
Abstract
The dry antibiotic development pipeline coupled with the emergence of multi-drug resistant Gram-negative ‘superbugs’ has driven the revival of the polymyxin lipopeptide antibiotics. Understanding the mode of action of antibiotics is an important precursor for optimizing their use and development. This chapter provides a concise treatise of the current knowledge-based on the primary mode of action of polymyxins as well as recent developments in understanding of bacterial cell responses and secondary modes of action.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
The outer membrane of the Gram-negative cell acts as a permeability barrier that protects the cell from various noxious substances, including numerous antimicrobials [1]. Polymyxins exert their antimicrobial action via direct interaction with the lipid A component of the lipopolysaccharide (LPS) which leads to a disruption of this critical barrier function. Accordingly, understanding the mechanism of polymyxin antibacterial activity requires a brief review of the architecture of LPS and the outer membrane. The complex asymmetrical structure of the outer membrane comprises an inner phospholipid leaflet, as well as an outer leaflet that predominantly contains LPS, proteins and phospholipids [1]. LPS is composed of three domains, a conserved inner core 2-keto-3-deoxyoctonoic acid (Kdo) bound to lipid A and a variable O-antigen composed of repeating units of various polysaccharides [1,2,3,4]. The structure of lipid A consists of aβ-10-6-linked D-glucosamine (GlcN) disaccharide that is phosphorylated at the 1- and 4′-positions and decorated by a variable number of saturated hydrocarbon chains, generally C10-C14 in length [2, 4]. Lipid A is intercalated within the outer leaflet, where in the saturated lipid A hydrocarbon chains are tightly packed together within the membrane through van der Waals forces, thereby acting as an anchor for the entire LPS structure [1, 5, 6]. Divalent cations (Mg2+ and Ca2+) associate with the lipid A phosphoresters and function to bridge adjacent LPS molecules [1, 5, 6]. The barrier function of the outer membrane is further accentuated by a highly repulsive anionic charge conveyed by lipid A phosphorester moieties, as well as phosphate and carboxylate functionalities decorating the core and O-antigen sugars [1, 5, 6].
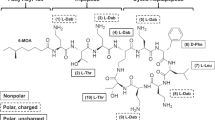
Amphipathicity of the polymyxins is critical for their outer membrane permeabilizing action. The conserved elements in the chemical structure of polymyxins that contribute to this amphipathicity includes the two hydrophobic domains (the N-terminal fatty acyl chain and the hydrophobic position 6–7 segment) separated by segments of polar (Thr) and cationic Dab side chains. The elucidation of the three-dimensional NMR solution state structure of polymyxin B in complex with LPS revealed the polymyxin B molecule is folded such that the polar and hydrophobic domains form two distinct faces, thereby conferring structural amphipathicity (Fig. 4.1) [7, 8].

4.1 Primary Mode of Action of Polymyxins
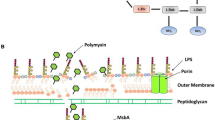
Polymyxins are believed to exert their primary antimicrobial mode of action by permeabilizing the outer membrane via a direct interaction with LPS . Polymyxins zone-into their primary cellular target, LPS through the initial electrostatic interaction of the cationic L-α,γ-diaminobutyric acid (Dab) side-chains with the phosphate groups of the lipid A component of LPS, displacing divalent cations (Ca2+ and Mg2+) that bridge adjacent LPS molecules (Fig. 4.2, Stage 1) [1, 5, 6, 9,10,11]. This initial electrostatic interaction allows the N-terminal fatty acyl chain and hydrophobic position 6–7 motif (Polymyxin B: D-Phe6-L-Leu7 and colistin: D-Leu6-L-Leu7) of the polymyxin molecule to insert into the fatty acyl chain layer of the lipid A molecules. The insertion of the hydrophobic domains of polymyxins possibly act to weaken the packing of adjacent lipid A fatty acyl chains causing expansion of the outer membrane monolayer. The fact that polymyxin B nonapeptide (derived by proteolytic cleavage of the fatty acyl-Dab1 from the N-terminus of the polymyxin) is devoid of antibacterial activity highlights the importance of the hydrophobic interactions for the mechanism of polymyxin action [12]. Subsequently, the polymyxin molecule inserts and disrupts the physical integrity of the phospholipid bilayer of the inner membrane leaflet via membrane thinning by straddling the interface of the hydrophilic head groups and fatty acyl chains or transient poration [6, 7, 10, 13, 14]. This ‘self-promoted’ uptake mechanism is believed to produce disruption of the outer membrane structures, which leads to bacterial cell death (Fig. 4.2, Stage 2) [5, 6, 9,10,11]. It has also been proposed that the MOA of polymyxins involves producing contacts between the periplasmic leaflets of the inner and outer membranes that promotes phospholipid exchange between the inner and outer membrane leaflets. This in turn would result in the loss of phospholipid compositional specificity, potentially to an osmotic imbalance that contributes to lytic cell death [15, 16]. This postulate is based on evidence that polymyxin B when bound to anionic phospholipid vesicles is capable of forming vesicle-to-vesicle contacts [13, 15,16,17]. The ability of polymyxins to disrupt the inner membrane structure is coincident with their inhibition of the inner membrane respiratory enzyme the alternative type 2 nicotinamide adenine dinucleoti dedehydrogenase (NDH-2) in Mycobacterium smegmatis and in a number of pathogenic Gram-negative bacteria [18, 19], which intuitively leads us to the next section that covers secondary modes of action of polymyxin lipopeptides.

Schematic diagram depicting the putative mode of action of polymyxins. Stage 1. Electrostatic attraction between the positively charges polymyxin molecule and the negatively charges bacterial outer membrane surface Fig. 4.2 (continued) leads to the displacement of divalent cations that help stabilize the outer membrane structure by bridging adjacent LPS molecules. Stage 2. The positively charged polymyxins displace divalent cations that bridge adjacent LPS molecules. The hydrophobic insertion destabilizes the outer membrane. Stage 3. The polymyxin molecule penetrates into the inner membrane and inhibits the respiratory enzyme NDH-2
4.2 Secondary Mode of Action of Polymyxins
Although cationic peptides such as the polymyxins are traditionally thought of as outer membrane-active agents [20], the bacterial outer membrane is not necessarily the sole target for their mode of action [21,22,23]. Secondary targets involved in the bactericidal activity of polymyxins remain poorly characterized. Based on available evidence, one possible secondary mode of action of polymyxin B and colistin in Gram-negative bacteria involves the inhibition of bacterial respiration [24, 25].
Instead of the multi-subunit complex I found in mammalian cells, protozoa, bacteria and plants possess a single sub-unit non-proton pumping, rotenone insensitive alternative type II NADH-menaquinone oxidoreductase (NDH-2) [26,27,28,29]. The NDH-2 enzyme contains a single non-covalently bound flavin adenine dinucleotide (FAD) cofactor and catalyzes the oxidation of NADH with menaquinone [28,29,30,31]. In general, the bacterial respiratory chain consists of three complexes with quinones and reduced nicotinamide adenine dinucleotide (NADH) acting as the carriers that shuttle electrons and protons between large protein complexes [32,33,34,35,36]. The exact organization of enzymes varies among different bacteria [32,33,34]. In Complex 1, three inner membrane respiratory enzymes of the NADH oxidase family have been identified: proton-translocating NADH-quinone (Q) oxidoreductase (NDH-1), NADH-Q oxidoreductase which lacks an energy-coupling site (NDH-2) and the sodium-translocating NADH-Q oxidoreductase [32,33,34, 36].
We have shown that polymyxin B, B1, B2 and colistin can inhibit NDH-2 activity in the inner membranes of three different Gram-negative bacterial species (Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii) in a concentration-dependent manner (Fig. 4.2, Stage 3). The mechanism of NDH-2 inhibition by polymyxin B was investigated in detail with E. coli inner membrane preparations and conformed to a mixed inhibition model with respect to ubiquinone-1 and a non-competitive inhibition model with respect to NADH. The structure of the polymyxins (cyclic peptides) being distinct from those of the NDH-2 substrates NADH and Q1 is supportive of the inhibition kinetic data, in that they are unlikely to compete for the same sites on the enzyme. Our kinetic data are in line with the reported data for Gluconobacter oxydans which showed that the inhibition by gramicidin S and scopafungin was non-competitive with respect to NADH [37]. Scopafungin, which like polymyxin B and colistin possesses a cyclic ring and a long acyl chain in its structure, displayed a mixed inhibition mode with respect to ubiquinone, whereas gramicidin S was a competitive inhibitor [37].
The IC50 values for the inhibition by polymyxin B and colistin of NDH-2 activity in inner membrane of the three different Gram-negative bacterial species were in most part comparable, indicating that inter-species differences in NDH-2 do not impact the inhibitory activity of the polymyxins. Polymyxin B was a better inhibitor compared to colistin, which is in line with reported results with the Gram-positive M. smegmatis NDH-2 [37]. Although polymyxin B and colistin display high IC50 values (polymyxin B IC50 = 50 μM; colistin IC50 = 251 μM) for NDH-2 inhibition, under in vivo conditions there remains the possibility that very high local concentrations of the antibiotic can accumulate at the site of infection that fall within these IC50 value ranges. Coincidently, we have garnered in vitro evidence that suggests that polymyxins can accumulate in the inner membrane of Gram-negative bacteria [38]. Therefore, the high IC50 values do not dismiss the possibility that NDH-2 represents one of the secondary pathways that is targeted once the polymyxin penetrates the outer membrane. Notably also colistin inhibited NADH-quinone oxidoreductase activity in the polymyxin-susceptible strain of K. pneumoniae with a comparable IC50 to that of the polymyxin-resistant strain, suggesting polymyxin resistance in these strains is not at the level of the inner membrane respiratory enzymes. Our previous study had indicated that the resistant derivative of K. pneumoniae exhibited less negative charge than the wild type that lead to failure of polymyxin interaction at the outer membrane [39]. The NDH-2 activity was not inhibited by CMS, polymyxin B nonapeptide and colistin nonapeptide. The loss of inhibitory activity seen with the polymyxin nonapeptide and CMS suggests that the N-terminal fatty acyl chain and the positive charges of the polymyxin molecule are critical for NDH-2 inhibitory activity [40].
The fact that NDH-2 enzymes are not found in mammalian mitochondria and are mainly expressed by protozoa, bacteria and plants makes them very attractive drug targets [41]. Our data suggest that one of the secondary target sites of polymyxins is the type II NADH-quinone oxidoreductase respiratory enzyme that forms an integral part of the bacterial electron transport pathway. In view of the dry antibiotic pipe-line, together with the increasing incidence of multidrug resistant in Gram-negative bacteria , NDH-2 represents an important target that can be exploited for the development of new antibiotics against these problematic pathogens. Notably, energy metabolism and NDH-2 in particular, is emerging as an important drug target in Mycobacterium tuberculosis and Plasmodium falciparum [28, 29, 31, 42,43,44,45].
A recent preliminary biochemical study reported that rapid killing of A. baumannii by polymyxins is mediated by a hydroxyl radical death pathway, which although under explored, potentially represents another secondary mode of action whereby polymyxin kill Gram-negative bacterial cells [46]. Coincidently, it has been proposed that most antibiotics cause bacterial cell death via a common mechanism whereby they disrupt bacterial metabolism leading to the generation of reactive oxygen species (ROS) that eventually kills the bacterial cell [47].
4.3 Stress Responses to Polymyxin Treatment in Gram-negative Bacteria
The antibacterial activity of bactericidal antibiotic is not solely governed by its mode of action and its ability to interact with targets [47,48,49,50,51]. There are a number of bacterial response factors associated with exposure to sub-lethal concentrations of polymyxins which will be reviewed in this section. These factors include activation of adaptive resistance mechanisms, the stimulation of protective changes to cell physiology, and even induction of resistance mutations.
The protective stress response, also named adaptive resistance, is an auto-regulated antibiotic induced phenomenon and reversal to the sensitive phenotype in the absence of inducer [52, 53]. Extensive studies in recent years have provided significant insight into the outer membrane remodelling mechanisms responsible for adaptive resistance to polymyxins, perhaps best studied in Salmonella [54,55,56,57].
In response to polymyxin exposure, the outer membrane undergoes extensive remodelling of structural alterations contributing to adaptive resistance to polymyxins which is triggered by two-component systems (Fig. 4.3) [52, 54, 58]. In Salmonella, the response to polymyxins is mediated by the PhoPQ two-component system, where polymyxins interact with and activate the sensor PhoQ by displacing divalent cations from their metal binding sites in the sensor domain [59] and then activates the PhoP response regulator to up-regulate a variety of target genes and ultimately promote adaptation to the stress. Specific changes in OM regulated by activated PhoP include: increasing O-antigen chain length, acylating, inhibiting deacylating, hydroxylating lipid A , derivitizing lipid A and LPS core phosphates with cationic groups, palmitoylating OM phosphatidylglycerols, and increasing the level of OM cardiolipins [54, 56]. Therefore, upon PhoPQ activation an extensive alteration of LPS, GPLs, and proteins elaborates the OM barrier more impermeable to polymyxins , thereby promoting bacterial survival (Fig. 4.3) [54, 56].

(a) Glycerophospholipid and lipid A modifications that ensue with Gram-negative membrane remodelling associated with the development of polymyxin resistance. (b) Polymyxin-induced outer membrane remodelling, intracellular biochemical perturbations and resistance pathways. The initial outer membrane disorganisation caused by polymyxin exposure is followed by intracellular redox perturbations of NDH-2 activity. These events are accompanied by activation of repair pathways and outer membrane remodelling. The PhoP/PhoQ two component regulatory system activates the lipid A deacylase PagL and PmrD which in turn activates PmrA. PmrA activates the expression of arnBCADTEF which are a collective of enzymes modifying lipid A with cationic groups (e.g. 4-amino-4-deoxy-L-arabinose or phosphoethanolamine) to repel the polymyxin. PmrA also activates PmrR responsible for the repression of LpxT (phosphorylation of lipid A) and LpxR (deacylation of lipid A) genes
4.4 Regulation of LPS Remodelling
Upon sensing polymyxins , PhoPQ increases transcription of pmrD and the pmrCAB operon to activate the response regulator PmrA (Fig. 4.3) [60,61,62]. The activated PmrA induces expression of a short membrane peptide, PmrR, which binds to and inhibits the lipid A phosphorylase LpxT, an enzyme responsible for increasing the negative charge of the outer membrane leaflet [56, 63]. The activated PmrA induces transcription of genes encoding enzymes that covalently modify lipid A and core sugar phosphates with positively charged aminoarabinose (L-Ara4N) and phosphoethanolamine (pEtN) [54]. The initial step for L-Ara4N moiety modification begins with the oxidation of UDP-glucose in the cytosol by the PhoP and PmrA-regulated UDP-glucose dehydrogenase (PagA, Ugd, or pmrE) [54, 64]. The remaining steps responsible for L-Ara4N moiety modification are encoded within an operon pmrHFIJKLM (also known as arnBCADTEF), which is activated by PhoPQ through PmrA [54, 64]. When the biosynthesis proceeds to complete UDP formylated-L-Ara4N synthesis, the formylated-L-Ara4N residue is transfer from UDP to undecaprenyl phosphate carrier lipid on the inner leaflet of the inner membrane and then deformylated to form undecaprenyl phosphate-L-Ara4N [54, 64]. Next, the undecaprenyl phosphate-L-Ara4N is flipped into the outer leaflet of the IM where the membrane protein ArnT (also known as PmrK) transfers the L-Ara4N moiety to nascent lipid A phosphates [54, 64]. Finally, O-antigen is loaded onto the core structure and then the assembled LPS molecules are moved from the inner membrane through the periplasm to the OM of cell surface by the lipopolysaccharide transport (Lpt) proteins complex acquired driving powers from cytoplasmic ATP hydrolysis [54, 65]. The PmrAB-controlled pEtN transferases encoded by eptA (also known as lptA or pmrC) and cptA (or eptB) also contribute to polymyxins resistance via their modification of lipid A and LPS core respectively, with positively charged pEtN [54, 56]. Decreased negative charge conferred by cationic groups L-Ara4N and pEtN on lipid A molecules diminishing binding sites plays a significant role in polymyxins resistance while modification of cationic groups on LPS core plays modest effect on resistance [54, 56, 66]. Though varies in details across Gram-negative species, the positively charged modification of lipid A mediated by two-component systems is the most commonly seen mechanism of polymyxins resistance [52, 56, 67, 68].
An abundance of uniquely hydroxylated myristoyl groups has been detected in lipid A structures under PhoPQ activation in S. typhimurium [69]. The modification of hydroxylation of myristoyl groups is catalyzed by the inner membrane dioxygenase, LpxO, which act as part of a coordinated stress response [54, 56, 70].
PhoPQ activation regulates the composition proportions of O-antigen repeats through the action of the PmrA-regulated inner membrane protein complex Wzzst and WzzfepE [71,72,73]. Wzzst selectively determines the formation of ‘long’ (L) O-chain lengths around 16–35 repeat units, while WzzfepE is responsible for the ‘very long’ (VL) lengths with over than one hundred subunits. Activation of pmrA increased the fraction of such L-type and VL-type O-antigen in LPS molecules [71,72,73]. Raising the proportions of L-type and VL-type O-antigen leads to heightened resistance to serum [71]; moreover, VL-type enhanced two-fold increase in polymyxin B resistance [73]. Thus, it is plausible that such types of O-antigen somehow promote barrier function and contribute to polymyxins resistance [54].
Three outer membrane enzymes PagP (or CrcA), PagL and LpxR with active-site exposed to the outer leaflet of OM are related to acylation or deacylation in barrier remodelling [54, 56]. In response to PhoPQ activation, the transcription of pagP is stimulated and thus upregulated the encoded proteins that modify lipid A with palmitate [74]. Palmitoyl group transferring from phospholipid (GPL) donors to lipid A occurs on the extracellular active-site of PagP and results in hepta-acylated lipid A species [54, 56, 64, 74]. The other two outer membrane enzymes, PagL and LpxR, acting as deacylation of lipid A are subjected to post-translational inhibition by L-Ara4N modified lipid A as their active sites are found on the extracellular surface and in close proximity to lipid A [56, 64]. Increased palmitoylation and inhibited deacylation enhance the hydrophobicity of the OM and prevent penetration of the amphipathic polymyxin molecules [54, 56].
4.5 Regulation of Glycerophospholipids (GPL) Remodelling
Recent research indicates that glycerophospholipids (GPL) are also regulated components of the OM barrier in Gram-negative bacteria [55, 75]. Penetration of polymyxin molecules can cause LPS layer to become displaced and shed from the outer leaflet and activated the PhoPQ system [59, 68, 75]. To maintain bilayer barrier integrity, GPL from the inner leaflet migrate into the outer leaflet of the OM to replace the breached areas of LPS with GPL as a consequence of locally weakened barrier which are only detectable in stressed cells [54, 55, 75, 76]. The outer membrane protein PagP with dual substrate specificity activated by the PhoPQ system can function as a membrane-intrinsic probe to restoration of the permeability barrier [55, 64]. In addition to lipid A , PagP transfers palmitoyl groups from GPL to the polar head group of phosphatidylglycerol (PG) that have flipped onto the surface of OM forming palmitoyl-PG [54, 55]. Therefore, once LPS layer disrupted and PhoPQ system activated, GPL may be increasingly translocated to the outer leaflet and further acylated by pagP to enhance barrier hydrophobicity [54, 55, 64]. Also, the research work detected modest yet significantly increases in cardiolipin (CL) amount of the OM on activation of PhoPQ, which was speculated to form functional micro-domains that promote OM lipid re-modelling [54, 55]. The phospholipid (PL) bilayer structure of OM is more permeable than the asymmetrical LPS-PL bilayer barrier, so the OM phospholipase A (OMPLA) and inter-membrane transport system Mla pathway are functioned to prevent surface exposure of PLs and maintain lipid asymmetry of the OM if necessary [75, 77]. Thus, coordinate regulation of LPS and GPL forms a remodelled OM barrier critical for bacterial protective responses and survival to polymyxins.
4.6 Other Responses to Polymyxin Treatment in Gram-negative Bacteria
In Salmonella, antimicrobial peptides and cations occupy the overlapping binding site of the sensor PhoQ [59]. Divalent cations are bound to the acidic surface region of PhoQ sensor under normal conditions, while the displacement of these cations by antimicrobial peptides results in a conformational change that activates PhoQ and triggers the hierarchical regulation [59]. To date, various two-component systems have been reported to associate with the adaption to sub-inhibitory concentrations of antimicrobial peptides in Pseudomonas aeruginosa , including the widespread PhoPQ and PmrAB systems, and the ParRS, ColRS and CprRS systems [52, 58, 78, 79]. In contrast to Salmonella, P. aeruginosa senses divalent cations and cationic peptides via different mechanisms. Divalent cations are detected by PhoQ and PmrB but not peptides [58, 79]. Whereas ParS and CprS can detect cationic antimicrobial peptides regardless of Mg2+ concentrations which are independent two-component systems that might recognize different properties of peptides or the different effects of peptides on cell at specific concentrations. As for polymyxins, both participated at all concentrations, with a greater involvement of ParRS which is likely to be the key component [78, 79]. The occurrence of at least two direct polymyxins response systems and three associated response regulatory systems in P. aeruginosa highlights the complexity of the adaptive resistant pathways in this organism.
Several tripartite efflux systems play considerable roles in the intrinsic and acquired resistance in P. aeruginosa. Each system consists of three proteins with presumed functions: a cytoplasmic membrane component of the resistance-nodulation-division family acting as a transporter, an outer membrane component forming channels, and a membrane fusion protein linking the two membranes [80, 81]. At the gene level, a constitutively expressed operon, mexAB-oprM, coding for an efflux system (MexAB-OprM) which contributes intrinsic resistance in P. aeruginosa produced at a basal level in wild-type bacteria [80]; however, it has been observed that the MexAB-OprM efflux system is overexpressed in the metabolically active subpopulations of P. aeruginosa biofilm , conferring an unspecific adaptive resistant phenotype to polymyxins [52, 82]. Another outer membrane efflux pump system MexXY lacking OM protein in its own operon utilises OprM of the MexAB-OprM system and forms MexXY-OprM system that has been shown to provide natural to aminoglycosides and various unrelated antibiotics [80, 81]. It has been demonstrated that polymyxins can promote expression of the mexXY operons besides pmrAB and arnBCDTEF-ugd, and to coordinately downregulate the oprD gene that promoting β-Lactams resistance through the activation of the two-component systems ParRS [78]. These researches indicate that polymyxins are able to induce multidrug adaptive resistance of cells through the activation of distinct mechanisms (efflux, porin loss, and LPS modification) in P. aeruginosa [78, 82].
Microarray and high-throughput RNA-seq analysis revealed a global change pattern of gene expression leading to adaptive responses. When P. aeruginosa cells are exposed to sub-inhibitory concentrations of colistin, approximately 0.5% of 5500 genes showed significantly changed expression levels in the colistin-treated sample. Among them, 13 were upregulated and 17 were downregulated. The upregulated genes are involved in quorum sensing (QS) and biofilm formation besides well-known LPS modification, while the downregulated genes are involved in motility (swarming and swimming motility) and osmotolerance [83]. Upon exposure to a much higher concentration of polymyxin (10 × MIC) treatment, a wider profile of global changes was identified from protective responses of Yersinia pestis to survive the stressful environments. A total of 291 genes were differentially expressed and 158 of them were induced. Among the 158 upregulated genes, 22 were regulatory genes including 8 two-component systems that globally or locally governing a wide set of stress-protective functions, 19 genes were involved in remodelling of cell envelope encoding membrane components or polysaccharide surface structures, 4 operons including 9 genes were essential for dissimilation of sn-glycerol 3-phosphate which is a direct precursor for phospholipid biosynthesis [84], 10 were heat shock proteins that play important roles in preventing aggregation of proteins and repairing misfolded or damaged proteins caused by environmental stresses, 5 were related to drug resistance (3 of tellurium resistance and 2 of multidrug transport system), 11 genes organized into four operons were components of siderophore-based iron acquisition systems which known as high-pathogenicity island shared by three pathogenic yersiniae [85]. Our recent high-throughput RNA-seq study of the transcriptomic response of Acinetobacter baumannii to colistin under conditions that able to kill partial cells revealed hundreds of genes differentially expressed including those of two-component systems, glycerophospholipid metabolism, lipopolysaccharide biosynthesis, biofilm synthesis, drug resistant proteins, heat shock proteins as discovered from other Gram-negative strains (data unpublished). Moreover, genes involved in nucleotide excision repair and peroxisome were also significantly induced by killing concentration of colistin. Two genes encoding the peroxisome superoxide dismutase SOD1 and catalase KatE (belonging to the antioxidant system) were upregulated after colistin treatment.
4.7 Mutations and Death
Recently, it has been demonstrated that a number of bactericidal antibiotic classes trigger the endogenous production of lethal active forms of hydroxyl radicals in bacteria through the Fenton reaction [46, 47, 51, 86] which depends on the availability of hydrogen peroxide, an iron species and reducing equivalents [86,87,88]. Hydrogen peroxide is generated within cells as a by-product of oxidative metabolism when molecular oxygen accidentally acquires electrons from the reduced cofactors of flavoproteins [50, 88] and bactericidal antibiotics elevated the generation of deleterious reactive species [48]. Iron is necessary for bacteria to survive and acquired from environment by biosynthetic iron chelators known as siderophores [86]. Hydrogen peroxide is capable of interacting with intracellular ferrous form iron unincorporated or associated with biological molecules including iron-sulphur-dependent dehydratases and mononuclear iron proteins, oxidizing the iron and forming hydroxyl radicals in the process [50, 86]. This is a cyclical process in vivo since intracellular reductants can reduce the oxidized iron back to ferrous form [86]. The event of iron disintegration from proteins eliminates a variety of enzyme activity and the hydroxyl radical is an extremely powerful oxidant that reacts with virtually all organic molecules including giving a wide variety of DNA damage [50]. Ultimately, the oxidative damage of hydroxyl radical to DNA, lipids, and proteins eventually reaches levels that cannot be controlled and thus contribute to cell death [47, 48].
It has been demonstrated that the rapid killing of Gram-negative species A. baumannii, Escherichia coli and Francisella novicida by polymyxins is in part mediated by a hydroxyl radical death pathway [46]. In addition, this mechanism of killing occurs in polymyxin-susceptible A. baumannii isolates including multidrug-resistant clinical isolates but this response is not induced in a polymyxin-resistant isolate [46]. The mechanism by which polymyxin treatment induces the production of hydroxyl radicals in Gram-negative bacteria is not clear yet. Polymyxin-induced hydroxyl radical death does not occur in polymyxin-resistance isolates of A. baumannii but in susceptible isolates [46], and polymyxin resistance in A. baumannii is commonly due to blocking the entries through remodelling of the outer membrane [67, 68, 89]. Therefore, it seems that the event of the hydroxyl radical death pathway induced by polymyxins happened after the initial drug-target interactions on the outer membrane. The secondary MOA of polymyxins that they can inhibit NDH-2 activity in the inner membranes of detected Gram-negative species [90] may result in accumulation of nicotinamide adenine dinucleotide (NADH) which can be utilized as a source of reducing equivalents that contribute to Fenton reaction (Fig. 4.3b, bottom left part) [88]. In vitro the NADH-Fe (III)-EDTA-H2O2 system drives an ongoing Fenton reaction and DNA ensue breaks in such system while NAD+ is ineffective. Furthermore, the rate of DNA nicking corresponds to the rate of NADH oxidation [88].
Exposure of Gram-negative species to low concentrations of hydrogen peroxide resulted in DNA damage that causes mutagenesis and kills the cell [87, 88]. It has been proposed that antibiotic-induced ROS may provide a mechanism of acquiring beneficial mutations when stresses are small but induce lethality when stresses are large [48]. A recent study revealed that treatments with sub-lethal levels of bactericidal antibiotics resulting in up to 10 times increases in the mutation rate relative to an untreated control, a strong correlation between ROS (reactive oxygen species) formation and fold change in mutation rate, and heterogeneous increases in the minimum inhibitory concentration for a range of antibiotics irrespective of the drug target [49]. Colistin was bactericidal in a concentration-dependent manner [91]. Although colistin resistance is rare, colistin-resistant A. baumannii, P. aeruginosa and K. pneumoniae isolates have been reported world-wide. The emergence of colistin resistance or heteroresistance after colistin treatment can be easily selected in vitro and in vivo with mutations of key genes for protective responses such as the initial two-component regulatory systems [91,92,93]. However, a recent study of colistin mutant prevention concentrations (MPCs) for A. baumannii, P. aeruginosa and K. pneumoniae were shown to be very high (>64 μg/mL) [93], which recommended polymyxin combination therapy to prevent the emergency of resistant mutants and the risk of toxicity at high concentrations [93].
Accordingly, bacterial cells contain scavenging enzymes to prevent the accumulation of reactive oxygen species. Two well-known antioxidant system enzymes, catalase and SOD, can transform active radicals into oxygen and water [72, 73]. Our unpublished work of transcriptomic response of A. baumannii to colistin supported such protective response as 6 genes encoding peroxidases (including catalase and SOD) were significantly unregulated. Moreover, nucleotide excision repair genes for DNA damage repairing in our unpublished data, heat shock proteins [85] for repairing mis-folded or damaged proteins, iron acquisition systems [85] for recovering the function of iron disintegrated proteins were up-regulated significantly which might partially arise from the response to hydroxyl radical damage.
In summary, polymyxins are capable of inducing cell damage and death by interfering with their primary targets which trigger stress responses that induce outer membrane remodelling preventing polymyxins from entering. While downstream secondary target-polymyxin interactions might trigger redox-related physiological alterations that result in the formation of toxic reactive oxygen species as well as stimulating repairing responses which further contribute to cellular damage, mutations and death [48]. The formidable challenge of sub-lethal polymyxin treatment leading to multidrug resistance rather than completely killing the bacteria required us to expand our understanding of the polymyxin stress responses in Gram-negative bacteria on a detailed system-wide level. Such understanding helps in providing a foundation for finding key protective responses molecules. Those molecules can be utilised as therapy targets in combination with polymyxin-treatments to improve current therapeutic options and avoid resistant mutations.
4.8 Imaging Polymyxin Penetration and Localization in the Gram-negative Bacterial Outer Membrane
The unavailability of valid imaging probes with native activities is a significant barrier to examine the intra-cellular localization of polymyxins in Gram-negative bacterial cells [94]. Most reports of polymyxin probes either employed inactive nonapeptide derivatives or dansylated polymyxin B, which was derived by non-specifically reacting polymyxin B with dansyl-chloride [95,96,97,98]. We have previously highlighted the deficiencies of directly amine-coupling dansyl groups onto the Dab side chains in semi-synthetic preparations of dansyl-polymyxin B [99]. Analysis of these semi-synthetic dansyl-polymyxin B preparations revealed the existence of mono-, di-, tri-, and tetra-dansyl substituted species [99]. Furthermore, as polymyxin B is comprised of two major components (B1 and B2), the potential for either of these components to be substituted at any of the five Dab side chains with up to four dansyl molecules results in a highly variable mixture of dansylated derivatives [99]. Commercial preparations of 4-bora-3a,4a-diaza-s-indacene (BODIPY) labelled polymyxin B displayed a markedly reduced antibacterial activities compared to polymyxin B [100]. Moreover, our group previously reported a fully synthetic [dansyl-Lys]1polymyxin B3 probe that was devoid of antibacterial activity [99]. These findings are consistent with our understanding of polymyxin SAR wherein the dansyl modification of the Dab side chains inactivates antibacterial activity [99]. Clearly, there is very little value in using these semi-synthetic preparations as imaging probes since they lack native antibacterial activityand pharmacological properties of the parent compound, polymyxin B.
We recently reported the regio-selective modification of the polymyxin B core scaffold at the N-terminus with the dansyl fluorophore to generate an active probe (probe (1)) that mimics polymyxin B pharmacologically (Fig. 4.4, bottom left panel) [38]. The design and synthesis of a dansyl molecular probe through the regio-selective modification of the polymyxin B core structure was undertaken with the aforementioned lipid A interaction principles in mind; in order to mimic the polymyxin B structure as closely as possible and to maintain its native antibacterial activity. The dansyl group was utilized for the fluorescent probe as it has suitable spectral properties and its relative small size would reduce the chance of steric effects. The regio-selective incorporation of the dansyl group into the hydrophobic N-terminal centre of the polymyxin B core scaffold is prudent as it has a minimal impact on the native antibacterial activity of the polymyxin B scaffold. Therefore, the strategy we employed was to replace the N-terminal fatty acyl group of polymyxin B with the amino acid L-octylglycine, where the eight-carbon fatty acyl chain emulated the N-terminal fatty acyl chain of polymyxin B, whilst the N α-amino group would provide a convenient point of attachment for the dansyl group (eliminating the need for additional orthogonal protection during the synthesis).

Bottom left panel. Chemical structure of the probe (1). Top left panel. NMR-based model of the probe (1)-Kdo2 lipid A complex. Right panel. Laser scanning confocal microscopy image of K. pneumoniae ATCC 13883 cells treated with probe (1)
The antimicrobial activity of probe (1) was screened against a panel of ATCC and recent clinical isolates of polymyxin-susceptible and -resistant strains of P. aeruginosa, A. baumannii and K. pneumoniae. The probe showed antibacterial activity against polymyxin-susceptible strains (MIC 4–16 mg/L), compared to colistin (MIC 0.125–2 mg/L) and polymyxin B (MIC <0.125–2 mg/L). Probe (1) also displayed activity against polymyxin-resistant P. aeruginosa and A. baumannii strains (MIC 4–8 mg/L); colistin and polymyxin B (MIC >32 mg/L). It is understood that Gram-negative pathogens resist the action of polymyxins by introducing cationic modifications onto the phosphate groups on the lipid A component of LPS [101,102,103,104,105,106]. The most common mechanism involves esterification of lipid A phosphates with aminoarabinose, or ethanolamine [101,102,103,104,105,106]. The molecular tailoring serves to reduce the net negative charge of the outer membrane surface, thereby repelling the electrostatic attraction with positively charged polymyxin molecules [107]. The NMR-based molecular model of the probe (1)-Kdo2 Lipid A complex implies that the combination of the L-octylglycine and the dansyl substituent at the N-terminus provides additional hydrophobic interactive forces that compensate for the electrostatic repulsion of the aminoarabinose phosphate modifications (Fig. 4.4, top left panel). The molecular model indicates that electrostatic interactions with the 1-phosphorester group on lipid A are not hampered by the dansyl group. The model further suggests that the hydrophobic dansyl group interacts with the apolar environment formed by the fatty acyl chains of lipid A. TEM imaging of K. pneumoniae ATCC 13883 cells treated with probe (1) at 0.5 × MIC revealed the formation of numerous protrusions or blebs extending from the outer membrane of the cells that possibly represent outer membrane fragments. A similar blebbing effect was observed with Gram-negative bacterial cells treated with polymyxin B and colistin [108].
Time-lapse laser scanning confocal microscopy imaging of the penetration of probe (1) into K. pneumoniae ATCC 13883 cells revealed that the probe initially accumulates in the outer membrane and subsequently penetrates into the inner membrane and finally becomes homogenously distributed into the cytoplasm (Fig. 4.4, right panel). Intriguingly, confocal imaging and spectrophotometric lysis assay experiments with spheroplasts isolated from K. pneumoniae ATCC 13883 revealed that probe (1) also accumulated within and disrupted the inner membrane structure. Coincidently, our group has recently show that polymyxin B and colistin inhibit the NDH-2 oxidoreductase inner membrane respiratory enzyme, which also may contribute towards their bactericidal effect (cf. preceding discussions; Fig. 4.1) [19]. Furthermore, the imaging experiments revealed that at sub-MIC concentrations, probe (1) tends to accumulate on the surface of the bacterial cell and partly penetrates into the outer membrane. Whereas at <MIC concentrations, probe (1) accumulated on the surface of the bacterial cell and entered into the cytoplasm. Fundamentally, the localization studies from the time-lapse laser scanning confocal microscopy imaging results helps validate the mechanistic model of polymyxin action (Fig. 4.1). Based on the imaging data, it is evident that polymyxins initially accumulate in the outer membrane, followed by a gradual penetration into the inner membrane and finally enter the cytoplasm in K. pneumoniae. These findings are consistent with the secondary mode of action of polymyxin B which involves an inhibitory activity against the inner membrane NDH-2 enzyme [19]. It will be most interesting to elucidate the intracellular bacterial cell target(s) for the polymyxins, which to date remains uncharacterized.
The commercial availability of this probe would greatly facilitate molecular imaging studies on both the mode of action and pharmacokinetics ; and contribute towards the development of a new generation of polymyxin lipopeptides with superior activity against polymyxin-resistant Gram-negative ‘superbugs’.
In summary, polymyxins are bioactive natural products with beneficial pharmacological activities and their antimicrobial properties have been investigated for decades. Albeit, their precise mode of action remains unknown and much progress has been made towards understanding their bacterial killing effect. The compendium of data has highlighted that their bacterial killing mechanism is more complex than simply the long-standing notion that they impart a membrane disorganising effect. Secondary, pathways such as the bacterial redox chain have been implicated in their killing effect. Clearly, much work needs to be done to comprehensively elucidate the precise mode-of-action of these valuable lipopeptide antibiotics.
References
Nikaido H (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67(4):593–656
Rietschel ET, Brade H (1987) Lipopolysaccharides, endotoxins and O-antigens of gram-negative bacteria: chemical structure, biologic effect and serologic properties. Infection 15(2):133–141
Caroff M, Karibian D (2003) Structure of bacterial lipopolysaccharides. Carbohydr Res 338(23):2431–2447
Rietschel ET, Brade H, Brade L, Brandenburg K, Schade U, Seydel U et al (1987) Lipid A, the endotoxic center of bacterial lipopolysaccharides: relation of chemical structure to biological activity. Prog Clin Biol Res 231:25–53
Hancock RE (1997) The bacterial outer membrane as a drug barrier. Trends Microbiol 5(1):37–42
Hancock RE (1997) Antibacterial peptides and the outer membranes of gram-negative bacilli. J Med Microbiol 46(1):1–3
Pristovsek P, Kidric J (2004) The search for molecular determinants of LPS inhibition by proteins and peptides. Curr Top Med Chem 4(11):1185–1201
Pristovsek P, Kidric J (1999) Solution structure of polymyxins B and E and effect of binding to lipopolysaccharide: an NMR and molecular modeling study. J Med Chem 42(22):4604–4613
Hancock RE (1997) Peptide antibiotics. Lancet 349(9049):418–422
Hancock RE, Lehrer R (1998) Cationic peptides: a new source of antibiotics. Trends Biotechnol 16(2):82–88
Velkov T, Thompson PE, Nation RL, Li J Structure--activity relationships of polymyxin antibiotics. J Med Chem 53(5):1898–1916
Ofek I, Cohen S, Rahmani R, Kabha K, Tamarkin D, Herzig Y et al (1994) Antibacterial synergism of polymyxin B nonapeptide and hydrophobic antibiotics in experimental gram-negative infections in mice. Antimicrob Agents Chemother 38(2):374–377
Clausell A, Garcia-Subirats M, Pujol M, Busquets MA, Rabanal F, Cajal Y (2007) Gram-negative outer and inner membrane models: insertion of cyclic cationic lipopeptides. J Phys Chem B 111(3):551–563
Powers JP, Hancock RE (2003) The relationship between peptide structure and antibacterial activity. Peptides 24(11):1681–1691
Cajal Y, Ghanta J, Easwaran K, Surolia A, Jain MK (1996) Specificity for the exchange of phospholipids through polymyxin B mediated intermembrane molecular contacts. Biochemistry 35(18):5684–5695
Cajal Y, Rogers J, Berg OG, Jain MK (1996) Intermembrane molecular contacts by polymyxin B mediate exchange of phospholipids. Biochemistry 35(1):299–308
Cajal Y, Berg OG, Jain MK (1995) Direct vesicle-vesicle exchange of phospholipids mediated by polymyxin B. Biochem Biophys Res Commun 210(3):746–752
Mogi T, Murase Y, Mori M, Shiomi K, Omura S, Paranagama MP et al (2009) Polymyxin B identified as an inhibitor of alternative NADH dehydrogenase and malate: quinone oxidoreductase from the gram-positive bacterium Mycobacterium smegmatis. J Biochem 146(4):491–499
Deris ZZ, Akter J, Sivanesan S, Roberts KD, Thompson PE, Nation RL et al (2013) A secondary mode of action of polymyxins against gram-negative bacteria involves the inhibition of NADH-quinone oxidoreductase activity. J Antibiot. 67(2):147–151
Hancock REW (1997) Peptide antibiotics. Lancet 349(9049):418–422
Hancock REW, Scott MG (2000) The role of antimicrobial peptides in animal defenses. Proc Natl Acad Sci U S A 97(16):8856–8861
Cho J, Kim S (2010) Non-membrane targets of antimicrobial peptides: novel therapeutic opportunities? Advances in Molecular and Cellular Microbiology No 18. CABI, Wallingford, pp 128–140
Otvos L (2005) Antibacterial peptides and proteins with multiple cellular targets. J Pept Sci 11(11):697–706
Storm DR, Rosenthal KS, Swanson PE (1977) Polymyxin and related peptide antibiotics. Annu Rev Biochem 46(1):723–763
Teuber M (1974) Action of polymyxin B on bacterial membranes. Arch Microbiol 100(1):131–144
Boshoff HI, Barry CE 3rd (2005) Tuberculosis – metabolism and respiration in the absence of growth. Nat Rev Microbiol 3(1):70–80
Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE 3rd (2004) The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem 279(38):40174–40184
Weinstein EA, Yano T, Li LS, Avarbock D, Avarbock A, Helm D et al (2005) Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc Natl Acad Sci U S A 102(12):4548–4553
Fisher N, Warman AJ, Ward SA, Biagini GA (2009) Chapter 17 Type II NADH: quinone oxidoreductases of Plasmodium falciparum and Mycobacterium tuberculosis kinetic and high-throughput assays. Methods Enzymol 456:303–320
Biagini GA, Viriyavejakul P, O’Neill PM, Bray PG, Ward SA (2006) Functional characterization and target validation of alternative complex I of Plasmodium falciparum mitochondria. Antimicrob Agents Chemother 50(5):1841–1851
Fisher N, Bray PG, Ward SA, Biagini GA (2007) The malaria parasite type II NADH:quinone oxidoreductase: an alternative enzyme for an alternative lifestyle. Trends Parasitol 23(7):305–310
Kim MS, Kim YJ (2004) Enzymatic properties of the membrane bound NADH oxidase system in the aerobic respiratory chain of Bacillus cereus. J Biochem Mol Biol 37(6):753–756
Kerscher S, Dröse S, Zickermann V, Brandt U (2008) The three families of respiratory NADH dehydrogenases. In: Schäfer G, Penefsky H (eds) Bioenergetics. Results and problems in cell differentiation, vol 45. Springer, Berlin/Heidelberg, pp 185–222
Yagi T, Yano T, Di Bernardo S, Matsuno-Yagi A (1998) Procaryotic complex I (NDH-1), an overview. Biochim Biophys Acta 1364(2):125–133
Yagi T (1991) Bacterial NADH-quinone oxidoreductases. J Bioenerg Biomembr 23(2):211–225
Yagi T, Matsuno-Yagi A (2003) The proton-translocating NADH-quinone oxidoreductase in the respiratory chain: the secret unlocked. Biochemistry 42(8):2266–2274
Mogi T, Murase Y, Mori M, Shiomi K, Ōmura S, Paranagama MP et al (2009) Polymyxin B identified as an inhibitor of alternative NADH dehydrogenase and malate: quinone oxidoreductase from the gram-positive bacterium Mycobacterium smegmatis. J Biochem 146(4):491–499
Deris ZZ, Swarbrick JD, Roberts KD, Azad MA, Akter J, Horne AS et al (2014) Probing the penetration of antimicrobial polymyxin lipopeptides into gram-negative bacteria. Bioconjug Chem 25(4):750–760
Velkov T, Deris ZZ, Huang JX, Azad MA, Butler M, Sivanesan S et al (2013) Surface changes and polymyxin interactions with a resistant strain of Klebsiella pneumoniae. Innate Immun. 20(4):350–363. https://doi.org/10.1177/1753425913493337
Velkov T, Thompson PE, Nation RL, Li J (2010) Structure−activity relationships of polymyxin antibiotics. J Med Chem 53(5):1898–1916
Melo AM, Bandeiras TM, Teixeira M (2004) New insights into type II NAD(P)H:quinone oxidoreductases. Microbiol Mol Biol Rev 68(4):603–616
Dong CK, Patel V, Yang JC, Dvorin JD, Duraisingh MT, Clardy J et al (2009) Type II NADH dehydrogenase of the respiratory chain of Plasmodium falciparum and its inhibitors. Bioorg Med Chem Lett 19(3):972–975
Koul A, Arnoult E, Lounis N, Guillemont J, Andries K (2011) The challenge of new drug discovery for tuberculosis. Nature 469(7331):483–490
Fisher N, Bray PG, Ward SA, Biagini GA (2008) Malaria-parasite mitochondrial dehydrogenases as drug targets: too early to write the obituary. Trends Parasitol 24(1):9–10
Biagini GA, Fisher N, Shone AE, Mubaraki MA, Srivastava A, Hill A et al (2012) Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc Natl Acad Sci 109:8298–8303
Sampson TR, Liu X, Schroeder MR, Kraft CS, Burd EM, Weiss DS (2012) Rapid killing of Acinetobacter baumannii by polymyxins is mediated by a hydroxyl radical death pathway. Antimicrob Agents Chemother 56(11):5642–5649
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130(5):797–810
Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N et al (2014) Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci U S A 111(20):E2100–E2109
Kohanski MA, DePristo MA, Collins JJ (2010) Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell 37(3):311–320
Imlay JA (2013) The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11(7):443–454
Wright GD, Hung DT, Helmann JD (2013) How antibiotics kill bacteria: new models needed? Nat Med 19(5):544–545
Skiada A, Markogiannakis A, Plachouras D, Daikos GL (2011) Adaptive resistance to cationic compounds in Pseudomonas aeruginosa. Int J Antimicrob Agents 37(3):187–193
Poole K (2012) Stress responses as determinants of antimicrobial resistance in gram-negative bacteria. Trends Microbiol 20(5):227–234
Dalebroux ZD, Miller SI (2014) Salmonellae PhoPQ regulation of the outer membrane to resist innate immunity. Curr Opin Microbiol 17:106–113
Dalebroux ZD, Matamouros S, Whittington D, Bishop RE, Miller SI (2014) PhoPQ regulates acidic glycerophospholipid content of the Salmonella Typhimurium outer membrane. Proc Natl Acad Sci U S A 111(5):1963–1968
Needham BD, Trent MS (2013) Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol 11(7):467–481
Poole K (2012) Bacterial stress responses as determinants of antimicrobial resistance. J Antimicrob Chemother 67(9):2069–2089
Fernandez L, Gooderham WJ, Bains M, McPhee JB, Wiegand I, Hancock RE (2010) Adaptive resistance to the “last hope” antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob Agents Chemother 54(8):3372–3382
Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W et al (2005) Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell 122(3):461–472
Gunn JS, Miller SI (1996) PhoP-PhoQ activates transcription of pmrAB, encoding a two-component regulatory system involved in Salmonella typhimurium antimicrobial peptide resistance. J Bacteriol 178(23):6857–6864
Luo SC, Lou YC, Rajasekaran M, Chang YW, Hsiao CD, Chen C (2013) Structural basis of a physical blockage mechanism for the interaction of response regulator PmrA with connector protein PmrD from Klebsiella pneumoniae. J Biol Chem 288(35):25551–25561
Kato A, Groisman EA (2004) Connecting two-component regulatory systems by a protein that protects a response regulator from dephosphorylation by its cognate sensor. Genes Dev 18(18):2302–2313
Kato A, Chen HD, Latifi T, Groisman EA (2012) Reciprocal control between a bacterium’s regulatory system and the modification status of its lipopolysaccharide. Mol Cell 47(6):897–908
Raetz CR, Reynolds CM, Trent MS, Bishop RE (2007) Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem 76:295–329
Okuda S, Freinkman E, Kahne D (2012) Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science 338(6111):1214–1217
Tamayo R, Choudhury B, Septer A, Merighi M, Carlson R, Gunn JS (2005) Identification of cptA, a PmrA-regulated locus required for phosphoethanolamine modification of the Salmonella enterica serovar typhimurium lipopolysaccharide core. J Bacteriol 187(10):3391–3399
Beceiro A, Llobet E, Aranda J, Bengoechea JA, Doumith M, Hornsey M et al (2011) Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrob Agents Chemother 55(7):3370–3379
Velkov T, Roberts KD, Nation RL, Thompson PE, Li J (2013) Pharmacology of polymyxins: new insights into an ‘old’ class of antibiotics. Future Microbiol 8(6):711–724
Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, Hackett M et al (1997) Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP-phoQ. Science 276(5310):250–253
Gibbons HS, Reynolds CM, Guan Z, Raetz CR (2008) An inner membrane dioxygenase that generates the 2-hydroxymyristate moiety of Salmonella lipid A. Biochemistry 47(9):2814–2825
Delgado MA, Mouslim C, Groisman EA (2006) The PmrA/PmrB and RcsC/YojN/RcsB systems control expression of the Salmonella O-antigen chain length determinant. Mol Microbiol 60(1):39–50
Larue K, Kimber MS, Ford R, Whitfield C (2009) Biochemical and structural analysis of bacterial O-antigen chain length regulator proteins reveals a conserved quaternary structure. J Biol Chem 284(11):7395–7403
Pescaretti Mde L, Lopez FE, Morero RD, Delgado MA (2011) The PmrA/PmrB regulatory system controls the expression of the wzzfepE gene involved in the O-antigen synthesis of Salmonella enterica serovar Typhimurium. Microbiology 157(Pt 9):2515–2521
Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CR (2000) Transfer of palmitate from phospholipids to lipid A in outer membranes of gram-negative bacteria. EMBO J 19(19):5071–5080
Malinverni JC, Silhavy TJ (2009) An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc Natl Acad Sci U S A 106(19):8009–8014
Jia W, El Zoeiby A, Petruzziello TN, Jayabalasingham B, Seyedirashti S, Bishop RE (2004) Lipid trafficking controls endotoxin acylation in outer membranes of Escherichia coli. J Biol Chem 279(43):44966–44975
Dekker N (2000) Outer-membrane phospholipase A: known structure, unknown biological function. Mol Microbiol 35(4):711–717
Muller C, Plesiat P, Jeannot K (2011) A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and beta-lactams in Pseudomonas aeruginosa. Antimicrob Agents Chemother 55(3):1211–1221
Fernandez L, Jenssen H, Bains M, Wiegand I, Gooderham WJ, Hancock RE (2012) The two-component system CprRS senses cationic peptides and triggers adaptive resistance in Pseudomonas aeruginosa independently of ParRS. Antimicrob Agents Chemother 56(12):6212–6222
Li XZ, Nikaido H (2004) Efflux-mediated drug resistance in bacteria. Drugs 64(2):159–204
Masuda N, Sakagawa E, Ohya S, Gotoh N, Tsujimoto H, Nishino T (2000) Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob Agents Chemother 44(12):3322–3327
Pamp SJ, Gjermansen M, Johansen HK, Tolker-Nielsen T (2008) Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol Microbiol 68(1):223–240
Cummins J, Reen FJ, Baysse C, Mooij MJ, O’Gara F (2009) Subinhibitory concentrations of the cationic antimicrobial peptide colistin induce the pseudomonas quinolone signal in Pseudomonas aeruginosa. Microbiology 155.(Pt 9:2826–2837
Yang B, Larson TJ (1996) Action at a distance for negative control of transcription of the glpD gene encoding sn-glycerol 3-phosphate dehydrogenase of Escherichia coli K-12. J Bacteriol 178(24):7090–7098
Zhou D, Han Y, Qiu J, Qin L, Guo Z, Wang X et al (2006) Genome-wide transcriptional response of Yersinia pestis to stressful conditions simulating phagolysosomal environments. Microbes Infect/Inst Pasteur 8(12–13):2669–2678
Yeom J, Imlay JA, Park W (2010) Iron homeostasis affects antibiotic-mediated cell death in Pseudomonas species. J Biol Chem 285(29):22689–22695
Imlay JA, Chin SM, Linn S (1988) Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240(4852):640–642
Imlay JA, Linn S (1988) DNA damage and oxygen radical toxicity. Science 240(4857):1302–1309
Pelletier MR, Casella LG, Jones JW, Adams MD, Zurawski DV, Hazlett KR et al (2013) Unique structural modifications are present in the lipopolysaccharide from colistin-resistant strains of Acinetobacter baumannii. Antimicrob Agents Chemother 57(10):4831–4840
Deris ZZ, Akter J, Sivanesan S, Roberts KD, Thompson PE, Nation RL et al (2014) A secondary mode of action of polymyxins against gram-negative bacteria involves the inhibition of NADH-quinone oxidoreductase activity. J Antibiot 67(2):147–151
Li J, Rayner CR, Nation RL, Owen RJ, Tan KE, Spelman D (2006) Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 50(9):2946–2950
Hawley JS, Murray CK, Jorgensen JH (2008) Colistin heteroresistance in acinetobacter and its association with previous colistin therapy. Antimicrob Agents Chemother 52(1):351–352
Choi MJ, Ko KS (2014) Mutant prevention concentrations of colistin for Acinetobacter baumannii, Pseudomonas aeruginosa and Klebsiella pneumoniae clinical isolates. J Antimicrob Chemother 69(1):275–277
Velkov T, Thompson PE, Nation RL, Li J (2010) Structure-activity relationships of polymyxin antibiotics. J Med Chem 53(5):1898–1916
Moore RA, Bates NC, Hancock RE (1986) Interaction of polycationic antibiotics with Pseudomonas aeruginosa lipopolysaccharide and lipid A studied by using dansyl-polymyxin. Antimicrob Agents Chemother 29(3):496–500
Tsubery H, Ofek I, Cohen S, Eisenstein M, Fridkin M (2002) Modulation of the hydrophobic domain of polymyxin B nonapeptide: effect on outer-membrane permeabilization and lipopolysaccharide neutralization. Mol Pharmacol 62(5):1036–1042
Tsubery H, Ofek I, Cohen S, Fridkin M (2000) Structure-function studies of polymyxin B nonapeptide: implications to sensitization of gram-negative bacteria. J Med Chem 43(16):3085–3092
Tsubery H, Ofek I, Cohen S, Fridkin M (2001) N-terminal modifications of polymyxin B nonapeptide and their effect on antibacterial activity. Peptides 22(10):1675–1681
Soon RL, Velkov T, Chiu F, Thompson PE, Kancharla R, Roberts K et al (2011) Design, synthesis, and evaluation of a new fluorescent probe for measuring polymyxin-lipopolysaccharide binding interactions. Anal Biochem 409(2):273–283
Benincasa M, Pacor S, Gennaro R, Scocchi M (2009) Rapid and reliable detection of antimicrobial peptide penetration into gram-negative bacteria based on fluorescence quenching. Antimicrob Agents Chemother 53(8):3501–3504
Gunn JS (2008) The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol 16(6):284–290
Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M et al (1998) PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol 27(6):1171–1182
Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI (2000) Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect Immun 68(11):6139–6146
Schurek KN, Sampaio JL, Kiffer CR, Sinto S, Mendes CM, Hancock RE (2009) Involvement of pmrAB and phoPQ in polymyxin B adaptation and inducible resistance in non-cystic fibrosis clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother 53(10):4345–4351
Shi Y, Cromie MJ, Hsu FF, Turk J, Groisman EA (2004) PhoP-regulated Salmonella resistance to the antimicrobial peptides magainin 2 and polymyxin B. Mol Microbiol 53(1):229–241
Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CR (2001) An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J Biol Chem 276(46):43122–43131
Soon RL, Nation RL, Cockram S, Moffatt JH, Harper M, Adler B et al (2011) Different surface charge of colistin-susceptible and -resistant Acinetobacter baumannii cells measured with zeta potential as a function of growth phase and colistin treatment. J Antimicrob Chemother 66(1):126–133
Koike M, Iida K, Matsuo T (1969) Electron microscopic studies on mode of action of polymyxin. J Bacteriol 97(1):448–452
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Li, Z., Velkov, T. (2019). Polymyxins: Mode of Action. In: Li, J., Nation, R., Kaye, K. (eds) Polymyxin Antibiotics: From Laboratory Bench to Bedside. Advances in Experimental Medicine and Biology, vol 1145. Springer, Cham. https://doi.org/10.1007/978-3-030-16373-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-16373-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-16371-6
Online ISBN: 978-3-030-16373-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)