
Abstract
The treatment landscape for non-small-cell lung cancer (NSCLC) has dramatically shifted over the past two decades. Targeted or precision medicine has primarily been responsible for this shift. Older paradigms of treating metastatic NSCLC with cytotoxic chemotherapy, while still important, have given way to evaluating tumor tissues for specific driver mutations that can be treated with targeted agents. Patients treated with targeted agents frequently have improved progression-free survival and overall survival compared to patients without a targetable driver mutation, highlighting the clinical benefit of precision medicine. In this chapter, we explore the historic landmark trials, the current state of the field, and potential future targets under investigation, in this exciting, rapidly evolving discipline of precision medicine in lung cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Even though the incidence of lung cancer is decreasing, unfortunately over 160,000 people still die from the disease every year and it remains the leading cause of cancer death in the USA. Lung cancer has been traditionally classified as small-cell lung cancer and non-small-cell lung cancer. Non-small-cell lung cancer can further be classified into adenocarcinoma, squamous cell carcinoma, and large-cell carcinoma. Histologically, small-cell lung cancer and non-small-cell lung cancer have different natural histories and therapeutic approaches. Prior to 2004, there was no need of distinguishing various subtypes of non-small-cell lung cancer as the therapeutic management was similar within all subtypes.
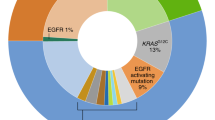
However, management and treatment of lung cancer has transformed in the past decade. This changed particularly in 2004 when a small percentage of lung adenocarcinomas were identified with mutations in the EGFR gene that rendered those tumors sensitive to the EGFR tyrosine kinase inhibitors. Since then, there has been a surge of other actionable mutations in lung cancer. Up to 69% of the patients with advanced lung cancer have actionable mutations. The majority of them are KRAS (25%), EGFR sensitizing (17%), ALK (7%), MET (3%), HER-2 (2%), ROS1 (2%), BRAF (2%), RET (2%), NTRK1 (1%), PIK3CA (1%), and MEK1 (1%) [1]. In addition, 31% patients are found to have unknown oncogenic driver mutations for which we currently do not have any targets [1]. Because of these advances, the current guidelines such as American Society of Clinical Oncology (ASCO) or National Comprehensive Cancer Network (NCCN) guidelines recommend molecular profiling or next-generation sequencing to determine the best treatment options for the individual patients or small subsets of newly diagnosed lung cancer patients. There is also growing understanding that there are increased genomic alterations through treatment lines, and there is more emphasis on better understanding of mechanisms of resistance and clonal evolvement of tumor. Thus, precision or personalized medicine has become an emerging approach for treatment and research of lung cancer taking into account personalized genetic landscape, tumor microenvironment, and available therapeutics.
In this current chapter, we will review the available options for next-generation sequencing and the importance of value-based genomics. We will then attempt to define the molecular abnormalities in lung cancer with standards of care and also potential diagnostic platforms such as proteomics and genomics. Then, we will individually discuss the current actionable mutations, genomic alterations with emphasis on the underlying mechanism of abnormality with the potential to therapy and available therapeutic options with evidence of clinical trials.
2 Value-Based Genomic Profiling
In the past few years, there has been advancement in next-generation sequencing techniques which have led to the development of biomarker-driven cancer therapies. The NGS techniques have now become more commercialized and affordable with various different platforms. These sequencing data has not only impacted clinical decision making but also allocation of resources in research and development of therapeutics and lung cancer. However, the current challenge is the standardization of these recommended NGS across different academic and community practices in a more value-based approach to obtain a greater clinical benefit with minimizing cost and risk of genomic profiling in cancer care [2].
The human genome sequencing project was initially completed in 2003 and led to further investigations and understanding of the genomics of various mutations and alterations in development of cancer [1]. First-generation Sanger sequencing technique was used for sequencing of human genome which required a decade of multi-central collaboration, automated analysis, and roughly $3 billion [3]. However, there is an exponential decline in the sequencing cost since then. James Watson’s genome was completed for less than $1 million. By 2009, the cost of genome sequencing dropped to $100,000 [4,5,6]. Since then, there has been development of several next-generation sequencers by different companies such as Roche, Life Technologies (SOLiD), Illumina, Pacific Biosciences, and Ion Torrent, all of which provide platform for faster and cheaper next-generation sequencing of cancer genome [4, 7,8,9]. The next-generation sequencing can be further subdivided into more affordable, interpretable, and commonly used targeted sequencing of a panel of recognized or putative cancer-associated genes versus whole-exome or whole-genome sequencing which provide comprehensive profiling of all protein-encoding genes of the genome giving more information and long-term cost-effectiveness [10,11,12].
In this rapidly progressing era, it is important to practice value-based medicine focusing not only on cancer drugs but also on value of genomic profiling in cancer clinic. Most of the next-generation sequencing platforms aim at detecting somatic mutations using formalin-fixed paraffin-embedded tissue tumor. Other rarely use samples include malignant fluid, blood samples, and salivary swabs. Common NGS platforms are FoundationOne that covers 315 genes costing $5800 with 14 day turnaround time; Caris Molecular Intelligence covering more than 600 genes with a cost of $6500 and similar 14 day turnaround; OncoDeep, 75 gene panel, costing $3500 with 7 day turnaround time; Paradigm cancer diagnostic with 186 genes and the cost of $4800 and 5-day return as well as Oncomine Dx Target Test (NSCLC only) with 23 genes and quick 4-day return [13,14,15,16,17].
There are multiple aspects of cost including sample collection, experimental design, sample sequencing, data management, and downstream analysis [18]. Even though the cost for the DNA sequencing may be reducing rapidly due to the advancements in NGS techniques, data management and downstream analysis costs still remain a challenge [19]. To date, no randomized controlled trials have investigated the cost-effectiveness of NGS and there is limited health economic evidence for genomic sequencing and a comprehensive calculation of genomic sequencing containing multiple aspects of the cost is needed.
Despite the high cost, it would be beneficial to use precision oncology if it shows clinical effectiveness. A large retrospective study of 143 single-agent phase II trials from Year 2000–2009 in over 7000 advanced non-small-cell lung cancer patients showed superior median overall response rate, progression-free survival, and overall survival in trials enriched for the presence of molecular targets compared to studies with non-selective patients [20]. Similarly another meta-analysis of 112 registration trials from 1998 to 2013 comparing efficacy outcomes between therapies employing a personalized treatment approach versus general non-selective treatment showed higher response rate, longer progression-free survival, and longer overall survival in patients treated based on precision medicine [21].
At the same time, it is important to keep in mind the treatment-related toxicity and financial burden for this personalized treatment approach. The same meta-analyses did not show any increased treatment-related mortality compared to the non-personalized treatment strategy [22]. Other meta-analyses have shown that cytotoxic agents have higher treatment-related or adverse effects compared to targeted therapies. In addition, a recent meta-analysis of 41 randomized clinical trials evaluating 28 targeted agents for solid tumors approved by the Food and Drug Administration (FDA) evaluating the rate of treatment discontinuation due to toxicity and grade 3–4 adverse effects showed that targeted therapies with companion diagnostic tests were associated with improved safety and tolerability [23].
The financial burden associated with the cancer care has increased rapidly with the cost of out-of-pocket expenses, copayments, and insurance premiums. Since most of these NGS diagnostic tests are associated with targeted therapies which are considered “experimental or investigational,” insurance companies often do not reimburse for these agents which are considered “off label.” Therefore, NGS has not yet become standard of care across most practices in USA.
However, undoubtedly precision medicine and value-based care may be the future of cancer medicine. There needs to be happy medium with guidelines or standardization in NGS so that insurance payers allow for coverage for appropriate and most cost-effective NGS testing which is in best interest of the patient.
3 Defining the Molecular Abnormalities in Lung Cancer
3.1 Molecular Abnormalities in Lung Cancer
In 2017, FDA approved immune checkpoint inhibitor pembrolizumab for treatment of cancer patients with high microsatellite instability or mismatch repair deficient markers, regardless of the tumor locations or tissue types. This is a milestone in the development of molecular profiling of cancer and its implications of cancer treatments. The future direction of precision medication in oncology will rely more on the molecular features of a tumor than the tissue types. Lung cancer has been histologically classified as small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC) which includes lung adenocarcinoma, large-cell carcinoma, and squamous cell carcinoma (SCC). With the advancement of technology especially next-generation sequencing, genetic and molecular profiling has identified different subtypes of lung cancer with specific molecular characteristics, which are associated with the clinical/pathological features, prognosis, and treatment responses. Molecular targeted therapy and immunotherapy based on specific somatic genetic mutations/alterations and molecular markers of lung cancer have been changing the paradigm of lung cancer management drastically.
The Cancer Genome Atlas Research Network analyzed 230 lung adenocarcinoma using messenger RNA, micro-RNA, and DNA sequencing integrated with copy number, methylation, and proteomic analyses [24]. The whole-exome sequencing had revealed high rates of somatic mutation (mean 8.9 mutations per mega base), and 18 genes were statistically significantly mutated. TP53 was the commonly mutated (46%), followed by mutations in KRAS (33%), EGFR (14%), BRAF (10%), as were PIK3CA (7%), MET (7%) and the small GTPase gene, RIT1 (2%). Mutations in tumor suppressor genes including STK11 (17%), KEAP1 (17%), NF1 (11%), RB1 (4%), and CDKN2A (4%) were observed. Mutations in chromatin modifying genes SETD2 (9%), ARID1A (7%), and SMARCA4 (6%) and the RNA splicing genes RBM10 (8%) and U2AF1 (3%) were also common. EGFR mutations were more frequent in female patients, whereas mutations in RBM10 were more common in males. Aberrations in NF1, MET, ERBB2, and RIT1 occurred in 13% of cases and were enriched in samples otherwise lacking an activated oncogene, suggesting a driver role for these events in certain tumors. By sequencing the DNA and mRNA sequence from the same sample, splicing alterations driven by somatic genomic changes such as exon 14 skipping in MET mRNA was found in 4% of cases.
When measured at the protein level, recurrent aberrations in multiple key pathways were characterized. Such as RTK/RAS/RAF pathway activation (76% of cases), PI3K-mTOR pathway activation (25%), p53 pathway alteration (63%), cell cycle regulation alteration (64%), and mutation of various chromatin and splicing factors (49%). There are mechanisms other than genetic mutations suggested for activations of signaling pathways. For example, the KRAS-mutated lung adenocarcinoma had higher levels of phosphorylated MAPK than KRAS wild-type tumors on average; however, a lot of KRAS wild-type tumors also have significant MAPK activation. MAPK and PI(3)K pathway activation can be explained by known mutations in only a fraction of cases. The somatic alterations involve key pathway components for RTK signaling, mTOR signaling, oxidative stress response, proliferation and cell cycle progression, nucleosome remodeling, histone methylation, and RNA splicing/processing [24].
Genetic analysis of lung adenocarcinoma is the standard of care for treatment selection nowadays. The Lung Cancer Mutation Consortium (LCMC) did a multi-institutional analysis of 10 potential oncogenic driver mutations in at least one of the 8 genes (EGFR, KRAS, ERBB2, AKT1, BRAF, MEK1, NRAS, and PIK3CA) in 1007 specimens and 733 specimens had all 10 markers tested (including ALK and MET) [25]. KRAS mutations are the most commonly found with a frequency of around 25% followed by EGFR mutations in 22% of the samples. In this cohort, EGFR mutations were highly associated with female sex, Asian race, and never-smoking status; and less strongly associated with stage IV disease, the presence of bone metastases, and absence of adrenal metastases. ALK rearrangements were strongly associated with never-smoking status and more weakly associated with the presence of liver metastases. ERBB2 mutations were strongly associated with Asian race and never-smoking status. Two mutations were seen in 2.7% of samples (27/1007), all but one of which involved one or more of PIK3CA, ALK, or MET, including 14 with two small mutations and 13 with either a small mutation and ALK rearrangement (4); a small mutation and MET amplification (7); or concurrent ALK rearrangement and MET amplification (2). Of 14 cases with two small mutations, 13 (92%) had a PIK3CA mutation in addition to another mutation, including 9 with EGFR, 2 with BRAF, 1 with KRAS, and 1 with MEK1 mutation. One case had EGFR ex19del and AKT1 c.49G>A (p.E17K) mutations.
Unlike non-small-cell lung cancer (NSCLC), targeted therapy and molecular profiling are less utilized in small-cell lung cancer (SCLC) in clinical practice [26]. The most common genetic alterations in SCLC are inactivation of the tumor suppressor genes TP53 and RB1. In a study which sequenced 108 SCLC tumors without chromothripsis, TP53 and RB1 had bi-allelic losses in 100% and 93% of the cases, which included mutations, translocations, homozygous deletions, hemizygous losses, copy-neutral losses of heterozygosity (LOH), and LOH at higher ploidy [27]. The other common genetic alterations found in SCLC include copy-number gains of genes encoding MYC family members, mutations in enzymes involved in chromatin remodeling, receptor tyrosine kinases, and Notch pathway [27]. Around 98% of SCLC cases are associated with smoking and only 2% occur in non-smokers [28]. SCLCs have extremely high mutation rates (around 8.62 non-synonymous mutations per million base pairs), and C:G > A:T transversions were found in 28% of all mutations on average, a pattern indicative of heavy smoking [27]. The high mutational burden of SCLC might provide opportunities for immunotherapy.
According to the NCCN guidelines (Version 4.2018), molecular testing of EGFR mutation, ALK, ROS1, BRAF, and programmed death ligand 1 (PD-L1) is recommended in metastatic lung adenocarcinoma, large-cell lung cancer, and NSCLC not otherwise specified (NOS). For SCC, consider molecular testing of EGFR and ALK in never smokers or small biopsy or mixed histology, and consider ROS1, BRAF testing as part of broad molecular profiling. PD-L1 testing was also suggested for SCC. PD-L1 immunohistochemistry (IHC) testing is approved for formalin-fixed, paraffin-embedded (FFPE) surgical pathology specimens and helps select patients most likely to respond to immune checkpoint inhibitors. PD-L1 expression level ≥50% is indicated for first-line pembrolizumab therapy of NSCLC.
Various methods have been utilized for molecular profiling of lung cancer. Mutations can be detected by next general sequencing as well as various methods including direct Sanger sequencing and pyrosequencing, mutation-specific PCR, multiplex PCR assay followed by single base extension sequencing (SNaPshot, Life Technologies, Grand Island, NY) or matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MassARRAY, Sequenom, San Diego, CA), high-performance liquid chromatography (HPLC), etc. [29]. Multiple commercial next-generation sequencing platforms are available now as summarized in a recent review [2]. Liquid biopsy which refers to testing mutations on circulating tumor DNA (ctDNA) in blood samples is a promising method to detect genomic alterations and can potentially be used a surrogate method for tissue biopsy testing and even complementary approach [30]. Due to the tumor heterogeneity, a single tissue biopsy may not reflect complete genomic mutations and there are discordance between tissue and ctDNA sequencing results, so both approaches are recommended to enhance mutation detection [31]. Fluorescence in situ hybridization (FISH) analysis is utilized for detecting copy number, amplification, and structural alterations such as gene arrangements. FISH is commonly used for ALK/ROS1 rearrangement and MET amplification.
4 Epidermal Growth Factor Receptor (EGFR)
4.1 EGFR Mutation in Lung Cancer
The epidermal growth factor receptor (EGFR) is a transmembrane signaling receptor that was discovered in the early 2000s [32]. Under normal conditions, once stimulated by epidermal growth factor, EGFR monomers on the cell surface dimerize to activate the intracellular tyrosine kinase. This activates the RAS, RAF, MEK, ERK pathway and the PI3K, AKT, mTOR pathway to increase expression of genes promoting cell growth and proliferation. In non-small-cell lung cancer (NSCLC), the EGFR gene can become mutated leading to constitutive activation of the EGFR tyrosine kinase. This permits increased tumor growth and proliferation uninhibited by extracellular or intracellular signals.
Approximately 15–20% of patients with NSCLC adenocarcinoma in the USA have mutations in the EGFR tyrosine kinase domain in their tumors [33]. The EGFR mutation frequency is highest in Asian populations. In a meta-analysis of 151 worldwide studies, the Asia-Pacific NSCLC subgroup had the highest EGFR mutation frequency at 47% but there was a wide range between studies from 20 to 76% [34]. The European subgroup in this meta-analysis had an overall EGFR mutation frequency of 15% [34]. EGFR mutations are also more prevalent in females and never smokers. However, EGFR mutations are not restricted to patients with Asian ethnicity, female gender, or never smoker status. The PIONEER study performed in Asia revealed that more than 50% of patients with EGFR mutations were not female non-smokers [35]. This highlights the need to test all patients with NSCLC adenocarcinoma for EGFR mutations regardless of clinical characteristics.
The two most common activating mutations in the tyrosine kinase domain of the EGFR are deletion of exon 19 (EGFR del19) and a point mutation in exon 21 (EGFR L858R) which substitutes an arginine for a leucine at position 858. Other possible mutations include T790M (substitution of a methionine for a threonine at position 790 in exon 20), S768I, L861Q, G719X, and many others. In a meta-analysis of studies from China, L858R accounted for 38.3% of all EGFR mutations and del19 accounted for 37% [36]. T790M occurred at a rate of 1.5% in treatment-naïve patients [36]. Of note, the T790M mutation rate increases with exposure to EGFR tyrosine kinase inhibitors, which will be discussed later. The rate of EGFR mutations sensitive to tyrosine kinase inhibitors was 88.5% [36].
The presence of an EGFR mutation is a key piece of clinical information because it is associated with a high response rate to therapy with EGFR tyrosine kinase inhibitors (TKIs). First-generation EGFR TKIs include erlotinib and gefitinib; these drugs compete with ATP to reversibly bind the intracellular catalytic domain of EGFR tyrosine kinase, thus blocking downstream signaling and reducing cell growth [37]. The second-generation EGFR TKIs include afatinib and dacomitinib; these drugs irreversibly inhibit the catalytic domain of the EGFR tyrosine kinase [37]. The third-generation EGFR TKI is osimertinib. Osimertinib is an irreversible EGFR TKI that inhibits both EGFR TKI sensitizing mutations and EGFR T790M resistance mutations. It is currently recommended for frontline treatment of advanced EGFR-mutant NSCLC based on the FLAURA trial [38].
In the FLAURA trial, 556 patients with previously untreated EGFR mutated advanced NSCLC were randomly assigned to either osimertinib 80 mg, PO, QD, or a standard EGFR TKI such as gefitinib 250 mg, PO, QD, or erlotinib 150 mg, PO, QD. The median PFS was longer with osimertinib at 18.9 months versus 10.2 months for the standard EGFR TKIs (HR 0.46; CI 0.37–0.57). The ORR was 80% with osimertinib and 76% with standard EGFR TKIs. The median duration of response was 17.2 months with osimertinib versus 8.5 months with standard EGFR TKIs. Data on OS is not yet mature. Grade 3 or higher adverse events were less common with osimertinib (34% vs. 45%). Osimertinib is now recommended in the first-line setting for EGFR mutated advanced NSCLC due to its improved PFS and lower rate of serious adverse events [38].
Prior to this trial, the standard of care was erlotinib, gefitinib, or afatinib in the frontline setting. After progression, the presence or absence of the resistance mechanism T790M was evaluated by liquid biopsy or tissue biopsy. This resistance mechanism develops in approximately 50% of cases [39]. If T790M is present, the patient is eligible for second-line therapy with osimertinib. This is based on the AURA3 trial; 419 patients with T790M-positive advanced NSCLC who had disease progression after standard EGFR TKI therapy received either osimertinib 80 mg PO QD or pemetrexed plus either carboplatin or cisplatin every 3 weeks for up to 6 cycles. Pemetrexed maintenance was permitted. The median PFS was significantly longer with osimertinib at 10.1 months versus 4.4 months (HR 0.30; CI 0.23–0.41). The ORR was 71% with osimertinib versus 31% with combination chemotherapy. Among patients with CNS disease, the median PFS was 8.5 months with osimertinib versus 4.2 months with chemotherapy. Furthermore, grade 3 or higher adverse events were lower with osimertinib (23% vs. 47%) [40]. As many patients have been on standard EGFR TKI therapy, this strategy for using second-line osimertinib remains relevant.
Erlotinib, gefitinib, and afatinib have all been evaluated by clinical trials comparing these agents to platinum-based chemotherapy doublets in patients with advanced NSCLC and EGFR activating mutations. One meta-analysis looked at 13 phase III trials including 2620 patients and concluded that the PFS was significantly prolonged with EGFR TKIs (HR 0.43; CI 0.38–0.49) compared to chemotherapy. Overall survival was not prolonged (HR 1.01; CI 0.87–1.18), but it was hypothesized that this is due to significant crossover between the treatment arms [41].
Three large trials assessed erlotinib versus chemotherapy. The OPTIMAL trial assigned 154 patients to erlotinib or gemcitabine plus carboplatin. Erlotinib increased PFS (13.1 vs. 4.6 months, HR 0.16; CI 0.10–0.26) and increased the ORR (83% vs. 36%) [42]. OS was not significantly different [43]. The EURTAC trial assigned 174 patients to erlotinib or a platinum-based chemotherapy doublet and found erlotinib increased PFS (9.7 vs. 5.2 months, HR 0.37; CI 0.25–0.54) but did not increase OS [44]. The ENSURE trial assigned 275 patients to erlotinib or gemcitabine plus cisplatin and found erlotinib increased PFS (11 vs. 5.5 months, HR 0.34; CI 0.22–0.51) but did not increase OS [45]. The most common side effects of erlotinib include rash, diarrhea, and less commonly interstitial pneumonitis and hepatic toxicity. The most common grade 3 or higher adverse event was rash (6.4–13%) in the erlotinib group, which had a favorable toxicity profile compared to chemotherapy [44, 45].
The IPASS trial assessed gefitinib versus chemotherapy. In this trial, 1217 Asian patients who were never or former light smokers with advanced NSCLC were assigned to gefitinib or carboplatin plus paclitaxel. Gefitinib improved PFS (12 month progression-free rate 25% vs. 7%, HR 0.74) but did not change overall survival in the cohort [46, 47]. Subgroup analysis revealed that patients with an EGFR mutation had a significantly improved PFS (9.5 vs. 6.3 months, HR 0.48). Patients without an EGFR mutation had a significantly shorter PFS (1.5 vs. 6.5 months, HR 2.85). This highlighted the importance of testing for the presence of EGFR mutation rather than relying on clinical characteristics to determine therapy [46, 47]. Further trials, such as the North East Japan Study Group 002 trial conducted in patients with known EGFR mutations, reported similar results to the IPASS trial [48]. The most common adverse events with gefitinib were rash (71%) and elevated liver function tests (55.3%). The rate of grade 3 or higher adverse events was approximately 41% in the gefitinib group and 71% in the chemotherapy group [48].
The LUX-Lung 3 and the LUX-Lung 6 trial assessed afatinib versus chemotherapy. The LUX-Lung 3 trial assigned 345 patients with EGFR mutated NSCLC to afatinib 40 mg PO QD or cisplatin plus pemetrexed for up to 6 cycles. Afatinib increased PFS compared with chemotherapy (11.1 months vs. 6.9 months, HR 0.58; CI 0.43–0.78). The ORR was increased with afatinib (56% vs. 23%), and time to symptom progression and quality of life were improved with afatinib [49, 50]. The most common side effects included diarrhea (95%), rash (89%), stomatitis (72%), nail changes (57%), and dry skin (29%) [49]. The LUX-Lung 6 trial assigned 364 Asian patients to afatinib or cisplatin plus gemcitabine. Afatinib increased PFS compared with chemotherapy (11 vs. 5.6 months) and afatinib increased the ORR (67% vs. 23%) [51]. When these two trials were combined, the median OS was not significantly different between the two therapy groups. However, there was a significant increase in OS in the subgroup of patients with the exon 19 deletion [52].
Of note, patients with NSCLC with uncommon EGFR mutations such as S768I, L861Q, or G719X can be treated with afatinib in the first-line setting based on analysis of the LUX-Lung trials, but afatinib is less active in other uncommon mutation types [53]. Dacomitinib is another second-generation EGFR TKI that was compared to gefitinib as first-line treatment for patients with EGFR mutation-positive NSCLC (ARCHER 1050). Dacomitinib did have a longer PFS, but it had greater toxicity and is not currently approved in the USA [54].
Erlotinib, gefitinib, and afatinib are considered to all have similar efficacy in EGFR mutated NSCLC and are all generally well tolerated. Some data suggests that afatinib may be slightly more efficacious but may also cause the most side effects, and many clinicians start at a lower dose than used in the LUX-Lung trials. Some data suggests gefitinib may be the best tolerated of the three agents, but the data is inconsistent. One study randomized 256 patients to either erlotinib 150 mg PO QD or gefitinib 250 mg PO QD and found no significant difference in PFS, ORR, OS, and grade 3 or 4 toxicities. The ORR was 56.3% versus 52.3% (P = 0.53), and the median OS was 22.9 versus 20.1 months (P = 0.25) [55]. The LUX-Lung 7 trial assessed afatinib 40 mg PO QD versus gefitinib 250 mg PO QD and found that median OS was 27.9 months with afatinib versus 24.5 months with gefitinib (HR 0.86, CI 0.62–1.36) [56]. In this trial, although there was no significant difference in OS with afatinib, PFS was improved with afatinib versus gefitinib [56].
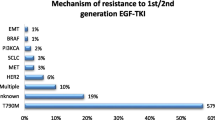
The majority of patients who initially respond to an EGFR TKI eventually develop resistance to the drug and have progression of disease. We have already discussed using osimertinib in patients who develop T790M resistance after treatment with a first- or second-generation EFGR TKI. There are other mechanisms of resistance that can develop. One mechanism of resistance is the amplification of the MET oncogene. This has been linked to resistance in 5–20% of patients taking erlotinib or gefitinib [57]. This has been linked to resistance in up to 30% of patients taking osimertinib [58]. Another interesting but less common mechanism of resistance is histologic transformation of EGFR mutated NSCLC into small-cell lung cancer [59]. In one analysis of 37 tumor biopsies taken after progression on EGFR TKI therapy, 5 resistant tumors (14%) transformed from NSCLC into small-cell lung cancer; these tumors were sensitive to standard small-cell lung cancer chemotherapy regimens [59]. Although it is not standard of care, it may be reasonable to biopsy a site of progressive disease to determine if another targetable mutation is present or if there has been a transformation in histology.
There has been some investigation into whether or not continuing an EGFR TKI after progression has benefit. One retrospective analysis looked at Japanese patients with EGFR mutations who progressed on first- or second-line EGFR TKI and compared those who continued EGFR TKI beyond progression (39 patients) and those who were switched to cytotoxic chemotherapy alone (25 patients). The median OS was 32.2 months in the group receiving the EGFR TKI beyond progression and 23 months in the group receiving chemotherapy (HR 0.42, CI 0.21–0.83, p = 0.013) [60]. However, a prospective study is needed to confirm these results. Due to anecdotal evidence that EGFR-positive lung cancer can progress more rapidly even after progression when discontinuing EGFR TKI therapy, some clinicians elect to continue the EGFR TKI therapy until the next line of therapy can be initiated.
There has also been investigation into whether adding bevacizumab to EGFR TKI therapy adds benefit. During the JO25567 trial, 154 patients in Japan with EGFR mutations and no prior therapy were assigned to either erlotinib 150 mg PO QD alone or erlotinib plus bevacizumab 15 mg/kg every 3 weeks until disease progression or unacceptable toxicity. Median PFS with erlotinib plus bevacizumab was 16 months versus 9.7 months with erlotinib alone (HR 0.54, CI 0.36–0.79) [61]. Serious adverse events occurred at a similar frequency in both groups (~25%) [61]. The overall survival data is not yet mature. The combination of erlotinib plus bevacizumab is approved by the European Medicines Agency in Europe.
Finally, there has been investigation into whether adding chemotherapy to EGFR TKI therapy adds benefit. In the FASTACT-2 trial, 451 patients with were assigned to either chemotherapy (gemcitabine plus platinum) plus erlotinib or chemotherapy plus placebo. In the patients with an EGFR activating mutation, PFS was improved with chemotherapy plus erlotinib (7.6 vs. 6.0 months) and OS was improved with chemotherapy plus erlotinib (18.3 vs. 15.2 months) [62]. Another study evaluated gefitinib with and without pemetrexed in chemotherapy-naïve patients with EGFR-positive NSCLC. Median PFS was longer with gefitinib with pemetrexed (15.8 vs. 10.9 months, HR 0.68, CI 0.48–0.96) [63]. Overall survival data is immature.
Although these studies have shown a possible benefit of combining chemotherapy with EGFR TKI, four large randomized clinical trials using gefitinib or erlotinib all failed to show a survival benefit from the combination with chemotherapy [64,65,66,67]. However, these trials did not select patients based on the presence of an EGFR driver mutation so further investigation is needed in this area. In the IMPRESS trial, 265 patients with an EGFR mutation who had disease progression on gefitinib were assigned to cisplatin, pemetrexed, gefitinib or to cisplatin, pemetrexed, placebo. Patients completed 6 cycles of chemotherapy and then were continued on gefitinib or placebo for maintenance. There was no significant difference in median PFS (5.4 vs. 5.4 months, HR 0.86, CI 0.65–1.13) [68]. There was a decrease in median OS in those on chemotherapy plus gefitinib versus chemotherapy alone (13.4 vs. 19.5 months, HR 1.44, CI 1.07–1.94) [68]. At this time, patients with advanced EGFR-positive NSCLC generally do not receive combination chemotherapy with an EGFR TKI as initial therapy outside of a clinical trial.
5 Anaplastic Lymphoma Kinase (ALK)
Anaplastic lymphoma kinase (ALK) driver mutations are found in a variety of solid tumors. ALK receptor tyrosine kinase gene, located on chromosome 2p23, encodes a receptor that belongs to the insulin receptor superfamily. The protein is made up of an extracellular, transmembrane, and intracellular domain. It is believed that ALK plays a role in the development of neurons in the central nervous system.
Activation of ALKs kinase catalytic domains has been implicated in the growth and development of cancer. Multiple pathways are involved, including phospholipase Cγ (PLCγ), Janus kinase (JAK)–signal transducer and activator of transcription (STAT), PI3K–AKT, mTOR, sonic hedgehog (SHH), JUNB, CRKL–C3G–RAP1 GTPase, and MAPK. The most common mechanisms that are involved in ALK mutations are chromosomal translocations or rearrangements. The resultant oncogenic ALK fusion gene results in constitutive ALK activity.
The FDA has approved testing to identify ALK rearrangements with immunohistochemistry (IHC) and fluorescent in situ hybridization (FISH). In addition, ALK rearrangements and their resultant fusion proteins can also be identified via reverse transcription polymerase chain reaction (RT-PCR).
EML4-ALK is identified in 2–7% of all non-small-cell lung cancers, most prevalent in non-smokers, light smokers, and adenocarcinomas. These patients with ALK fusion lung cancers are relatively younger than typical NSCLC patients. Histologically almost all ALK fusion oncogenes are adenocarcinoma. In addition, signet ring cells, which portend for a poor prognosis and are associated with a more aggressive clinical course, have been identified to be more common.
The optimal approach to treat advanced NSCLC with an ALK fusion variant first line is an ALK inhibitor. The first ALK inhibitor approved by the FDA to treat metastatic NSCLC with an ALK rearrangement was crizotinib in 2011 under the accelerated approval process.
5.1 Crizotinib
Crizotinib, a first-generation ALK inhibitor, is a small-molecule tyrosine kinase inhibitor. Crizotinib was approved by the FDA after a phase I trial [69] with confirmatory trials in phases II [70] and III [71]. PROFILE 1014 compared crizotinib to a platinum doublet with pemetrexed for first-line treatment in advanced ALK rearranged NSCLC. Crizotinib had improved PFS, RR, and duration of response compared to traditional cytotoxic therapy. Crizotinib unfortunately has poor CSF penetration with the second- and next-generation ALK inhibitors shown to have better response rates intracranially [72].
5.2 Ceritinib
Ceritinib, a second-generation ALK inhibitor, initially received accelerated approval in 2014 for advanced NSCLC patients who progressed or who were intolerant to crizotinib based on the phase I study ASCEND-1 [73]. The phase III study ASCEND-4 compared ceritinib to front doublet platinum therapy and was found to be superior. Ceritinib has proven in preclinical studies to have activity against crizotinib-resistant cells including gatekeeper mutation L1196M. ASCEND-5 has also been evaluated in those who progressed on crizotinib to either ceritinib or single-agent chemotherapy with improvements in PFS and RR [74]. Ceritinib is currently approved to be used in either treatment-naïve ALK rearranged advanced NSCLC patients or those who have progressed on crizotinib.
5.3 Alectinib
Alectinib, also a second-generation ALK inhibitor, received accelerated approval in 2015 for ALK-positive metastatic NSCLC who progressed or who are intolerant to crizotinib after two single-arm clinical trials [75,76,77]. Alectinib is active against gatekeeper mutation L1196M and other crizotinib-resistant mutations such as C1156Y and F1174L. ALEX, an open-label phase III trial, compared alectinib and crizotinib in treatment-naïve advanced NSCLC. The primary end point, PFS, was superior in alectinib (25.7 months) compared to 10.4 months with crizotinib (HR 0.53; p < 0.0001) [78]. For CNS progression, a secondary end point in this study, alectinib showed superior aversion to progression in CNS in comparison with crizotinib (12% vs. 45%, respectively) [78]. The results of this study led to the approval of alectinib for use in treatment-naïve, ALK rearranged advanced NSCLC and is the treatment of choice in this setting.
5.4 Brigatinib
Brigatinib is a second-generation tyrosine kinase inhibitor, with potent activity against active ALK, developed to treat advanced NSCLC for those patient who have progressed or intolerant to crizotinib with activity against active ALK, and mutant L1196M. In 2017, the drug received accelerated approval based on the randomized, open-label, non-comparative, phase II ALTA study designed to evaluate anti-tumor activity of brigatinib in patients with metastatic ALK-positive NSCLC who have previously received crizotinib demonstrated improved PFS compared to historical data for patients who progressed on crizotinib (9.2 mo 90 mg/d vs. 12.9 mo 180 mg/d) [79]. Further investigation in the frontline setting is currently taking place with ALTA-1L. Brigatinib is approved to be used in crizotinib refractory or intolerant, advanced ALK rearranged NSCLC setting.
5.5 Lorlatinib
Lorlatinib is a novel third-generation, ALK inhibitor designed to overcome ALK-resistant mutations including G1202R, and improved CNS penetration was granted FDA breakthrough designation in 2017. It has shown promise (46% ORR, 9.6 mo PFS) in its first in human open-label phase I study in advanced ALK-positive NSCLC [80]. It is unclear in the sequence of available therapies where lorlatinib will lie but currently considered to be used in patients’ refractory to second-generation inhibitors or multiple TKIs. An investigation for its efficacy in the first-line setting is being investigated currently (NCT03052608).
6 ROS Proto-oncogene 1 (ROS-1)
6.1 ROS-1 (Reactive Oxygen Species-1)
ROS-1 was initially discovered as the cellular homolog of the chicken c-ros gene. This gene is the proto-oncogene for v-ros which is the transforming sequence of UR2 sarcoma virus [81]. It is located at chromosome 6q22 and encodes for a receptor tyrosine kinase belonging to the insulin receptor family closely related to anaplastic lymphoma kinase (ALK) and leukocyte receptor tyrosine kinase (LTK) [82]. ROS1 protein expression in adult humans appears to be the highest in kidney but is also found in cerebellum, peripheral neural tissue, stomach, small intestine, and colon, with lower expression in several other tissues and absent in lungs. In mouse model studies, mice that lack the receptor appear to be healthy. No ligands for the receptor have been found.
Constitutive activation of ROS1 signaling leads to the phosphorylation of SHP-2 (tyrosine phosphatase, non-receptor type 11) and activation of downstream signaling pathways such as MEK/ERK, JAK/STAT, or PI3K/AKT [83]. Chromosomal rearrangements involving the ROS1 gene were originally described in glioblastomas, where ROS1 (chromosome 6q22) is fused to the FIG gene located on chromosome 6q22 immediately adjacent to ROS1. Known ROS1 fusion partners in lung cancer include FIG, CD74, SLC34A2, and SDC. Expression of the FIG-ROS1 and SDC4-ROS1 fusions in murine Ba/F3 cells has been demonstrated to result in IL3 independent proliferation, and this proliferation was sensitive to treatment of small-molecule ROS1 inhibitors [84].
Crizotinib is approved as the first-line therapy for advanced ROS1 fusion-positive NSCLC. ROS1-positive NSCLC demonstrated a 72% response rate and 19.2-month median progression-free survival in a phase I expansion cohort [73]. To date, acquired resistance to crizotinib has been reported in clinical studies because of the secondary S1986Y/F, G2032R, and D2033N mutations in ROS1. Preclinical data has suggested that missense mutations within the ROS1 kinase domain can drive acquired resistance to crizotinib. A patient with a crizotinib-sensitive NSCLC harboring a CD74-ROS1 fusion was found to have an acquired ROS1 G2032R mutation at the time of progression. However, preclinical data suggests that the next-generation ALK/MET/ROS1 inhibitors cabozantinib, foretinib, and PF-06463922 are capable of overcoming this resistance mutation [85].
7 V-Raf Murine Sarcoma Viral Oncogene Homolog B1 (BRAF)
7.1 BRAF in Non-small-Cell Lung Cancer
Mutations in the BRAF proto-oncogene were first described in 2002 with an incidence of 8% across all cancers and 3% in lung cancer [86]. The BRAF proto-oncogene encodes the intracellular B-Raf protein. The B-Raf protein phosphorylates and activates downstream MEK, which in turn phosphorylates and activates downstream ERK. This signaling pathway leads to the upregulation of genes promoting cell proliferation and survival. In healthy cells, this signaling pathway is modulated by extracellular signals such as growth factors transmitting information to the cell via transmembrane receptors. However, in cancer cells with a BRAF mutation, this regulation is lost due to constitutive activation of the B-Raf protein. This leads to increased cell proliferation and survival independent of extracellular factors [87].
BRAF mutations occur in 1–3% of patients with NSCLC [88,89,90]. The most common mutation is V600E, which is an amino acid substitution at position 600 from a valine to a glutamic acid. Other described mutations include G469A, D594G, K601E, G464E, G596R, A598T, G606R, and G469V [88, 89]. The BRAF V600E mutation is generally cited to be approximately 50% of all BRAF mutations in NSCLC, although various studies have documented rates from 30 to 80% [88, 91]. Compared to BRAF V600E mutations in melanoma, BRAF V600E mutations in NSCLC are less common.
To date, NSCLC patients with BRAF mutations are not statistically more likely to belong to a particular gender, sex, or race [91]. There is no unique histologic subtype that is more likely to harbor a BRAF mutation [89, 91]. There is no association of BRAF mutations with stage of disease at diagnosis [91]. Only smoking history consistently correlates with BRAF mutation status [89, 91, 92]. The majority of patients with BRAF mutations are former or current smokers. This signal is particularly strong with non-V600E mutations since patients with V600E mutations are more likely to be light/never smokers [92].
In general, concurrent mutations with other driver mutations in patients with BRAF mutations are rare. However, concurrent mutations have been reported with BRAF V600E or BRAF non-V600E mutations and mutations in other genes including EGFR, KRAS, ALK, and PIK3CA [88, 89, 91, 92].
Multiple studies have evaluated the prognosis and overall survival of patients with a BRAF mutation in NSCLC. Compared to patients with NSCLC with other driver mutations, patients with BRAF mutations had no statistically different overall survival [89]. In a study with 63 patients with BRAF mutations, their overall survival was intermediate between patients with EGFR and KRAS mutations but this was not statistically different [92]. In early-stage disease, there is no difference in overall survival between mutation types [92]. Overall survival may be slightly better with BRAF V600E mutations compared to non-V600E mutations, which may reflect the lighter smoking history of the former [92]. Finally, patients with concurrent driver mutations do have shorter overall survival compared to those with a single driver mutation [91].
Two BRAF inhibitors, vemurafenib and dabrafenib, have shown clinical activity in metastatic BRAF V600E mutated lung cancers. Vemurafenib works by inhibiting the active form of the B-Raf kinase by attaching to the ATP-binding site. In 2015, Hyman et al. evaluated vemurafenib in multiple non-melanoma cancers with BRAF V600 mutations in a histology-independent phase II “basket” trial. A total of 122 patients with BRAF V600E mutations with various histologies received vemurafenib (960 mg, PO, BID) until progression of disease or unacceptable toxicity. In the 19 patients with NSCLC, there was a response rate (RR) of 42% (95% CI 20–67) with 8 patients having a partial response. Tumor regression was observed in most patients (14 of 19 patients). The median progression-free survival (PFS) was 7.3 months (95% CI 3.5–10.8). The 12-month overall survival (OS) was 66%. The majority of these patients received prior platinum-based chemotherapy. Common side effects included rash (68%), fatigue (56%), and arthralgia (40%) [93].
In 2016, Planchard et al. evaluated another BRAF inhibitor dabrafenib in patients with BRAF V600E mutations in advanced NSCLC in a phase II trial. Dabrafenib is an adenosine-triphosphate competitive inhibitor of B-Raf kinase that is selective for the V600E mutant. In this study, 84 patients with metastatic BRAF V600E NSCLC received dabrafenib (150 mg, PO, BID). Out of 6 patients who had no prior treatment, 4 (66%) had a partial response. Out of the remaining 78 patients who had prior treatment, 26 (33% [95% CI 23–45]) had an overall response (CR + PR). There was disease control in 58% of patients. The response was quick with 73% of responses occurring at 6 weeks. The side effects were mostly skin related with 42% of patients experiencing some adverse events. Grade 3 or 4 adverse reactions included cutaneous squamous cell carcinoma (12%), basal cell carcinoma (5%), and asthenia (5%). One patient died from an intracranial hemorrhage while concurrently taking a factor Xa inhibitor [94].
Unfortunately, treatment with a BRAF inhibitor alone can lead to resistance within 6–7 months in other tumor types. The BRAF inhibitor dabrafenib plus the MEK inhibitor trametinib has shown synergistic anti-tumor activity in BRAF mutant human cancer cell lines. Planchard et al. evaluated dabrafenib (150 mg, PO, BID) plus trametinib (2 mg, PO, QD) in patients with previously treated BRAF V600E mutant metastatic NSCLC. Out of 57 patients, 36 patients responded (63% [95% CI 49.3–75.6]). Approximately 79% of patients obtained disease control (CR + PR + stable disease). The median PFS was 9.7 months (95% CI 6.9–19.6). The median duration of treatment was 10.6 months with 30% of patients receiving treatment for more than 12 months. Common adverse events included fever, nausea, vomiting, diarrhea, asthenia, and anorexia. Grade 3–4 toxicity included neutropenia (9%), hyponatremia (7%), and anemia (5%). The combination of dabrafenib and trametinib had a high overall response rate, often a prolonged duration of response and manageable toxicity [95].
Planchard et al. also evaluated dabrafenib and trametinib in patients with previously untreated BRAF V600E mutant NSCLC. The study included 36 patients with an overall response rate of 64% (95% CI 46–79) and PFS of 10.9 months (95% CI 7.0–16.6). The median duration of response was 10.4 months, and the median overall survival was 24.6 months. The 2-year overall survival was 51%. Of note, there were similar response rates between patients who had been previously treated and those who had not (63% vs. 64%). Furthermore, PFS was similar between the previously treated and untreated groups (9.7 vs. 10.9 months). This suggests that treating clinicians have flexibility to treat patients with dabrafenib and trametinib in either the first-line metastatic setting or in the second line following chemotherapy [96].
8 MET Proto-oncogene (MET)
8.1 MET in Lung Cancer
MET proto-oncogene, located on chromosome 7q31, was identified in early 1980s. Its protein product is a transmembrane tyrosine kinase, which binds to the ligand scatter factor/hepatocyte growth factor (HGF). The downstream signaling activates the mitogen-activated protein kinase (MAPK), phosphoinositide-3-kinase (PI3K)/AKT, signal transducer and activator of transcription proteins (STATs), and nuclear factor kappa B (NF-kB) pathways, thus promoting proliferation, escaping apoptosis, and increasing cell motility [97,98,99,100].
MET pathway abnormality is commonly found in lung cancer. The mechanisms include protein phosphorylation (p-MET), overexpression, amplification, rearrangement, and mutations [101]. Mutations in the splicing sites that cause MET exon 14 skipping are the most studied MET abnormality which occurs in around 3–4% of lung adenocarcinoma and 2% of SCC [102, 103]. Exon 14 of MET encodes the juxtamembrane domain of the protein, which is the binding site for E3 ubiquitin ligase for protein degradation; thus, skipping of exon 14 causes prolonged signal transduction of the MET pathway, which leads to cell proliferation and migration, and subsequently facilitates oncogenesis, cancer invasion, and metastasis [104, 105]. MET gene rearrangement is less reported, but the kinase fusion KIF5B-MET has been reported in lung adenocarcinoma [106]. Overexpression of MET is found in around 35–72% of the NSCLC, and p-MET can be found in 67% of the NSCLC, while amplification of MET is around 2–5% of newly diagnosed adenocarcinoma [107, 108]. The MET gene copy number (GCN) is associated with worse prognosis in surgically resected NSCLC, with overall survival (OS) of 25.5 months for patients with MET ≥5 copies/cell compared with 47.5 months for patients with MET < 5 copies/cell (P = 0.0045) [109].
Studies of targeting MET pathway in cancer have been ongoing for decades. The available agents and clinical trials have been summarized in recent reviews [108, 110]. There are small molecular tyrosine kinase inhibitors such as selective inhibitor tivantinib (targets MET), capmatinib (targets MET), savolitinib (targets MET), tepotinib (targets MET), SAR125844 (targets MET), sitravatinib (targets MET), AMG 337 (targets MET), non-selective inhibitor crizotinib (targets ALK/ROS/MET), cabozantinib (targets MET/RET/others), glesatinib (targets MET/AXL/others), merestinib (targets MET/ROS1/AXL/FLTs/others), S49076 (targets MET/AXL/FGFR1-3), as well as monoclonal antibodies including emibetuzumab (anti-MET), onartuzumab (anti-MET), rilotumumab (anti-HGF), and ficlatuzumab (anti-HGF) [108, 110].
Various clinical trials have investigated the efficacy of MET inhibition in lung cancer. Responses to non-selective MET inhibitors crizotinib and cabozantinib were reported in lung adenocarcinoma with MET abnormalities; however, cabozantinib also has activity against RET and had 28% overall response rate in a phase II clinical trial of 26 patients with RET-rearranged lung adenocarcinoma [111, 112]. So far the results of clinic trials for the selective MET inhibitors are not satisfactory, which may be partially due to lack of valid predictive biomarkers for patient selection as discussed previously [108, 110]. In the phase III OAM4971g (METLung) trial comparing erlotinib plus onartuzumab versus erlotinib plus placebo in patients with locally advanced or metastatic NSCLC with MET overexpressing defined by MET IHC staining, there is no difference in clinical outcomes (median OS was 6.8 vs. 9.1 months for onartuzumab vs. placebo, P = 0.067), with shorter OS in the onartuzumab arm, compared with erlotinib in patients with MET-positive non-small-cell lung cancer, and the median progression-free survival was 2.7 versus 2.6 months (stratified HR, 0.99; 95% CI 0.81–1.20; P = 0.92) with overall response rate of 8.4 and 9.6% for onartuzumab versus placebo, respectively [113]. A phase III trial of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated locally advance or metastatic non-squamous NSCLC reported by Scagliotti et al. in 2015 [114] showed OS did not improve, although PFS increased (median PFS, 3.6 vs. 1.9 months; HR, 0.74; 95% CI 0.62–0.89; P < 0.001). Subgroup analyses suggested OS improvement in patients with high MET expression (HR, 0.70; 95% CI 0.49–1.01). Most common adverse events occurring were rash (33.1% vs. 37.3%, respectively), diarrhea (34.6% vs. 41.0%), asthenia or fatigue (43.5% vs. 38.1%), and neutropenia (grade 3–4; 8.5% vs. 0.8%). It has been reported that MET amplification can increase to 5–22% after treatment with EGFR TKI erlotinib or gefitinib [115]. A phase II study of erlotinib plus tivantinib in 45 patients with locally advanced or metastatic EGFR mutation-positive non-small-cell lung cancer just after progression on EGFR TKI erlotinib or gefitinib did not prove clinical benefit of tivantinib in patients with acquired resistance to EGFR TKIs; however, the patients having high activated MET signaling have longer survival by tivantinib/erlotinib (c-Met high vs. low: median PFS 4.1 vs. 1.4 months; median OS 20.7 vs. 13.9 months) [116]. The ATTENTION study [117], a phase III trial of erlotinib plus tivantinib versus erlotinib in stage IIIB/IV Asian non-squamous NSCLC with wild-type EGFR, was prematurely terminated due to the increased interstitial lung disease (ILD) incidence in the tivantinib group. ILD developed in 14 patients (3 deaths) and 6 patients (0 deaths) in the tivantinib and the placebo groups, respectively, in total of 307 patients enrolled. Median OS was 12.7 and 11.1 months in the tivantinib and the placebo groups, respectively [hazard ratio (HR) = 0.891, P = 0.427]. Median PFS was 2.9 and 2.0 months in the tivantinib and the placebo groups, respectively (HR = 0.719, P = 0.019). Although this study lacked statistical power because of the premature termination and did not demonstrate an improvement in OS, the results suggest that tivantinib plus erlotinib might improve PFS compared to erlotinib alone in non-squamous NSCLC patients with WT-EGFR. The overexpression or phosphorylation of MET is less predictive for the response to MET inhibitors; according to the above-mentioned negative data and correlations between MET germline mutations, MET amplification, somatic mutation, overexpression, and activation with treatment responses have not been confirmed yet [113]. Targeting MET pathway is promising; however, we need better strategies to select the patients who are going to benefit from it [108].
MET mutations are frequently associated with other gene mutations (around 44%), and there are cross talks between MET pathway and other pathways such as mTOR, PI3K/AKT, STATs, MEK pathways, which may blunt the clinical benefit of MET inhibition [118]. Based on the synthetic lethality [119], combination of MET inhibitor and other pathway inhibitors could be an effective strategy. In a phase I study of tivantinib plus the mTOR inhibitor temsirolimus, the pharmacokinetic analysis showed no interaction in the plasma concentrations of the two drugs and the combination appears to be well tolerated with clinical activity [120]. MET abnormality is also common in NSCLC patients with brain metastases [121]. MET-amplified recurrent glioblastoma have been shown to respond to crizotinib treatment [122]. Intracranial activity of cabozantinib has also been shown in MET exon 14 skipping NSCLC patient with brain metastasis [123]. MET inhibitors penetrate the blood–brain barrier and could be effective for brain metastasis, especially for patients who failed the EGFR inhibitors due to brain metastasis.
9 Tropomyosin-Related Kinase (TRK) and (Rearranged During Transfection Kinase) RET
9.1 TRK
Tropomyosin receptor kinase (Trk) receptor family comprises 3 transmembrane proteins referred to as Trk A, B, and C receptors (TrkA, TrkB, and TrkC) that are encoded by the NTRK1, NTRK2, and NTRK3 genes [124]. These receptor tyrosine kinases are expressed in human neuronal tissue, activate neurotrophin (NTs), and play an role in nervous system. The NTRK1 gene is located on chromosome 1q21-q22, and mutation of which disrupts the function of the TrkA protein which can cause congenital insensitivity to pain with anhidrosis. The NTRK2 gene is on chromosome 9q22.1 (codes for TrkB receptor). The NTRK3 gene is located on chromosome 15q25 (TrkC) expressed in the human hippocampus, in the cerebral cortex, and in the granular cell layer of the cerebellum. Reported gene fusion SQSTM1-NTRK1, NTRK1-SQSTM1, CD74-NTRK 1, MPRIP-NTRK1, TRIM24-NTRK2, RFWD2-NTRK1. Gene fusions involving NTRK genes lead to the transcription of chimeric Trk proteins which elevates kinase function, resulting in oncogenic potential. Entrectinib is an orally bioavailable inhibitor of the tyrosine kinase TrkA, TrkB, and TrkC as well as of c-ros oncogene 1 (ROS1) and anaplastic lymphoma kinase (ALK). Entrectinib can cross the blood–brain barrier and could thus potentially be effective in the treatment of brain metastases and GBM with activating gene fusions of NTRK, ROS1, or ALK. In the subgroup of NTRK-rearranged cancers, 100% of patients (n = 5) with various tumor histologies and fusion types responded to entrectinib treatment and had a good intracranial activity [125].
There are phase I trials investigating Altiratinib (DCC-2701) and sitravatinib (MGCD516) which are multi-kinase inhibitors with reported in vitro inhibitory activity against TrkA and TrkB. Other Trk inhibitors that are being investigated in phase I/II trial include TSR-011, PLX7486, DS-6051b, F17752, and cabozantinib (XL184). Larotrectinib is a selective small-molecule pan-TRK inhibitor currently being investigated in an adult/adolescent phase II trial. Primary objective of the trial was investigator-assessed overall response rate which was 78%, and duration of response has not reached [126]. NTRK gene fusions are emerging as novel target; however, due to the low incidence of Trk alterations across multiple histologies, it is challenging to study the various targets.
9.2 RET
RET (rearranged during transfection) is a proto-oncogene which through cytogenetic rearrangement and activating point mutations can undergo oncogenic activation. RET is localized to human chromosome 10q11.2. The expression of RET is the highest during development and the lowest in normal adult tissues. It is predominately expressed in neural crest-derived cells and urogenital cells. RET is required for the development of the enteric nervous system, kidney morphogenesis, and spermatogenesis [127]. Distinct chromosomal translocations produce different RET fusions which occur in 1–2% of NSCLCs and are mutually exclusive of mutations in EGFR, KRAS, ALK, HER2, and BRAF.
RET-rearranged lung adenocarcinomas (LUADs) are often found in never smokers (82%) and overall younger patients (≤60 years; 73%), more poorly differentiated (64%), solid subtype (64%), have a smaller size (≤3 cm) with N2 disease (54%). RET has been shown to form fusions with eight different genes in NSCLC: KIF5B (most common), CCDC6, NCOA, TRIMM33, CUX1, KIAA1468, KIAA1217, and FRMD4A. Reverse transcriptase polymerase chain reaction (RT-PCR) is both sensitive and specific for the detection of known fusions, but it is not reliable for the detection of new fusion partners or isoforms. Currently, a few drugs have been investigated in phase II studies for RET-positive lung adenocarcinoma. Cabozantinib, an oral multi-kinase inhibitor of RET, was investigated in a phase II study (n = 25) which showed an objective response rate (ORR) of 28%. The median progression-free survival (PFS) was 5.5 months (95% CI [3.8, 8.4]), and the median overall survival (OS) was 9.9 months (95% CI [8.1, not reached]). Vandetanib, an oral RET, VEGFR-2, and EGFR kinase inhibitor, demonstrated an ORR of 18%, and a disease control rate (DCR) of 65% in patients with advanced/refractory RET-rearranged NSCLC. The PFS was 4.5 months, and the OS was 11.6 months. The 1-year OS rate was 33%. Ten out of 18 patients (56%) had died at the data cutoff. Lenvatinib, a multi-kinase inhibitor, achieved an ORR of 16% (four patients with partial responses), and a DCR of 76% with 48% of patients showed a durable response. Other multi-kinase inhibitors currently being investigated include Alectinib, a tyrosine kinase inhibitor of ALK that is also active against RET in vitro, and ponatinib. Studies showed that different multi-kinase inhibitors may produce variable responses depending on the type of RET fusion. RET inhibitor resistance was also found and investigated. Huang et al. recently identified cabozantinib-resistant KIF5B-RETV804L and the vandetanib-resistant KIF5B-RETG810A mutations in lung adenocarcinoma cells. Interestingly, vandetanib-resistant KIF5B-RETG810A mutant cells displayed gain of sensitivity to ponatinib and lenvatinib, suggesting that the different RET inhibitors can overcome vandetanib-induced mechanisms of resistance. A recent study of lung cancer cells that had CCDC6-RET genes suggested that activation of EGFR signaling may allow the cells to become resistant to RET inhibition via a bypass survival signaling through ERK and AKT. The combination of vandetanib and the mTOR inhibitor everolimus has demonstrated higher anti-tumor activity than either single agent alone. The combination is being further studied. At present, RET-mutated patients are a very small subgroup, which poses a challenge to develop a targeted therapy; however, identifying biomarkers in patients with NSCLC may result in clinical benefit from RET inhibitors and continues to be an active area of investigation [128].
10 Checkpoint Inhibitors
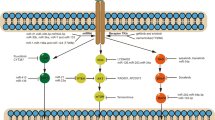
Immunotherapy has had drastic impacts on the treatment of some types of tumors including melanoma, renal cell cancer, and non-small-cell lung cancer. Immunotherapy in lung cancer has been used either as a single-agent or in combination with chemotherapy in a first- or second-line treatment setting in the recent times. Our own immune system consisting of the adaptive and innate immunity is one of the mechanisms of defense against tumor cells. Specifically, the immune response is initiated when T cell receptor recognizes and binds to major histocompatibility complex (MHC) on the surface off the antigen presenting cell (APC) or the tumor cells, which leads to the interaction between cytokines and stimulatory signals causing T lymphocyte activation, proliferation, and differentiation (Fig. 1.1).

Mechanism of action of checkpoint inhibitors
However, the activation and proliferation of T cells are affected by inhibitory immune checkpoint molecules such as the cytotoxic T lymphocyte-associated protein 4 (CTLA-4), programmed cell death 1 (PD1), and programmed death ligand 1 and 2 (PD-L1, PDL2). For example, the interaction between CD28 on T cells and B7 on the APCs is a key step in activation of T cells; however, CTLA-4 competes with CD28 for binding to B7 and transmits an inhibitory signal that suppresses T cell activation. PD-L1/2 is expressed on the surface of multiple cell types including tumor cells and helps evade anti-tumor immune response. The interaction between PD-L1 with APC and PD1 on T cells inhibits apoptosis in tumor cells, promotes peripheral T effector cell exhaustion, and promotes the conversion of T effector cells to Treg cells [129, 130]. Other checkpoints which act as inhibitory receptors expressed by T cells or NK cells such as T cell Ig and T cell immunoglobulin mucin domain 3 (TIM3) and lymphocyte activation gene 3 (LAG3) and killer cell immunoglobulin-like receptor (KIR) have been discovered as well [131, 132]. Therefore, targeted treatments that inhibit these checkpoint proteins could restore and augment cytotoxic T cell responses, leading to potentially resilient responses and prolonged overall survival (OS) with tolerable toxicity.
10.1 PD1/PD-L1/2 Inhibitors
10.1.1 Nivolumab
Nivolumab is a fully humanized IgG4-blocking antibody against PD-1 checkpoint protein that disrupts interactions with PDL1/2. In an early phase I study of 39 patients with advanced metastatic melanoma, colon cancer, castrate resistant prostate cancer, renal cell cancer, and non-small-cell lung cancer (NSCLC), nivolumab was well tolerated with no dose-limiting toxicity. There was evidence of anti-tumor activity in 6/39 patients in the dose escalating and expansion phase up to the dose of 10 mg/kg [133].
Several phase II and III clinical trials have been performed with nivolumab to improve the outcomes of patients with NSCLC. Currently, nivolumab is FDA approved for the treatment of patients with advanced NSCLC who experience progression of disease on or after standard platinum-based chemotherapy. CheckMate 057, a phase III randomized control trial compared nivolumab versus docetaxel in a second-line treatment of advanced non-squamous NSCLC, showed a median OS of 12.2 months (95% CI: 9.7–15.0) among 292 patients in the nivolumab group and 9.4 months (95% CI: 8.1–10.7) among 290 patients in the docetaxel group (hazard ratio: 0.73; 96% CI: 0.59–0.89; P = 0.002) [134]. ORR and median durations of response were higher in nivolumab arm (19 vs. 12% and 18.3 vs. 5.6 months, respectively) [134]. The 3-year OS rates for the nivolumab and docetaxel arms were 18% and 9%, respectively [135]. Similarly, another phase III trial (CheckMate 017) compared nivolumab (3 mg/kg, IV, Q2W) with docetaxel (75 mg/m2, IV, Q3W) in 272 patients with advanced, squamous NSCLC who had progressive disease on platinum-based doublet chemotherapy. OS was prolonged with nivolumab compared to docetaxel (median OS: 9.2 vs. 6.0 months) [136]. The two- and three-year OS rates for nivolumab versus docetaxel were 23% versus 8%, and 16% versus 6%, respectively [135, 137]. ORR was higher with nivolumab (20% vs. 9%), as was the duration of response (25.2 vs. 8 months) [136]. In terms of toxicity, when compared to docetaxel, nivolumab had fever severe grade 3–4 treatment-related adverse effects (7–10% vs. 54%) [134, 136]. In the subgroup analysis, OS benefit was only seen in non-squamous patients with increased tumor PD-L1 expression. There was no OS improvement in PD-L1-negative tumors in the non-squamous cohort and PD-L1 expressing tumors in the squamous cohort. However, better side effect and toxicity profile of nivolumab makes it a better choice than docetaxel [134, 136].
Currently, nivolumab as a single agent is not approved by FDA for the frontline setting in treatment-naïve patients regardless of PD-L1 level. The CheckMate 026 trial which sought to compare the activity of nivolumab versus platinum doublet chemotherapy in 541 treatment-naïve, PD-L1 positive (at least 1% of tumor cells with PD-L1 staining) NSCLC patients did not show any prolongation of OS or PFS with nivolumab [138] (Table 1.1). The combination of nivolumab plus ipilimumab in lung cancer is currently being tested in the CheckMate 227 trial (Table 1.1). In this trial, patients with PD-L1 expression level of at least 1% were randomly assigned in 1:1:1 ratio to receive nivolumab plus ipilimumab, nivolumab monotherapy, or chemotherapy. Additionally, patients with tumor PD-L1 expression less than 1% were randomly assigned in 1:1:1 ratio to receive nivolumab plus ipilimumab, nivolumab plus chemotherapy or chemotherapy alone. In the recently reported part 1 data (n = 299) of this trial, there was improvement in median PFS with frontline nivolumab plus ipilimumab compared to chemotherapy among patients with high tumor mutational burden (defined as >10 mutations per megabase) irrespective of PD-L1 expression level (7.2 months (95% CI: 5.5–13.2) vs. 5.5 months (95% CI: 4.4–5.8) [139]. The objective response rate was 45.3% with nivolumab plus ipilimumab and 26.9% with chemotherapy [139]. The OS data is not mature, but it is likely that after completion of this clinical trial, nivolumab will gain FDA approval in certain patients with lung cancer in the first-line setting.
Nivolumab has also been studied in untreated patients with surgically resectable early stage (stage I, II, or IIIA) NSCLC. In a phase I study with primary end point of safety and feasibility, nivolumab (at a dose of 3 mg/kg) was given IV every 2 weeks, with surgery planned approximately 4 weeks after the first dose. The study showed that neoadjuvant nivolumab was associated with few side effects, did not delay surgery, and induced a major pathological response in 45% of resected tumors. The tumor mutational burden was predictive of the pathological response to PD1 blockade [140].
10.1.2 Pembrolizumab
Pembrolizumab is another IgG monoclonal antibody that targets PD1 on T cells and inhibits the interaction between PD1 and PD-L1 on the tumor cells. Currently, it is approved by FDA for the frontline treatment of EGFR/ALK wild-type NSCLC with at least 50% of tumor cells expressing PD-L1. This approval came after completion of the phase III KEYNOTE024 trial which compared pembrolizumab monotherapy to standard platinum-based chemotherapy in EGFR/ALK wild-type NSCLC with at least 50% of tumor cells expressing PD-L1. There were both OS (6 month OS rate of 80.2% vs. 72.4% with HR: 0.60; 95% CI: 0.41–0.89; P = 0.005) and PFS (10.3 months vs. 6 months; HR: 0.50; 95% CI: 0.37–0.68; P < 0.001) benefit in the pembrolizumab arm [141].
Recently, pembrolizumab in combination with pemetrexed and carboplatin also received accelerated FDA approval for treatment of metastatic non-squamous NSCLC, irrespective of the PD-L1 expression. This approval was based on the KEYNOTE 021 phase II trial which compared chemotherapy alone or with pembrolizumab in 123 patients with advanced untreated non-squamous NSCLC without any EGFR or ALK alterations. This study showed that patients who receive progressive map had better ORR (55 vs. 29%, 95% CI 8–42) and PFS (13 vs. 6 months; HR 0.53, 95% CI 0.31–0.91) [142]. These findings were later confirmed in a larger phase III trial (KEYNOTE-189) which was reported this year and also showed improvement in OS (12 month OS rate: 69% vs. 49%; HR: 0.49; 95% CI: 0.38–0.64), PFS (8.8 month vs. 4.9 months; HR: 0.52; 95% CI: 0.43–0.64) and ORR (48% vs. 19%) in the platinum doublet plus pembrolizumab arm compared to the chemotherapy plus placebo arm [143] (Table 1.1).
Pembrolizumab has also been approved for treatment of advanced NSCLC as a second-line therapy after disease progression on platinum-based chemotherapy. This approval was based on the phase II/III KEYNOTE-010 study in which patients with disease progression on or after platinum-containing chemotherapy and had >1% tumor cell PD-L1 expression as determined by the 22C3 pharmDx test received either pembrolizumab (2 mg/kg or 10 mg/kg via IV) or docetaxel (75 mg/m2) every 3 weeks. The HR and p value for OS was 0.71 (95% CI: 0.58–0.88) and <0.001 comparing pembrolizumab (2 mg/kg) with chemotherapy and 0.61 (95% CI: 0.49–0.75) and <0.001 comparing pembrolizumab (10 mg/kg) with chemotherapy [144].
10.1.3 Atezolizumab
Unlike nivolumab and pembrolizumab, atezolizumab is an antibody against PD-L1, the ligand for PD-1. By binding to the PD-L1 receptor present on tumor cells atezolizumab activates antibody depended cell mediated toxicity which enhances immune system to fight tumor cells. Currently, it is approved for the management of patients with metastatic NSCLC who are EGFR- or ALK-negative and have disease progression on platinum-containing chemotherapy. This approval was based on a phase III randomized controlled trial (the OAK trial) comparing atezolizumab at 1200 mg, IV, every three weeks (n = 425) to docetaxel at 75 mg/m2, every 3 weeks (n = 425). OS was significantly better in the atezolizumab arm than the docetaxel arm (13.8 months [95% CI: 11.8–15.7] vs. 9.6 months [95% CI: 8.6–11.2]; HR: 0.73; 95% CI: 0.62–0.87; p = 0.0003). Patients with low PD-L1 or undetectable expression levels also benefited with improvement in survival in the atezolizumab arm [145].
Although atezolizumab has not been approved by FDA for the treatment of NSCLC in a frontline setting, there are ongoing clinical trials that show promising results. Specifically interim results of the phase III IMpower 131 trial showed improvement in PFS (6.3 vs. 5.6 months; HR: 0.7; 95% CI: 0.60–0.85) when patients were given platinum-based chemotherapy combined with atezolizumab versus chemotherapy alone. The improvement in PFS was the most significant in PD-L1-high (expression in ≥50% of tumor cells) group (10.1 vs. 5.5 months; HR: 0.44; 95% CI: 0.27–0.71), but benefits were seen in all PD-L1-positive subgroups and not in the PD-L1-negative subgroup [146] (Table 1.1). Final OS data is still pending.
Recently another phase III trial, IMpower150, showed that addition of atezolizumab to bevacizumab plus platinum-based chemotherapy can lead to improved PFS (8.3 vs. 6.8 months; HR: 0.62; 95% CI: 0.52–0.74) and OS (19.2 vs. 14.7 months; HR: 0.78; 95% CI: 0.64–0.96) in patients with metastatic non-squamous NSCLC, regardless of PD-L1 expression and EGFR or other genetic alterations [147] (Table 1.1).
Overall, pembrolizumab currently remains the drug of choice in the frontline setting for patients with NSCLC until other checkpoint inhibitors are approved by FDA.
10.1.4 Durvalumab
Durvalumab, a humanized immunoglobulin G1 kappa monoclonal antibody that blocks the binding of programmed cell death ligand 1 (PD-L1) to PD-1 and CD80 (B7.1), has been approved for treatment of unresectable stage III NSCLC that has not progressed following concurrent platinum-based chemotherapy and radiation therapy. This approval came on the basis of a large phase III trial (the PACIFIC trial) in which patients with unresectable stage III NSCLC without any progression after at least 2 lines of platinum-based chemotherapy were randomly assigned to the PD-L1 antibody or placebo. The immunotherapy group had improved PFS (16.8 vs. 5.6 months; HR: 0.52; 95% CI: 0.42–0.65), response rate (28% vs. 16%; relative risk [RR]: 1.78; 95% CI: 1.27–2.51), and median time to death or distant metastasis (23.2 vs. 14.6 months; HR: 0.52: 95% CI: 0.39–0.69) [148]. However, OS data of the study is still pending and therefore some investigators recommend against using immunotherapy in patients with stage III lung cancer at this point.
10.2 CTLA-4 Antagonists
The combination of a CTLA 4 inhibitor (ipilimumab) and a PD-1 blocker (nivolumab) has shown promising results in chemotherapy-naïve patients with metastatic NSCLC. However, without PD-1 blockade ipilimumab may not be as effective. There was one phase II randomized control trial that compared ipilimumab plus paclitaxel and carboplatin with paclitaxel and carboplatin alone as first-line treatment. However, there was no statistical difference in primary end point of progression-free survival between the two arms. In addition, there was no OS benefit in the ipilimumab arm [149].
10.3 Toxicity Associated with Immune Checkpoint Inhibitors
Although the immune checkpoint inhibitors are in general less toxic than chemotherapies, there have been several reports of immune-related adverse events that can occur occasionally. These immune-related adverse events include inflammatory reactions against normal cells apart from the tumor cells. Common side effects include rash (33%), colitis (14%), endocrinopathies (8%), hepatitis (4%), pneumonitis (2%), and acute kidney injury (2%) [150]. Other rare ones including pancreatitis, Guillain-Barré syndrome or myasthenia gravis, myocarditis or venous thromboembolism, thrombocytopenias or neutropenia, ocular inflammation, and inflammatory arthritis. Management of grade 1 toxicities includes close monitoring of patients and continuation of immune checkpoint inhibitors with the exception of some toxicities such as neurotoxicity, cardiotoxicity, and hematological toxicity. For grade 2 toxicities, immune checkpoint inhibitor treatment should be suspended with resumption when symptoms revert to grade 1 or less. Corticosteroids may be administered to help in reducing the inflammation. For grade 3 toxicities, high-dose steroids should be initiated along with suspension of immunotherapy. Corticosteroids should be tapered slowly over the course of at least 4–6 weeks. If patients are refractory to corticosteroids, then other forms of immunosuppressive agents such as infliximab could be used. For any grade 4-related toxicity permanent discontinuation of checkpoint inhibitors is recommended with the exception of endocrinopathies which can be controlled with hormone replacement therapy. Specific recommendations for the management of adverse events with checkpoint inhibitors have been published by American Society of Clinical Oncology [151].
10.4 Immunotherapy Biomarkers in Lung Cancer
With the approval of multiple checkpoint inhibitors, it is important to select the appropriate patients who might benefit from this class of therapies. In majority of the trials that led to the approval of these agents, it was noted that patients with tumors expressing high levels of PD-L1 benefit the most. For example in the KEYNOTE 189 trial with chemotherapy-naïve patients, pembrolizumab showed the highest OS benefit in patients with PD-L1 expression in ≥50% of tumor cells (12-month OS rate: 73.0% vs. 48.1%; HR: 0.42; 95% CI: 0.26–0.68) [143]. The clinical trial CheckMate 227 showed improved PFS in patients with higher tumor mutational burden which may be a new biomarker for immunotherapy.
One of the challenges facing these biomarkers is that PD-L1 and tumor mutational burden in tumor samples may be heterogeneous and repetitive tumor biopsies may be needed but is not always feasible [152]. Additionally, companion tests for evaluating PD-L1 expression as a biomarker of response use a variety of detection platforms for different forms (protein or mRNA), employ diverse biopsy and surgical samples, and have disparate positivity cutoff points and scoring systems, all of which complicate the standardization of clinical decision-making process [153]. Currently, four immunohistochemistry (IHC)-based assays using diagnostic monoclonal antibodies, 22C3 (pembrolizumab), 28-8 (nivolumab), SP142 (atezolizumab), or SP263 (durvalumab), to detect PD-L1 expression have been approved by FDA. Finally, the expression levels of PD-L1 may change after treatment with chemotherapy or immunotherapy and the patterns of resistance need to be better studied. In case of clear progressive disease upon immunotherapy, chemotherapy should be offered to the patients. However, if there are only 1 or 2 sites of progressive disease local modalities such as radiation or surgery with continuation of immunotherapy may be considered. It is important to develop a feasible, predictive, and reproducible biomarker which can predict patient response and help improve the overall survival among all these immunotherapies.
11 Other Potential Immune Therapy Targets
11.1 HER2
ERBB2 gene, a proto-oncogene on chromosome 17q12, encodes the ERBB2 protein which is also known as human epidermal growth factor receptor 2 (Her2), a tyrosine kinase receptor of the EGFR family. Her2 alterations have been detected in 1–4% of NSCLC tumors by multiplex testing and next-generation sequencing [154, 155]. These tumors are most commonly found in never smokers, adenocarcinomas, and women. Aberrations on exon 20 lead to phosphorylation of Her2 and activation of downstream pathways including RAS/RAF/MEK/ERK and PI3K/AKT/mTOR. These pathways have been implicated in the cancer cell proliferation, survival, growth, and tumor angiogenesis. The mutations most frequently found in Her2 gene are in-frame insertions in exon 20. There are currently no approved targeted agents for these patients. Her-2 targeted agents studied include: dacomitinib, neratinib, neratinib in combination with temsirolimus, afatinib, trastuzumab in combination with pertuzumab, and ado-trastuzumab emtansine. Response rates for these regimens have been meager at best (0–19%) [156,157,158] with ado-trastuzumab emtansine showing some of the most promise with a response rate up to 44% [159]. Another drug under investigation, poziotinib has been shown to have robust responses in preclinical models, and currently, there is a phase II study evaluating Her2 exon 20 insertion mutant advanced NSCLC with poziotinib as a treatment (NCT 03318939). Her2 amplification and Her2 exon 20 mutated advanced NSCLC represent two distinct subsets of disease in an ever-expanding field of potential clinically relevant targets.
11.2 Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS)
The KRAS gene (located on chromosome 12p12.1) is primarily involved in regulating cell division. It is a member of the RAS family of genes that encodes four proteins that are highly related mediators of the mitogen-activated protein kinase (MAPK) pathway: HRAS, KRAS4a, KRAS4b, and NRAS. These proteins function as guanosine triphosphatases (GTPases) binary switches that turn on or turn off multiple pathways involved in cancer cell survival, proliferation, angiogenesis, and differentiation via effector proteins. KRAS mutations have been found to be one of the most common oncogenic drivers in NSCLC (20–30%). The most common KRAS mutations are G12C with KRAS transversions typical for smokers and transitions typical for never smokers. Targeting RAS mutations remains elusive with most of the research focusing on RAF/MAPK pathway or novel approaches to RAS inhibition. MEK inhibitors such as trametinib and selumetinib have yet to show any survival benefit in patients with KRAS mutations compared to non-mutated patients [160,161,162,163,164]. Prior to activation of downstream effectors of RAS, RAS attaches to the cell membrane via farnesyl transferase. Investigations on utilizing farnesyl transferase inhibition have also failed to yield any promising results [165, 166]. The RAF/MEK inhibitor, RO5126766 (CH5127566), has shown some promising results in a basket trial with KRAS mutant NSCLC showing a 60% response rate [167].
11.3 Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha (PIK3CA)
PI3K/AKT signaling promotes carcinogenesis and development of NSCLC. PIK3CA encodes for PI3K which promotes cell survival. Its activation triggers downstream AKT. Mutations in the pathway include gain of function mutations in PIK3CA and AKT1 or loss of function mutations in the negative regulator protein PTEN, which occur in 16% of cancer cases. These mutations are found predominantly in SCC and smokers. Investigation is ongoing to determine if these are passenger or driver mutations. A variety of small molecules have been or currently being investigated as single agents or in combination with others but thus far failed to show improved efficacy over standard approaches [168,169,170,171]. Pictilisib in combination with standard of care did show some encouraging anti-tumor activity as a first-line treatment in a phase IB dose-escalation study [172], but this activity did not translate into improved PFS in the phase II FIGARO study [173].
11.4 Ephrin Type-B Receptor 4 (EPHB4)
EphB4 is a receptor found on venous endothelial cells, while its companion ligand Ephrin B2 is often expressed on arterial endothelial cells. EphB4 is overexpressed in epithelial tumors, which has been shown to be associated with poor prognosis in a variety of tumor types. This induces bidirectional signaling between EphB4 and EphB2 incudes the activation of PI3K/AKT/mTOR, Rho, Ras, Abl, Src, and MAPK signaling pathways, leading to increased cancer cell migration, proliferation, and adhesion. A biologic drug, sEphB4-HSA, which interferes the interaction between EphB4 and its ligand, has shown promising results in a dose finding phase IA study [174]. sEphB4-HSA in combination with pembrolizumab is currently under investigation in a phase II clinical trial (NCT03049618).
11.5 Fibroblast Growth Factor Receptor (FGFR)
Fibroblast growth factor receptor (FGFR) and its associated pathway are important in cell cycle progression, survival, and proliferation and can activate RAS and MAPK signaling cascades. FGFR mutations have been detected in 3–19% of non-small-cell lung cancer cases. These aberrations are mostly gene amplifications as well as nucleotide sequence alterations. FGFR inhibitors are mostly still investigational and their clinical significance remains to be seen [175,176,177,178].
12 Conclusions
In summary, the treatment of the lung cancer has advanced rapidly with the emergence of targeted therapy, immunotherapy, biomarker-based treatments, and availability of new clinical trials. Precision medicine may become increasingly important in the future of lung cancer treatment. Currently, testing for PD-L1 expression levels, EGFR mutations, ALK and ROS1 translocations are essential in selecting the best therapy for lung cancer patients, especially those with lung adenocarcinoma, large-cell histology, and non-small-cell lung cancer. Those tests can also be extended to patients who are never smokers or light smokers with squamous cell histology. Furthermore, broad genomic profiling to identify molecular alterations such as HER2 insertions, BRAF mutations, MET, TRK and RET alterations can help to identify investigational targeted agents that are in clinical trials.
For patients who are EGFR-positive, EGFR TKIs remain standard of care in the first-line metastatic setting. Patients with a ROS1 fusions should consider crizotinib as the initial treatment. For patients with ALK mutations, recent trials have shown that the next-generation ALK inhibitor such as alectinib may be superior to crizotinib in the first-line setting. Patients whose tumors have high PD-L1 expression (≥50%) should receive pembrolizumab as the first-line therapy. Ongoing clinical trials are evaluating the benefits of combining immunotherapy or targeted therapy with chemotherapy. Patients whose tumors harbor other mutations should be encouraged to participate in clinical trials for corresponding targeted agents.
References
Wheeler DA, Wang L (2013) From human genome to cancer genome: the first decade. Genome Res 23(7):1054–1062
Gong J et al (2018) Value-based genomics. Oncotarget 9(21):15792–15815
National Human Genome Research Institute (NHGRI) (2003) Human genome project completion: frequently asked questions. In: National Human Genome Research Institute (ed) 2003 release: international consortium completes HGP
El-Metwally S, Ouda OM, Helmy M (2014) Next generation sequencing technologies and challenges in sequence assembly. SpringerBriefs in Systems Biology, vol 7. XII, 118 11 b/w illustrations, 1 illustrations in colour. Springer-Verlag, New York
Mardis ER (2011) A decade’s perspective on DNA sequencing technology. Nature 470(7333):198–203
Wheeler DA et al (2008) The complete genome of an individual by massively parallel DNA sequencing. Nature 452(7189):872–876
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17(6):333–351
Heather JM, Chain B (2016) The sequence of sequencers: the history of sequencing DNA. Genomics 107(1):1–8
Quail MA et al (2012) A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom 13:341
Feliubadalo L et al (2017) Benchmarking of whole exome sequencing and ad hoc designed panels for genetic testing of hereditary cancer. Sci Rep 7:37984
Khotskaya YB, Mills GB, Mills Shaw KR (2017) Next-generation sequencing and result interpretation in clinical oncology: challenges of personalized cancer therapy. Annu Rev Med 68:113–125
Schram AM, Berger MF, Hyman DM (2017) Precision oncology: charting a path forward to broader deployment of genomic profiling. PLoS Med 14(2):e1002242
Cubiella J et al (1999) Prognostic factors in nonresectable pancreatic adenocarcinoma: a rationale to design therapeutic trials. Am J Gastroenterol 94(5):1271–1278
Frampton GM et al (2013) Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31(11):1023–1031
Herzog TJ et al (2016) Impact of molecular profiling on overall survival of patients with advanced ovarian cancer. Oncotarget 7(15):19840–19849
Radovich M et al (2016) Clinical benefit of a precision medicine based approach for guiding treatment of refractory cancers. Oncotarget 7(35):56491–56500
Weiss GJ et al (2015) Evaluation and comparison of two commercially available targeted next-generation sequencing platforms to assist oncology decision making. Onco Targets Ther 8:959–967
Sboner A et al (2011) The real cost of sequencing: higher than you think! Genome Biol 12(8):125
Frank M et al (2013) Genome sequencing: a systematic review of health economic evidence. Health Econ Rev 3(1):29
Jardim DL et al (2015) Impact of a biomarker-based strategy on oncology drug development: a meta-analysis of clinical trials leading to FDA approval. J Natl Cancer Inst 107(11)
Schwaederle M et al (2015) Impact of precision medicine in diverse cancers: a meta-analysis of phase II clinical trials. J Clin Oncol 33(32):3817–3825
Schwaederle M et al (2016) Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: a meta-analysis. JAMA Oncol 2(11):1452–1459
Ocana A et al (2015) Influence of companion diagnostics on efficacy and safety of targeted anti-cancer drugs: systematic review and meta-analyses. Oncotarget 6(37):39538–39549
Cancer Genome Atlas Research Network (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511(7511):543–550
Sholl LM et al (2015) Multi-institutional oncogenic driver mutation analysis in lung adenocarcinoma: the Lung Cancer Mutation Consortium experience. J Thorac Oncol 10(5):768–777
Sabari JK et al (2017) Unravelling the biology of SCLC: implications for therapy. Nat Rev Clin Oncol 14(9):549–561
George J et al (2015) Comprehensive genomic profiles of small cell lung cancer. Nature 524(7563):47–53
Pesch B et al (2012) Cigarette smoking and lung cancer—relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int J Cancer 131(5):1210–1219
Ellison G et al (2013) EGFR mutation testing in lung cancer: a review of available methods and their use for analysis of tumour tissue and cytology samples. J Clin Pathol 66(2):79–89
Mayo-de-Las-Casas C et al (2017) Large scale, prospective screening of EGFR mutations in the blood of advanced NSCLC patients to guide treatment decisions. Ann Oncol 28(9):2248–2255
Toor OM et al (2018) Correlation of somatic genomic alterations between tissue genomics and ctDNA employing next-generation sequencing: analysis of lung and gastrointestinal cancers. Mol Cancer Ther 17(5):1123–1132
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2(2):127–137
Rosell R et al (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361(10):958–967
Midha A, Dearden S, McCormack R (2015) EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII). Am J Cancer Res 5(9):2892–2911
Shi Y et al (2014) A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol 9(2):154–162
Wang S, Wang Z (2014) EGFR mutations in patients with non-small cell lung cancer from mainland China and their relationships with clinicopathological features: a meta-analysis. Int J Clin Exp Med 7(8):1967–1978
Information NCfB, NIH. PubChem. NIH
Soria JC et al (2018) Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med 378(2):113–125
Xu J, Wang J, Zhang S (2017) Mechanisms of resistance to irreversible epidermal growth factor receptor tyrosine kinase inhibitors and therapeutic strategies in non-small cell lung cancer. Oncotarget 8(52):90557–90578
Mok TS et al (2017) Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 376(7):629–640
Lee CK et al (2013) Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: a meta-analysis. J Natl Cancer Inst 105(9):595–605
Zhou C et al (2011) Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 12(8):735–742
Zhou C et al (2015) Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann Oncol 26(9):1877–1883
Rosell R et al (2012) Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13(3):239–246
Wu YL et al (2015) First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 26(9):1883–1889
Fukuoka M et al (2011) Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol 29(21):2866–2874
Mok TS et al (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361(10):947–957
Maemondo M et al (2010) Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 362(25):2380–2388
Sequist LV et al (2013) Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 31(27):3327–3334
Yang JC et al (2013) Symptom control and quality of life in LUX-Lung 3: a phase III study of afatinib or cisplatin/pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations. J Clin Oncol 31(27):3342–3350
Wu YL et al (2014) Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 15(2):213–222
Yang JC et al (2015) Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol 16(2):141–151
Yang JC et al (2015) Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol 16(7):830–838
Wu YL et al (2017) Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol 18(11):1454–1466
Yang JJ et al (2017) A phase III randomised controlled trial of erlotinib vs gefitinib in advanced non-small cell lung cancer with EGFR mutations. Br J Cancer 116(5):568–574
Paz-Ares L et al (2017) Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: overall survival data from the phase IIb LUX-Lung 7 trial. Ann Oncol 28(2):270–277
Chabon JJ et al (2016) Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 7:11815
Piotrowska Z et al (2017) MET amplification (amp) as a resistance mechanism to osimertinib. J Clin Oncol 35:6
Sequist LV et al (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3(75):75ra26
Nishie K et al (2012) Epidermal growth factor receptor tyrosine kinase inhibitors beyond progressive disease: a retrospective analysis for Japanese patients with activating EGFR mutations. J Thorac Oncol 7(11):1722–1727
Seto T et al (2014) Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomised, multicentre, phase 2 study. Lancet Oncol 15(11):1236–1244
Wu YL et al (2013) Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non-small-cell lung cancer (FASTACT-2): a randomised, double-blind trial. Lancet Oncol 14(8):777–786
Cheng Y et al (2016) Randomized phase II trial of gefitinib with and without pemetrexed as first-line therapy in patients with advanced nonsquamous non-small-cell lung cancer with activating epidermal growth factor receptor mutations. J Clin Oncol 34(27):3258–3266
Gatzemeier U et al (2007) Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol 25(12):1545–1552
Giaccone G et al (2004) Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial—INTACT 1. J Clin Oncol 22(5):777–784
Herbst RS et al (2004) Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial—INTACT 2. J Clin Oncol 22(5):785–794
Herbst RS et al (2005) TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol 23(25):5892–5899
Mok TSK et al (2017) Gefitinib plus chemotherapy versus chemotherapy in epidermal growth factor receptor mutation-positive non-small-cell lung cancer resistant to first-line gefitinib (IMPRESS): overall survival and biomarker analyses. J Clin Oncol 35(36):4027–4034
Kwak EL et al (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363(18):1693–1703
Crino L et al (2011) Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol 29(15):1
Solomon BJ et al (2014) First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 371(23):2167–2177
Awad MM, Shaw AT (2014) ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol 12(7):429–439
Shaw AT et al (2014) Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 371(21):1963–1971
Shaw AT et al (2017) Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 18(7):874–886
Yang JC et al (2017) Pooled systemic efficacy and safety data from the pivotal phase II studies (NP28673 and NP28761) of alectinib in ALK-positive non-small cell lung cancer. J Thorac Oncol 12(10):1552–1560
Shaw AT et al (2016) Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet Oncol 17(2):234–242
Bendell JC et al (2016) Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J Clin Oncol 34(15):2
Peters S et al (2017) Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med 377(9):829–838
Kim DW et al (2017) Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 35(22):2490–2498
Shaw AT et al (2017) Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol 18(12):1590–1599
Davies KD, Doebele RC (2013) Molecular pathways: ROS1 fusion proteins in cancer. Clin Cancer Res 19(15):4040–4045
Rossi G et al (2017) Detection of ROS1 rearrangement in non-small cell lung cancer: current and future perspectives. Lung Cancer (Auckl) 8:45–55
Ogura H et al (2017) TKI-addicted ROS1-rearranged cells are destined to survival or death by the intensity of ROS1 kinase activity. Sci Rep 7(1):5519
Bergethon K et al (2012) ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 30(8):863–870
Sporn JR (1999) Practical recommendations for the management of adenocarcinoma of the pancreas. Drugs 57(1):69–79
Davies H et al (2002) Mutations of the BRAF gene in human cancer. Nature 417(6892):949–954
Ascierto PA et al (2012) The role of BRAF V600 mutation in melanoma. J Transl Med 10:85
Kinno T et al (2014) Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 25(1):138–142
Paik PK et al (2011) Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 29(15):2046–2051
Sequist LV et al (2011) Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol 22(12):2616–2624
Villaruz LC et al (2015) Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 121(3):448–456
Litvak AM et al (2014) Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 9(11):1669–1674
Hyman DM et al (2015) Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 373(8):726–736
Planchard D et al (2016) Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 17(5):642–650
Planchard D et al (2016) Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 17(7):984–993
Planchard D et al (2017) Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 18(10):1307–1316
Giordano S et al (1989) Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 339(6220):155–156
Bottaro DP et al (1991) Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 251(4995):802–804
Naldini L et al (1991) Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J 10(10):2867–2878
Ponzetto C et al (1994) A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 77(2):261–271
Sadiq AA, Salgia R (2013) MET as a possible target for non-small-cell lung cancer. J Clin Oncol 31(8):1089–1096
Ma PC et al (2008) Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer 47(12):1025–1037
Sattler M, Salgia R (2016) MET in the driver’s seat: exon 14 skipping mutations as actionable targets in lung cancer. J Thorac Oncol 11(9):1381–1383
Maulik G et al (2002) Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev 13(1):41–59
Krishnaswamy S et al (2009) Ethnic differences and functional analysis of MET mutations in lung cancer. Clin Cancer Res 15(18):5714–5723
Stransky N et al (2014) The landscape of kinase fusions in cancer. Nat Commun 5:4846
Watermann I et al (2015) Improved diagnostics targeting c-MET in non-small cell lung cancer: expression, amplification and activation? Diagn Pathol 10:130
Salgia R (2017) MET in lung cancer: biomarker selection based on scientific rationale. Mol Cancer Ther 16(4):555–565
Cappuzzo F et al (2009) Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 27(10):1667–1674
Raghav K et al (2018) Untying the gordion knot of targeting MET in cancer. Cancer Treat Rev 66:95–103
Waqar SN, Morgensztern D, Sehn J (2015) MET mutation associated with responsiveness to crizotinib. J Thorac Oncol 10(5):e29–e31
Drilon A et al (2016) Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 17(12):1653–1660
Spigel DR et al (2017) Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: METLung. J Clin Oncol 35(4):412–420
Scagliotti G et al (2015) Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol 33(24):2667–2674
Bean J et al (2007) MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 104(52):20932–20937
Azuma K et al (2016) Phase II study of erlotinib plus tivantinib (ARQ 197) in patients with locally advanced or metastatic EGFR mutation-positive non-small-cell lung cancer just after progression on EGFR-TKI, gefitinib or erlotinib. ESMO Open 1(4):e000063
Yoshioka H et al (2015) A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-Met inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study). Ann Oncol 26(10):2066–2072
Zenali M et al (2015) Retrospective review of MET gene mutations. Oncoscience 2(5):533–541
McLornan DP, List A, Mufti GJ (2014) Applying synthetic lethality for the selective targeting of cancer. N Engl J Med 371(18):1725–1735
Kyriakopoulos CE et al (2017) A phase I study of tivantinib in combination with temsirolimus in patients with advanced solid tumors. Invest New Drugs 35(3):290–297
Preusser M et al (2014) Amplification and overexpression of CMET is a common event in brain metastases of non-small cell lung cancer. Histopathology 65(5):684–692
Chi AS et al (2012) Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J Clin Oncol 30(3):e30–e33
Klempner SJ et al (2017) Intracranial activity of cabozantinib in MET exon 14-positive NSCLC with brain metastases. J Thorac Oncol 12(1):152–156
Amatu A, Sartore-Bianchi A, Siena S (2016) NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 1(2):e000023
Broderick JM (2017) Entrectinib granted breakthrough designation by FDA for NTRK + solid tumors. Targeted Oncology News article
Hyman DM et al (2017) The efficacy of larotrectinib (LOXO-101), a selective tropomyosin receptor kinase (TRK) inhibitor, in adult and pediatric TRK fusion cancers. J Clin Oncol 35(18_suppl):LBA2501
Bagheri-Yarmand R et al (2015) A novel dual kinase function of the RET proto-oncogene negatively regulates activating transcription factor 4-mediated apoptosis. J Biol Chem 290(18):11749–11761
Cascone T, Subbiah V, Heymach JV (2017) Targeting RET rearrangements in non-small cell lung cancer
Amarnath S et al (2011) The PDL1-PD1 axis converts human T H1 cells into regulatory T cells. Sci Transl Med 3(111)
Francisco LM et al (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206(13):3015–3029
Shin DS, Ribas A (2015) The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr Opin Immunol 33:23–35
Kusnierczyk P (2013) Killer cell immunoglobulin-like receptor gene associations with autoimmune and allergic diseases, recurrent spontaneous abortion, and neoplasms. Front Immunol 4:8
Brahmer JR et al (2010) Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 28(19):3167–3175
Borghaei H et al (2015) Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 373(17):1627–1639
Vokes EE et al (2018) Nivolumab versus docetaxel in previously treated advanced non-small-cell lung cancer (CheckMate 017 and CheckMate 057): 3-year update and outcomes in patients with liver metastases. Ann Oncol 29(4):959–965
Brahmer J et al (2015) Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 373(2):123–135
Horn L et al (2017) Nivolumab versus docetaxel in previously treated patients with advanced non-small-cell lung cancer: two-year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057). J Clin Oncol 35(35):3924–3933
Carbone DP et al (2017) First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med 376(25):2415–2426
Hellmann MD et al (2018) Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 378(22):2093–2104
Forde PM et al (2018) Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med 378(21):1976–1986
Reck M et al (2016) Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med 375(19):1823–1833
Langer CJ et al (2016) Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol 17(11):1497–1508
Gandhi L et al (2018) Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med 378(22):2078–2092
Herbst RS et al (2016) Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387(10027):1540–1550
Rittmeyer A et al (2017) Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389(10066):255–265
Jotte RM et al (2018) IMpower131: primary PFS and safety analysis of a randomized phase III study of atezolizumab + carboplatin + paclitaxel or nab-paclitaxel vs carboplatin + nab-paclitaxel as 1L therapy in advanced squamous NSCLC. J Clin Oncol 36(18_suppl):LBA9000
Socinski MA et al (2018) Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med 378(24):2288–2301
Antonia SJ et al (2017) Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med 377(20):1919–1929
Lynch TJ et al (2012) Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol 30(17):2046–2054
Weber JS et al (2017) Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol 35(7):785–792
Brahmer JR, Lacchetti C, Thompson JA (2018) Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American society of clinical oncology clinical practice guideline summary. J Oncol Pract 14(4):247–249
The Lancet Respiratory Medicine (2018) Lung cancer immunotherapy biomarkers: refine not reject. Lancet Respir Med 6(6):403
Liu D, Wang S, Bindeman W (2017) Clinical applications of PD-L1 bioassays for cancer immunotherapy. J Hematol Oncol 10(1):110
Stephens P et al (2004) Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431(7008):525
Shigematsu H et al (2005) Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Can Res 65(5):1642–1646
Kris M et al (2015) Targeting HER2 aberrations as actionable drivers in lung cancers: phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann Oncol 26(7):1421–1427
Gandhi L et al (2017) MA04. 02 neratinib ± temsirolimus in HER2-mutant lung cancers: an international, randomized phase II study. J Thorac Oncol 12(1): S358–S359
Lai WCV et al (2017) Afatinib in patients with metastatic HER2-mutant lung cancers: an international multicenter study. Am Soc Clin Oncol
Li BT et al (2018) Ado-trastuzumab emtansine for patients with HER2-mutant lung cancers: results from a phase II basket trial. J Clin Oncol. https://doi.org/10.1200/JCO.2018.77.9777
Blumenschein G Jr et al (2015) A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann Oncol 26(5):894–901
Gandara DR et al (2013) Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with docetaxel in KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): a phase I/Ib trial. Am Soc Clin Oncol
Kelly K et al (2013) Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with pemetrexed for KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): a phase I/Ib trial. Am Soc Clin Oncol
Jänne PA et al (2013) Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol 14(1):38–47
Jänne PA et al (2017) Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with kras-mutant advanced non-small cell lung cancer: the select-1 randomized clinical trial. JAMA 317(18):1844–1853
Aviel-Ronen S et al (2006) K-ras mutations in non-small-cell lung carcinoma: a review. Clin Lung Cancer 8(1):30–38
Reuter CW, Morgan MA, Bergmann L (2000) Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood 96(5):1655–1669
Chenard-Poirier M et al (2017) Results from the biomarker-driven basket trial of RO5126766 (CH5127566), a potent RAF/MEK inhibitor, in RAS- or RAF-mutated malignancies including multiple myeloma. Am Soc Clin Oncol
Vansteenkiste JF et al (2015) Safety and efficacy of buparlisib (BKM120) in patients with PI3K pathway-activated non-small cell lung cancer: results from the phase II BASALT-1 study. J Thorac Oncol 10(9):1319–1327
Soria J-C et al (2015) Phase I dose-escalation study of pilaralisib (SAR245408, XL147), a pan-class I PI3K inhibitor, in combination with erlotinib in patients with solid tumors. Oncologist 20(3):245–246
Levy B et al (2014) A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic non-small-cell lung cancer. J Thorac Oncol 9(7):1031–1035
Wade JL et al (2017) A phase II study of GDC-0032 (taselisib) for previously treated PI3K positive patients with stage IV squamous cell lung cancer (SqNSCLC): LUNG-MAP sub-study SWOG S1400B. Am Soc Clin Oncol
Soria J-C et al (2017) A phase IB dose-escalation study of the safety and pharmacokinetics of pictilisib in combination with either paclitaxel and carboplatin (with or without bevacizumab) or pemetrexed and cisplatin (with or without bevacizumab) in patients with advanced non-small cell lung cancer. Eur J Cancer 86:186–196
Besse B et al (2015) A phase II trial of pictilisib with chemotherapy in first-line non-squamous NSCLC. J Thorac Oncol. Elsevier
Thomas JS et al (2018) A first-in-human phase I study of sEphB4-HSA (sEphB4) with expansion in hepatocellular (HCC) and cholangiocarcinoma (CCA). Am Soc Clin Oncol
Nogova L et al (2017) Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J Clin Oncol 35(2):157–165
Tabernero J et al (2015) Phase I dose-escalation study of JNJ-42756493, an oral pan–fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol 33(30):3401–3408
Paik PK et al (2014) A phase 1b open-label multicenter study of AZD4547 in patients with advanced squamous cell lung cancers: preliminary antitumor activity and pharmacodynamic data. Am Soc Clin Oncol
Smyth EC et al (2015) Phase II multicenter proof of concept study of AZD4547 in FGFR amplified tumours. Am Soc Clin Oncol
Acknowledgements
Special thanks to Sam Garcia, Library Assistant, and Andrea Lynch, Scholarly Communications Librarian at the City of Hope Graff Library in Duarte, California.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hill, A., Gupta, R., Zhao, D., Vankina, R., Amanam, I., Salgia, R. (2019). Targeted Therapies in Non-small-Cell Lung Cancer. In: Von Hoff, D., Han, H. (eds) Precision Medicine in Cancer Therapy . Cancer Treatment and Research, vol 178. Springer, Cham. https://doi.org/10.1007/978-3-030-16391-4_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-16391-4_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-16390-7
Online ISBN: 978-3-030-16391-4
eBook Packages: MedicineMedicine (R0)