction of CsgB with CsgF (Hammer et al. 2012). Deletion of R4 or R5 in CsgB results in secretion of CsgA and CsgB to the exterior without any association to the cell surface (Hammer et al. 2012). Although CsgB is required for nucleation of CsgA in vivo, both CsgA and CsgB can form fibers independently in vitro (Shu et al. 2012).
Experimental structures of the CsgA or CsgB subunits and the assembled curli are currently lacking. Staining with amyloid-responsive dyes (Congo Red and thioflavin T), circular dichroism spectra and X-ray diffraction experiments on CsgA fibers point to the presence of a cross-β structure with nucleation-dependent growth characteristics, typical of amyloids (Chapman et al. 2002; Shewmaker et al. 2009). Solid state NMR studies, however, indicate that unlike classical amyloids, CsgA does not have an in-register parallel β sheet architecture (Shewmaker et al. 2009). Using calculations of amino acid contacts based on covariation of amino acids in various CsgA homologues, a model has been proposed. According to this model CsgA is expected to have a β helical architecture, where each pseudo-repeat forms one helical turn (Tian et al. 2015). This model is consistent with results of simulations designed to find the lowest energy conformation of a single molecule. Another model proposed by Louros et al. (2016) based on the Salmonella CsgA homology model depicts the curli fibers as two-start filaments made of stacked β-helical solenoids (Collinson et al. 1993). However, there is ambiguity in the handedness of the helix. Experimental evidence supporting these models is still missing. In vitro studies of CsgA self-assembly using high speed atomic force microscopy has revealed polarity in growth of the fiber, with one end growing visibly faster than the other. Also, they exhibit stop-and-go dynamics in growth where periods of stagnations are interrupted by steady bursts of elongation (Sleutel et al. 2017).
The Curli Assembly Machinery
Secretion and assembly of curli requires a dedicated transport channel in the outer membrane, CsgG, and two accessory proteins CsgE and CsgF (Loferer et al. 1997; Nenninger et al. 2009; Robinson et al. 2006). In E. coli CsgG is a 262-residue long lipoprotein that forms a nonameric transport complex that traverses the OM through a 36-stranded β-barrel (Cao et al. 2014; Goyal et al. 2014) (Fig. 12.3). Prior to insertion in the OM, CsgG is found as a soluble monomeric protein. The transmembrane β-barrel is formed upon circular oligomerization of nine subunits, each contributing two β-hairpins to the channel (Goyal et al. 2014). The pore volume is separated into two 3–4 nm wide cavities on the periplasmic and extracellular side of the outer membrane, separated by a diaphragm-like 1 nm wide constriction formed by the lateral packing of a conserved 12-residue ‘constriction loop’ (CL) in the nine subunits. The luminal lining of the constriction is composed of three stacked concentric rings formed by the side chains of residues Y51, N55, and F56 (Goyal et al. 2014). The latter is strictly conserved and was found important for curli secretion. A structurally equivalent phenylalanine “clamp” is observed in the Anthrax protective antigen, where it facilitates recruitment and translocation of unfolded protein substrate to the secretion channel (Krantz et al. 2005). Single channel conductance experiments and structural data suggest CsgG operates as an ungated peptide diffusion channel (Goyal et al. 2014). The driving force for protein translocation in the curli pathway is currently unknown. Under physiological concentrations, secretion of curli subunits requires the 12 kDa periplasmic accessory protein CsgE (Nenninger et al. 2011). CsgE provides specificity to the CsgG channel and has been found to form a periplasmic plug to the channel (Nenninger et al. 2011; Goyal et al. 2014, #122). In vitro, CsgE maintains equilibrium between a monomeric and nonameric species. The latter forms a dynamic complex with CsgG, forming a cap-like structure that closes off the periplasmic vestibule of the channel (Goyal et al. 2014). CsgE inhibits CsgA polymerization when added in stoichiometric amounts, indicating it can directly interact with the secretion substrate (Nenninger et al. 2011). The solution NMR structures of CsgE W48A/F79A, a mutant that stabilizes the CsgE monomer, reveal a globular mixed α/β fold with three distinct electrostatic surfaces, (i) a positively charged “head,” (ii) a negatively charged “stem” and (iii) a negatively charged “tail” region (Klein et al. 2018; Shu et al. 2016). The positively charged head region has been implicated in CsgE-CsgA interaction. However, how and at what time during the secretion process CsgE interacts with the secretion substrates is currently unknown. It is speculated that CsgE recruits CsgA and CsgB to the secretion complex. A second accessory factor to the curli secretion-assembly machinery is CsgF, a 13 kDa protein that is localized on the extracellular surface (Nenninger et al. 2009). CsgF acts a coupling factor between secretion and assembly of curli subunits. Although csgF null mutants secrete CsgA and CsgB, they lack surface-attached curli fibers (Chapman et al. 2002; Nenninger et al. 2009). Instead, secreted CsgA is found as non-polymerized monomers and dispersed curli fibers in the extracellular milieu (Nenninger et al. 2009). In vivo, CsgB surface attachment and activity as curli nucleator depend on CsgF, and available data suggest CsgF acts as a curli assembly chaperone through direct interaction with the curli nucleator CsgB, an interaction that is found to depend on CsgB’s C-terminal repeats R4 and R5 (Nenninger et al. 2009). A recently determined solution NMR structure of CsgF shows a small α/β domain, preceded by a long, highly disordered N-terminus (Schubeis et al. 2018). How the protein interacts with the secretion channel and the secretion substrates is unknown.

The biogenesis pathways of the bacterial amyloids curli and Fap. a Schematic representation of the curli assembly pathway and the E. coli csgBAC and csgDEFG gene clusters. Blue and dark blue: major curli subunit CsgA and nucleator subunit CsgB, respectively. CsgG forms the OM curli translocation channel (green; PDB entry: 4UV3), bound on the outer side by the accessory protein CsgF (dark green; PDB entry: 5M1U), and the on the periplasmic side by the secretion factor CsgE (yellow; PDB entry: 2NA4). In the periplasm, the amyloid chaperone CsgC (orange, PDB entry: 2Y2Y) prevents premature curli fiber formation. b Schematic representation and operon structure of the Fap assembly pathway. The major Fap subunit FapC and presumed Fap nucleator subunit FapB are shown in blue and dark blue, respectively. FapF (dark green, PDB entry: 5O65)
A last component in the curli assembly-secretion pathway is the CsgC (also referred to as CsgH outside γ-proteobacteria), an 11 kDa periplasmic protein (Hammar et al. 1995; Dueholm et al. 2012; Evans et al. 2015; Taylor et al. 2016). CsgC is dispensable for curli assembly, and for long, the physiological role of the protein remained obscure (Gibson et al. 2007; Taylor et al. 2011). A recent study showed the protein protects bacterial cells from toxicity associated with an accidental periplasmic accumulation of curli subunits (Evans et al. 2015). It does so by inhibiting CsgA and CsgB fiber formation at sub-stoichiometric concentrations. The crystal structure of CsgC reveals a 7-stranded β-sandwich protein with a striking CxC motif similar to its structural homolog DsbD (Taylor et al. 2011). However, this CXC motif does not appear to be involved in CsgC’s amyloid inhibitory activity, which relies on electrostatic interaction with the curli subunits (Taylor et al. 2016). The exact mode of action of the amyloid inhibitor remains unclear. Whilst Taylor and co-workers suggested CsgC to act on an amyloid precursor (Taylor et al. 2016), a study that followed CsgA fibrillation in presence of CsgC using atomic force microscopy indicated the protein acts primarily by inhibiting fiber elongation, presumably by capping the fiber growth poles (Sleutel et al. 2017). Strikingly, the inhibitor is also able to attenuate fibrillation in other amyloids such as α-synuclein, albeit at higher concentrations (Evans et al. 2015).
Fap Fibers
Fap is another functional bacterial amyloid system found in proteobacteria (β-, δ-, and γ-proteobacteria). Although evolutionarily unrelated to curli, it too uses a dedicated pathway for secretion and assembly of cell-surface localized amyloid fibers. Similar to curli, Fap are part of the extracellular matrix of biofilm-associated cells, where they increase colony hydrophobicity and stiffness to enhance survival under various harsh environments (Zeng et al. 2015). Fap was initially identified in pathogenic and non-pathogenic Pseudomonas species, and subsequently found in the genomes of at least 39 additional genera of proteobacteria (Dueholm et al. 2010; Rouse et al. 2018a). The Fap pathway is encoded by the fapABCDEF operon, which includes the structural fiber proteins (FapB, FapC and FapE), as well as the proteins required for safe guidance across the periplasm (FapD, FapA) and secretion across the outer membrane (FapF) (Dueholm et al. 2010). Fap components are exported to periplasm via the SecYEG translocon where the signal sequence is cleaved off. Mature FapB, FapC and FapE are then secreted through FapF, a trimeric outer membrane pore composed of three 12-stranded β-barrels, each plugged by a 13-residue α-helix that join into a periplasmic asymmetric triple coiled-coil via a 40–50-residue long flexible connector (Rouse et al. 2017, 2018b). The N terminal coiled coil domain and helical plug are thought to present a mechanism to regulate substrate transport, although if and how the plugged channels are gated remains unknown. FapC is the major structural component of the amyloid fiber, analogous to CsgA in curli. The protein is composed of three amyloid-prone pseudo-repeat regions of approximately 30 residues, interrupted by two linker regions (Bleem et al. 2018; Dueholm et al. 2010, 2013). FapB is a minor structural component of Fap, has shorter repeats compared to FapC, and is proposed to act as nucleator, analogous to CsgB (Dueholm et al. 2010). FapD and FapF are two periplasmic accessory proteins (Rouse et al. 2017, 2018b). FapD is a C39-family protease that is proposed to play a key role in selectivity of the FapF pore (Rouse et al. 2017). Deletion of FapD or mutation in its cysteine active site resulted in the loss of FapC secretion, suggesting FapD is indeed a peptidase and has a regulatory role (Rouse et al. 2017). FapA is speculated to be a periplasmic inhibitor of Fap assembly, countering cytotoxic effects of premature amyloid polymerization of FapC amyloid, similar to the role of CsgC in the curli system (Rouse et al. 2018a).
Type IV Secretion Pilus
Type IV secretion(T4SS) pili are the extracellular, tubular filaments of Type IV secretion machineries, capable of transporting DNA, proteins and nucleoprotein complexes in and out bacteria and archaea. These systems serve three major purposes in the bacteria, (i) conjugation, (ii) DNA uptake (competence) and release, and (iii) delivery of effector proteins (Grohmann et al. 2018). Conjugation is the process by which a donor cell delivers genetic material (plasmids, Integrative and Conjugative Elements (ICEs)) into a recipient cell in a contact dependent manner (Waksman 2019). Two well-known examples are the F plasmid transfer system (Tra operon) in E. coli, mediating horizontal gene transfer and propagating antibiotic resistance genes and the Ti plasmid injection system in Agrobacterium tumefaciens, associated with crown gall disease in plant hosts. The second function of the type IV secretion systems is DNA uptake, also known as bacterial competence (ComB system in Helicobacter pylori) and release (Tra system in N. gonorrhoeae), for fitness, pathogenicity and virulence of the species. A third group of T4SSs found in bacterial pathogens functions to deliver protein effectors into eukaryotic host cells. Examples include the VirB/VirD4 and Trw T4SSs in Bartonella species, and the Cag secretion system in the gastrointestinal pathogen H. pylori, which mediates the injection of the oncogenic effector protein CagA (Backert et al. 2017; Wagner and Dehio 2019).
Type IV secretion systems are broadly distributed and diverse in composition and function. In this review, we will focus on the VirB/VirD4 T4SS of A. tumefaciens and the F plasmid conjugative system of E. coli.
Architecture of Type IV Secretion Pili
Recent cryo-EM structures of F-family pili encoded by the pOX38 and pED208 conjugative plasmids have greatly enhanced our understanding of the build-up of these surface appendages (Costa et al. 2016). In this chapter, we will use these conjugative systems as the point of reference to describe the architecture of Type IV secretion pili. However, T4SSs differ strongly in the composition and complexity of their associated pili, as well as in the nature of the translocation substrates. Additional structures will thus be needed to determine how representative the conjugative pilus structures are for the architecture of Type IV secretion pili in general. F pili are tubular appendages on the outer membrane responsible for conjugation. The F pilus establishes contact between the donor and recipient cell and acts as a conduit for the exchange of genetic material between the cells, in the form of ssDNA (Waksman 2019). The pili are composed of thousands of TraA subunits, which can assemble and disassemble to extend or retract the appendage (Clarke et al. 2008a). Both the pOX38 and pED208 conjugative pili are hollow cylinders of comparable dimension. The pED208 encoded pilus is 87 Å thick with a lumenal diameter of 28 Å. Both conjugative pili show as five-start helical filaments (Fig. 12.4). The pED208 pilus shows twist angle of 28.2° and a rise of 12.1 Å, while the F pilus (encoded by pOX38) is found two distinctive conformational populations, one with twist of 27.9° and a rise of 13.2 Å, and a second with a twist of 28.1° and rise of 12.5 Å. The conjugative pili are homopolymers of the TraA pilin, which is a ~64 AA long peptide that folds into an all helical structure comprising three helices (α1, α2, and α3). TraA is synthesized as a pro-pilin with an unusual leader peptide of ~50 AA (Majdalani et al. 1996). The pro-pilin is inserted into the inner membrane with the help of TraQ, a transmembrane protein encoded by the F plasmid, in a process that requires ATP and proton motive force. After its insertion, the 5.5 kDa leader peptide is cleaved off by the periplasmic peptidase LepB, resulting in arrangement where the α2–α3 loop is pointing towards the cytoplasm while N- and C-termini of α1 and α3 are in the periplasm (Majdalani and Ippen-Ihler 1996). The N-terminus of mature TraA is then acetylated by TraX, another protein encoded by the F plasmid (Maneewannakul et al. 1995).

Structure and assembly pathway of conjugative Type IV secretion pili. The shown structures are a collage of different studies and systems. a Ribbon representation of the pED208 TraA monomer (yellow) shown in isolation and docked into the cyoEM density of the pED208 F-pilus (PDB entry: 5LEG; EMDB entry: EMD-4042; the TraA bound lipid is shown in red). CryoEM structure of the R388 conjugative T4SS complex (green, EMDB entry: EMD-2567), and ribbon representation of the TraI relaxase of the F/R1 plasmid (grey, PDB entry: 5N8O). b Segmented cryoEM volume for the F conjugative pilus in side and top cross-sectional view (EMDB entry EMD-4042). The individual filaments of the five-start helices are differentiated in colour. c Ribbon representation of the pED208 F-pilus shown in top view (PDB entry: 5LEG). d Segmented cryoEM volume of the R388 conjugative T4SS complex (EMDB entry: EMD-2567) with a colour assignment of the different components, making use of the Vir nomenclature
A remarkable feature that has been discovered during helical reconstruction of the conjugative pili is the a stoichiometric binding of a phospholipids by the TraA subunits. Mass spectrometry analysis confirmed these to be phosphatidylglycerol (PG) species, primarily PG 32:1, PG33:1 and PG 34:1. Interestingly the cell membrane also contains these two phospholipids. However, other constituents of the membrane such as phosphatidylethanolamine (PE) and cardiolipin were absent, indicating TraA pilin subunits might have special preference for PGs. The head groups of the lipids are exposed to the lumen, whilst the acyl chains are completely buried between the helical segments of the TraA subunits. This coating of the pilus lumen with phospholipid head groups makes it electronegative, which thought to help in transport of the ssDNA. Mutation in the phospholipid binding sites (i.e. Y37V and A28F in pED208) switched off pilus assembly at the membrane surface, suggesting that the lipids have an additional role of maintaining integrity of the pilus assembly (Costa et al. 2016). The bound lipids may also assist in insertion of the appendage into the host membrane during establishment of initial contact, and may lower the energy barrier for subunit extraction during retraction of conjugative pili (Clarke et al. 2008b) (Hospenthal et al. 2017). Further studies are required to prove or disprove these speculative roles of lipids in the pilus.
Structure and Mechanism of the Type IV Secretion Machinery
The assembly and disassembly of conjugative pili and other Type IV secretion pili, as well as the passage of the secreted substrate depends on the trans-envelope spanning TypeIV secretion machinery. One of the canonical T4SS is the tDNA injecting system in A. tumefaciens. The components of this system are arranged in two operons, the VirB operon encoding 11 structural genes (VirB1–VirB11) and the VirD operon encoding 4 (VirD1–VirD4) genes with regulatory and accessory functions. Although different amongst species and from the type of system, a typical T4SS generally has the following building blocks (i) the transmembrane secretion complex composed of energy components (VirB4 and VirB11) and structural components (VirB3, VirB6, and VirB8 in the IM, and VirB7, VirB9, and VirB10 in the outer membrane (OM)), Hydrolase VirB1, (ii) the type 4 coupling protein(T4CP), (iii) the pilus (Waksman 2019).
X-ray structures of T4SS subcomplexes and the recent cryo-EM structure of the nearly complete T4SS secretion complex of conjugative plasmid R388 has vastly enhanced our understanding of the arrangement of various components in the 3 Mda complex (Low et al. 2014). It consists of an outer membrane embedded core complex (OMC), an inner membrane core complex (IMC) with 2 barrel-like legs inserting into the cytoplasm (Fig. 12.4). These components span the full Gram-negative cell envelope, going from the cytoplasmic side of the inner membrane to the extracellular side outer membrane, together forming the T4SS complex. The 1 Mda OMC is connected to the 2.6 Mda IMC through a connecting stalk. The OMC is composed of 14 copies each of VirB7, VirB9 and VirB10 while the IMC is made of 12 copies each of VirB3, VirB4, VirB5, VirB8 and 24 copies of VirB6. A segment of VirB10 is inserted into the inner membrane likely to regulate the conformational changes induced by various ATPases present in the inner membrane. The OMC can be further divided into two layers, an outer membrane O layer and an Inner membrane I layer (Marlovits et al. 2004; Rivera-Calzada et al. 2013). The O layer forms a channel in the outer membrane that is composed of an α helical barrel, where each α hairpin motif is contributed by 14 VirB10 units (Chandran et al. 2009). The IMC in the periplasm is composed of two arches connected to two barrel-like leg structures span the inner membrane and project into the cytoplasm. The leg like structures are made of 6 copies each of VirB4, an ATPase, constituting trimer of dimers (12 VirB4/TrwK subunits in total) (Low et al. 2014). VirB11, another ATPase, belonging to the large family of AAA+ hexameric traffic ATPases is responsible for energising the secretion process (Rashkova et al. 2000) along with VirB4. It is found embedded in the cytoplasmic membrane and has nucleoside triphosphatase activity that gets enhanced in the presence of lipids (Planet et al. 2001; Rivas et al. 1997). VirB11 of H pylori and Brucella suis is reported to form double hexameric rings (Hare et al. 2006; Savvides et al. 2003; Yeo et al. 2000). Although it has been structurally well characterized, its exact location in the assembled complex and its structure-function relationship in it is not clear. VirD4 ATPase is the type IV coupling proteins (T4CP) located between the two barrel like legs of the complex (Redzej et al. 2017). It recruits the relaxosome processed substrates(DNA/protein) that is to be transported across the secretion machinery (Cabeźn et al. 1997; Lang et al. 2010; Vergunst et al. 2000). Two copies of VirD4 dimers are located opposite to each other in the cleft formed by VirB4 hexamers in the cytoplasmic side of the inner membrane. As known from its crystal structure (Gomis-Rüth et al. 2001). It has a N-terminal transmembrane domain anchored to the cell membrane that provides stability to the cytoplasmic C terminal domain. The C terminal domain comprises of a catalytic DNA binding domain(NDB) and all helical α domain(AAD) (Chandran Darbari and Waksman 2015).
VirD4 can sense both intracellular and extracellular signal to regulate the opening/closing of the T4SS machinery to allow exchange of substrates (Berry and Christie 2011). It plays the crucial role of recruiting substrates for secretion through the machinery. For translocation of DNA substrate it first undergoes pre-processing by a relaxosome complex composed of 3–4 cytoplasmic proteins (Ilangovan et al. 2017) one of them being the relaxase. Relaxase is an enzyme that can execute two activities namely trans-esterase and helicase. Two relaxase molecules attaches to the origin of Transfer region (oriT) of the plasmid DNA with the help of other proteins of the relaxase complex. A first relaxase molecule makes a single strand nick in the transfer strand of the plasmid DNA (T strand) at the nic site of the oriT with its trans-esterase activity, and gets covalently linked to the free 5′ phosphate. A second relaxase molecule then attaches to the 3′ end of the other strand and unwinds the DNA by its helicase activity (Ilangovan et al. 2017). VirD4 subsequently captures the transfer complex through the relaxase attached to the nic site (Atmakuri et al. 2004). Notably, several T4SSs such as that of Bartonella tribocorum, Brucella sp., and Bordetella pertussis lack ViD4, suggesting VirD4 might have a redundant role in pilus biogenesis. The T4SS is thought be responsible for both pilus biogenesis and conjugation, however mechanistic details of pilus biogenesis are still not clear. Likely, after assembling the pilus it switches to DNA transfer mode (Ilangovan et al. 2017; Zechner et al. 2012). VirD4 and the relaxase complex remain in a dormant state until some contact has been established with the recipient cell through the conjugation pilus. Upon contact the ATPases and the relaxases are activated which creates a ssDNA bubble on the 3′ side exposing the nic site that is further processed by the relaxase. The pathway of the transfer DNA through the secretion machinery, and the mechanism of transfer into the recipient cell remain unknown in molecular detail.
Sortase Mediated Pili of Gram-Positive Bacteria
Gram-positive bacteria lack the diderm cell envelope of Gram-negative bacteria, so that secretion and attachment of pili to the cell surface can be expected to be fundamentally different. In these monoderm systems, pilus subunits are secreted through the general secretory pathway, but require an alternative mechanism for polymerization and attachment to the cell surface. Both these steps rely on the transpeptidation activity a family of enzymes called sortases. Sortase mediated pili were first identified in Gram-positive bacteria in 1968 (Yanagawa et al. 1968), however, their detailed molecular characterization started relatively recently (Ton-That et al. 2004). They lack homology to pili of Gram negative bacteria and are usually much thinner and flexible (Hae et al. 2007). Sortase mediated pili are linear polymers of covalently associated subunits, and are attached to the cell surface by covalent association to the peptidoglycan layer. The intersubunit contacts and cell wall association are formed by a transpeptidase reaction catalysed by surface-localized sortases. Typically, the pili contain two to three subunit types, referred to as basal, backbone and auxiliary pilins, each with their own specific function (Pansegrau and Bagnoli 2017). The structural data available for sortase mediated pili from various bacterial species have shown the presence of intramolecular isopeptide bonds in the pilins (Hae et al. 2007). These isopeptide bonds are formed autocatalytically and are thought to be the functional equivalent of disulphide bonds in other systems, i.e. providing extra stability to the proteins by crosslinking (Kang and Baker 2009). Another interesting feature of these systems is the formation of thioester bonds between the tip pilin and host receptor proteins (Linke-Winnebeck et al. 2014; Pointon et al. 2010).
Pilus Architectures
The pili of Gram-positive bacteria are composed of two or three types of multidomain pilins, called backbone pilin, basal pilin, and tip pilin. Corynebacterium diphtheria, for example, has three distinct pilus systems, SpaABC, SpaDEF, and SpaHIG (Spa for sortase‐mediated pilus assembly), each with three distinct subunits. In these trimeric systems SpaA, SpaD and SpaH are the major or the backbone pilins, while SpaB, SpaE, SpaI, and SpaC, SpaF, SpaG are basal and tip pilins, respectively (Ton-That and Schneewind 2003). The Spa operon model has been found conserved across many other Gram-positive bacteria such as Streptococcus agalactiae, S. pyogenes, S. pneumonia, Enterococcus faecalis, E. faecium, Bacillus cereus, and Actinomyces naeslundii (Mandlik et al. 2008b). The structures of a large number of pilins have now been studied, a detailed review of which can be found in (Krishnan 2015). Despite having limited primary sequence homology most pilins have a similar core tertiary structure. The pilins are modular multidomain proteins that are composed of multiple immunoglobulin like β-sandwich domains, which can be grouped into two main types: CnaA and CnaB domains. The CnaA domain was first identified and coined in the ligand‐binding region A of the collagen adhesin CNA of S. aureus (Symersky et al. 1997). In addition to the CnaA and CnaB domains, the pilins can also include a von Willebrand A (VWA)‐like domain(vAPD) or thioester containing adhesin pilin domain(TAPD) (Fig. 12.5).

Schematic representation of sortase-mediated pilus assembly. a Representative structures of adhesins and pilus subunits. From left to right: the tip pilin or pilus adhesin RrgA adhesin from S. pneumoniae (blue, PDB entry: 2WW8), the basal pilin FctB (cyan, PDB entry: 3KLQ) and backbone pilin Spy0128 (magenta, PDB entry: 3B2M) from S. pyogenes, and the sortase C of S. pneumoniae (dark grey, PDB entry: 2W1J) and sortase A (red, PDB entry:1T2P) of S. aureus. b Schematic diagram of sortase-mediated crosslinking and extension of Gram-positive pili (tip, backbone and basal pilins are coloured as in (a). The SecYEG translocase is shown in grey (PDB entry 5AWW). c Archetypical domain organization in tip, backbone and basal pilins
Backbone pilins. The backbone pilins are the major pilin subunit forming the Gram-positive pili, present in multiple tens to hundreds of copies. These subunits generally consist of two to four CnaA or CnaB domains that form intramolecular isopeptide bonds between the ε-amino group of a lysine and the carbonyl of a glutamic or aspartic acid, or the amide group of an asparagine residue (Kang et al. 2007; Kang and Baker 2011). These bonds are often made such that they link the first and last β strand of the domains, thereby resulting in a tightly packed β sheet structure with high thermal stability and protease resistance (Hae et al. 2007). The N terminal domain of backbone pilins, called D1, often holds a YPKN motif, of which the lysine is involved in the intermolecular transpeptide reaction during polymerization. Backbone pilins of several Gram-positive bacteria have been structurally characterized (Krishnan 2015), showing different architectures comprising pilins with two domains like Spy012 of Streptococcus pyogenes (Kang et al. 2007), three domains like SpaA from Corynebacterium diphtheriae (Kang et al. 2009), or 4 domains such as in BcpA in Bacillus cereus (Budzik et al. 2009).
Basal pilins. The basal pilins are responsible for anchoring of the pilus to the peptidoglycan layer. They are the last to be incorporated into the pilus and along with the housekeeping sortase (SrtF) act as the termination signal (Mandlik et al. 2008a). Basal pilins are often made of one to three CnaB type domains with or without isopeptide bonds. Basal pilins have a conserved proline rich C terminal tail that anchors to the cell wall (Fig. 12.5). The C terminal sorting motif of basal pilins have a triple glycine (TG) motif which possibly is required for sortase specificity, as demonstrated in L. rhamnosus GG (Douillard et al. 2014). The first basal pilin structure to be reported was GBS52 (Krishnan et al. 2007). It has two CnaB like domains linked head to tail through a hydrophobic interface. It has a IYPK pilin motif, of which lysine is thought to be responsible for incorporation into the pilus shaft by forming transpeptide bond with a corresponding backbone pilus C terminal. The crystal structure of another basal pilin FctB reveals the C terminal proline rich tail to fold into a polyproline‐II helix (Linke et al. 2010). And again, FctB also has a lysine in the last strand that is responsible for the transpeptide bond formation. Interestingly, RrgC, a basal pilin of Streptococcus pneumoniae has three CnaB type domains instead of two (Shaik et al. 2014). It has two isopeptide bonds in domain 2 and domain 3 and a short polyproline C terminal tail. In all the reported basal pilin structures there is no isopeptide bond in the domain 1, presumably making it more flexible, which might be a necessity for cell wall anchoring of the pilus.
Tip pilins. Tip pilins are responsible for adhesion of the pilin to various host surfaces. These pilins are usually the largest among the pilus subunits, with 4 domains, which can include both CnaA and CnaB domains, as well as a specialized domain for adhesion: vAPD or TAPD, present at the pilus tip. CnaA and CnaB domains provide support to the adhesive domain and connect it to the backbone pilin (Fig. 12.5). The first tip pilin to be structurally characterized was RrgA (Izoré et al. 2010). It has 4 domains with D3 being a vAPD domain that is located at the tip and has adhesive properties. The D4 domain is responsible for connecting to the pilus shaft. The typical vAPD domain consists of a six stranded β sheets surrounded by α helices and resembles collagen‐binding eukaryotic integrin I‐domains and the MIDAS‐containing vWFA‐like subdomains of complement convertases. The MIDAS motif in RrgA is surrounded by two arm-like structures made of β hairpins and long loops that are implicated in collagen and metal ion binding, and can provide a binding site for anionic extracellular matrix molecules. Similar to RrgA two other tip pilins GBS104 (Krishnan et al. 2013) and Cpa (Linke-Winnebeck et al. 2014) have been partially characterized. Both have 4 domains, though while Cpa differs markedly in term of supertertiary organisation of its domains and its adhesive domain into Y shaped molecule with 2 thioester containing adhesion domains. The Y shape is formed by the centrally located D2 connecting D1 and D3 that form the binding arms and the D4 acts as the stalk. The adhesive domains, D1 and D3, share a common fold comprising of a β sandwich in a helical bundle where the β sandwich contains the thioester binding domain. Apart from that, thioester bonds can be found in adhesion domains of some tip pilins, e.g. Cpa from S. pyogenes. These bonds are formed between the side chains of cysteine and glutamine and are believed to mediate interaction with ligands present on the host cells.
Biogenesis and Secretion of Sortase Mediate Pili
Pilus biogenesis in Gram-positive bacteria is a biphasic process. A class of transpeptidases called sortases are responsible for biogenesis and housekeeping of the pili. Gram-positive bacteria encode several sortase types, each with diverse roles. Based on sequence homology, sortases have been classified into 6 main classes, sortase A–F (Spirig et al. 2011). Among these, class A and class C sortases are crucial from the perspective of pilus biogenesis and maintenance. Class C sortases catalyse the covalent linking reactions during initiation and elongation of pilus assembly (Budzik et al. 2007; Marraffini et al. 2006). Class A sortases are housekeeping enzymes that are responsible for covalent linking of the pili to the peptidoglycan layer (Bradshaw et al. 2015). Apart from the above-mentioned classes, in some species (e.g. Corynebacterium diphtheriae) Class E sortases act as the housekeeping enzyme instead of Class A (Comfort and Clubb 2004; Ton-That and Schneewind 2003). In general, substrates of sortases are transported to the cell surface via the Sec translocon to eventually get anchored in the cell membrane through its C-terminal part. The C-terminal part contains a cell wall targeting signal sequence (CWSS), which consists of a pentapeptide sorting motif followed by a hydrophobic domain which precedes a positively charged short chain (Schneewind and Missiakas 2014). The sorting motif is generally LPXTG that gets cleaved between threonine and glycine by a suitable sortase and produces an acyl intermediate which later gets incorporated in the growing pili.
Proteins that are targets of sortases often have an N-terminal signal peptide and C-terminal cell wall targeting signal sequence (CWSS) with LPXTG motif. In pilins, there is an additional conserved motif in the N-terminal region with sequence YPKN, also known as the pilin motif. After transport through the SecYEG translocon, the N-terminal signal sequence gets cleaved off and the pre-pilin subunit gets inserted into the cell membrane through the hydrophobic domain of the CWSS while the LPXTG motif is exposed to the exterior. A sortase enzyme present on the cell membrane recognizes the anchored pre-pilins through this LPXTG motif. Upon recognition, the cysteine of the TLXTC carries out a nucleophilic attack on the peptide bond between threonine and glycine in the LPXTG motif to break the pre-pilin into two parts (i) a thioester acyl intermediate containing the long N-terminal fragment and (ii) a short C-terminal cleavage product that is released (Marraffini et al. 2006). The resultant acyl intermediate forms a stable product by a nucleophilic substitution reaction, the nucleophile for which is variable depending upon the respective sortase in action. This nucleophile can either be an amine-containing residue in the peptide moiety of a lipid II molecule (class A sortase), meso-2,6-diaminopimelic acid (mDap) in the peptidoglycan cross-bridge (class A, D and E sortases), a lysine residue in a peptidoglycan cross-bridge (class B sortases), or a lysine residue that is part of the “YPKN” motif of a pilin precursor (class C sortases and certain class B sortases) (Frankel et al. 2005; Ton-That et al. 2004). The nucleophilic substitution marks the completion of the transpeptidation reaction, which either results in elongation of the polymerising pili or its covalent attachment to the peptidoglycan layer. These two processes are catalysed by two classes of sortases, the Class C type sortase for initiating and elongating the pilus and the housekeeping class A type sortase for terminating and anchoring of pili to the cell wall. These reactions require that both sortases and pilins are inserted in the cytoplasmic membrane. For this purpose, the pilins have a C-terminal CWSS, whilst sortases have a transmembrane domain in their N terminal regions (Schneewind and Missiakas 2014). The pilus assembly reaction starts when the LPXTG motif of a tip pilin is recognized and cleaved by a class C sortase present in the membrane. The resulting acyl intermediate then undergoes nucleophilic attack by the ε amine group of a specific lysine residue in a second pilus subunit, most often from the conserved YPKN motif in the N-terminus of a backbone pilin, resulting in the formation of an isopeptide bond between the tip pilin and the backbone pilin (Ton-That and Schneewind 2004). The backbone pilin is recognized by a separate C type sortase, which catalyses the transpeptidation reaction with subsequent backbone pilins until it reaches a termination signal by transpeptidation to a basal pilin. The latter is recognized and coupled to a class A sortase, which catalyses transpeptidation to the peptidoglycan precursor lipid II. In a second group of organisms such as in Bacillus cereus there is no distinct basal pilin (Budzik et al. 2007). In such cases the termination occurs when sortase A recognizes a backbone pilin and gets coupled to it by cleaving the LPXTG motif. This happens stochastically after a few rounds of polymerization. The acyl intermediate formed in this transpeptidation reaction can only be resolved by a nucleophilic reaction with the N-terminal glycine of peptide side chains in lipid II molecules. Therefore, addition of a sortase A coupled backbone pilin acts as the termination signal. As a final step, the pilus filaments coupled to lipid II are then incorporated into the peptidoglycan layer, resulting in their covalent attachment to the cell wall.
References
Alaei SR, Park JH, Walker SG, Thanassi DG (2019) Peptide-based inhibitors of fimbrial biogenesis in Porphyromonas gingivalis. Infect Immun. https://doi.org/10.1128/iai.00750-18
Alteri CJ (2005) Novel pili of Mycobacterium tuberculosis. PhD thesis, The University of Arizona
Anderson KL, Billington J, Pettigrew D, Cota E, Simpson P, Roversi P, Chen HA, Urvil P, du Merle L, Barlow PN, Medof ME, Smith RAG, Nowicki B, Le Bouguénec C, Lea SM, Matthews S (2004) An atomic resolution model for assembly, architecture, and function of the Dr. Adhesins. Mol Cell 15(4):647–657. https://doi.org/10.1016/j.molcel.2004.08.003
Atmakuri K, Cascales E, Christie PJ (2004) Energetic components VirD4, VirB11 and VirB4 mediate early DNA transfer reactions required for bacterial type IV secretion. Mol Microbiol 54(5):1199–1211. https://doi.org/10.1111/j.1365-2958.2004.04345.x
Backert S, Haas R, Gerhard M, Naumann M (2017) The helicobacter pylori type IV secretion system encoded by the cag Pathogenicity Island: architecture, function, and signaling, vol 413, pp 187–220. https://doi.org/10.1007/978-3-319-75241-9_8
Bann JG, Pinkner JS, Frieden C, Hultgren SJ (2004) Catalysis of protein folding by chaperones in pathogenic bacteria. PNAS 101(50):17389–17393. https://doi.org/10.1073/pnas.0408072101
Bao R, Nair MKM, W-k Tang, Esser L, Sadhukhan A, Holland RL, Xia D, Schifferli DM (2013) Structural basis for the specific recognition of dual receptors by the homopolymeric pH 6 antigen (Psa) fimbriae of Yersinia pestis. Proc Natl Acad Sci USA 110(3):1065–1070. https://doi.org/10.1073/pnas.1212431110
Bečárová Z (2015) Mechanism of FimI, the assembly termination subunit of the type 1 pili from uropathogenic Escherichia coli. Doctoral thesis, ETH Zurich,
Berry J-L, Pelicic V (2015) Exceptionally widespread nanomachines composed of type IV pilins: the prokaryotic Swiss Army knives. FEMS Microbiol Rev 39(1):134–154. https://doi.org/10.1093/femsre/fuu001
Berry J-L, Phelan MM, Collins RF, Adomavicius T, Tønjum T, Frye SA, Bird L, Owens R, Ford RC, Lian L-Y, Derrick JP (2012) Structure and assembly of a trans-periplasmic channel for type IV Pili in Neisseria meningitidis. PLoS Pathog 8(9):e1002923. https://doi.org/10.1371/journal.ppat.1002923
Berry TM, Christie PJ (2011) Caught in the act: the dialogue between bacteriophage R17 and the type IV secretion machine of plasmid R1. Mol Microbiol 82(5):1039–1043. https://doi.org/10.1111/j.1365-2958.2011.07870.x
Bitter W, Koster M, Latijnhouwers M, de Cock H, Tommassen J (1998) Formation of oligomeric rings by XcpQ and PilQ, which are involved in protein transport across the outer membrane of Pseudomonas aeruginosa. Mol Microbiol 27(1):209–219
Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR (2012) Diversity, biogenesis and function of microbial amyloids. Trends Microbiol 20(2):66–73. https://doi.org/10.1016/j.tim.2011.11.005
Bleem A, Christiansen G, Madsen DJ, Maric H, Strømgaard K, Bryers JD, Daggett V, Meyer RL, Otzen DE (2018) Protein engineering reveals mechanisms of functional amyloid formation in Pseudomonas aeruginosa biofilms. J Mol Biol 430(20):3751–3763. https://doi.org/10.1016/j.jmb.2018.06.043
Bradshaw WJ, Davies AH, Chambers CJ, Roberts AK, Shone CC, Acharya KR (2015) Molecular features of the sortase enzyme family. FEBS J 282(11):2097–2114. https://doi.org/10.1111/febs.13288
Branda SS, Vik Å, Friedman L, Kolter R (2005) Biofilms: the matrix revisited. Trends Microbiol 13(1):20–26. https://doi.org/10.1016/j.tim.2004.11.006
Brombacher E, Dorel C, Zehnder AJB, Landini P (2003) The curli biosynthesis regulator CsgD co-ordinates the expression of both positive and negative determinants for biofilm formation in Eschericia coli. Microbiology 149(10):2847–2857. https://doi.org/10.1099/mic.0.26306-0
Budzik JM, Marraffini LA, Schneewind O (2007) Assembly of pili on the surface of Bacillus cereus vegetative cells. Mol Microbiol 66(2):495–510. https://doi.org/10.1111/j.1365-2958.2007.05939.x
Budzik JM, Poor CB, Faull KF, Whitelegge JP, He C, Schneewind O (2009) Intramolecular amide bonds stabilize pili on the surface of bacilli. Proc Natl Acad Sci USA 106(47):19992–19997. https://doi.org/10.1073/pnas.0910887106
Cabeźn E, Ignacio Sastre J, De La Cruz F (1997) Genetic evidence of a coupling role for the TraG protein family in bacterial conjugation. Mol Gen Genet 254(4):400–406. https://doi.org/10.1007/s004380050432
Cao B, Zhao Y, Kou Y, Ni D, Zhang XC, Huang Y (2014) Structure of the nonameric bacterial amyloid secretion channel. PNAS 111(50):E5439–E5444. https://doi.org/10.1073/pnas.1411942111
Chai L, Romero D, Kayatekin C, Akabayov B, Vlamakis H, Losick R, Kolter R (2013) Isolation, characterization, and aggregation of a structured bacterial matrix precursor. J Biol Chem 288(24):17559–17568. https://doi.org/10.1074/jbc.M113.453605
Chandran Darbari V, Waksman G (2015) Structural biology of bacterial type IV secretion systems. Annu Rev Biochem 84(1):603–629. https://doi.org/10.1146/annurev-biochem-062911-102821
Chandran V, Fronzes R, Duquerroy S, Cronin N, Navaza J, Waksman G (2009) Structure of the outer membrane complex of a type IV secretion system. Nature 462(7276):1011–1015. https://doi.org/10.1038/nature08588
Chang Y-W, Rettberg LA, Treuner-Lange A, Iwasa J, Søgaard-Andersen L, Jensen GJ (2016) Architecture of the type IVa pilus machine. Science 351(6278):aad2001. https://doi.org/10.1126/science.aad2001
Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ (2002) Role of fiber Escherichia in coli curli operons directing formation amyloid. Science 295:851–855. https://doi.org/10.1126/science.1067484
Chiang P, Habash M, Burrows LL (2005) Disparate subcellular localization patterns of Pseudomonas aeruginosa Type IV pilus ATPases involved in twitching motility. J Bacteriol 187(3):829–839. https://doi.org/10.1128/JB.187.3.829-839.2005
Childers SE, Ciufo S, Lovley DR (2002) Geobacter metallireducens accesses insoluble Fe(III) oxide by chemotaxis. Nature 416:767–769
Chirwa NT, Herrington MB (2003) CsgD, a regulator of curli and cellulose synthesis, also regulates serine hydroxymethyltransferase synthesis in Escherichia coli K-12. Microbiology 149(2):525–535. https://doi.org/10.1099/mic.0.25841-0
Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75(1):333–366. https://doi.org/10.1146/annurev.biochem.75.101304.123901
Choudhury D, Thompson A, Stojanoff V, Langermann S, Pinkner J, Hultgren SJ, Knight SD (1999) X-ray structure of the FimC-FimH chaperone-adhesin complex from uropathogenic Escherichia coli. Science 285(5430):1061–1066
Clarke M, Maddera L, Harris RL, Silverman PM (2008a) F-pili dynamics by live-cell imaging. Proc Natl Acad Sci USA 105(46):17978–17981. https://doi.org/10.1073/pnas.0806786105
Clarke M, Maddera L, Harris RL, Silverman PM (2008b) F-pili dynamics by live-cell imaging. PNAS 105(46):17978–17981. https://doi.org/10.1073/pnas.0806786105
Collins R, Karuppiah V, Siebert CA, Dajani R, Thistlethwaite A, Derrick JP (2018) Structural cycle of the Thermus thermophilus PilF ATPase: the powering of type IVa pilus assembly. Sci Rep 8(1):14022. https://doi.org/10.1038/s41598-018-32218-3
Collinson SK, Doig PC, Doran JL, Clouthier S, Trust TJ, Kay WW (1993) Thin, aggregative fimbriae mediate binding of Salmonella enteritidis to fibronectin. J Bacteriol 175(1):12–18. https://doi.org/10.1128/jb.175.1.12-18.1993
Comfort D, Clubb RT (2004) A comparative genome analysis identifies distinct sorting pathways in Gram-positive bacteria. Infect Immun 72(5):2710–2722. https://doi.org/10.1128/IAI.72.5.2710-2722.2004
Conover Matt S, Ruer S, Taganna J, Kalas V, De Greve H, Pinkner Jerome S, Dodson Karen W, Remaut H, Hultgren Scott J (2016) Inflammation-induced adhesin-receptor interaction provides a fitness advantage to uropathogenic E. coli during chronic infection. Cell Host Microbe 20(4):482–492. https://doi.org/10.1016/j.chom.2016.08.013
Costa TRD, Ilangovan A, Ukleja M, Redzej A, Santini JM, Smith TK, Egelman EH, Waksman G (2016) Structure of the bacterial sex F pilus reveals an assembly of a stoichiometric protein-phospholipid complex. Cell 166(6):1436–1444.e1410. https://doi.org/10.1016/j.cell.2016.08.025
Craig L, Li J (2008) Type IV pili: paradoxes in form and function. Curr Opin Struct Biol 18(2):267–277. https://doi.org/10.1016/j.sbi.2007.12.009
Craig L, Pique ME, Tainer JA (2004) Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol 2(5):363–378. https://doi.org/10.1038/nrmicro885
Craig L, Taylor RK, Pique ME, Adair BD, Arvai AS, Singh M, Lloyd SJ, Shin DS, Getzoff ED, Yeager M, Forest KT, Tainer JA (2003) Type IV pilin structure and assembly: X-ray and EM analyses of Vibrio cholerae toxin-coregulated pilus and Pseudomonas aeruginosa PAK pilin. Mol Cell 11(5):1139–1150
Craig L, Volkmann N, Arvai AS, Pique ME, Yeager M, Egelman EH, Tainer JA (2006) Type IV pilus structure by cryo-electron microscopy and crystallography: implications for pilus assembly and functions. Mol Cell 23(5):651–662. https://doi.org/10.1016/j.molcel.2006.07.004
Cusumano CK, Pinkner JS, Han Z, Greene SE, Ford BA, Crowley JR, Henderson JP, Janetka JW, Hultgren SJ (2011) Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci Transl Med 3(109):109ra115. https://doi.org/10.1126/scitranslmed.3003021
Danelishvili L, Yamazaki Y, Selker J, Bermudez LE (2010) Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c proteins participate in the suppression of macrophage apoptosis. PLoS ONE 5(5). https://doi.org/10.1371/journal.pone.0010474
Depas WH, Chapman MR, Hufnagel DA (2015) The biology of the Escherichia coli extracellular matrix. In: Microbial biofilms, 2nd edn, pp 249–267. https://doi.org/10.1128/microbiolspec.mb-0014-2014
Dodson KW, Jacob-Dubuisson F, Striker RT, Hultgren SJ (1993) Outer-membrane PapC molecular usher discriminately recognizes periplasmic chaperone-pilus subunit complexes. Proc Natl Acad Sci USA 90(8):3670–3674
Dodson KW, Pinkner JS, Rose T, Magnusson G, Hultgren SJ, Waksman G (2001) Structural basis of the interaction of the Pyelonephritic E. coli adhesin to its human kidney receptor. Cell 105(6):733–743. https://doi.org/10.1016/s0092-8674(01)00388-9
Douillard FP, Rasinkangas P, Von Ossowski I, Reunanen J, Palva A, De Vos WM (2014) Functional identification of conserved residues involved in Lactobacillus rhamnosus strain GG sortase specificity and pilus biogenesis. J Biol Chem 289(22):15764–15775. https://doi.org/10.1074/jbc.M113.542332
Dueholm MS, Albertsen M, Otzen D, Nielsen PH (2012) Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure. PLoS ONE 7(12):1–10. https://doi.org/10.1371/journal.pone.0051274
Dueholm MS, Petersen SV, Sønderkær M, Larsen P, Christiansen G, Hein KL, Enghild JJ, Nielsen JL, Nielsen KL, Nielsen PH, Otzen DE (2010) Functional amyloid in pseudomonas. Mol Microbiol 77(4):1009–1020. https://doi.org/10.1111/j.1365-2958.2010.07269.x
Dueholm MS, Søndergaard MT, Nilsson M, Christiansen G, Stensballe A, Overgaard MT, Givskov M, Tolker-Nielsen T, Otzen DE, Nielsen PH (2013) Expression of Fap amyloids in Pseudomonas aeruginosa, P. fluorescens, and P. putida results in aggregation and increased biofilm formation. MicrobiologyOpen 2(3):365–382. https://doi.org/10.1002/mbo3.81
Evans ML, Chorell E, Taylor JD, Åden J, Götheson A, Li F, Koch M, Sefer L, Matthews SJ, Wittung-Stafshede P, Almqvist F, Chapman MR (2015) The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol Cell 57(3):445–456. https://doi.org/10.1016/j.molcel.2014.12.025
Fällman E, Schedin S, Jass J, Uhlin B-E, Axner O (2005) The unfolding of the P pili quaternary structure by stretching is reversible, not plastic. EMBO Rep 6(1):52–56. https://doi.org/10.1038/sj.embor.7400310
Filman DJ, Marino SF, Ward JE, Yang L, Mester Z, Bullitt E, Lovley DR, Strauss M (2018). Structure of a cytochrome-based bacterial nanowire. 492645
Fowler DM, Koulov AV, Balch WE, Kelly JW (2007) Functional amyloid—from bacteria to humans. Trends Biochem Sci 32(5):217–224. https://doi.org/10.1016/j.tibs.2007.03.003
Frankel BA, Kruger RG, Robinson DE, Kelleher NL, McCafferty DG (2005) Staphylococcus aureus sortase transpeptidase SrtA: insight into the kinetic mechanism and evidence for a reverse protonation catalytic mechanism. Biochemistry 44(33):11188–11200. https://doi.org/10.1021/bi050141j
Gerstel U, Römling U (2003) The csgD promoter, a control unit for biofilm formation in Salmonella typhimurium. Res Microbiol 154(10):659–667. https://doi.org/10.1016/j.resmic.2003.08.005
Gibson DL, White AP, Rajotte CM, Kay WW (2007) AgfC and AgfE facilitate extracellular thin aggregative fimbriae synthesis in Salmonella Enteritidis. Microbiology 153(4):1131–1140. https://doi.org/10.1099/mic.0.2006/000935-0
Giltner CL, Nguyen Y, Burrows LL (2012) Type IV pilin proteins: versatile molecular modules. Microbiol Mol Biol Rev 76(4):740–772. https://doi.org/10.1128/MMBR.00035-12
Gold VAM, Salzer R, Averhoff B, Kühlbrandt W (2015) Structure of a type IV pilus machinery in the open and closed state. eLife
Gomis-Rüth FX, Moncalián G, Pérez-Luque R, González A, Cabezón E, De La Cruz F, Coll M (2001) The bacterial conjugation protein TrwB resembles ring helicases and F1-atpase. Nature 409(6820):637–641. https://doi.org/10.1038/35054586
Goyal P, Krasteva PV, Van Gerven N, Gubellini F, Van Den Broeck I, Troupiotis-Tsaïlaki A, Jonckheere W, Péhau-Arnaudet G, Pinkner JS, Chapman MR, Hultgren SJ, Howorka S, Fronzes R, Remaut H (2014) Structural and mechanistic insights into the bacterial amyloid secretion channel CsgG. Nature 516(7530):250–253. https://doi.org/10.1038/nature13768
Grohmann E, Christie PJ, Waksman G, Backert S (2018) Type IV secretion in Gram-negative and Gram-positive bacteria. Mol Microbiol 107(4):455–471. https://doi.org/10.1111/mmi.13896
Grund S, Weber A (1988) A new type of fimbriae on Salmonella typhimurium. J Vet Med Ser B 35(1–10):779–782. https://doi.org/10.1111/j.1439-0450.1988.tb00560.x
Hae JK, Coulibaly F, Clow F, Proft T, Baker EN (2007) Stabilizing isopeptide bonds revealed in Gram-positive bacterial pilus structure. Science 318(5856):1625–1628. https://doi.org/10.1126/science.1145806
Hall-Stoodley L, Stoodley P (2009) Evolving concepts in biofilm infections. Cell Microbiol 11(7):1034–1043. https://doi.org/10.1111/j.1462-5822.2009.01323.x
Hamada N, Sojar HT, Cho MI, Genco RJ (1996) Isolation and characterization of a minor fimbria from Porphyromonas gingivalis. Infect Immun 64(11):4788–4794
Hammar M, Arnqvist A, Bian Z, Olsen A, Normark S (1995) Expression of two csg operons is required for production of fibronectin- and congo red-binding curli polymers in Escherichia coli K-12. Mol Microbiol 18(4):661–670
Hammer ND, McGuffie BA, Zhou Y, Badtke MP, Reinke AA, Brännström K, Gestwicki JE, Olofsson A, Almqvist F, Chapman MR (2012) The C-terminal repeating units of CsgB direct bacterial functional amyloid nucleation. J Mol Biol 422(3):376–389. https://doi.org/10.1016/j.jmb.2012.05.043
Hammer ND, Schmidt JC, Chapman MR (2007) The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. PNAS 104(30):12494–12499. https://doi.org/10.1073/pnas.0703310104
Hare S, Bayliss R, Baron C, Waksman G (2006) A large domain swap in the VirB11 ATPase of Brucella suis leaves the hexameric assembly intact. J Mol Biol 360(1):56–66. https://doi.org/10.1016/j.jmb.2006.04.060
Hirota S, Hattori Y, Nagao S, Taketa M, Komori H, Kamikubo H, Wang Z, Takahashi I, Negi S, Sugiura Y, et al (2010) Cytochrome c polymerization by successive domain swapping at the Cterminal helix. Proc Nat Acad Sci US 107:12854–12859
Holmes DE, Chaudhuri SK, Nevin KP, Mehta T, Methé BA, Liu A, Ward JE, Woodard TL, Webster J, Lovley DR (2006) Microarray and genetic analysis of electron transfer to electrodes in Geobacter sulfurreducens. Environ Microbiol 8:1805–1815
Hospenthal MK, Costa TRD, Waksman G (2017) A comprehensive guide to pilus biogenesis in Gram-negative bacteria. Nat Rev Microbiol 15(6):365–379. https://doi.org/10.1038/nrmicro.2017.40
Hospenthal Manuela K, Redzej A, Dodson K, Ukleja M, Frenz B, Rodrigues C, Hultgren Scott J, DiMaio F, Egelman Edward H, Waksman G (2016) Structure of a chaperone-usher pilus reveals the molecular basis of rod uncoiling. Cell 164(1–2):269–278. https://doi.org/10.1016/j.cell.2015.11.049
Hultgren SJ, Abraham S, Caparon M, Falk P, St Geme JW, Normark S (1993) Pilus and nonpilus bacterial adhesins: assembly and function in cell recognition. Cell 73(5):887–901
Hung C-S, Bouckaert J, Hung D, Pinkner J, Widberg C, DeFusco A, Auguste CG, Strouse R, Langermann S, Waksman G, Hultgren SJ (2002) Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol Microbiol 44(4):903–915. https://doi.org/10.1046/j.1365-2958.2002.02915.x
Ilangovan A, Kay CWM, Roier S, El Mkami H, Salvadori E, Zechner EL, Zanetti G, Waksman G (2017) Cryo-EM structure of a relaxase reveals the molecular basis of DNA unwinding during bacterial conjugation. Cell 169(4):708–721.e712. https://doi.org/10.1016/j.cell.2017.04.010
Imam S, Chen Z, Roos DS, Pohlschröder M (2011) Identification of surprisingly diverse type IV Pili, across a broad range of Gram-positive bacteria. PLoS ONE 6(12). https://doi.org/10.1371/journal.pone.0028919
Izoré T, Contreras-Martel C, El Mortaji L, Manzano C, Terrasse R, Vernet T, Di Guilmi AM, Dessen A (2010) Structural basis of host cell recognition by the pilus adhesin from Streptococcus pneumoniae. Structure 18(1):106–115. https://doi.org/10.1016/j.str.2009.10.019
Jeter C, Matthysse AG (2005) Characterization of the binding of diarrheagenic strains of E. coli to plant surfaces and the role of curli in the interaction of the bacteria with alfalfa sprouts. Mol Plant-Microbe Interact 18(11):1235–1242. https://doi.org/10.1094/mpmi-18-1235
Kalas V, Hibbing ME, Maddirala AR, Chugani R, Pinkner JS, Mydock-McGrane LK, Conover MS, Janetka JW, Hultgren SJ (2018) Structure-based discovery of glycomimetic FmlH ligands as inhibitors of bacterial adhesion during urinary tract infection. Proc Natl Acad Sci USA 115(12):E2819–E2828. https://doi.org/10.1073/pnas.1720140115
Kang HJ, Baker EN (2009) Intramolecular isopeptide bonds give thermodynamic and proteolytic stability to the major Pilin protein of Streptococcus pyogenes. J Biol Chem 284(31):20729–20737. https://doi.org/10.1074/jbc.M109.014514
Kang HJ, Baker EN (2011) Intramolecular isopeptide bonds: Protein crosslinks built for stress? Trends Biochem Sci 36(4):229–237. https://doi.org/10.1016/j.tibs.2010.09.007
Kang HJ, Paterson NG, Gaspar AH, Ton-That H, Baker EN (2009) The Corynebacterium diphtheriae shaft pilin SpaA is built of tandem Ig-like modules with stabilizing isopeptide and disulfide bonds. PNAS 106(40):16967–16971. https://doi.org/10.1073/pnas.0906826106
Karuppiah V, Derrick JP (2011) Structure of the PilM-PilN inner membrane type IV pilus biogenesis complex from Thermus thermophilus. J Biol Chem 286(27):24434–24442. https://doi.org/10.1074/jbc.M111.243535
Keller R, Ordoñez JG, de Oliveira RR, Trabulsi LR, Baldwin TJ, Knutton S (2002) Afa, a diffuse adherence fibrillar adhesin associated with enteropathogenic Escherichia coli. Infect Immun 70(5):2681–2689. https://doi.org/10.1128/IAI.70.5.2681-2689.2002
Kim K, Oh J, Han D, Kim EE, Lee B, Kim Y (2006) Crystal structure of PilF: functional implication in the type 4 pilus biogenesis in Pseudomonas aeruginosa. Biochem Biophys Res Commun 340(4):1028–1038. https://doi.org/10.1016/j.bbrc.2005.12.108
Klein RD, Shu Q, Cusumano ZT, Nagamatsu K, Gualberto NC, Lynch AJL, Wu C, Wang W, Jain N, Pinkner JS, Amarasinghe GK, Hultgren SJ, Frieden C, Chapman MR (2018) Structure-function analysis of the curli accessory protein CsgE defines surfaces essential for coordinating amyloid fiber formation. mBio 9(4). https://doi.org/10.1128/mbio.01349-18
Kloppsteck P, Hall M, Hasegawa Y, Persson K (2016) Structure of the fimbrial protein Mfa4 from Porphyromonas gingivalis in its precursor form: implications for a donor-strand complementation mechanism. Sci Rep 6. https://doi.org/10.1038/srep22945
Kolappan S, Coureuil M, Yu X, Nassif X, Egelman EH, Craig L (2016) Structure of the Neisseria meningitidis Type IV pilus. Nat Commun 7:13015. https://doi.org/10.1038/ncomms13015
Korhonen TK, Parkkinen J, Hacker J, Finne J, Pere A, Rhen M, Holthofer H (1986) Binding of Escherichia coli S fimbriae to human kidney epithelium. Infect Immun 54(2):322–327
Krantz BA, Melnyk RA, Zhang S, Juris SJ, Lacy DB, Wu Z, Finkelstein A, Collier RJ (2005) A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 309(5735):777–781. https://doi.org/10.1126/science.1113380
Krishnan V (2015) Pilins in Gram-positive bacteria: a structural perspective. IUBMB Life 67(7):533–543. https://doi.org/10.1002/iub.1400
Krishnan V, Dwivedi P, Kim BJ, Samal A, MacOn K, Ma X, Mishra A, Doran KS, Ton-That H, Narayana SVL (2013) Structure of Streptococcus agalactiae tip pilin GBS104: a model for GBS pili assembly and host interactions. Acta Crystallogr D Biol Crystallogr 69(6):1073–1089. https://doi.org/10.1107/S0907444913004642
Krishnan V, Gaspar AH, Ye N, Mandlik A, Ton-That H, Narayana SVL (2007) An IgG-like domain in the minor pilin GBS52 of Streptococcus agalactiae mediates lung epithelial cell adhesion. Structure 15(8):893–903. https://doi.org/10.1016/j.str.2007.06.015
Lang S, Gruber K, Mihajlovic S, Arnold R, Gruber CJ, Steinlechner S, Jehl MA, Rattei T, Fröhlich KU, Zechner EL (2010) Molecular recognition determinants for type IV secretion of diverse families of conjugative relaxases. Mol Microbiol 78(6):1539–1555. https://doi.org/10.1111/j.1365-2958.2010.07423.x
Langermann S, Möllby R, Burlein JE, Palaszynski SR, Auguste CG, DeFusco A, Strouse R, Schenerman MA, Hultgren SJ, Pinkner JS, Winberg J, Guldevall L, Söderhäll M, Ishikawa K, Normark S, Koenig S (2000) Vaccination with FimH adhesin protects cynomolgus monkeys from colonization and infection by uropathogenic Escherichia coli. J Infect Dis 181(2):774–778. https://doi.org/10.1086/315258
Langermann S, Palaszynski S, Barnhart M, Auguste G, Pinkner JS, Burlein J, Barren P, Koenig S, Leath S, Jones CH, Hultgren SJ (1997) Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science 276(5312):607–611
Larsen P, Nielsen JL, Otzen D, Nielsen PH (2008) Amyloid-like adhesins produced by floc-forming and filamentous bacteria in activated sludge. Appl Environ Microbiol 74(5):1517–1526. https://doi.org/10.1128/AEM.02274-07
Leang C, Malvankar NS, Franks AE, Nevin KP, Lovley DR (2013) Engineering Geobacter sulfurreducens to produce a highly cohesive conductive matrix with enhanced capacity for current production. Energy Environ Sci 6:1901–1908
Leighton TL, Buensuceso RNC, Howell PL, Burrows LL (2015a) Biogenesis of Pseudomonas aeruginosa type IV pili and regulation of their function. Environ Microbiol 17(11):4148–4163. https://doi.org/10.1111/1462-2920.12849
Leighton TL, Dayalani N, Sampaleanu LM, Howell PL, Burrows LL (2015b) Novel role for PilNO in type IV pilus retraction revealed by alignment subcomplex mutations. J Bacteriol 197(13):2229–2238. https://doi.org/10.1128/JB.00220-15
Linke C, Young P, Kang H, Bunker R, Middleditch M, Caradoc-Davies T, Proft T, Baker E (2010) Crystal structure of the minor pilin FctB reveals determinants of Group A streptococcal pilus anchoring. J Biol Chem 285(26):20381–20389. https://doi.org/10.1074/jbc.M109.089680
Linke-Winnebeck C, Paterson NG, Young PG, Middleditch MJ, Greenwood DR, Witte G, Baker EN (2014) Structural model for covalent adhesion of the Streptococcus pyogenes pilus through a thioester bond. J Biol Chem 289(1):177–189. https://doi.org/10.1074/jbc.M113.523761
Liu X, Zhuo S, Rensing C, Zhou S (2018) Syntrophic growth with direct interspecies electron transfer between pili-free Geobacter species. ISME J 12:2142–2151
Lo AWH, Van de Water K, Gane PJ, Chan AWE, Steadman D, Stevens K, Selwood DL, Waksman G, Remaut H (2014) Suppression of type 1 pilus assembly in uropathogenic Escherichia coli by chemical inhibition of subunit polymerization. J Antimicrob Chemother 69(4):1017–1026. https://doi.org/10.1093/jac/dkt467
Loferer H, Hammer M, Normark S (1997) Availability of the fibre subunit CsgA and the nucleator protein CsgB during assembly of fibronectin-binding curli is limited by the intracellular concentration of the novel lipoprotein CsgG. Mol Microbiol 26(1):11–23. https://doi.org/10.1046/j.1365-2958.1997.5231883.x
Lory S, Strom MS (1997) Structure-function relationship of type-IV prepilin peptidase of Pseudomonas aeruginosa—a review. Gene 192(1):117–121
Louros NN, Bolas GMP, Tsiolaki PL, Hamodrakas SJ, Iconomidou VA (2016) Intrinsic aggregation propensity of the CsgB nucleator protein is crucial for curli fiber formation. J Struct Biol 195(2):179–189. https://doi.org/10.1016/j.jsb.2016.05.012
Low HH, Gubellini F, Rivera-Calzada A, Braun N, Connery S, Dujeancourt A, Lu F, Redzej A, Fronzes R, Orlova EV, Waksman G (2014) Structure of a type IV secretion system. Nature 508(7497):550–553. https://doi.org/10.1038/nature13081
Maier B, Potter L, So M, Seifert HS, Sheetz MP (2002) Single pilus motor forces exceed 100 pN. PNAS 99(25):16012–16017. https://doi.org/10.1073/pnas.242523299
Majdalani N, Ippen-Ihler K (1996) Membrane insertion of the F-pilin subunit is Sec independent but requires leader peptidase B and the proton motive force. J Bacteriol 178(13):3742–3747. https://doi.org/10.1128/jb.178.13.3742-3747.1996
Majdalani N, Moore D, Maneewannakul S, Ippen-Ihler K (1996) Role of the propilin leader peptide in the maturation of F pilin. J Bacteriol 178(13):3748–3754. https://doi.org/10.1128/jb.178.13.3748-3754.1996
Malvankar NS, Lovley DR (2014) Microbial nanowires for bioenergy applications. Curr Opin Biotechnol 27:88–95
Malvankar NS, Vargas M, Nevin KP, Franks AE, Leang C, Kim B-C, Inoue K, Mester T, Covalla SF, Johnson JP, et al (2011) Tunable metallic-like conductivity in microbial nanowire networks. Nat Nanotechnol 6:573
Mandlik A, Das A, Ton-That H (2008a) The molecular switch that activates the cell wall anchoring step of pilus assembly in Gram-positive bacteria. Proc Natl Acad Sci USA 105(37):14147–14152. https://doi.org/10.1073/pnas.0806350105
Mandlik A, Swierczynski A, Das A, Ton-That H (2008b) Pili in Gram-positive bacteria: assembly, involvement in colonization and biofilm development. Trends Microbiol 16(1):33–40. https://doi.org/10.1016/j.tim.2007.10.010
Maneewannakul K, Maneewannakul S, Ippen-Ihler K (1995) Characterization of traX, the F plasmid locus required for acetylation of F-pilin subunits. J Bacteriol 177(11):2957–2964. https://doi.org/10.1128/jb.177.11.2957-2964.1995
Marlovits TC, Kubori T, Sukhan A, Thomas DR, Galán JE, Unger VM (2004) Structural insights into the assembly of the type III secretion needle complex. Science 306(5698):1040–1042. https://doi.org/10.1126/science.1102610
Marraffini LA, DeDent AC, Schneewind O (2006) Sortases and the art of anchoring proteins to the envelopes of Gram-positive bacteria. Microbiol Mol Biol Rev 70(1):192–221. https://doi.org/10.1128/MMBR.70.1.192-221.2006
Matias J, Berzosa M, Pastor Y, Irache JM, Gamazo C (2017) Maternal vaccination. Immunization of sows during pregnancy against ETEC Infections. Vaccines (Basel) 5(4). https://doi.org/10.3390/vaccines5040048
Mattick JS (2002) Type IV pili and twitching motility. Annu Rev Microbiol 56:289–314. https://doi.org/10.1146/annurev.micro.56.012302.160938. Epub 2002 Jan 30
Mehta T, Coppi MV, Childers SE, Lovley DR (2005) Outer membrane c-type cytochromes required for Fe(III) and Mn(IV) oxide reduction in Geobacter sulfurreducens. Appl Environ Microbiol 71:8634–8641
Merz AJ, So M, Sheetz MP (2000) Pilus retraction powers bacterial twitching motility. Nature 407(6800):98–102. https://doi.org/10.1038/35024105
Misic AM, Satyshur KA, Forest KT (2010) P. aeruginosa PilT structures with and without nucleotide reveal a dynamic type IV pilus retraction motor. J Mol Biol 400(5):1011–1021. https://doi.org/10.1016/j.jmb.2010.05.066. Epub 2010 June 1
Moonens K, Remaut H (2017) Evolution and structural dynamics of bacterial glycan binding adhesins. Curr Opin Struct Biol 44:48–58. https://doi.org/10.1016/j.sbi.2016.12.003
Moonens K, Van den Broeck I, De Kerpel M, Deboeck F, Raymaekers H, Remaut H, De Greve H (2015) Structural and functional insight into the carbohydrate receptor binding of F4 fimbriae-producing enterotoxigenic Escherichia coli. J Biol Chem 290(13):8409–8419. https://doi.org/10.1074/jbc.M114.618595
Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ (1998) Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 282(5393):1494–1497
Nakayama K, Yoshimura F, Kadowaki T, Yamamoto K (1996) Involvement of arginine-specific cysteine proteinase (Arg-gingipain) in fimbriation of Porphyromonas gingivalis. J Bacteriol 178(10):2818–2824
Nenninger AA, Robinson LS, Hammer ND, Epstein EA, Badtke MP, Hultgren SJ, Chapman MR (2011) CsgE is a curli secretion specificity factor that prevents amyloid fibre aggregation. Mol Microbiol 81(2):486–499. https://doi.org/10.1111/j.1365-2958.2011.07706.x
Nenninger AA, Robinson LS, Hultgren SJ (2009) Localized and efficient curli nucleation requires the chaperone-like amyloid assembly protein CsgF. PNAS 106(3):900–905. https://doi.org/10.1073/pnas.0812143106
Nishiyama M, Horst R, Eidam O, Herrmann T, Ignatov O, Vetsch M, Bettendorff P, Jelesarov I, Grütter MG, Wüthrich K, Glockshuber R, Capitani G (2005) Structural basis of chaperone-subunit complex recognition by the type 1 pilus assembly platform FimD. EMBO J 24(12):2075–2086. https://doi.org/10.1038/sj.emboj.7600693
Nishiyama M, Ishikawa T, Rechsteiner H, Glockshuber R (2008) Reconstitution of pilus assembly reveals a bacterial outer membrane catalyst. Science 320(5874):376–379. https://doi.org/10.1126/science.1154994
Nishiyama M, Vetsch M, Puorger C, Jelesarov I, Glockshuber R (2003) Identification and characterization of the chaperone-subunit complex-binding domain from the type 1 pilus assembly platform FimD. J Mol Biol 330(3):513–525. https://doi.org/10.1016/S0022-2836(03)00591-6
Nuccio S-P, Bäumler AJ (2007) Evolution of the chaperone/usher assembly pathway: fimbrial classification goes Greek. Microbiol Mol Biol Rev 71(4):551–575. https://doi.org/10.1128/MMBR.00014-07
Ohlsson J, Jass J, Uhlin BE, Kihlberg J, Nilsson UJ (2002) Discovery of potent inhibitors of PapG adhesins from uropathogenic Escherichia coli through synthesis and evaluation of galabiose derivatives. ChemBioChem 3(8):772–779. https://doi.org/10.1002/1439-7633(20020802)3:8%3c772:AID-CBIC772%3e3.0.CO;2-8
Olsén A, Jonsson A, Normark S (1989) Fibronectin binding mediated by a novel class of surface organelles on Escherichia coli. Nature 338(6217):652–655. https://doi.org/10.1038/338652a0
Omattage NS, Deng Z, Pinkner JS, Dodson KW, Almqvist F, Yuan P, Hultgren SJ (2018) Structural basis for usher activation and intramolecular subunit transfer in P pilus biogenesis in Escherichia coli. Nat Microbiol 3(12):1362. https://doi.org/10.1038/s41564-018-0255-y
Otter JA, Vickery K, Walker JT, deLancey Pulcini E, Stoodley P, Goldenberg SD, Salkeld JA, Chewins J, Yezli S, Edgeworth JD (2015) Surface-attached cells, biofilms and biocide susceptibility: implications for hospital cleaning and disinfection. J Hosp Infect 89(1):16–27. https://doi.org/10.1016/j.jhin.2014.09.008
Pansegrau W, Bagnoli F (2017) Pilus assembly in Gram-positive bacteria, vol 404, pp 203–233. https://doi.org/10.1007/82_2015_5016
Parge HE, Forest KT, Hickey MJ, Christensen DA, Getzoff ED, Tainer JA (1995) Structure of the fibre-forming protein pilin at 2.6 A resolution. Nature 378(6552):32–38. https://doi.org/10.1038/378032a0
Pelicic V (2008) Type IV pili: e pluribus unum? Mol Microbiol 68(4):827–837. https://doi.org/10.1111/j.1365-2958.2008.06197.x
Phan G, Remaut H, Wang T, Allen WJ, Pirker KF, Lebedev A, Henderson NS, Geibel S, Volkan E, Yan J, Kunze MBA, Pinkner JS, Ford B, Kay CWM, Li H, Hultgren S, Thanassi DG, Waksman G (2011) Crystal structure of the FimD usher bound to its cognate FimC:FimH substrate. Nature 474(7349):49–53. https://doi.org/10.1038/nature10109
Pinkner JS, Remaut H, Buelens F, Miller E, Aberg V, Pemberton N, Hedenström M, Larsson A, Seed P, Waksman G, Hultgren SJ, Almqvist F (2006) Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc Natl Acad Sci USA 103(47):17897–17902. https://doi.org/10.1073/pnas.0606795103
Planet PJ, Kachlany SC, DeSalle R, Figurski DH (2001) Phylogeny of genes for secretion NTPases: identification of the widespread tadA subfamily and development of a diagnostic key for gene classification. PNAS 98(5):2503–2508. https://doi.org/10.1016/j.jad.2013.02.029
Pointon JA, Smith WD, Saalbach G, Crow A, Kehoe MA, Banfield MJ (2010) A highly unusual thioester bond in a pilus adhesin is required for efficient host cell interaction. J Biol Chem 285(44):33858–33866. https://doi.org/10.1074/jbc.M110.149385
Proft T, Baker EN (2009) Pili in Gram-negative and Gram-positive bacteria—structure, assembly and their role in disease. Cell Mol Life Sci 66(4):613–635. https://doi.org/10.1007/s00018-008-8477-4
Puorger C, Eidam O, Capitani G, Erilov D, Grütter MG, Glockshuber R (2008) Infinite kinetic stability against dissociation of supramolecular protein complexes through donor strand complementation. Structure 16(4):631–642. https://doi.org/10.1016/j.str.2008.01.013
Puorger C, Vetsch M, Wider G, Glockshuber R (2011) Structure, folding and stability of FimA, the main structural subunit of type 1 pili from uropathogenic Escherichia coli strains. J Mol Biol 412(3):520–535. https://doi.org/10.1016/j.jmb.2011.07.044
Ramboarina S, Fernandes PJ, Daniell S, Islam S, Simpson P, Frankel G, Booy F, Donnenberg MS, Matthews S (2005) Structure of the bundle-forming pilus from enteropathogenic Escherichia coli. J Biol Chem 280(48):40252–40260. https://doi.org/10.1074/jbc.M508099200
Ramsugit S, Pillay M (2015) Pili of Mycobacterium tuberculosis: current knowledge and future prospects. Arch Microbiol 197(6):737–744. https://doi.org/10.1007/s00203-015-1117-0
Rashkova S, Zhou XR, Chen J, Christie PJ (2000) Self-assembly of the Agrobacterium tumefaciens VirB11 traffic ATPase. J Bacteriol 182(15):4137–4145. https://doi.org/10.1128/JB.182.15.4137-4145.2000
Redzej A, Ukleja M, Connery S, Trokter M, Felisberto-Rodrigues C, Cryar A, Thalassinos K, Hayward RD, Orlova EV, Waksman G (2017) Structure of a VirD4 coupling protein bound to a VirB type IV secretion machinery. EMBO J 36(20):e201796629–e201796629. https://doi.org/10.15252/embj.201796629
Reguera G, McCarthy KD, Mehta T, Nicoll JS, Tuominen MT, Lovley DR (2005) Extracellular electron transfer via microbial nanowires. Nature 435:1098–1101
Reichhardt C, Cegelski L (2014) Solid-state NMR for bacterial biofilms. Mol Phys 112(7):887–894. https://doi.org/10.1080/00268976.2013.837983
Remaut H, Rose RJ, Hannan TJ, Hultgren SJ, Radford SE, Ashcroft AE, Waksman G (2006) Donor-strand exchange in chaperone-assisted pilus assembly proceeds through a concerted beta strand displacement mechanism. Mol Cell 22(6):831–842. https://doi.org/10.1016/j.molcel.2006.05.033
Remaut H, Tang C, Henderson NS, Pinkner JS, Wang T, Hultgren SJ, Thanassi DG, Waksman G, Li H (2008) Fiber formation across the bacterial outer membrane by the chaperone/usher pathway. Cell 133(4):640–652. https://doi.org/10.1016/j.cell.2008.03.033
Richter LV, Sandler SJ, Weis RM (2012) Two isoforms of Geobacter sulfurreducens PilA have distinct roles in pilus biogenesis, cytochrome localization, extracellular electron transfer, and biofilm formation. J Bacteriol 194:2551–2563
Rivas S, Bolland S, Cabezón E, Goñi FM, De La Cruz F (1997) TrwD, a protein encoded by the IncW plasmid R388, displays an ATP hydrolase activity essential for bacterial conjugation. J Biol Chem 272(41):25583–25590. https://doi.org/10.1074/jbc.272.41.25583
Rivera-Calzada A, Fronzes R, Savva CG, Chandran V, Lian PW, Laeremans T, Pardon E, Steyaert J, Remaut H, Waksman G, Orlova EV (2013) Structure of a bacterial type IV secretion core complex at subnanometre resolution. EMBO J 32(8):1195–1204. https://doi.org/10.1038/emboj.2013.58
Roberts JA, Marklund BI, Ilver D, Haslam D, Kaack MB, Baskin G, Louis M, Mollby R, Winberg J, Normark S (1994) The Gal(alpha 1–4)Gal-specific tip adhesin of Escherichia coli P-fimbriae is needed for pyelonephritis to occur in the normal urinary tract. Proc Natl Acad Sci USA 91(25):11889–11893
Robinson LS, Ashman EM, Hultgren SJ, Chapman MR (2006) Secretion of curli fibre subunits is mediated by the outer membrane-localized CsgG protein. Mol Microbiol 59(3):870–881. https://doi.org/10.1111/j.1365-2958.2005.04997.x
Rose RJ, Verger D, Daviter T, Remaut H, Paci E, Waksman G, Ashcroft AE, Radford SE (2008) Unraveling the molecular basis of subunit specificity in P pilus assembly by mass spectrometry. PNAS 105(35):12873–12878. https://doi.org/10.1073/pnas.0802177105
Rouse SL, Hawthorne WJ, Berry JL, Chorev DS, Ionescu SA, Lambert S, Stylianou F, Ewert W, Mackie U, Morgan RML, Otzen D, Herbst FA, Nielsen PH, Dueholm M, Bayley H, Robinson CV, Hare S, Matthews S (2017) A new class of hybrid secretion system is employed in Pseudomonas amyloid biogenesis. Nat Commun 8(1):263–263. https://doi.org/10.1038/s41467-017-00361-6
Rouse SL, Matthews SJ, Dueholm MS (2018a) Ecology and biogenesis of functional amyloids in pseudomonas. J Mol Biol 430(20):3685–3695. https://doi.org/10.1016/j.jmb.2018.05.004
Rouse SL, Stylianou F, Wu HYG, Berry JL, Sewell L, Morgan RML, Sauerwein AC, Matthews S (2018b) The FapF amyloid secretion transporter possesses an atypical asymmetric coiled coil. J Mol Biol 430(20):3863–3871. https://doi.org/10.1016/j.jmb.2018.06.007
Ruer S, Pinotsis N, Steadman D, Waksman G, Remaut H (2015) Virulence-targeted antibacterials: concept, promise, and susceptibility to resistance mechanisms. Chem Biol Drug Des 86(4):379–399. https://doi.org/10.1111/cbdd.12517. Epub 2015 Feb 6
Ryu JH, Beuchat LR (2005) Biofilm formation by Escherichia coli O157:H7 on stainless steel: effect of exopolysaccharide and curli production on its resistance to chlorine. Appl Environ Microbiol 71(1):247–254. https://doi.org/10.1128/AEM.71.1.247-254.2005
Sauer FG, Fütterer K, Pinkner JS, Dodson KW, Hultgren SJ, Waksman G (1999) Structural basis of chaperone function and pilus biogenesis. Science 285(5430):1058–1061
Sauer FG, Pinkner JS, Waksman G, Hultgren SJ (2002) Chaperone priming of pilus subunits facilitates a topological transition that drives fiber formation. Cell 111(4):543–551
Sauer FG, Remaut H, Hultgren SJ, Waksman G (2004) Fiber assembly by the chaperone–usher pathway. Biochimica et Biophysica Acta (BBA)—Mol Cell Res 1694(1–3):259–267. https://doi.org/10.1016/j.bbamcr.2004.02.010
Savvides SN, Yeo HJ, Beck MR, Blaesing F, Lurz R, Lanka E, Buhrdorf R, Fischer W, Haas R, Waksman G (2003) VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO J 22(9):1969–1980. https://doi.org/10.1093/emboj/cdg223
Schneewind O, Missiakas D (2014) Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochimica et Biophysica Acta—Mol Cell Res 1843(8):1687–1697. https://doi.org/10.1016/j.bbamcr.2013.11.009
Schubeis T, Spehr J, Viereck J, Köpping L, Nagaraj M, Ahmed M, Ritter C (2018) Structural and functional characterization of the Curli adaptor protein CsgF. FEBS Lett 592(6):1020–1029. https://doi.org/10.1002/1873-3468.13002
Shaik MM, Maccagni A, Tourcier G, Di Guilmi AM, Dessen A (2014) Structural basis of pilus anchoring by the ancillary pilin RrgC of Streptococcus pneumoniae. J Biol Chem 289(24):16988–16997. https://doi.org/10.1074/jbc.M114.555854
Shewmaker F, McGlinchey RP, Thurber KR, McPhie P, Dyda F, Tycko R, Wickner RB (2009) The functional curli amyloid is not based on in-register parallel β-sheet structure. J Biol Chem 284(37):25065–25076. https://doi.org/10.1074/jbc.M109.007054
Shoji M, Naito M, Yukitake H, Sato K, Sakai E, Ohara N, Nakayama K (2004) The major structural components of two cell surface filaments of Porphyromonas gingivalis are matured through lipoprotein precursors. Mol Microbiol 52(5):1513–1525. https://doi.org/10.1111/j.1365-2958.2004.04105.x
Shu Q, Crick SL, Pinkner JS, Ford B, Hultgren SJ, Frieden C (2012) The E. coli CsgB nucleator of curli assembles to -sheet oligomers that alter the CsgA fibrillization mechanism. PNAS 109(17):6502–6507. https://doi.org/10.1073/pnas.1204161109
Shu Q, Krezel AM, Cusumano ZT, Pinkner JS, Klein R, Hultgren SJ, Frieden C (2016) Solution NMR structure of CsgE: structural insights into a chaperone and regulator protein important for functional amyloid formation. PNAS 113(26):7130–7135. https://doi.org/10.1073/pnas.1607222113
Siewering K, Jain S, Friedrich C, Webber-Birungi MT, Semchonok DA, Binzen I, Wagner A, Huntley S, Kahnt J, Klingl A, Boekema EJ, Søgaard-Andersen L, van der Does C (2014) Peptidoglycan-binding protein TsaP functions in surface assembly of type IV pili. Proc Natl Acad Sci USA 111(10):E953–E961. https://doi.org/10.1073/pnas.1322889111
Sleutel M, Van Den Broeck I, Van Gerven N, Feuillie C, Jonckheere W, Valotteau C, Dufrêne YF, Remaut H (2017) Nucleation and growth of a bacterial functional amyloid at single-fiber resolution. Nat Chem Biol 13(8):902–908. https://doi.org/10.1038/nchembio.2413
Spaulding CN, Iv HLS, Zheng W, Dodson KW, Hazen JE, Conover MS, Wang F, Svenmarker P, Luna-Rico A, Francetic O, Andersson M, Hultgren S, Egelman EH (2018) Functional role of the type 1 pilus rod structure in mediating host-pathogen interactions. eLife
Spaulding CN, Klein RD, Ruer S, Kau AL, Schreiber HL, Cusumano ZT, Dodson KW, Pinkner JS, Fremont DH, Janetka JW, Remaut H, Gordon JI, Hultgren SJ (2017) Selective depletion of uropathogenic E. coli from the gut by a FimH antagonist. Nature 546(7659):528–532. https://doi.org/10.1038/nature22972. Epub 2017 June 14
Spirig T, Weiner EM, Clubb RT (2011) Sortase enzymes in Gram-positive bacteria. Mol Microbiol 82(5):1044–1059. https://doi.org/10.1111/j.1365-2958.2011.07887.x
Steadman D, Lo A, Waksman G, Remaut H (2014) Bacterial surface appendages as targets for novel antibacterial therapeutics. Future Microbiol 9(7):887–900. https://doi.org/10.2217/fmb.14.46
Summers ZM, Fogarty HE, Leang C, Franks AE, Malvankar NS, Lovley DR (2010) Direct exchange of electrons within aggregates of an evolved syntrophic coculture of anaerobic bacteria. Science 330:1413–1415
Symersky J, Patti JM, Carson M, House-Pompeo K, Teale M, Moore D, Jin L, Schneider A, Delucas LJ, Hook M, Narayana SVL (1997) Structure of the collagen-binding domain from a Staphylococcus aureus adhesin. Nat Struct Biol 4(10):833–838. https://doi.org/10.1038/nsb1097-833
Szabó Z, Stahl AO, Albers S-V, Kissinger JC, Driessen AJM, Pohlschröder M (2007) Identification of diverse archaeal proteins with Class III signal peptides cleaved by distinct archaeal prepilin peptidases. J Bacteriol 189(3):772–778. https://doi.org/10.1128/JB.01547-06
Takhar HK, Kemp K, Kim M, Howell PL, Burrows LL (2013) The platform protein is essential for type IV pilus biogenesis. J Biol Chem 288(14):9721–9728. https://doi.org/10.1074/jbc.M113.453506. Epub 2013 Feb 14
Tammam S, Sampaleanu LM, Koo J, Manoharan K, Daubaras M, Burrows LL, Howell PL (2013) PilMNOPQ from the Pseudomonas aeruginosa Type IV pilus system form a transenvelope protein interaction network that interacts with PilA. J Bacteriol 195(10):2126–2135. https://doi.org/10.1128/JB.00032-13
Taylor JD, Hawthorne WJ, Lo J, Dear A, Jain N, Meisl G, Andreasen M, Fletcher C, Koch M, Darvill N, Scull N, Escalera-Maurer A, Sefer L, Wenman R, Lambert S, Jean J, Xu Y, Turner B, Kazarian SG, Chapman MR, Bubeck D, De Simone A, Knowles TPJ, Matthews SJ (2016) Electrostatically-guided inhibition of Curli amyloid nucleation by the CsgC-like family of chaperones. Sci Rep 6:1–11. https://doi.org/10.1038/srep24656
Taylor JD, Zhou Y, Salgado PS, Patwardhan A, McGuffie M, Pape T, Grabe G, Ashman E, Constable SC, Simpson PJ, Lee WC, Cota E, Chapman MR, Matthews SJ (2011) Atomic resolution insights into curli fiber biogenesis. Structure 19(9):1307–1316. https://doi.org/10.1016/j.str.2011.05.015
Thanassi DG, Bliska JB, Christie PJ (2012) Surface organelles assembled by secretion systems of Gram-negative bacteria: diversity in structure and function. FEMS Microbiol Rev 36(6):1046–1082. https://doi.org/10.1111/j.1574-6976.2012.00342.x
Thanassi DG, Saulino ET, Hultgren SJ (1998) The chaperone/usher pathway: a major terminal branch of the general secretory pathway. Curr Opin Microbiol 1(2):223–231
Tian P, Boomsma W, Wang Y, Otzen DE, Jensen MH, Lindorff-Larsen K (2015) Structure of a functional amyloid protein subunit computed using sequence variation. J Am Chem Soc 137(1):22–25. https://doi.org/10.1021/ja5093634
Ton-That H, Marraffini LA, Schneewind O (2004) Sortases and pilin elements involved in pilus assemly of Corynebacterium diphtheriae. Mol Microbiol 53(1):251–261. https://doi.org/10.1111/j.1365-2958.2004.04117.x
Ton-That H, Schneewind O (2003) Assembly of pili on the surface of Corynebacterium diphtheriae. Mol Microbiol 50(4):1429–1438. https://doi.org/10.1046/j.1365-2958.2003.03782.x
Ton-That H, Schneewind O (2004) Assembly of pili in Gram-positive bacteria. Trends Microbiol 12(5):228–234. https://doi.org/10.1016/j.tim.2004.03.004
Tonjum T, Freitag NE, Namork E, Koomey M (1995) Identification and characterization of pilG, a highly conserved pilus-assembly gene in pathogenic Neisseria. Mol Microbiol 16(3):451–464
Turner LR, Lara JC, Nunn DN, Lory S (1993) Mutations in the consensus ATP-binding sites of XcpR and PilB eliminate extracellular protein secretion and pilus biogenesis in Pseudomonas aeruginosa. J Bacteriol 175(16):4962–4969
Uhlich GA, Cooke PH, Solomon EB (2006) Analyses of the red-dry-rough phenotype of an Escherichia coli O157:H7 strain and its role in biofilm formation and resistance to antibacterial agents. Appl Environ Microbiol 72(4):2564–2572. https://doi.org/10.1128/AEM.72.4.2564
Van Gerven N, Van der Verren SE, Reiter DM, Remaut H (2018) The role of functional amyloids in bacterial virulence. J Mol Biol 430(20):3657–3684. https://doi.org/10.1016/j.jmb.2018.07.010
Verger D, Miller E, Remaut H, Waksman G, Hultgren S (2006) Molecular mechanism of P pilus termination in uropathogenic Escherichia coli. EMBO Rep 7(12):1228–1232. https://doi.org/10.1038/sj.embor.7400833
Vergunst AC, Schrammeijer B, Den Dulk-Ras A, De Vlaam CMT, Regensburg-Tuink TJG, Hooykaas PJJ (2000) VirB/D4-dependent protein translocation from Agrobacterium into plant cells. Science 290(5493):979–982. https://doi.org/10.1126/science.290.5493.979
Vetsch M, Puorger C, Spirig T, Grauschopf U, Weber-Ban EU, Glockshuber R (2004) Pilus chaperones represent a new type of protein-folding catalyst. Nature 431(7006):329–333. https://doi.org/10.1038/nature02891
Virdi V, Coddens A, De Buck S, Millet S, Goddeeris BM, Cox E, De Greve H, Depicker A (2013) Orally fed seeds producing designer IgAs protect weaned piglets against enterotoxigenic Escherichia coli infection. Proc Natl Acad Sci USA 110(29):11809–11814. https://doi.org/10.1073/pnas.1301975110. Epub 2013 June 25
Wagner A, Dehio C (2019) Role of distinct Type-IV-secretion systems and secreted effector sets in host adaptation by pathogenic Bartonella species. Cell Microbiol e13004–e13004. https://doi.org/10.1111/cmi.13004
Waksman G (2019) From conjugation to T4S systems in Gram‐negative bacteria: a mechanistic biology perspective. EMBO Rep e47012–e47012. https://doi.org/10.15252/embr.201847012
Wang X, Smith DR, Jones JW, Chapman MR (2007) In vitro polymerization of a functional Escherichia coli amyloid protein. J Biol Chem 282(6):3713–3719. https://doi.org/10.1074/jbc.M609228200
Wang F, Gu Y, O’Brien JP, Yi SM, Yalcin SE, Srikanth V, Shen C, Vu D, Ing NL, Hochbaum AI, et al (2019) Structure of microbial nanowires reveals stacked hemes that transport electrons over micrometers. Cell 177:361–369.e310
Whitchurch CB, Hobbs M, Livingston SP, Krishnapillai V, Mattick JS (1991) Characterisation of a Pseudomonas aeruginosa twitching motility gene and evidence for a specialised protein export system widespread in eubacteria. Gene 101(1):33–44
Winther-Larsen HC, Wolfgang M, Dunham S, van Putten JP, Dorward D, Lovold C, Aas FE, Koomey M (2005) A conserved set of pilin-like molecules controls type IV pilus dynamics and organelle-associated functions in Neisseria gonorrhoeae. Mol Microbiol 56(4):903–917. https://doi.org/10.1111/j.1365-2958.2005.04591.x
Xu Q, Shoji M, Shibata S, Naito M, Sato K, Elsliger M-A, Grant JC, Axelrod HL, Chiu H-J, Farr CL, Jaroszewski L, Knuth MW, Deacon AM, Godzik A, Lesley SA, Curtis MA, Nakayama K, Wilson IA (2016) A distinct type of pilus from the human microbiome. Cell 165(3):690–703. https://doi.org/10.1016/j.cell.2016.03.016
Yanagawa R, Otsuki K, Tokui T (1968) Electron microscopy of fine structure of Corynebacterium renale with special reference to pili. Jpn J Vet Res 16(1):31–37
Yeo HJ, Savvides SN, Herr AB, Lanka E, Waksman G (2000) Crystal structure of the hexameric traffic ATPase of the Helicobacter pylori type IV secretion system. Mol Cell 6(6):1461–1472. https://doi.org/10.1016/S1097-2765(00)00142-8
Yoshimura F, Takahashi K, Nodasaka Y, Suzuki T (1984) Purification and characterization of a novel type of fimbriae from the oral anaerobe Bacteroides gingivalis. J Bacteriol 160(3):949–957
Zav’yalov VP (2013) Polyadhesins: an armory of Gram-negative pathogens for penetration through the immune shield
Zavialov AV, Berglund J, Pudney AF, Fooks LJ, Ibrahim TM, MacIntyre S, Knight SD (2003) Structure and biogenesis of the capsular F1 antigen from Yersinia pestis: preserved folding energy drives fiber formation. Cell 113(5):587–596
Zavialov AV, Tischenko VM, Fooks LJ, Brandsdal BO, Aqvist J, Zav’yalov VP, Macintyre S, Knight SD (2005) Resolving the energy paradox of chaperone/usher-mediated fibre assembly. Biochem J 389(Pt 3):685–694. https://doi.org/10.1042/BJ20050426
Zechner EL, Lang S, Schildbach JF (2012) Assembly and mechanisms of bacterial type IV secretion machines. Philos Trans R Soc B: Biol Sci 367(1592):1073–1087. https://doi.org/10.1098/rstb.2011.0207
Zeng G, Vad BS, Dueholm MS, Christiansen G, Nilsson M, Tolker-Nielsen T, Nielsen PH, Meyer RL, Otzen DE (2015) Functional bacterial amyloid increases Pseudomonas biofilm hydrophobicity and stiffness. Front Microbiol 6:1099–1099. https://doi.org/10.3389/fmicb.2015.01099
Zhang HZ, Lory S, Donnenberg MS (1994) A plasmid-encoded prepilin peptidase gene from enteropathogenic Escherichia coli. J Bacteriol 176(22):6885–6891
Acknowledgements
This work was supported by VIB and the ERC under consolidator grant BAS-SBBT.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Addendum on e-pili
Addendum on e-pili
Geobacter species such as Geobacter sulfurreducens possess micrometer long conductive filaments or “nanowires” that mediate extracellular electron transport (Reguera et al. 2005), and play a role in respiration (Malvankar et al. 2011) and interspecific electron exchange (Summers et al. 2010). Nanowire-expressing bacteria are associated with many important redox processes such as carbon and mineral recycling in soil, metal corrosion, conversion of organic waste to methane and electricity (Malvankar and Lovley 2014). The molecular nature of these nanowires has been obscure, however, and for a long time they were considered to be composed of Type IV pilus-like PilA subunits (Childers et al. 2002), with the conductive properties proposed to be coming from stacked aromatic residues in the pilus subunits, or through association with the C-type cytochrome OmcS (Leang et al. 2013).
The recent cryoEM structures of isolated conductive pili or “e-pili” from G. sulfurreducens have now revealed their unique structure, composition and electron transport mechanism (Filman et al. 2018; Wang et al. 2019). This study shows Geobacter e-pili are composed of a unique noncovalent linear polymer of the cytochrome OmcS (Fig. 12.6). OmcS was previously known to be essential for bacterial growth on insoluble electron acceptors such as Fe (III) oxide and electrodes, but had not been directly implicated as the main nanowire component (Holmes et al. 2006; Mehta et al. 2005). The structures show each OmcS subunit contains six stacked hemes that are placed within 3.5–6 Å to each other in a trajectory that spans the length of the nanowire and results in electric coupling within and across subunits. The hemes are found in parallel pairs, each pair oriented perpendicular to the next. In the parallel arrangement, the hemes are present within a range of 3.4–4.1 Å while in the perpendicular arrangement they are within a range of 5.4–6.1 Å (Fig. 12.6). The parallel arrangement is expected to maximize electron coupling while the perpendicular arrangement is thought to enhance the structural stability of the subunit. For each heme group, two histidines situated axially form coordination bonds with the iron atom present at the center, and the vinyl groups form covalent thioester bonds with cysteine. In the filament, subunits associate in a head-to-tail arrangement, with each subunit in contact with only one preceding and succeeding subunit, through an inter-subunit contact comprising ~2,600 Å2 surface area. Histidine 16 of each subunit coordinates an adjacent heme group present in the succeeding subunit. These inter-subunit coordination bonds are proposed to maximize the structural stability of the nanowire. This cross-interface heme coordination is speculated to result from a domain swapping of the C-terminal helix, a process that has been observed in induced cytochrome polymerization (Hirota et al. 2010; Wang et al. 2019). Little is known about the secretion and in vivo assembly pathway of OmcS e-pili. Although it is now evident that PilA is not the main structural subunit of the nanowires, previous genetic studies clearly associate it with the presence of Geobacter’s e-pili and the secretion of OmcS to the extracellular space (Liu et al. 2018; Richter et al. 2012). Deletion or mutation of PilA leads to the absence of OmcS on the surface (Reguera et al. 2005) while overexpression of PilA leads to overproduction of nanowires (Leang et al. 2013; Summers et al. 2010). The cryoEM structures now clearly demonstrate PilA is not found as a component of e-pili (Filman et al. 2018; Wang et al. 2019). Whether PilA is found as an additional extracellular fiber or acts as a pseudopilus of a T2SS, and where and how OmcS subunits polymerize and associate with the cell surface will require further study.

a 3.7 Å cryo-EM map of Geobacter sulfurreducens e-pili or nanowires (EMD_9046). b atomic model of OmcS subunits arranged in the e-pili (width 55 Å, PDB id: 6ef8), and c atomic model of a single OmcS subunit, with its chain of six stacked heme groups shown in pink and the iron atom in green
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lukaszczyk, M., Pradhan, B., Remaut, H. (2019). The Biosynthesis and Structures of Bacterial Pili. In: Kuhn, A. (eds) Bacterial Cell Walls and Membranes . Subcellular Biochemistry, vol 92. Springer, Cham. https://doi.org/10.1007/978-3-030-18768-2_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-18768-2_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-18767-5
Online ISBN: 978-3-030-18768-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)