
Abstract
The calcium-sensing receptor (CaSR) is a G protein-coupled receptor that plays a key role in calcium homeostasis, by sensing free calcium levels in blood and regulating parathyroid hormone secretion in response. The CaSR is highly expressed in parathyroid gland and kidney where its role is well characterised, but also in other tissues where its function remains to be determined. The CaSR can be activated by a variety of endogenous ligands, as well as by synthetic modulators such as Cinacalcet, used in the clinic to treat secondary hyperparathyroidism in patients with chronic kidney disease. The CaSR couples to multiple G proteins, in a tissue-specific manner, activating several signalling pathways and thus regulating diverse intracellular events. The multifaceted nature of this receptor makes it a valuable therapeutic target for calciotropic and non-calciotropic diseases. It is therefore essential to understand the complexity behind the pharmacology, trafficking, and signalling characteristics of this receptor. This review provides an overview of the latest knowledge about the CaSR and discusses future hot topics in this field.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Extracellular calcium
- Parathyroid hormone
- G protein-coupled receptor
- G proteins
- Biased signalling
- Calcimimetics
- Calcilytics
- Allosteric modulators
- Orthosteric ligands
- Cellular trafficking
41.1 Introduction
Calcium (Ca2+) is a macro element representing 1.5–2% of an adult’s total body weight and is mostly found in bones and teeth. Only 1% of the body’s Ca2+ is located in cells and tissues, where it regulates numerous critical cellular responses. The extracellular calcium concentration [Ca2+]o is much higher (20,000-fold) than in the cytosol and changes in this balance trigger various signalling pathways. This gradient allows Ca2+ to act as a second messenger in intracellular signalling [1]. Many tissues are equipped with a cell-surface sensor for Ca2+ that extends the signalling properties of Ca2+ to being an extracellular first messenger also. This receptor is known as the extracellular calcium-sensing receptor (CaSR) and is a G protein-coupled receptor (GPCR).
CaSR is a member of the class C GPCRs which also includes the metabotropic glutamate (mGlu) receptors, the gamma-aminobutyric acid (GABA) receptors, the taste 1 receptors (TAS1R) and 8 orphan receptors [2]. The class C orphan receptor GPRC6 shares the highest sequence similarity with the CaSR (Fig. 41.1).

Phylogenetic tree of the class C GPCRs generated with the neighbour-joining method of the full receptor sequences; The different colors represent the receptor families based on the endogenous ligand affiliation: CaSR family: calcium-sensing receptor (green), GPR family: class C orphan receptors with unknown endogenous ligands (blue), GABA family: gamma-aminobutyric acid receptors (yellow), mGlu family: metabotropic glutamate receptors (red), TAS1R family: taste 1 receptors (orange)
The CaSR plays an essential role in calcium homeostasis and its existence was confirmed by cloning in 1993 [3]. CaSR senses changes in [Ca2+]o and also interacts with other multivalent cations (Mg2+, Gd3+), organic cations (neomycin), polyamines (spermine, polyarginine and polylysine) and possibly even beta amyloid [4]. It is highly expressed in the parathyroid glands, pancreas, duodenum and kidney and less in the digestive system, stomach and respiratory system [5]. This review will give an overview of the (patho)physiological roles, structure, ligands, trafficking, signalling pathways and tissue specific functions of the CaSR.
41.1.1 Physiological Role of the CaSR in Calcium Regulation
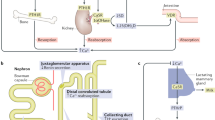
The blood [Ca2+]o is influenced by parathyroid hormone (PTH), 1α,25-dihydroxyvitamin D3 (1,25D3) and calcitonin [4, 6]. PTH is expressed by and secreted from chief cells of the parathyroid gland and acts upon the kidneys, bones, and intestines. In the kidney, CaSR stimulates Ca2+ and Mg2+ reabsorption in the distal tubules whereas in the proximal tubules it promotes the excretion of hydrogen phosphate and dihydrogen phosphate. PTH induces the 25-hydroxyvitamin D3 1α-hydroxylase to produce 1,25D3 which is essential for intestinal Ca2+ absorption [7]. Hypercalcaemia is prevented by CaSR, which inhibits PTH secretion and suppresses the transcription of PreProPTH and cell proliferation (Fig. 41.2) [8]. PTH stimulates bone remodelling. Calcitonin protects against hypercalcaemia and inhibits osteoclast activity and consequently the release of Ca2+ from bones. The transcription of calcitonin in thyroidal C-cells is inhibited by increasing 1,25D3 concentrations [6]. However, the impact of calcitonin in maintaining systemic blood Ca2+ is still contradictory because its absence or excess does not result in any significant metabolic abnormalities.

Overview of the calcium homeostasis. (a) Location of the parathyroid glands (red dots); (b) Chief cell of the parathyroid gland, 1) CaSR is inactive at low [Ca2+]o and PTH is secreted; 2) PTH stimulates Ca2+ release from bone, the reabsorption from the kidney and the 1,25D3 synthesis which induces Ca2+ uptake from the intestine (c); 3) The resulting increase in blood Ca2+ activates CaSR and at high concentrations calcitonin secretion; (d) Calcitonin inhibits the osteoclast activity and transiently the Ca2+ release from bone; 4) The active CaSR inhibits the PTH expression and secretion and consequently lowers the blood Ca2+ level; 5) Until the process starts again at 1)
The physiological range of serum [Ca2+]o is tightly regulated between 2.2 and 2.4 mM by the CaSR, facilitated by the high cooperativity of Ca2+ on the receptor. About half of the [Ca2+]o is free, and the rest of it is bound mainly to albumin. PTH secretion is induced when the free [Ca2+]o drops below 1.2 mM (to ∼2.2 mM total Ca2+) and it is effectively suppressed by CaSR activation when free [Ca2+]o rises above 1.2 mM (towards 2.5 mM) [9]. High free [Ca2+]o activates renal CaSR leading to inhibition of Ca2+ reabsorption resulting in elevated renal Ca2+ excretion [10, 11].
41.1.2 Pathophysiological Role of the CaSR
After the successful cloning of the bovine parathyroid CaSR [3], a number of diseases were identified which are caused by CaSR mutations.
Heterozygous loss-of-function mutations in CaSR are associated with familial hypocalciuric hypercalcaemia (FHH1) and homozygous CaSR mutations to neonatal severe hyperparathyroidism (NSHPT) [12]. FHH1 is characterised by disabled Ca2+ reabsorption causing hypocalciuria, moderate hypercalcaemia, hypermagnesaemia and a disabled inhibition of PTH secretion which leads to an elevated steady-state [Ca2+]o level [4, 13]. Usually this disease remains asymptomatic over one’s lifetime, but a few patients show signs of pancreatitis or chondrocalcinosis. FHH2 and FHH3 are the result of mutations in the G protein GNA11 and APS1 gene, respectively [13], these proteins acting downstream of CaSR signalling.
NSHPT is characterised by severe hypercalcaemia and very high PTH levels. The defective feedback regulation of the CaSR leads to bone demineralisation and to pathological fractures [4]. It is currently treated with bisphosphonates, dialysis, calcimimetics or by total parathyroidectomy [13, 14].
In contrast, autosomal dominant hypocalcaemia type 1 (ADH1) and type 2 (ADH2) are caused by gain-of-function mutations of the CaSR and the GNA11 gene, respectively. ADH1 results in a reduced steady-state of blood [Ca2+]o and causes low PTH levels, hypercalciuria, hypomagnesaemia, and hyperphosphataemia. Symptoms of type 1 are paraesthesia, tetany, epilepsy, severe hypocalcaemia and basal ganglia calcification which are the same for ADH2 but without hypercalciuria and hypomagnesaemia [12]. Another gain-of-function disease is connected to a renal salt-wasting form called Bartter Syndrome type-5. It is the result of unrestrained CaSR activity which leads to dysfunctional Na-K-Cl cotransporter (NKCC2)-dependent NaCl reabsorption [4].
Autoimmune diseases of the CaSR have also been described in rare cases due to the presence of anti-CaSR antibodies. These antibodies can have CaSR-stimulating or CaSR-blocking effects causing a form of acquired autoimmune hypoparathyroidism or autoimmune hypocalciuric hypercalcaemia, respectively [13].
Mutations of the CaSR are also observed in a variety of non-calciotropic diseases, for example the R990G variant is associated with an elevated risk for hypercalciuria and nephrolithiasis [15]. Other diseases are connected to changed expression levels of the receptor. In colorectal and parathyroid cancer CaSR expression is decreased or lost, attenuating its tumour preventive effect. In breast and prostate tumours, CaSR is overexpressed which correlates with an increasing risk for metastases to the bone [16]. There is also evidence that changes in CaSR activity or expression are associated with alterations in cardiac function, insulin secretion, postprandial blood glucose regulation, lipolysis and inhibition of myocardial cell proliferation. In the digestive tract, CaSR shows anti-inflammatory, anti-secretory, pro-absorbent, and obstructive properties while in the respiratory tract CaSR activation is associated with inflammation and nonspecific hyperresponsiveness in asthma [13, 17].
41.2 Structure of the CaSR
The CaSR functions as a disulphide-tethered homodimer composed of three main domains: an extracellular domain (ECD), a heptahelical transmembrane domain (TMD) and an intracellular C-terminal domain (ICD) [18] (Fig. 41.3).

Structure of the CaSR including the crystal structure of the ECD from the human CaSR, formed by LB1, LB2 and CR (PDB: 5k5s), and a schematic representation of the TMD formed by the seven transmembrane helixes followed by the ICD. Calcium ions are represented as red spheres in the ECD
The human CaSR ECD contains 612 amino acids and consists of two lobe-shaped domains (LB1 and LB2) that form the N-terminal Venus Fly Trap (VFT) domain, and the cysteine-rich (CR) domain [19]. The VFT is the ligand-binding region, reminiscent of gram-negative bacterial periplasmic binding proteins [20]. Both LB domains are formed by typical β-sheets and α-helices, where the central parallel β-sheets are sandwiched by α-helices [21]. The CR region, located between the ECD and the TMD contains nine-conserved cysteines. It transmits and amplifies signals from the VFT domain to the intracellular loops of the TMD [22]. The CR region is present in all class C GPCRs except in GABA B receptors and is required for receptor activation [21,22,23].
CaSR is expressed on the cell surface as a homodimer formed by direct interactions involving the ECD and the TMD. The ECDs of both monomers interact in a side-by-side fashion by a covalent disulphide bridge involving residues Cys-129 and Cys-131, whereas the TMDs establish hydrophobic interactions between them [24, 25]. However, there is also evidence suggesting heterodimerisation with other class C GPCRs. These heterodimers are considered new types of receptors that lead to changes in CaSR expression, signalling and sensitivity. For instance, CaSR may form dimers with mGlu1a or mGlu5, in hippocampal and cerebellar neurons, and with GABA B receptors [26, 27].
The CaSR ECD also includes 20–40 kDa of either high mannose or complex carbohydrates. These glycosylations are believed to be important for cell-surface localisation of the CaSR, intracellular trafficking, protein folding and secretion [25].
Recently, two different groups have simultaneously resolved the crystal structures of the human CaSR ECD in resting and active conformations [28, 29]. Zhang and partners crystallised the ECD in the active conformation and identified two Ca2+ binding sites plus an additional orthosteric binding site for L-Trp. The Ca2+ binding sites can also be occupied by other divalent metals such as Mg2+ whereas, the additional orthosteric binding site was occupied by a L-Trp derivate, L-1,2,3,4,-tetrahydronorharman-3-carboxylic acid and it was located in the hinge region between the two subdomains. The L-Trp binding site was described as crucial for receptor activation and stabilisation of the active conformation [29]. Meanwhile, Geng and colleagues crystallised the receptor in its active and inactive conformations. The active structure was obtained in the presence of 10 mM Ca2+ and 10 mM L-Trp, when the receptor is in its closed conformation (active state, closed-closed) (Fig. 41.4 left). They also identified the same orthosteric binding site described by Zhang and partners, located in the ligand-binding cleft of each protomer and also occupied by L-Trp. In this model, the authors defined four different Ca2+-binding sites in the active structure, including one Ca2+-binding site in each protomer that is common in the active and inactive structures suggesting an integral part of the receptor. On the other hand, the inactive CaSR ECD structure was obtained in the presence and absence of 2 mM Ca2+, when the receptor is in open conformation (inactive state, open-open) and the interdomain cleft is empty (Fig. 41.4 right). In this model, they also defined three anion-binding sites. The authors proposed a CaSR activation model where L-Trp facilitates the CaSR-ECD closure by contacting LB1 and LB2 domains of the VFT module to bring the CR domains closer together. These interactions form a large homodimer interface that is unique for the active state, reduce the distance between the C-terminal tails and might cause a rearrangement of the TMD [28] (Fig. 41.4).

Crystal structure of the human CaSR ECD in its active (left) and inactive (right) conformations. In the active conformation the VFT is closed and LB1 and LB2 interact to bring the CR domains closer together. In the inactive conformation the VFT is open and the interactions between LB1 and LB2 are minimal, therefore the CR domains do not interact. Calcium ions are shown as red spheres. PDB accession numbers: 5k5s and 5k5t [28]
Results from both groups suggest that the CaSR follows a universal activation mechanism similar for all class C GPCRs, despite the low sequence similarity (20–30%) [30]. This mechanism can be summarised in three steps. First, agonist binding causes the closure of the VFT. Second, membrane-proximal domains associate forming a homodimer interface between LB2 and CR domains. Third, agonist binding is accompanied by an approach between the C-terminal ends of ECDs of both protomers suggesting rearrangement of the TMD [31].
The ICD allows accurate receptor-specific control of diverse downstream signalling pathways. It represents the most diverse region of the class C GPCRs and determines selectivity of CaSR by coupling to different G proteins through the intracellular loops [32]. The ICD is exposed to the cytoplasm and begins with Lys-863 [33]. The amino acid sequence of the ICD is well conserved among species, although amino acids in the C-terminal tail are quite diverse [32]. Until now, two residues (Phe-706 and Leu-703) in intracellular loop two and eight residues (including Leu-797 and Phe-801) in intracellular loop three have been shown to be important for activating phospholipase C (PLC), the major pathway of the CaSR intracellular signalling [34]. Furthermore, there are several well-defined phosphorylation sites, especially Thr-888, in the ICD that are important for protein kinase C (PKC)-dependent inhibition of CaSR [22, 35, 36]. Also, this inhibitory effect may be counteracted by a protein phosphatase (most likely PP2A) that dephosphorylates Thr-888, restoring CaSR responsiveness [37].
41.3 CaSR Modulation
GPCRs can recognise diverse extracellular stimuli and are one of the most successful pharmaceutical target classes for different disorders. The ligands for GPCRs are typically polypeptides, amino acids and/or other small biological molecules that bind in well-defined pockets [38]. The CaSR, as a multifaceted receptor, is able to bind a broad range of molecules in addition to Ca2+, its primary ligand. CaSR modulators can be divided into two groups: type I or orthosteric modulators, which bind to the active site, and type II or allosteric modulators, which bind elsewhere in the receptor.
41.3.1 Orthosteric Modulators of the CaSR
Orthosteric modulators are type I CaSR agonists and include all ligands that are thought to compete with Ca2+ for the same binding sites on the receptor. In addition, they are sufficient to activate the CaSR on their own, in the absence of Ca2+.
Although Ca2+ is crucial for CaSR function, many other organic cations can activate CaSR in vitro for instance the divalent cations Mg2+ and Sr2+ and trivalent cations such as Gd3+, as well as heavy metals such as Pb2+ and Co2+ which are more potent than Ca2+ [39]. In fact, the order of agonist potency for inositol metabolism in bovine parathyroid cells depends on two factors; the charge of the ion and the ionic radii. Thus, among ions with the same charge, those with greater radius have a greater potency and among ions of a similar size those with greater charge have a greater potency [40]. The order of potency for the main orthosteric modulators is as follows: Gd3+ > La3+ > Ca2+ = Ba2+ > Sr2+ > Mg2+ [39]. Many organic polycations, such as the poly-amino acids poly-L-lysine or poly-arginine and aminoglycoside antibiotics such as neomycin, are also orthosteric modulators of the CaSR [41,42,43]. Polyamines produced in the gut and in the synaptic cleft in vivo, are also CaSR agonists. Spermine is the most potent polyamine followed by spermidine and putrescine. In this case, potency is linked to the number of amine groups in the ligand [44].
41.3.2 Allosteric Modulators of the CaSR
In addition to orthosteric agonists, the CaSR can also be activated by allosteric modulators, sometimes referred to as type II CaSR agonists, and these do not compete for the same binding sites as Ca2+, instead they allosterically modify the endogenous affinity of the receptor for Ca2+ o [45]. The allosteric modulators affect the conformational equilibrium of the receptor and they can be divided into two groups: activators or positive allosteric modulators (PAM) if they shift the equilibrium towards the active state, and inhibitors or negative allosteric modulators (NAM), if they stabilise the inactive state. Both types of modulators include compounds that can be found in the body under physiological conditions like L-aromatic amino acids, glutathione, ionic strength and alkalinisation [46], but also synthetic compounds like calcimimetic drugs [47].
L-aromatic amino acids were the first endogenous PAMs identified and these include L-Phe, L-Tyr, L-His and L-Trp, with the short aliphatic amino acids L-Thr and L-Ala also effective [48]. L-amino acids increase CaSR sensitivity in the presence of other agonists, such as Ca2+ or Gd3+. This demonstrates that CaSR is able to sense a broad range of nutrients having special relevance in the gastrointestinal tract where the CaSR has been identified as an L-amino acid sensor for macronutrient-dependent hormone secretion [49, 50]. In addition, increased aromatic L-amino acid concentration suppresses PTH secretion stereoselectively by activating endogenous CaSR [51]. Therefore, L-amino acids may play an important role as physiological regulators of PTH secretion and calcium metabolism via CaSR modulation.
Interestingly, pH and ionic strength play a double role in modulating CaSR sensitivity. In the case of pH, CaSR sensitivity can be enhanced when pH is elevated (>7.5), but also reduced when pH is low (<7.3) [52]. Decreasing blood pH by only 0.2–0.4 units significantly increases PTH secretion, suggesting a functionally less active CaSR [53]. In contrast, moderate alkalinisation equivalent to that seen in metabolic alkalosis significantly inhibits PTH secretion independently of a change in [Ca2+]o, suggestive of a more sensitive CaSR [54]. This has been confirmed in vitro, whereby small pathophysiologic pH changes (0.2 units) significantly inhibit CaSR-induced intracellular calcium Ca2+ i mobilisation (and also extracellular signal-regulated kinase (ERK1/2) phosphorylation and actin polymerisation [55]) in CaSR-HEK cells and in bovine parathyroid cells [34]. Similarly, increasing the ionic strength of the surrounding buffer can also reduce CaSR sensitivity, whereas reducing the buffer’s ionic strength enhances CaSR sensitivity [46]. This suggests that protons and Na+ can both act as NAMs of the CaSR.
41.3.3 Synthetic Modulators of the CaSR
Over the last 20 years, scientists have been looking for drugs to alleviate pathological abnormalities in plasma PTH and Ca2+ levels. As the secretion of PTH is mainly regulated by CaSR, compounds that affect this receptor are good candidates to treat PTH disorders. Thus, new synthetic allosteric modulators with higher potency and specificity have been developed.
Nemeth and colleagues at NPS Pharmaceuticals Inc. successfully identified two small organic molecules that caused a leftward shift in the concentration-response curve of the CaSR for [Ca2+]o. They named them calcimimetics. These compounds are able to potentiate the effects of [Ca2+]o probably by stabilising the active conformation of the receptor by binding to the TMD [56,57,58]. Calcimimetics are considered type II CaSR agonists and most of them are phenylalkylamines and derivatives of Ca2+ channel blockers [57, 59]. Some Ca2+ channel blockers can also activate the CaSR, worsening the effects in pulmonary arterial hypertension [60].
Cinacalcet, a calcimimetic molecule more easily absorbed than the initially identified analogue NPS R-568, was the first PAM acting on a GPCR to receive FDA approval and enter the clinic. It represents a targeted therapy for the treatment of disorders linked to hyperparathyroidism, including chronic kidney disease (CKD), life-threatening NSHPT, and parathyroid carcinoma [61,62,63]. In patients with end-stage CKD, treatment with Cinacalcet lowers PTH levels after 2–4 h [64]. However, calcimimetics can evoke significant side effects including adverse gastrointestinal effects, due to the fact that the CaSR is expressed in many other tissues, where it activates different signalling pathways [64, 65]. Apart from nausea, the main side effect of Cinacalcet is hypocalcaemia [66]. Recently, a new peptide calcimimetic called Etelcalcetide (Parsabiv) has just received FDA approval for the treatment of secondary hyperparathyroidism in adult haemodialysis patients with CKD [66, 67]. Other calcimimetics in use either as research tools or as potential clinical agents include Calindol (AC265347) and Velcalcetide (AMG416) [68].
In contrast, synthetic CaSR NAMs called calcilytics have opposite effects to calcimimetics. Calcilytics include the substituted phenyl-O-alkylamine NPS 2143 and NPS 89636 [56]. Their binding site is located within the CaSR TMD and is partly overlapping with the calcimimetic binding site. Two other structural types of compounds, amino alcohols (e.g. Ronacaleret) and quinazolinones (e.g. ATF936), were identified by high-throughput screening and shown to reduce CaSR affinity for [Ca2+]o [56, 69]. As calcilytics can increase endogenous PTH secretion by inhibiting CaSR they were initially developed to treat osteoporosis by delivering endogenous, anabolic pulses of PTH but they had insufficient efficacy [65]. Currently, calcilytics are being studied in different drug repurposing projects, including asthma and other lung-related diseases [70].
41.4 CaSR Trafficking
Receptor trafficking plays a critical role in GPCR activity through tight regulation of GPCR expression levels at the cellular surface. This regulation can be divided into two opposing routes: (1) trafficking of newly synthesised GPCRs to the cellular surface (i.e. exocytic trafficking) and (2) removal of GPCRs from the cell surface to intracellular compartments (i.e. endocytic trafficking) [71]. This section will focus on the processes and interacting partners involved in CaSR trafficking.
41.4.1 From Protein Synthesis to the Cellular Surface
To initiate downstream signalling, a GPCR is required to be present at the cellular surface where the agonist binding site is accessible to ligand stimulation and its intracellular part can interact with G proteins or other binding partners [71,72,73]. The outward motion of newly synthesised GPCRs to the cellular surface is driven by exocytic receptor trafficking. In this section, the term exocytic trafficking will be used in the broadest sense to refer to protein synthesis, protein maturation and the transport of newly synthesised GPCRs from the endoplasmic reticulum (ER) and Golgi system to the cellular surface [74].
To date, the processes and binding partners involved in exocytic CaSR trafficking are poorly understood. In humans, the gene that encodes for CaSR is located on chromosome 3q13.3-21 [75]. The CaSR protein is transcribed from six out of the eight mapped exons in this gene and transcription can be initiated from two different promoter sites (i.e. promoter P1 or P2) [76, 77]. In an investigation into the regulation of CaSR transcription, Canaff and Hendy have identified functional vitamin D and NF-κB response elements within both promoters of the CaSR gene [78, 79]. In agreement with these findings, vitamin D and several proinflammatory cytokines have been reported to upregulate rodent and human CaSR expression [79,80,81,82].
Correct protein folding and protein maturation through post-translational modifications are essential for cell-surface targeting of GPCRs. Protein folding into the GPCR’s functional three-dimensional conformation is assisted by chaperones. To date, a large number of chaperones or GPCR-interacting proteins with chaperone function have been identified, but none of these proteins have been associated with CaSR folding [72, 83]. CaSR maturation involves extensive N-linked glycosylation in the ECD. A total of 11 potential N-linked glycosylation sites have been identified in the CaSR protein. Glycosylation of at least three sites have been found crucial for cell-surface expression. Moreover, western blot analyses of cell lysates containing CaSR demonstrate immunoreactive bands at approximately 140–160 kDa corresponding to immature monomeric CaSR and fully mature monomeric CaSR respectively [25, 84,85,86].
As mentioned earlier, the CaSR predominantly exists on the cell surface as a homodimer, but with evidence suggesting potential heterodimerisation with other class C GPCR members including mGlu and GABA B receptors [26, 87]. The CaSR homodimerisation process takes place in the ER and is directed by the formation of disulphide linkages and non-covalent interactions at the dimer interface, as confirmed by the recently resolved crystal structures of the CaSR ECD [28, 29].
Protein synthesis and maturation are strictly regulated by the cell to ensure that only correctly folded and fully matured GPCRs are targeted for trafficking towards the cellular surface. This quality control system is proposed to be regulated by GPCR-interacting proteins such as the previously mentioned chaperones as well as by recognition of conserved retention or export motifs [71, 83]. Bouschet et al. have investigated the involvement of receptor activity-modifying proteins (RAMPs) in exocytic CaSR trafficking. According to Bouschet et al., CaSR interaction with RAMP subtype 1 or 3 facilitates delivery to the cellular surface [88]. This view is supported by Desai and co-workers who demonstrated direct interactions between CaSR and both RAMP subtypes at the cellular surface using FRET-based stoichiometry [89]. The CaSR-interacting protein dorfin mediates ER-associated degradation of the receptor, while filamin A, another interacting protein, protects CaSR from degradation [90,91,92,93]. Furthermore, an extended phosphorylation-regulated arginine-rich region was identified in the carboxyl terminus of CaSR which has been shown to be involved in intracellular retention through interaction with 14-3-3 proteins [94,95,96,97].
In general, the number of receptors expressed at the cellular surface influences the magnitude of downstream signalling responses. Multiple studies have demonstrated that differences in cell surface expression levels influence CaSR-mediated signalling. Cell surface expression levels of CaSR can be influenced by multiple factors [84, 98, 99]. First, CaSR expression is affected by numerous naturally occurring mutations and polymorphisms. Interestingly, cell surface expression levels of most CaSR mutants could be effectively rectified towards wild-type expression levels upon treatment with calcimimetics or calcilytics [90, 94, 100,101,102]. Second, phosphorylation at residue Ser-899, a protein kinase A (PKA) phosphorylation site located next to the extended arginine-rich region, has been reported to increase CaSR surface localisation by disruption of 14-3-3 protein binding [94, 95]. Third, a novel trafficking mechanism, referred to as agonist-driven insertional signalling, has been proposed to regulate cell surface expression in response to CaSR activation. According to this mechanism, agonist binding promotes an increase in the forward trafficking of newly synthesised CaSR to the cellular surface from a consistently present intracellular CaSR pool [95].
41.4.2 From the Cellular Surface to Protein Degradation
Endocytic receptor trafficking, also commonly referred to as receptor endocytosis or receptor internalisation, regulates the duration and magnitude of GPCR-activated G protein signalling responses by effective removal of GPCRs from the cellular surface. Besides its crucial role in the termination of GPCR activity, multiple studies have linked receptor endocytosis to the initiation of non-canonical G protein-independent signalling pathways [71, 103]. Endocytic trafficking of CaSR was described to play a role in parathyroid hormone-related protein (PTHrP) secretion and ERK1/2 activation [104, 105].
The molecular mechanism underlying endocytic CaSR trafficking is still poorly investigated. The reported experimental data is rather controversial, and there is no general agreement about the endocytic trafficking route of CaSR. One of the main findings related to CaSR endocytosis is the ability to initiate endocytic trafficking independently of ligand activation [95, 104, 106]. Pi and colleagues stated that phosphorylation preferentially by GRK4 promotes ß-arrestin binding [107]. However, Lorenz et al. argue that phosphorylation by PKC rather than GRKs mediates ß-arrestin recruitment [108]. This disagreement could potentially be linked to receptor origin as Pi et al. measured desensitisation of rat CaSR while the studies of Lorenz et al. were conducted with human CaSR.
The internalised CaSR can be either recycled or degraded [95, 104, 106, 109]. Similarly as for the exocytic trafficking pathway, the endocytic trafficking pathway is strongly regulated by GPCR-interacting proteins and conserved motifs [103, 110, 111]. In 2012, a presumed internalisation motif linked to lysosomal degradation has been discovered at the CaSR carboxyl terminus [106]. Interestingly, this motif shows an overlap with the filamin A binding site indicating that filamin A might be involved in both exocytic and endocytic trafficking of the CaSR [91]. This hypothesis is supported by the finding that filamin A contributes to the localisation of CaSR to caveolae, a specialised cell membrane region known to be involved in clathrin-independent endocytosis [92, 112, 113]. Furthermore, the CaSR-interacting protein AMSH-1 (associated molecule with the SH3 domain of STAM) has been reported to promote ubiquitin-mediated degradation of internalised CaSR [104, 114].
The ability to rectify the expression of disease-related mutants by calcimimetics and calcilytics highlights the therapeutic potential of modulating endocytic CaSR trafficking in the treatment of CaSR-related diseases. However, further research is needed to fully understand the molecular mechanism underlying CaSR trafficking and its potential as therapeutic target.
41.5 Overview of Signalling Pathways Activated by the CaSR
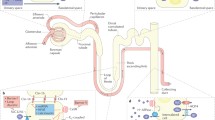
This section will focus on the signalling pathways mediated by the CaSR, with special attention to the diversity of responses elicited upon activation of the receptor in different tissues. Figure 41.5 shows a simplified overview of what is known to date about CaSR signalling.

General representation of the signalling pathways activated by CaSR. Arrows, bar-headed lines, and dashed arrows represent activation, inhibition, and a chemical reaction respectively. In green and blue, a CaSR homodimer; in light blue and yellow, the α and βγ subunits of the G proteins; in dark blue, kinases; in light green, phospholipases; in light brown, MAP kinases; in light yellow, Rho GTPases; in grey, second messengers and derivatives
41.5.1 G Proteins Activated by the CaSR
G proteins, also called guanine nucleotide-binding proteins, can bind the guanine nucleotides GDP and GTP. GTP-bound, active G proteins have GTPase activity, hydrolysing GTP into GDP and inorganic phosphate (Pi), returning the G protein to its inactive GDP-bound form. The equilibrium of GDP- and GTP-bound forms of the G proteins is a result of the activities of three groups of molecules. Guanine nucleotide exchange factors (GEFs) activate the G proteins by exchanging GDP for GTP. GTPase-activating proteins (GAPs) accelerate the GTPase activity of the G protein and thus terminate its activity. Guanine nucleotide dissociation inhibitors (GDIs) bind GDP-bound G proteins and inhibit activation by the GEFs. G proteins can be either heterotrimeric or monomeric. In this section we will refer to them as “G proteins” and “small GTPases” respectively.
G proteins are heterotrimers composed of α, β, and γ subunits. To date, a total of 21 α, 6 β, and 12 different γ subunits have been identified in humans. G proteins are activated by GPCRs, such as the CaSR, which act as GEFs. The exchange of GDP for GTP, bound to the α subunit, results in the dissociation of the α subunit from the βγ dimer. Subsequently, both the GTP-bound α subunit and the βγ heterodimer activate signalling pathways until the GTP is hydrolysed into GDP and the heterotrimer is reassembled. GAPs accelerate the hydrolysis and are also known as regulators of G protein signalling (RGS). These events are illustrated in Fig. 41.6.

Simplified representation of the cycles of activation and deactivation of G proteins
Among the well-accepted effects of the βγ dimer are the regulation of K+ and voltage-dependent Ca2+ channels, adenylyl cyclases (ACs), phospholipases C (PLCs), and phosphoinositide 3-kinases (PI3Ks) [115]. In addition, βγ heterodimers have been suggested to affect transcription, trafficking and signalling at different subcellular locations [116].
As for many GPCRs, the CaSR couples to more than one family of heterotrimeric G proteins, especially to Gq/11 and Gi/o. However several studies also suggest that the CaSR may couple to members of the G12/13 family, and also to Gs in cancer-derived cell lines [18].
41.5.1.1 Gq/11
Gq and G11 share 90% sequence homology, are ubiquitously expressed, and have similar functions. For historic reasons, most studies focus on Gq and to a lesser extent on G11 [117]. The α subunits of Gq/11 activate PLCβ, which cleaves membrane-located phosphatidylinositol 4,5-bisphosphate (PIP2) into the second messengers 1,2-diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). IP3 diffuses into the cytosol and binds the IP3 receptors that reside in the ER, causing the release of Ca2+ i. IP3 is then metabolised to IP2 and IP1. Increased [Ca2+]i together with DAG, localised in the plasma membrane, results in the recruitment and activation of multiple isoforms of PKC. PKC phosphorylates numerous other proteins [118], including CaSR at Thr-888 to regulate Ca2+ i oscillations [119]. CaSR-induced IP3 generation and Ca2+ i mobilisation was first shown in Xenopus laevis oocytes expressing the bovine parathyroid CaSR in the original cloning paper [3] but is more commonly investigated in HEK293 cells stably expressing the CaSR (HEK-CaSR) [18] [120] thus confirming the central role of the Gq/11-PLCβ pathway in CaSR signalling.
In addition to the classic Gq/11-PLCβ pathway, recent studies have shown that Gq/11 can also activate other signalling pathways via RhoGEFs such as RhoA [121,122,123], although the relevance of this pathway for the CaSR has not yet been determined.
41.5.1.2 Gi/o
The members of the Gi/o family are characterised by their sensitivity to pertussis toxin (PTx), which inhibits their interaction with the GPCR, the only exception being Gz [124]. Gi1, Gi2, Gi3 and Go subtypes share a high sequence homology and probably have overlapping functions, although Go is localised predominantly in the central nervous system [125]. Due to the relatively high abundance of this family of G proteins compared to the others, and since the majority of signalling events activated by βγ are sensitive to PTx [126], the signalling by βγ dimers is often attributed to activation of Gi/o [127].
Activation of Gi inhibits several types of ACs. ACs increase cytosolic levels of cAMP and therefore Gi activation lowers cAMP levels. Studies in HEK-CaSR cells show that increased [Ca2+]o decreases forskolin-induced increase in cAMP, suggesting the activation of Gi [120]. In bovine parathyroid and HEK-CaSR cells, CaSR stimulates ERK1/2 phosphorylation via Gq/11 and Gi pathways [128]. A later study suggested that Gi2 is the Gi/o subtype responsible for ERK1/2 activation [105].
41.5.1.3 G12/13
The activation of G12/13 proteins recruit to the membrane and activate RhoGEFs that specifically activate RhoA, such as p115-RhoGEF, which also acts as a GAP through its RGS domain terminating the activity of G12/13 [129]. Using Madin-Darby canine kidney cells stably overexpressing CaSR, Miller and collaborators found that [Ca2+]o activated phospholipase D via RhoA, and that this was mediated by G12/13 and independent of Gq/11 and Gi [130]. Another study, suggested a pathway specifically activated by L-Phe via G12/13 in mouse embryonic fibroblasts that resulted in Ca2+ i oscillations [131].
41.5.1.4 Gs
A few studies have reported Gs coupling to the CaSR. Wysolmerski and collaborators first showed that CaSR couples to Gi in healthy mammary epithelial cells, but then switches to Gs in both MCF-7 human breast cancer cells, and in Comma-D immortalised murine mammary cells. Surprisingly, no IP1 accumulation was observed, whereas the levels of active mitogen-activated protein kinases (MAPKs) were increased upon stimulation with high [Ca2+]o. They also found that cAMP regulated the secretion of PTHrP via PKA [132], which was corroborated in a recent study [133]. In mouse pituitary gland tumour derived AtT-20 cells, the same group showed that CaSR activation stimulated PTHrP via the same mechanism, Gs-cAMP-PKA, independently of PLC or PKC [134]. In a previous study using the same cell line, increases in IP1 concentrations were sensitive to PTx, showing simultaneous coupling both to Gi and Gs [135]. G protein switching has been observed also for the β2-adrenergic receptor, where PKA phosphorylates the receptor, increasing its affinity for Gi versus Gs. As a result, it switches signalling from cAMP/PKA to MAPK activation [136].
41.5.2 Rho GTPases
Rho GTPases belong to the Ras family of GTPases, which are the most known small monomeric GTPases. Among the Rho GTPases, the best characterised are RhoA, Rac1 and Cdc42. Rho GTPases play a central role in cell migration, cell polarity, and cell cycle progression, by regulating cell adhesion and actin cytoskeleton dynamics [137]. The activation of RhoA has been traditionally associated exclusively with G12/13 signalling, however there is increasing evidence of activation by Gq/11 via RhoGEFs and independent of PLCβ [138]. It has been suggested that the CaSR activates PI4-kinase via Rho [139]. CaSR activation produced actin stress fibre assembly in HEK-CaSR, in a Rho kinase-dependent mechanism. This phenomenon was PTx-insensitive and the PLCβ inhibitor U73122 showed no effect [140]. Since U73122 can activate ion channels at the concentrations used to inhibit PLCβ [141] the recently available potent and specific Gq/11 inhibitors FR900359 and YM-254890 may prove better reagents for the investigation of Gq/11 signalling [142].
A study in human keratinocytes showed that CaSR-dependent activation of RhoA plays a role in cell-cell adhesion [143], whereas experiments in human podocytes showed that CaSR activated RhoA via Ca2+ i mobilisation, in a mechanism dependent of the ion channel TRPC6 [144].
The activation of Rac and Cdc42 by G proteins is less clearly defined. In highly motile cells βγ-mediated activation of PI3K and the GEF PRex resulted in Rac1 activity. Whether these signalling modules play a role in CaSR signal transduction remains to be demonstrated [145]. In primary human monocyte-derived macrophages CaSR activated Rac and/or Cdc42, but no RhoA, to regulate membrane ruffling via a mechanism dependent on PI3K [146]. A study in a human T cell line found that CaSR can promote cell migration by activating Cdc42, also via a PI3K-dependent mechanism [147]. A study in HEK-CaSR cells showed that membrane ruffling is Gq/11-dependent and G12/13-independent, suggesting activation of Rho GTPases by Gq/11 [148].
41.5.3 β-Arrestins
In addition to their key role in terminating G protein signalling pathways activated by GPCRs, β-arrestins can also activate signalling events [149]. Specifically in HEK-CaSR cells, β-arrestin 1 is involved in CaSR-induced plasma membrane ruffling [150] while β-arrestins 1 and 2 are involved in CaSR-induced ERK1/2 activation [151].
41.5.4 CaSR-Induced Protein Kinase Activation
The CaSR activates a number of protein kinase families including glycogen synthase kinase-3 (GSK3), Akt, and the MAPKs, and these will be detailed in turn.
41.5.4.1 Akt and GSK-3β
Akt, or protein kinase B, is a protein kinase that regulates multiple functions such as growth, proliferation and transcription. The first step for Akt activation is binding to PIP3 in the membrane. PIP3-bound Akt is sequentially phosphorylated first at Thr-308 and then at Ser-473 for full activation [152]. GSK3 is involved in the phosphorylation of over a hundred substrates, and it interacts with multiple types of receptors. It exists in two isoforms, α and β, and it can be phosphorylated by PKA, PKC, and Akt, among others. Phosphorylation of GSK3-β at Ser-9 results in inhibition of the binding to certain substrates that require binding to a domain in the protein prior to phosphorylation [153].
Studies in fetal rat calvarial cells, murine osteoblast 2T3 cells, and human osteoblasts, show that CaSR activation results in phosphorylation of Akt at Thr-308 and Ser-473, and of GSK3-β at Ser-9 [154, 155]. Further, in proximal tubular opossum kidney cells, the CaSR ligands neomycin and gentamicin elicit phosphorylation of Akt and GSK3-β in a PI3K-dependent fashion [42].
41.5.4.2 MAPKs
Several studies have recently explored the role played by CaSR in the phosphorylation of protein kinases, both in healthy tissue and in disease models. The MAPKs include ERK, c-Jun amino-terminal kinases (JNK), and P38. These proteins are activated by phosphorylation and thus we will refer to the active phosphorylated forms as p-ERK1/2, p-JNK and p-P38.
Activation of ERK1/2 can be Ras- or PKC-dependent. Ras-dependent activation involves PI3K, Src family kinases, and receptor tyrosine kinases such as the epidermal growth factor receptor. A study in HEK-CaSR cells showed that ERK1/2 activation by CaSR was Ras-dependent, relied largely on PI3K activity, and was independent of tyrosine kinase activity [156]. In contrast, another study in HEK-CaSR cells and in bovine parathyroid cells, showed that the cytoplasmic tyrosine kinase inhibitor, herbimycin, inhibited ERK1/2 phosphorylation [128]. A similar result was observed for ERK1 in Rat-1 fibroblasts [157]. In proximal tubular opossum-kidney cells, activation of CaSR by neomycin induced P38 activation via a PI3K-mediated mechanism [41].
Across different tissues, increased CaSR expression and activation correlates positively with an increase in p-ERK1/2 levels [41, 154, 158,159,160,161,162], except for a study on hearts of a rat epilepsy model where p-ERK1/2 levels decreased [163]. A similar positive correlation was observed for p-JNK [161,162,163,164], whereas one study showed no effect [160]. As for p-P38, a similar number of studies show a positive correlation [41, 161, 163, 164] or no effect [158, 160, 162].
Overall, a prolonged exposure to CaSR agonists increases mRNA or protein expression levels of CaSR, and this phenomenon often correlates positively with an increase in active ERK1/2, JNK, and P38. Differences in phosphorylation are observed reflecting the different signalling profiles of CaSR in different tissues.
41.6 Ligand-Biased Signalling Through the CaSR
Ligand-biased signalling is a relatively new concept based on the idea that a receptor can exist in multiple active conformations, each stabilised by a specific ligand, with characteristic binding kinetics, and therefore with a particular signalling profile [165]. For GPCRs, this would translate into different coupling behaviours towards G proteins and β-arrestins [166]. For allosteric modulators, the concept extends to how these molecules affect positively or negatively each of the pathways activated by the orthosteric ligands. Exploiting this phenomenon offers great potential for the discovery and development of new drugs with increased efficacy and safety, which is of course of interest for the pharmaceutical industry [167].
Several studies have used the CaSR as a model to study ligand-biased signalling, given that it can activate multiple signalling pathways and it can be modulated by a wide range of different ligands [168]. This phenomenon has been studied using pharmacological assays, applied in a high-throughput manner, and using multiple agonists. The receptor readouts usually follow changes in Ca2+ i mobilisation, IP1 accumulation, cAMP levels, phosphorylation of ERK1/2, and plasma membrane ruffling. The first two provide information on the Gq/11-PLCβ pathway; cAMP on Gi and Gs activity; p-ERK1/2 on Gq, Gi, and β-arrestins; and PM ruffling on Rho GTPases and β-arrestins.
These studies often rely on obtaining concentration-response curves for different ligands, and comparing calculated values such as EC50, dissociation constant, maximum response, or cooperativity αβ for allosteric modulators. Additionally, receptor expression levels are often also determined, and in fact regulation of cell surface expression has been proposed as a mechanism of bias by allosteric modulators [100, 169]. Multiple studies in HEK-CaSR cells have addressed ligand-bias by orthosteric ligands, as well as positive and negative allosteric modulators [151, 170,171,172,173], including one that explored the effect of naturally occurring CaSR mutations on ligand-bias [102].
These systematic in vitro studies provide valuable information to understand the differences in the effects of the ligands in vivo. For example, Leach and collaborators found that the calcimimetic AC265347 tunes the effect of [Ca2+]o to favour phosphorylation of ERK1/2 and accumulation of IP1, as compared to Ca2+ i mobilisation. Interestingly, AC265347 did not increase trafficking of loss-of-expression CaSR mutants, an effect observed by other calcimimetics, suggesting that it may act via a new mechanism [170]. Figure 41.7 shows an example of the signalling bias caused by two of the allosteric modulators used in this study.

Representation of bias by the positive allosteric modulators R-568 and AC265347 in Ca2+ i mobilisation and phosphorylation of ERK1/2 upon activation of the CaSR by Ca2+ o
41.7 Tissue-Specific Signalling of the CaSR
The pleiotropy of the CaSR arises as a result of its ability to couple to various G proteins and thus to mediate distinct signalling pathways. Consequently, the CaSR may fine-tune several physiological processes in a tissue-specific manner. The ability of GPCRs to mediate tissue-specific signalling is dictated by the cellular environment, as evidenced by recombinant systems where the same GPCR can have different pharmacological profiles in different cellular backgrounds. This phenomenon is termed tissue-specific signalling or system bias. It arises when ligands favour the interaction of a receptor ensemble to auxiliary proteins, or when receptors form heterodimers with distinct pharmacological signatures [174]. The capacity of CaSR ligands to promote coupling to multiple G proteins and to differing extents was previously discussed in the context of biased signalling. Here, tissue-specific signalling is discussed in light of the evidence showing interaction of the CaSR with other proteins and the formation of heterodimers in different cellular environments.
The CaSR interacts with various proteins that influence its signalling signature. Several CaSR interacting proteins have been identified and these include inwardly-rectifying potassium channels [175] and the previously described filamin A [92, 93, 176] as well as the RAMPs [88]. In addition, the CaSR may form heterodimers with other class C GPCRs including mGlu1a, mGlu5 and GABA B receptors, as shown in endogenous and recombinant systems [87]. Such heterodimerisation could thus provide the CaSR another mechanism for tissue-specific signalling.
41.8 Future Topics and Concluding Remarks
The recently published crystallographic data of the CaSR ECD structure has shed some light on CaSR ligand recognition, receptor activation, allosteric modulation, as well as on the structural basis of dimerisation. The medicinal importance of CaSR modulation is clear and therefore obtaining the ECD structure will facilitate structure-based drug discovery and might open up further therapeutic approaches. These data also raise the question of whether L-aromatic amino acids and relevant anions should be added to the experimental buffers when studying CaSR function to preserve the receptor’s native conformation. The full structure of the CaSR has yet to be determined and thus obtaining the crystal structures of the CaSR’s TMD and ICD is a high priority in the field as this will help understanding effector interactions. In addition, the current structural data provides only a snapshot of the receptor in a fixed conformation, whereas in physiology the receptor is a dynamic molecule wobbling between multiple conformations. Thus, we need a new and more dynamic approach able to reveal a protein’s structure in its transition states, ideally allowing us to see conformational changes upon agonist/antagonist binding at the receptor.
The CaSR field would also benefit from new reagents such as a CaSR-selective radioligand, as calcium itself is a too low-affinity ligand to be of use in binding studies. Such a radioligand would allow researchers to investigate whether CaSR biased signalling might be driven by ligand binding kinetics. Moreover, the radioligand could be used to study CaSR expression in native tissues and cells as CaSR expression analysis is currently hampered by nonspecific staining of commercially available CaSR antibodies. Next, the newly emerging FRET-based biosensors can be used to observe G protein activation of the CaSR directly, providing dynamic information on the first step in the signalling cascade [177]. Indeed by taking advantage of the plethora of fluorescent biosensors available, given particular spectroscopic properties, it is possible to measure activation of multiple proteins simultaneously and in real-time [178]. This would be of particular value when studying biased signalling, as current protocols are susceptible to time- and assay-specific artefacts.
Finally, a recent publication suggested that internalised CaSR could have a role in sustained signalling [148]. This phenomenon has been proposed before for some class A GPCRs [179], and implies a new signalling mechanism by CaSR. The relevance of the observations for internalised CaSR signalling needs to be addressed in follow-up studies.
We can conclude therefore that CaSR activation results in a wide range of downstream signals and functions at different timescales and across a variety of tissues. To make sense of this complexity will require better understanding of a range of factors including differential ligand affinity and bias, receptor heterodimerisation, as well as downstream effector selection. The benefit of such information could be the rational development of novel drugs with improved efficacy and safety.
Abbreviations
- 1,25D3:
-
1α,25-dihydroxyvitamin D3
- AC:
-
Adenylate cyclase
- ADH:
-
Autosomal dominant hypocalcaemia
- AP2:
-
Adaptor protein-2
- cAMP:
-
Cyclic adenosine monophosphate
- Ca2+ :
-
Calcium
- CaSR:
-
Calcium-sensing receptor
- CR:
-
Cysteine rich domain
- [Ca2+]o :
-
Extracellular calcium concentration
- Ca2+ i :
-
Intracellular calcium
- [Ca2+]i :
-
Intracellular calcium concentration
- CKD:
-
Chronic kidney disease
- DAG:
-
Diacylglycerol
- ECD:
-
Extracellular domain
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- FHH:
-
Familial hypocalciuric hypercalcaemia
- GABA:
-
Gamma-aminobutyric acid
- GAPs:
-
GTPase-activating proteins
- GEFs:
-
Guanine nucleotide exchange factors
- GDIs:
-
Guanine nucleotide dissociation inhibitors
- GPCR:
-
G protein-coupled receptor
- GRKs:
-
G protein-coupled receptor kinases
- GSK3:
-
Glycogen synthase kinase-3
- HEK:
-
Human embryonic kidney
- HEK-CaSR:
-
HEK293 cells stably expressing the CaSR
- ICD:
-
Intracellular domain
- IGF-1:
-
Insulin-like growth factor 1
- IP3:
-
Inositol 1,4,5-trisphosphate
- JNK:
-
C-Jun amino-terminal kinases
- mGlu:
-
Metabotropic glutamate receptor
- NAM:
-
Negative allosteric modulator
- LB:
-
Lobe-shaped domain
- MAPKs:
-
Mitogen-activated protein kinases
- NAM:
-
Negative allosteric modulator
- NSHPT:
-
Neonatal severe hyperparathyroidism
- NKCC2:
-
Na-K-Cl cotransporter 2
- PA:
-
Phosphatidic acid
- PAM:
-
Positive allosteric modulator
- PDEs:
-
Phosphodiesterases
- Pi:
-
Inorganic phosphate
- PI3Ks:
-
Phosphoinositide 3-kinases
- PIP2:
-
Phosphatidylinositol 4,5-bisphosphate
- PKA:
-
Protein Kinase A
- PKB:
-
Protein Kinase B
- PKC:
-
Protein Kinase C
- PLA2:
-
Phospholipase A2
- PLC:
-
Phospholipase C
- PLD:
-
Phospholipase D
- PreProPTH:
-
Prepro-parathyroid hormone
- PT:
-
Parathyroid
- PTH:
-
Parathyroid hormone
- PTHrP:
-
Parathyroid hormone-related protein
- PTx:
-
Pertussis-toxin
- RGS:
-
Regulator of G protein signalling
- RAMPs:
-
Receptor activity-modifying proteins
- TMD:
-
Transmembrane domain
- TAS1R:
-
Taste 1 receptors
- VFD:
-
Venus flytrap domain
References
Crichton RR (2008) Biological inorganic chemistry. Biological inorganic chemistry
Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017) The concise guide to pharmacology 2017/18: G protein-coupled receptors. Br J Pharmacol 174:S17–S129
Brown EM, Gamba G, RIccardi D, Lombardi M, Butters R, Kifor O et al (1993) Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366:461–464
Conigrave AD (2016) The calcium-sensing receptor and the parathyroid: past, present, future. Front Physiol 7:563
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A et al (2015) Tissue-based map of the human proteome. Science 347(6220):1260419–1260419
Felsenfeld AJ, Levine BS (2015) Calcitonin, the forgotten hormone: does it deserve to be forgotten? Clin Kidney J 8(2):180–187
Mutschler E, Geisslinger G, Kroemer HK, Menzel S, Ruth P Mutschler Arzneimittelwirkungen – Pharmakologie, Klinische Pharmakologie, Toxikologie. Lehrbuch der Pharmakologie, der klinischen Pharmakologie und Toxikologie; mit einführenden Kapiteln in die Anatomie, Physiologie und Pathophysiologie; mit 257 Tabellen und 1417 Strukturformeln. 2013. XXIII, 1197 Seiten
Brown EM, MacLeod RJ (2001) Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 81(1):239–297
Conigrave AD, Quinn SJ, Brown EM (2000) Cooperative multi-modal sensing and therapeutic implications of the extracellular Ca2+ sensing receptor. Trends Pharmacol Sci 21:401–407
Kantham L, Quinn SJ, Egbuna OI, Baxi K, Butters R, Pang JL et al (2009) The calcium-sensing receptor (CaSR) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am J Physiol Endocrinol Metab 297(4):E915–E923
Loupy A, Ramakrishnan SK, Wootla B, Chambrey R, De La Faille R, Bourgeois S et al (2012) PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J Clin Invest 122(9):3355–3367
Ward BK, Magno AL, Davis EA, Hanyaloglu AC, Stuckey BGA, Burrows M et al (2004) Functional deletion of the calcium-sensing receptor in a case of neonatal severe hyperparathyroidism. J Clin Endocrinol Metab 89:3721–3730
Vahe C, Benomar K, Espiard S, Coppin L, Jannin A, Odou MF et al (2017) Diseases associated with calcium-sensing receptor. Orphanet J Rare Dis 12:19
Pallan S, Rahman MO, Khan AA (2012) Diagnosis and management of primary hyperparathyroidism. BMJ 344(7849):181–192
Assimos DG (2015) The G allele of CaSR R990G polymorphism increases susceptibility to urolithiasis and hypercalciuria: evidences from a comprehensive meta-analysis. J Urol 194:1014
Tennakoon S, Aggarwal A, Kallay E (2016) The calcium-sensing receptor and the hallmarks of cancer. Biochim Biophys Acta 1863(6 Pt B):1398–1407
Yarova PL, Stewart AL, Sathish V, Britt RD, Thompson MA, Lowe APP et al (2015) Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci Transl Med 7(284):284ra58
Conigrave AD, Ward DT (2013) Calcium-sensing receptor (CaSR): pharmacological properties and signaling pathways. Best Pract Res Clin Endocrinol Metab 27(3):315–331
Muto T, Tsuchiya D, Morikawa K, Jingami H (2007) Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc Natl Acad Sci 104(10):3759–3764
Silve C, Petrel C, Leroy C, Bruel H, Mallet E, Rognan D et al (2005) Delineating a Ca2+ binding pocket within the venus flytrap module of the human calcium-sensing receptor. J Biol Chem 280(45):37917–37923
Hendy GN, Canaff L, Cole DEC (2013) The CASR gene: alternative splicing and transcriptional control, and calcium-sensing receptor (CaSR) protein: structure and ligand binding sites. Best Pract Res Clin Endocrinol Metab 27:285–301
Bai M (2004) Structure-function relationship of the extracellular calcium-sensing receptor. Cell Calcium 35:197–207
Hauache OM, Hu J, Ray K, Spiegel AM (2000 Aug) Functional interactions between the extracellular domain and the seven-transmembrane domain in Ca2+ receptor activation. Endocrine 13(1):63–70
Ward DT, Brown EM, Harris HW (1998) Disulfide bonds in the extracellular calcium-polyvalent cation-sensing receptor correlate with dimer formation and its response to divalent cations in vitro. J Biol Chem 273(23):14476–14483
Ray K, Clapp P, Goldsmith PK, Spiegel AM (1998) Identification of the sites of N-linked glycosylation on the human calcium receptor and assessment of their role in cell surface expression and signal transduction. J Biol Chem 273(51):34558–34567
Gama L, Wilt SG, Breitwieser GE (2001) Heterodimerization of calcium sensing receptors with metabotropic glutamate receptors in neurons. J Biol Chem 276(42):39053–39059
Chang W, Tu C, Cheng Z, Rodriguez L, Chen T-H, Gassmann M et al (2007) Complex formation with the Type B gamma-aminobutyric acid receptor affects the expression and signal transduction of the extracellular calcium-sensing receptor. Studies with HEK-293 cells and neurons. J Biol Chem 282(34):25030–25040
Geng Y, Mosyak L, Kurinov I, Zuo H, Sturchler E, Cheng TC et al (2016) Structural mechanism of ligand activation in human calcium-sensing receptor. elife 5(July):1–25
Zhang C, Zhang T, Zou J, Miller CL, Gorkhali R, Yang J et al (2016) Structural basis for regulation of human calcium-sensing receptor by magnesium ions and an unexpected tryptophan derivative co-agonist. Sci Adv 2(5):e1600241
Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T et al (2000) Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 407(6807):971–977
Matsushita S, Nakata H, Kubo Y, Tateyama M (2010) Ligand-induced rearrangements of the GABAB receptor revealed by fluorescence resonance energy transfer. J Biol Chem 285(14):10291–10299
Tfelt-Hansen J, Brown EM (2005) The calcium-sensing receptor in normal physiology and pathophysiology: a review. Crit Rev Clin Lab Sci 42:35–70
Garrett JE, Capuano IV, Hammerland LG, Hung BC, Brown EM, Hebert SC et al (1995) Molecular cloning and functional expression of human parathyroid calcium receptor cDNAs. J Biol Chem 270(21):12919–12925
Chang W, Chen TH, Pratt S, Shoback D (2000) Amino acids in the second and third intracellular loops of the parathyroid Ca2+-sensing receptor mediate efficient coupling to phospholipase C. J Biol Chem 275(26):19955–19963
Brown EM, Lian JB (2008) New insights in bone biology: unmasking skeletal effects of the extracellular calcium-sensing receptor. Sci Signal 1:pe40
Bai M, Trivedi S, Brown EM (1998) Dimerization of the extracellular calcium-sensing receptor (CaR) on the cell surface of CaR-transfected HEK293 cells. J Biol Chem 273(36):23605–23610
McCormick WD, Atkinson-Dell R, Campion KL, Mun HC, Conigrave AD, Ward DT (2010) Increased receptor stimulation elicits differential calcium-sensing receptorT888 dephosphorylation. J Biol Chem 285(19):14170–14177
Granier S, Kobilka B (2012) A new era of GPCR structural and chemical biology. Nat Chem Biol 8:670–673
Handlogten ME, Shiraishi N, Awata H, Huang CF, Miller RT (2000) Extracellular Ca2+-sensing receptor is a promiscuous divalent cation sensor that responds to lead. Am J Physiol Physiol 279(6):F1083–F1091
Quinn SJ, Ye CP, Diaz R, Kifor O, Bai M, Vassilev P et al (1997) The Ca2+-sensing receptor: a target for polyamines. Am J Phys 273:C1315–C1323
Ward DT, McLarnon SJ, Riccardi D (2002) Aminoglycosides increase intracellular calcium levels and ERK activity in proximal tubular OK cells expressing the extracellular calcium-sensing receptor. J Am Soc Nephrol 13(6):1481–1489
Ward DT, Maldonado-Pérez D, Hollins L, Riccardi D (2005) Aminoglycosides induce acute cell signaling and chronic cell death in renal cells that express the calcium-sensing receptor. J Am Soc Nephrol 16(5):1236–1244
Brauner-Osborne H, Wellendorph P, Jensen A (2007) Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets 8(1):169–184
Chang W, Shoback D (2004) Extracellular Ca2+-sensing receptors – an overview. Cell Calcium 35(3):183–196
Nemeth EF (2004) Calcimimetic and calcilytic drugs: just for parathyroid cells? Cell Calcium 35(3):283–289
Bandyopadhyay S, Tfelt-Hansen J, Chattopadhyay N (2010) Diverse roles of extracellular calcium-sensing receptor in the central nervous system. J Neurosci Res 88:2073–2082
Ward DT, Riccardi D (2012) New concepts in calcium-sensing receptor pharmacology and signalling. Br J Pharmacol 165:35–48
Conigrave AD, Quinn SJ, Brown EM (2000 Apr) L-amino acid sensing by the extracellular Ca2+-sensing receptor. Proc Natl Acad Sci U S A 97(9):4814–4819
Liou AP, Sei Y, Zhao X, Feng J, Lu X, Thomas C et al (2011) The extracellular calcium-sensing receptor is required for cholecystokinin secretion in response to L-phenylalanine in acutely isolated intestinal I cells. Am J Physiol Gastrointest Liver Physiol 300(4):G538–G546
Feng J, Petersen CD, Coy DH, Jiang J-K, Thomas CJ, Pollak MR et al (2010 Oct) Calcium-sensing receptor is a physiologic multimodal chemosensor regulating gastric G-cell growth and gastrin secretion. Proc Natl Acad Sci U S A 107(41):17791–17796
Conigrave AD, Mun H-C, Delbridge L, Quinn SJ, Wilkinson M, Brown EM (2004 Sep) L-amino acids regulate parathyroid hormone secretion. J Biol Chem 279(37):38151–38159
Quinn SJ, Bai M, Brown EM (2004) pH sensing by the calcium-sensing receptor. J Biol Chem 279(36):37241–37249
López I, Aguilera-Tejero E, Estepa JC, Rodriguez M, Felsenfeld AJ (2004) Role of acidosis-induced increases in calcium on PTH secretion in acute metabolic and respiratory acidosis in the dog. Am J Physiol Endocrinol Metab 286(5):E780–E785
Lopez I, Rodriguez M, Felsenfeld AJ, Estepa JC, Aguilera-Tejero E (2003) Direct suppressive effect of acute metabolic and respiratory alkalosis on parathyroid hormone secretion in the dog. J Bone Miner Res 18(8):1478–1485
Campion KL, McCormick WD, Warwicker J, Khayat MEB, Atkinson-Dell R, Steward MC et al (2015) Pathophysiologic changes in extracellular pH modulate parathyroid calcium-sensing receptor activity and secretion via a histidine-independent mechanism. J Am Soc Nephrol 26(9):2163–2171
Nemeth EF (2002) The search for calcium receptor antagonists (calcilytics). J Mol Endocrinol 29(1):15–21
Nemeth EF, Steffey ME, Hammerland LG, Hung BC, Van Wagenen BC, DelMar EG et al (1998) Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc Natl Acad Sci U S A 95(March):4040–4045
Miedlich S, Gama L, Breitwieser GE (2002) Calcium sensing receptor activation by a calcimimetic suggests a link between cooperativity and intracellular calcium oscillations. J Biol Chem 277(51):49691–49699
Saidak Z, Brazier M, Kamel S, Mentaverri R (2009) Agonists and allosteric modulators of the calcium-sensing receptor and their therapeutic applications. Mol Pharmacol 76(6):1131–1144
Yamamura A (2016) Molecular mechanism of Dihydropyridine Ca2+ channel blockers in pulmonary hypertension. Yakugaku Zasshi 136(10):1373–1377
Block GA, Zaun D, Smits G, Persky M, Brillhart S, Nieman K et al (2010) Cinacalcet hydrochloride treatment significantly improves all-cause and cardiovascular survival in a large cohort of hemodialysis patients. Kidney Int 78(6):578–589
Gannon AW, Monk HM, Levine MA (2014) Cinacalcet monotherapy in neonatal severe hyperparathyroidism: a case study and review. J Clin Endocrinol Metab 99(1):7–11
Silverberg SJ, Rubin MR, Faiman C, Peacock M, Shoback DM, Smallridge RC et al (2007) Cinacalcet hydrochloride reduces the serum calcium concentration in inoperable parathyroid carcinoma. J Clin Endocrinol Metab 92(10):3803–3808
Nemeth EF, Goodman WG (2016) Calcimimetic and calcilytic drugs: feats, flops, and futures. Calcif Tissue Int 98:341–358
Steddon SJ, Cunningham J (2005) Calcimimetics and calcilytics – fooling the calcium receptor. Lancet 365(9478):2237–2239
Block GA, Bushinsky DA, Cheng S, Cunningham J, Dehmel B, Drueke TB et al (2017) Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism. JAMA 317(2):156–164
Eidman KE, Wetmore JB (2018) Managing hyperparathyroidism in hemodialysis: role of etelcalcetide. Int J Nephrol Renovasc Dis 11:69–80
Ma J-N, Owens M, Gustafsson M, Jensen J, Tabatabaei A, Schmelzer K et al (2011) Characterization of highly efficacious allosteric agonists of the human calcium-sensing receptor. J Pharmacol Exp Ther 337(1):275–284
Nemeth EF, Delmar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL et al (2001) Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther 299(1):323–331
Lembrechts R, Brouns I, Schnorbusch K, Pintelon I, Kemp PJ, Timmermans J-P et al (2013) Functional expression of the multimodal extracellular calcium-sensing receptor in pulmonary neuroendocrine cells. J Cell Sci 126(Pt 19):4490–4501
Drake MT, Shenoy SK, Lefkowitz RJ (2006) Trafficking of G protein-coupled receptors. Circ Res 99(6):570–582
Ritter SL, Hall RA (2009) Fine-tuning of GPCR activity by receptor-interacting proteins. Nat Rev Mol Cell Biol 10(12):819–830
Pierce KL, Premont RT, Lefkowitz RJ (2002) Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3(9):639–650
Tokarev AA, Alfonso A, Segev N (2009) Overview of intracellular compartments and trafficking pathways. In: Trafficking inside cells: pathways, mechanisms and regulation. Springer, New York, pp 3–14
Janicic N, S E, Pausova Z, Seldin MF, Rivi M, Szpirer J et al (1995) Mapping of the calcium-sensing receptor gene (CASR) to human chromosome 3q13. 3-21 by fluorescence in situ hybridization, vol 801. Springer, New York, pp 798–801
Chikatsu N, Fukumoto S, Takeuchi Y, Suzawa M, Obara T, Matsumoto T et al (2000) Cloning and characterization of two promoters for the human calcium-sensing receptor (CaSR) and changes of CaSR expression in parathyroid adenomas. J Biol Chem 275(11):7553–7557
Yun FHJ, Wong BYL, Chase M, Shuen AY, Canaff L, Thongthai K et al (2007) Genetic variation at the calcium-sensing receptor (CASR) locus: implications for clinical molecular diagnostics. Clin Biochem 40(8):551–561
Canaff L, Hendy GN (2002) Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J Biol Chem 277(33):30337–30350
Canaff L, Hendy GN (2005) Calcium-sensing receptor gene transcription is up-regulated by the proinflammatory cytokine, interleukin-1β: role of the NF-κB pathway and κB elements. J Biol Chem 280(14):14177–14188
Brown AJ, Zhong M, Finch J, Ritter C, McCracken R, Morrissey J et al (1996) Rat calcium-sensing receptor is regulated by vitamin D but not by calcium. Am J Phys 270(3 Pt 2):F454–F460
Yao JJ (2005) Regulation of renal calcium receptor gene expression by 1,25-dihydroxyvitamin D3 in genetic hypercalciuric stone-forming rats. J Am Soc Nephrol 16(5):1300–1308
Fetahu IS, Hummel DM, Manhardt T, Aggarwal A, Baumgartner-Parzer S, Kállay E (2014) Regulation of the calcium-sensing receptor expression by 1,25-dihydroxyvitamin D3, interleukin-6, and tumor necrosis factor alpha in colon cancer cells. J Steroid Biochem Mol Biol 144:228–231
Dong C, Filipeanu CM, Duvernay MT, Wu G (2007) Regulation of G protein-coupled receptor export trafficking. Biochim Biophys Acta Biomembr 1768(4):853–870
Ray K, Fan GF, Goldsmith PK, Spiegel AM (1997) The carboxyl terminus of the human calcium receptor. Requirements for cell-surface expression and signal transduction. J Biol Chem 272(50):31355–31361
Fan G, Goldsmith PK, Collins R, Dunn CK, Krapcho KJ, Rogers KV et al (1997) N-linked glycosylation of the human Ca2+ receptor is essential for its expression at the cell surface. Endocrinology 138(5):1916–1922
Aida K, Koishi S, Tawata M, Onaya T (1995) Molecular cloning of a putative Ca2+-sensing receptor cDNA from human kidney. Biochem Biophys Res Commun 214(2):524–529
Chang W, Tu C, Cheng Z, Rodriguez L, Chen TH, Gassmann M et al (2007) Complex formation with the type B γ-aminobutyric acid receptor affects the expression and signal transduction of the extracellular calcium-sensing receptor: studies with HEK-293 cells and neurons. J Biol Chem 282(34):25030–25040
Bouschet T, Stéphane M, Henley JM (2005) Receptor-activity-modifying proteins are required for forward trafficking of the calcium-sensing receptor to the plasma membrane. J Cell Sci 118(20):4709–4720
Desai AJ, Roberts DJ, Richards GO, Skerry TM (2014) Role of receptor activity modifying protein 1 in function of the calcium sensing receptor in the human TT thyroid carcinoma cell line. PLoS One 9(1):e85237
Huang Y, Breitwieser GE (2007) Rescue of calcium-sensing receptor mutants by allosteric modulators reveals a conformational checkpoint in receptor biogenesis. J Biol Chem 282(13):9517–9525
Zhang M, Breitwieser GE (2005) High affinity interaction with filamin A protects against calcium-sensing receptor degradation. J Biol Chem 280(12):11140–11146
Hjälm G, MacLeod RJ, Kifor O, Chattopadhyay N, Brown EM (2001) Filamin-A binds to the carboxyl-terminal tail of the calcium-sensing receptor, an interaction that participates in CaR-mediated activation of mitogen-activated protein kinase. J Biol Chem 276(37):34880–34887
Awata H, Huang C, Handlogten ME, Miller RT (2001) Interaction of the calcium-sensing receptor and filamin, a potential scaffolding protein. J Biol Chem 276(37):34871–34879
Stepanchick A, McKenna J, McGovern O, Huang Y, Breitwieser GE (2010) Calcium sensing receptor mutations implicated in pancreatitis and idiopathic epilepsy syndrome disrupt an arginine-rich retention motif. Cell Physiol Biochem 26(3):363–374
Grant MP, Stepanchick A, Cavanaugh A, Breitwieser GE (2011) Agonist-driven maturation and plasma membrane insertion of calcium-sensing receptors dynamically control signal amplitude. Sci Signal 4(200):1–9
Grant MP, Cavanaugh A, Breitwieser GE (2015) 14-3-3 proteins buffer intracellular calcium sensing receptors to constrain signaling. PLoS One 10(8):1–20
Arulpragasam A, Magno AL, Ingley E, Brown SJ, Conigrave AD, Ratajczak T et al (2012) The adaptor protein 14-3-3 binds to the calcium-sensing receptor and attenuates receptor-mediated Rho kinase signalling. Biochem J 441(3):995–1007
Brennan SC, Mun H-C, Leach K, Kuchel PW, Christopoulos A, Conigrave AD (2015) Receptor expression modulates calcium-sensing receptor mediated intracellular Ca2+ mobilization. Endocrinology 156(4):1330–1342
Chang W, Pratt S, Chen TH, Bourguignon L, Shoback D (2001) Amino acids in the cytoplasmic C terminus of the parathyroid Ca2+−sensing receptor mediate efficient cell-surface expression and phospholipase C activation. J Biol Chem 276(47):44129–44136
Leach K, Wen A, Cook AE, Sexton PM, Conigrave AD, Christopoulos A (2013) Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium-sensing receptor by positive and negative allosteric modulators. Endocrinology 154(3):1105–1116
White E, McKenna J, Cavanaugh A, Breitwieser GE (2009) Pharmacochaperone-mediated rescue of calcium-sensing receptor loss-of-function mutants. Mol Endocrinol 23(7):1115–1123
Leach K, Wen A, Davey AE, Sexton PM, Conigrave AD, Christopoulos A (2012) Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor. Endocrinology 153(9):4304–4316
Moore CAC, Milano SK, Benovic JL (2007) Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol 69(1):451–482
Reyes-Ibarra AP, García-Regalado A, Ramírez-Rangel I, Esparza-Silva AL, Valadez-Sánchez M, Vázquez-Prado J et al (2007) Calcium-sensing receptor endocytosis links extracellular calcium signaling to parathyroid hormone-related peptide secretion via a Rab11a-dependent and AMSH-sensitive mechanism. Mol Endocrinol 21(6):1394–1407
Holstein DM, Berg KA, Leeb-Lundberg LMF, Olson MS, Saunders C (2004) Calcium-sensing receptor-mediated ERK1/2 activation requires Gα i2 coupling and dynamin-independent receptor internalization. J Biol Chem 279(11):10060–10069
Zhuang X, Northup JK, Ray K (2012) Large putative PEST-like sequence motif at the carboxyl tail of human calcium receptor directs lysosomal degradation and regulates cell surface receptor level. J Biol Chem 287(6):4165–4176
Pi M, Oakley RH, Gesty-Palmer D, Cruickshank RD, Spurney RF, Luttrell LM et al (2005) β-arrestin- and G protein receptor kinase-mediated calcium-sensing receptor desensitization. Mol Endocrinol 19(4):1078–1087
Lorenz S, Frenzel R, Paschke R, Breitwieser GE, Miedlich SU (2007) Functional desensitization of the extracellular calcium-sensing receptor is regulated via distinct mechanisms: role of G protein-coupled receptor kinases, protein kinase C and β-arrestins. Endocrinology 148(5):2398–2404
Huang Y, Niwa JI, Sobue G, Breitwieser GE (2006) Calcium-sensing receptor ubiquitination and degradation mediated by the E3 ubiquitin ligase dorfin. J Biol Chem 281(17):11610–11617
Marchese A, Paing MM, Temple BRS, Trejo J (2008) G protein–coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol 48(1):601–629
Magalhaes AC, Dunn H, Ferguson SSG (2012) Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br J Pharmacol 165(6):1717–1736
Kifor O, Diaz R, Butters R, Kifor I, Brown EM (1998) The calcium-sensing receptor is localized in caveolin-rich plasma membrane domains of bovine parathyroid cells. J Biol Chem 273(34):21708–21713
Chini B, Parenti M (2004) G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there? J Mol Endocrinol 32(2):325–338
Herrera-Vigenor F, Hernández-García R, Valadez-Sánchez M, Vázquez-Prado J, Reyes-Cruz G (2006) AMSH regulates calcium-sensing receptor signaling through direct interactions. Biochem Biophys Res Commun 347(4):924–930
Khan SM, Sleno R, Gora S, Zylbergold P, Laverdure J-P, Labbe J-C et al (2013) The expanding roles of Gβɣ subunits in G protein-coupled receptor signaling and drug action. Pharmacol Rev 65(2):545–577
Khan SM, Sung JY, Hébert TE (2016) Gβγ subunits—different spaces, different faces. Pharmacol Res 111:434–441
Hubbard KB, Hepler JR (2006) Cell signalling diversity of the Gqα family of heterotrimeric G proteins. Cell Signal 18(2):135–150
Newton AC (2018) Protein kinase C: perfectly balanced. Crit Rev Biochem Mol Biol 53(2):208–230
Davies SL, Ozawa A, McCormick WD, Dvorak MM, Ward DT (2007) Protein kinase C-mediated phosphorylation of the calcium-sensing receptor is stimulated by receptor activation and attenuated by calyculin-sensitive phosphatase activity. J Biol Chem 282(20):15048–15056
Chang W, Pratt S, Chen TH, Nemeth E, Huang Z, Shoback D (1998) Coupling of calcium receptors to inositol phosphate and cyclic AMP generation in mammalian cells and Xenopus laevis oocytes and immunodetection of receptor protein by region-specific antipeptide antisera. J Bone Miner Res 13(4):570–580
Rojas RJ, Yohe ME, Gershburg S, Kawano T, Kozasa T, Sondek J (2007) Gαq directly activates p63RhoGEF and trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J Biol Chem 282(40):29201–29210
Lutz S, Shankaranarayanan A, Coco C, Ridilla M, Nance MR, Vettel C et al (2007) Structure of Gaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318:1923–1927
Van Unen J, Reinhard NR, Yin T, Wu YI, Postma M, Gadella TWJ et al (2015) Plasma membrane restricted RhoGEF activity is sufficient for RhoA-mediated actin polymerization. Sci Rep 5(October):1–16
Gagnon AW, Manning DR, Catani L, Gewirtz A, Poncz M, Brass LF (1991) Identification of Gz as a pertussis toxin-insensitive G protein in human platelets and megakaryocytes. Blood 78:1247–1254
Jiang M, Bajpayee NS (2009) Molecular mechanisms of go signaling. Neurosignals 17(1):23–41
Smrcka AV (2008) G protein βɣ subunits: central mediators of G protein-coupled receptor signaling. Cell Mol Life Sci 65:2191–2214
Wettschureck N, Offermanns S (2005) Mammalian G proteins and their cell type specific functions. Physiol Rev 85:1159–1204
Kifor O, MacLeod RJ, Diaz R, Bai M, Yamaguchi T, Yao T et al (2001) Regulation of MAP kinase by calcium-sensing receptor in bovine parathyroid and CaR-transfected HEK293 cells. Am J Physiol Renal Physiol 280(2):F291–F302
Kozasa T, Hajicek N, Chow CR, Suzuki N (2011) Signalling mechanisms of RhoGTPase regulation by the heterotrimeric G proteins G12 and G13. J Biochem 150(4):357–369
Huang CF, Hujer KM, Wu ZZ, Miller RT (2004) The Ca2+-sensing receptor couples to G alpha(12/13) to activate phospholipase D in Madin-Darby canine kidney cells. Am J Physiol Physiol 286(1):C22–C30
Rey O, Young SH, Yuan J, Slice L, Rozengurt E (2005) Amino acid-stimulated Ca2+ oscillations produced by the Ca2+-sensing receptor are mediated by a phospholipase C/inositol 1,4,5-trisphosphate-independent pathway that requires G12, Rho, filamin-A, and the actin cytoskeleton. J Biol Chem 280(24):22875–22882
Mamillapalli R, VanHouten J, Zawalich W, Wysolmerski J (2008) Switching of G-protein usage by the calcium-sensing receptor reverses its effect on parathyroid hormone-related protein secretion in normal versus malignant breast cells. J Biol Chem 283(36):24435–24447
Kim W, Takyar FM, Swan K, Jeong J, Vanhouten J, Sullivan C et al (2016) Calcium-sensing receptor promotes breast cancer by stimulating intracrine actions of parathyroid hormone-related protein. Cancer Res 76(18):5348–5360
Mamillapalli R, Wysolmerski J (2010) The calcium-sensing receptor couples to Galpha(s) and regulates PTHrP and ACTH secretion in pituitary cells. J Endocrinol 204(3):287–297
Emanuel RL, Adler GK, Kifor O, Quinn SJ, Fuller F, Krapcho K et al (1996) CaSR expression and regulation by extracellular calcium in the AtT-20 pituitary cell line. Mol Endocrinol 10(5):555–565
Daaka Y, Luttrell LM, Lefkowitz RJ (1997) Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 390(November):88–91
Hodge RG, Ridley AJ (2016) Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol 17(8):496–510
Vogt S, Grosse R, Schultz G, Offermanns S (2003) Receptor-dependent RhoA activation in G12/G13-deficient cells. Genetic evidence for an involvement of Gq/G11. J Biol Chem 278(31):28743–28749
Huang C, Handlogten ME, Tyler Miller R (2002) Parallel activation of phosphatidylinositol 4-kinase and phospholipase C by the extracellular calcium-sensing receptor. J Biol Chem 277(23):20293–20300
Davies SL, Gibbons CE, Vizard T, Ward DT, Sarah L, Gibbons CE et al (2006) CaSR induces Rho kinase-mediated actin stress fiber assembly and altered cell morphology, but not in response to aromatic amino acids. Am J Physiol Cell Physiol 290:1543–1551
Leitner MG, Michel N, Behrendt M, Dierich M, Dembla S, Wilke BU et al (2016) Direct modulation of TRPM4 and TRPM3 channels by the phospholipase C inhibitor U73122. Br J Pharmacol 173:2555–2569
Kukkonen JP (2016) G-protein inhibition profile of the reported Gq/11 inhibitor UBO-QIC. Biochem Biophys Res Commun 469(1):101–107
Tu CL, Chang W, Bikle DD (2011) The calcium-sensing receptor-dependent regulation of cell-cell adhesion and keratinocyte differentiation requires rho and filamin a. J Invest Dermatol 131(5):1119–1128
Zhang L, Ji T, Wang Q, Meng K, Zhang R, Yang H et al (2017) Calcium-sensing receptor stimulation in cultured glomerular podocytes induces TRPC6-dependent calcium entry and RhoA activation. Cell Physiol Biochem 43:1777–1789
Vazquez-Prado J, Bracho-Valdes I, Cervantes-Villagrana RD, Reyes-Cruz G (2016) G pathways in cell polarity and migration linked to oncogenic GPCR signaling: potential relevance in tumor microenvironment. Mol Pharmacol 90(5):573–586
Canton J, Schlam D, Breuer C, Gütschow M, Glogauer M, Grinstein S (2016) Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat Commun 7(11284):1–12
Chang F, Kim JM, Choi Y, Park K (2018) MTA promotes chemotaxis and chemokinesis of immune cells through distinct calcium-sensing receptor signaling pathways. Biomaterials 150:14–24
Gorvin CM, Rogers A, Hastoy B, Tarasov AI, Frost M, Sposini S et al (2018) AP2σ mutations impair calcium-sensing receptor trafficking and signaling, and show an endosomal pathway to spatially direct G-protein selectivity. Cell Rep 22(4):1054–1066
Lefkowitz R (2007) Introduction to special section on β-Arrestins. Annu Rev Physiol 69(1)
Bouschet T, Martin S, Kanamarlapudi V, Mundell S, Henley JM (2007) The calcium-sensing receptor changes cell shape via a beta-arrestin-1 ARNO ARF6 ELMO protein network. J Cell Sci 120(Pt):2489–2497
Thomsen ARB, Hvidtfeldt M, Bräuner-Osborne H (2012) Biased agonism of the calcium-sensing receptor. Cell Calcium 51(2):107–116
Hemmings BA, Restuccia DF, Wrana JL, Alto NM, Orth K, Kopan R et al (2012) PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 4:a011189
Beurel E, Grieco SF, Jope RS (2015) Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148:114–131
Dvorak MM, Siddiqua A, Ward DT, Carter DH, Dallas SL, Nemeth EF et al (2004) Physiological changes in extracellular calcium concentration directly control osteoblast function in the absence of calciotropic hormones. Proc Natl Acad Sci U S A 101(14):5140–5145
Rybchyn MS, Slater M, Conigrave AD, Mason RS (2011) An Akt-dependent increase in canonical Wnt signaling and a decrease in sclerostin protein levels are involved in strontium ranelate-induced osteogenic effects in human osteoblasts. J Biol Chem 286(27):23771–23779
Hobson SA, Wright J, Lee F, McNeil SE, Bilderback T, Rodland KD (2003) Activation of the MAP kinase cascade by exogenous calcium-sensing receptor. Mol Cell Endocrinol 200:189–198
McNeil SE, Hobson SA, Nipper V, Rodland KD (1998) Functional calcium-sensing receptors in rat fibroblasts are required for activation of SRC kinase and mitogen-activated protein kinase in response to extracellular calcium. J Biol Chem 273(2):1114–1120
Wang P, Wang L, Wang S, Li S, Li Y, Zhang L (2015) Effects of calcium-sensing receptors on apoptosis in rat hippocampus during hypoxia/reoxygenation through the ERK1/2 pathway. Int J Clin Exp Pathol 8(9):10808–10815
Mizumachi H, Yoshida S, Tomokiyo A, Hasegawa D, Hamano S, Yuda A et al (2017) Calcium-sensing receptor-ERK signaling promotes odontoblastic differentiation of human dental pulp cells. Bone 101:191–201
Li T, Sun M, Yin X, Wu C, Wu Q, Feng S et al (2013) Expression of the calcium sensing receptor in human peripheral blood T lymphocyte and its contribution to cytokine secretion through MAPKs or NF-κB pathways. Mol Immunol 53(4):414–420
Kong W-Y, Tong L-Q, Zhang H-J, Cao Y-G, Wang G-C, Zhu J-Z et al (2016) The calcium-sensing receptor participates in testicular damage in streptozotocin-induced diabetic rats. Asian J Androl 18:803–808
Qi H, Cao Y, Huang W, Liu Y, Wang Y, Li L et al (2013) Crucial role of calcium-sensing receptor activation in cardiac injury of diabetic rats. PLoS One 8(5):e65147
Li L, Chen F, Cao YG, Qi HP, Huang W, Wang Y et al (2015) Role of calcium-sensing receptor in cardiac injury of hereditary epileptic rats. Pharmacology 95:10–21
Zhen Y, Ding C, Sun J, Wang Y, Li S, Dong L (2016) Activation of the calcium-sensing receptor promotes apoptosis by modulating the JNK/p38 MAPK pathway in focal cerebral ischemia-reperfusion in mice. Am J Transl Res 8(2):911–921
Herenbrink CK, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J et al (2016) The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun 7:1–14
Kenakin T (2011) Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther 336(2):296–302
Kenakin T, Christopoulos A (2013) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12(3):205–216
Leach K, Conigrave AD, Sexton PM, Christopoulos A (2015) Towards tissue-specific pharmacology: insights from the calcium-sensing receptor as a paradigm for GPCR (patho)physiological bias. Trends Pharmacol Sci 36(4):215–225
Cavanaugh A, Huang Y, Breitwieser GE (2012) Behind the curtain: cellular mechanisms for allosteric modulation of calcium-sensing receptors. Br J Pharmacol 165(6):1670–1677
Cook AE, Mistry SN, Gregory KJ, Furness SGB, Sexton PM, Scammells PJ et al (2015) Biased allosteric modulation at the CaS receptor engendered by structurally diverse calcimimetics. Br J Pharmacol 172(1):185–200
Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A (2012) Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology 153(3):1232–1241
Leach K, Gregory KJ, Kufareva I, Khajehali E, Cook AE, Abagyan R et al (2016) Towards a structural understanding of allosteric drugs at the human calcium-sensing receptor. Cell Res 26(5):574–592
Thomsen ARB, Worm J, Jacobsen SE, Stahlhut M, Latta M, Brauner-Osborne H (2012) Strontium is a biased agonist of the calcium-sensing receptor in rat medullary thyroid carcinoma 6-23 cells. J Pharmacol Exp Ther 343(3):638–649
Kenakin T (2002) Drug efficacy at G protein–coupled receptors. Annu Rev Pharmacol Toxicol 42(1):349–379
Huang C, Sindic A, Hill CE, Hujer KM, Chan KW, Sassen M et al (2007) Interaction of the Ca2+-sensing receptor with the inwardly rectifying potassium channels Kir4.1 and Kir4.2 results in inhibition of channel function. Am J Physiol Renal Physiol 44106:1073–1081
Pi M, Spurney RF, Tu Q, Hinson T, Quarles LD (2018) Calcium-sensing receptor activation of Rho involves filamin and Rho-guanine nucleotide exchange factor. Endocrinology 143(January):3830–3838
Adjobo-Hermans MJW, Goedhart J, van Weeren L, Nijmeijer S, Manders EMM, Offermanns S et al (2011) Real-time visualization of heterotrimeric G protein Gq activation in living cells. BMC Biol 9(1):32
Depry C, Mehta S, Zhang J (2013) Multiplexed visualization of dynamic signaling networks using genetically encoded fluorescent protein-based biosensors. Pflugers Arch Eur J Physiol 465(3):373–381
Jean-Alphonse F, Bowersox S, Chen S, Beard G, Puthenveedu MA, Hanyaloglu AC (2014) Spatially restricted G protein-coupled receptor activity via divergent endocytic compartments. J Biol Chem 289(7):3960–3977
Acknowledgements
The authors thank Hans Bräuner-Osborne for critical review of the manuscript. The authors of this chapter have received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 675228 (CaSR Biomedicine).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Chavez-Abiega, S. et al. (2020). Sensing Extracellular Calcium – An Insight into the Structure and Function of the Calcium-Sensing Receptor (CaSR). In: Islam, M. (eds) Calcium Signaling. Advances in Experimental Medicine and Biology, vol 1131. Springer, Cham. https://doi.org/10.1007/978-3-030-12457-1_41
Download citation
DOI: https://doi.org/10.1007/978-3-030-12457-1_41
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-12456-4
Online ISBN: 978-3-030-12457-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)