
Abstract
Recently, a new class of small (19–25 nucleotides) noncoding RNAs, microRNAs (miRs or miRNAs), has been linked to several human diseases, including cancer. MicroRNAs are involved in temporal and tissue-specific eukaryotic gene regulation, either by translational inhibition or exonucleolytic mRNA decay, targeting through imperfect complementarity, the 3′-untranslated region (3′-UTR) of the mRNA. Since their ability to potentially target any human mRNA, it is likely that microRNAs are involved in almost every biological process, including cell cycle regulation, cell growth, apoptosis, cell differentiation, and stress response.
The involvement of microRNAs in the biology of human cancer is supported by an increasing body of experimental evidence, which has gradually switched from profiling studies, describing an aberrant microRNA expression in different tumor types, to biological demonstrations of the causal role of these small molecules in the tumorigenic process, and the possible implications as biomarkers or therapeutic tools. These more recent studies have widely demonstrated that microRNAs can modulate oncogenic or tumor suppressor pathways, and that, at the same time, their expression can be regulated by oncogenes or tumor suppressor genes.
The possibility to modulate microRNA expression either in vitro and in vivo, by developing synthetic pre-microRNA molecules or antisense oligonucleotides, has at the same time provided a powerful tool to a deeper comprehension of the molecular mechanisms regulated by these molecules, and suggested the intriguing and promising perspective of a possible use in therapy. Here, we review our current knowledge about the involvement of microRNAs in cancer and their potential role as diagnostic, prognostic, and therapeutic tools.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Acute Myelocytic Leukemia
- Chronic Lymphocytic Leukemia
- miRNA Gene
- Chronic Lymphocytic Leukemia Patient
- Caucasian American
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Definition and Biogenesis
After the initial discovery in 1993, when a small RNA encoded by the lin-4 locus was associated to the developmental timing of the nematode Caenorhabditis elegans by modulating the protein lin-14 [1], microRNAs have undergone a long period of silence. It took indeed several more years to realize that these small (19–22 nucleotides) RNA molecules are actually expressed in several organisms, including Homo sapiens, highly conserved across different species, highly specific for tissue and developmental stage, and playing crucial functions in the regulation of important processes, such as development, proliferation, differentiation, apoptosis, and stress response. In the last few years, microRNAs have indeed taken their place in the complex circuitry of cell biology, revealing a key role as regulators of gene expression.
MicroRNA genes represent approximately 1% of the genome of different species, and each of them has hundreds of different conserved or nonconserved targets: it has been estimated that about 30% of the genes are regulated by at least one microRNA [2].
MicroRNAs are transcribed for the most part by RNA polymerase II as long primary transcripts (pri-microRNAs) characterized by hairpin structures and containing typical eukaryotic mRNA features such as cap structures and poly(A) tail.
Most microRNAs localize in intergenic regions; however, some of them are located in intronic regions of known genes, in sense or antisense orientation. This finding supports the notion that at least a part of them is transcribed as distinct transcriptional units. Fifty percent of known microRNA genes are located nearby other microRNAs, supporting the hypothesis that clustered microRNAs can be transcribed from their own promoters as polycistronic pri-microRNAs [3].
MicroRNAs are mostly transcribed by RNA polymerase II (Pol II), although the possibility that a small number of miRNA genes might be transcribed by other RNA polymerases cannot be excluded. Pol II produces mRNAs and some of the noncoding RNAs, such as small nucleolar RNAs and some of the small nuclear RNAs present in the spliceosoma, the complex of specialized RNA and protein subunits that removes introns from a transcribed pre-mRNA.
Many microRNAs are differentially expressed during the development, as frequently observed with genes transcribed by Pol II.
According to their genomic localization, microRNAs can be classified in (a) exonic microRNAs located in noncoding transcripts, (b) intronic microRNAs located in noncoding transcripts, and (c) intronic microRNA located in protein-coding transcripts. Mixed miRNA genes can be assigned to one of the above groups depending on the given splicing pattern. Intronic microRNAs are transcribed within the mRNA of the host gene generating a hairpin structure, recognized and cleaved by the spliceosome machinery [4]. Exonic microRNAs are transcribed within the pri-miR (up to 1 kb long) containing both the 5′-cap and the 3′-poly(A) tail.
Processing of a microRNA consists of two phases, one taking place into the nucleus and operated by RNAse III Drosha and the second one in the cytoplasm, by RNAse III Dicer. Drosha is a highly conserved 160 kDa protein containing two RNAse III domains and one double-strand RNA-binding domain. Drosha forms a huge complex, 500 kDa in D. melanogaster and 650 kDa in H. sapiens, called microprocessor and containing the cofactor DiGeorge syndrome critical region 8 (DGCR8), also known as Pasha in D. melanogaster and C. elegans.
The hairpin structure present in the pri-miRNA (primary transcript) is recognized and cleaved by RNAse III Drosha into 70-nts-long pre-microRNAs (precursor molecule).
These precursor molecules are actively exported by a Ran-GTP and exportin 5-mediated mechanism to the cytoplasm, where an additional step is mediated by the RNAse III Dicer, which acts in complex with the transactivating response RNA-binding protein (TRBP) generating a dsRNA of approximately 22 nucleotides, named miRNA/miRNA*. Dicer is an extremely conserved protein through eukaryotes, first identified for its involvement in siRNA (small interfering RNAs) generation.
Dicer is a very large enzyme (∼200 kDa) conserved among the species and containing different domains: a double-strand RNA-binding domain (dsRBD), two RNAse III catalytic domains, one PAZ domain, which binds the 3′-end of small RNAs, and other domains with ATPasic and RNA-helicasic activity. Dicer recognizes the double-strand region of the pre-miR in association with different proteins: RDE-4 (RNAi defective 4) in C. elegans, R2D2 e FMR1 (fragile X mental retardation syndrome 1 homolog) in D. melanogaster, and members of the Argonaute family in other species. In particular, these proteins are not needed for the endonucleasic activity of Dicer, but they play a role in stabilizing the complex Dicer-miR [5]. In mammalians, the Argonaute 2 (AGO2) protein complex, characterized by RNAse H activity, cooperates in the Dicer-mediated processing of some pre-miRs, yielding to another intermediate processing product, called AGO2-cleaved precursor miR (ac-pre-miR) [4].
The mature single-stranded microRNA product is then incorporated in the complex known as miRNA-containing ribonucleoprotein complex (miRNP), miRgonaute, or miRNA-containing RNA-induced silencing complex (miRISC), which generally selects one of the two strands as guide strand (mature miR) according to thermodynamic properties, whereas the other strand is likely subjected to degradation. miRISC is a ribonucleoproteic complex containing Argonaute proteins, the mature miR, the star miR, and several additional factors, some of them necessary for the enzymatic activity. Argonaute proteins are conserved among species and containing the PAZ and PIWI domains. The PAZ domain is involved in the recognition of the microRNA [6], whereas the PIWI domain seems to be involved in releasing the mature microRNA through an interaction with Dicer [7]. In H. sapiens, miRISC complex is formed by the Argonaute homologue eIF2C2 protein, the glycine–tryptophan protein of 182 kDa (GW182), and the helicases Gemin3 and Gemin4 [8]. The choice of the pre-miR strand that will generate the active complex resides in the relative thermodynamic stability of the two strands forming the duplex: the strand with a more unstable 5′-end is included in the miRISC complex.
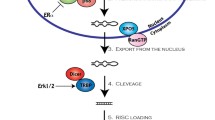
As part of this complex, the mature microRNA is able to regulate gene expression at posttranscriptional level, binding through partial complementarity to the 3′-UTR of target mRNAs, and leading to some degree of mRNA degradation and translation inhibition (Fig. 19.1).

Biogenesis, processing, and maturation of miRNAs. miRNAs are transcribed mainly by RNA polymerase II as long primary transcripts characterized by hairpin structures (pri-miRNAs) and processed in the nucleus by RNAse III Drosha in a 70-nucleotide-long pre-miRNA. This precursor molecule is exported by the exportin 5 to the cytoplasm, where RNAse III Dicer generates a dsRNA of approximately 22 nucleotides, named miRNA/miRNA*. The mature miRNA product is then incorporated in the complex known as miRISC, whereas the other strand is usually subjected to degradation. As part of this complex, the mature miRNA is able to regulate gene expression binding through partial homology the 3′-UTR of target mRNAs and leading to mRNA degradation in case of perfect matching or translation inhibition when there is partial complementarity. RISC RNA-induced silencing complex
MicroRNAs exert their function mostly binding a specific sequence within the 3′-UTR of target mRNAs. The 5′-end of microRNAs (seed site) is important in the target recognition mechanism [2]: nucleotides 2–8 (seed sites) of many miRNAs present a perfect match with the 3′-UTR seed regions involved in translational block and are also well conserved in homologue microRNAs.
MicroRNAs can regulate gene expression through the degradation of target mRNAs, concordantly with the evidence that mRNA levels can be reduced in presence of elevated levels of miRNAs. It has been demonstrated that a high complementary match between the microRNA and the target mRNA can lead to an Ago 2-mediated mRNA degradation. Recent studies suggested that also other processes are involved, like deadenylation, 5′-uncapping, and exonuclease activity. Indeed, mRNA degradation mechanism requires Ago 2 complex, GW182, and deadenylating and decapping enzymes [9].
Furthermore, the cleavage site does not depend on the match between miR and its target, but it is due to microRNA sequence only: the cleavage takes place between the corresponding mRNA residues of the 10th and the 11th nucleotide of the microRNA.
After the cleavage process, the microRNA remains intact and can drive the functioning of another miRISC complex [10]. However, the mechanisms underlying the mRNA target selection remain still unclear.
MicroRNAs can also inhibit gene expression blocking the translation of mRNAs target. The first evidence of this mechanism is the observation that many miR-targeted mRNAs maintain their level in presence of an abundance of the respective microRNAs, whereas the levels of the encoded protein are decreased [9]. The exact mechanism underlying the miRISC-mediated translational blockade remains still unknown: it is unclear whether the block overcomes at the beginning or in the next phases of the translational process. However, current models see the involvement of eIF4F, formed by eIF4A, eIF4E, and eIF4G. This proteic complex binds the 5′-cap of mRNAs and starts the translation initiation process. The translation initiation factor eIF3, interacting with eIF4G, contributes to the assembly of the ribosomal subunit 40S at the 5′-end of mRNAs and leads to the formation of the preinitiation complex. The elongation phase takes place when the ribosomal subunit 60S is assembled at the preinitiation complex in correspondence to the start codon AUG. eIF4G and eIF3 interact with the poly(A)-binding protein (PAPB1) resulting in mRNA circularization, phenomenon that leads to a higher translation efficiency [9]. A controversial body of evidence shows that microRNAs are able to inhibit both the preinitiation and the elongation phase. In 2006, Petersen et al. proposed a model in which miRISC acts as a repressor of the elongation phase, suggesting that miRISC can promote an early dissociation of the ribosome from the mRNA [11].
Controversially, three different models have been proposed to explain the initiation phase inhibition operated by miRISC:
In the first model, miRISC competes with eIF4E for the binding to the 5′-cap of the target mRNA, thus leading to the inhibition of the translational start; in the second model, miRISC blocks the mRNA circularization through the inhibition of the assembly of the 60S subunit with the 40S subunit, located on the target mRNA at the preinitiation complex.
The synergic action of multiple miRISC complexes leads to an efficient block of the translation process [12] explaining the presence of multiple seed regions within the same target.
MicroRNAs can also act through a different mechanism mediated by miRISC: the target mRNAs are seized into cytoplasmic foci called processing bodies (P-bodies), formed by mRNA and proteins [13]; since P-bodies lack of the translational machinery, this mechanism leads to a translational blockade of the sequestered mRNAs. In some instance, a deadenylation process coupled with the translational inhibition has been reported [9]. The deadenylation process is mediated by GW182 and Ago proteins. Whereas GW182 interacts with Ago through its glycine- and tryptophan-rich domains, it is also able to recruit through his C-terminus PAPB and the deadenylating enzymes CCR4 and CAF1 [4]. Furthermore, it has been observed that the number, the position, and the kind of nucleotide mismatches between the microRNA and the mRNA can play a role in the repression mechanism selection, deciding if the target mRNA would be degraded or translationally repressed.
MicroRNAs in Human Cancer: From Profiling Studies to Definition of a Functional Role as Oncogenes and Tumor Suppressors
Profiling of different cell types and tissues indicated that the pattern of miRNA expression is cell type and tissue specific, suggesting that the program regulating expression of miRNAs is exquisitely cell type dependent and tightly associated with cellular differentiation and development. Some of the most important miRNAs which are aberrantly expressed in tumors are listed in Table 19.1.
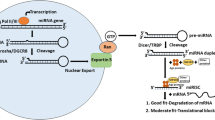
The first evidence of the involvement of microRNAs in human cancer derived from studies on chronic lymphocytic leukemia (CLL), the most common human leukemia in the Western world, particularly in an attempt to identify tumor suppressors at chromosome 13q14. Cytogenetic studies indicate deletions at chr.13q14 in approximately 50% of CLLs and loss of heterozygosity (LOH) in approximately 70% of CLLs. By taking advantage of chromosome translocations and small deletions, Dr. Croce’s group found that the critical region of 13q14 does not contain a protein-coding tumor suppressor gene but two microRNA genes, miR-15a and miR-16-1, that are expressed in the same polycistronic RNA. This result indicated that the deletion of chromosome 13q14 caused the loss of these two microRNAs, first evidence that microRNAs could be involved in the pathogenesis of human cancer [14]. Study of a large collection of CLLs showed knock down or knock out of miR-15a and miR-16-1 in approximately 69% of CLLs. Since such alteration is present in most indolent CLLs, they speculated that loss of miR-15a and miR-16-1 could be the initiating event or a very early event in the pathogenesis of this disease [14]. Immediately after these initial observations, they mapped all the known microRNA genes and found that many of them are located in regions of the genome involved in chromosomal alterations, such as deletion or amplification, in many different human tumors, in which the presumed tumor suppressor genes or oncogenes, respectively, failed to be discovered after many years of investigation [15]. Indeed, in cancer development chromosomal regions that encompass microRNAs involved in the negative regulation of a transcript encoding a known tumor suppressor may be amplified. This amplification would result in the increased expression of the microRNA and the consequent silencing of the tumor suppressor gene. Vice versa, microRNAs able to inhibit oncogenes are often located in fragile regions of the genome, where deletions or mutations can be responsible for their reduced levels and the resulting overexpression of the target oncogene (Fig. 19.2).

miRNAs as oncogenes or tumor suppressor genes. miRNAs can have oncogenic effects (oncomiRNA) when they target tumor suppressor genes. When an oncomiRNA is overexpressed, for example, because the encoding gene is located in an amplified region of the genome, this will lead to downregulation of the targets and to tumor formation (upper panel). Conversely, a miRNA can be characterized by tumor suppressor properties if the main target in that specific cellular context is an oncogene; in this case, if the miRNA expression is lost, for example, because the encoding gene is located in a deleted region of the genome, the resulting effect will be tumorigenic (lower panel). In summary, what usually happens in a tumor is the overexpression of an oncogenic miRNA and/or the loss of a miRNA with oncosuppressive properties
We can certainly affirm that alterations in microRNAs expression are not isolated, but the rule in human cancer. After these early studies indicating the role of microRNA genes in the pathogenesis of human cancer, Dr. Croce’s group and others have developed platforms to assess the global expression of microRNA genes in normal and diseased tissues, and have carried out profiling studies to assess microRNA dysregulation in human cancer. This was an attempt to establish whether microRNA profiling could be used for tumor classification, diagnosis, and prognosis [16].
Indeed, the predictive values of such microRNA signature have been validated for several types of tumors. Furthermore, the small size of miRNAs certainly contributes to a higher stability in comparison with mRNAs, allowing the study of their expression in fixed tissues or other biological material, and thus supporting their possible use as novel, minimally invasive, and robust biomarkers: indeed, it has been recently described how miRNAs can be reliably extracted and detected from paraffin-embedded tissues, from blood (either total blood, plasma, or serum) [17] and from circulating exosomes [18]. Moreover, it has been reported that the profile of circulating miRNA of individuals affected by different neoplasias can reflect the pattern observed in the tumor tissues, evidence suggesting the fascinating possibility of using circulating miRNAs as easily detectable tumor biomarkers [19], especially for early diagnosis: very recently, Sozzi’s group [20] has identified microRNA-expression signatures with strong predictive, diagnostic, and prognostic potential, analyzing plasma samples of lung cancer patients collected 1–2 years before the onset of disease.
Concerning breast cancer, for example, a pilot study performed by Roth et al. [21] provided the first evidence that tumor-associated circulating microRNAs are elevated in the blood of breast cancer patients and associated with tumor progression. In particular, the authors evaluated the relative concentrations of breast cancer-associated miR-10b, miR-34a, miR-141, and miR-155 in the blood serum of 89 patients with primary breast cancer and metastatic disease and 29 healthy women, finding that miR-10b, miR-34a, and miR-155 discriminated M1-patients from healthy controls.
Heneghan et al. [22] surveyed a panel of seven candidate miRNAs in whole blood RNAs from 148 breast cancer patients and 44 age-matched and disease-free controls. They found that the expression of miR-195 was significantly elevated in breast cancer patients and reduced in postoperative whole blood compared to the preoperative samples of the same patients. Zhao et al. [23] performed a microarray-based microRNA profiling in plasma samples from 20 women with early stage breast cancer (10 Caucasian American (CA) and 10 African American (AA) and 20 matched healthy controls (10 CAs and 10 AAs), demonstrating that the altered levels of circulating miRNAs might have great potential to serve as novel, noninvasive biomarkers for early detection of breast cancer.
Switching then from profiling studies to the definition of a functional role of microRNAs, it has been demonstrated that their aberrant expression in cancer is not just a random association, but the indication of a causal role exerted by these small RNA molecules in the tumorigenic process. Indeed, due to the role of microRNAs in regulating the expression of signaling molecules, such as cytokine, growth factors, proapoptotic and antiapoptotic genes, it has been demonstrated that miRNAs can act either as oncogenes or tumor suppressor, and more recently, it has been demonstrated that a microRNA can exploit both functions according to the cellular context of its target genes. Another important issue concerns the role of miRNAs in regulating the interaction between cancer cells and the microenvironment, particularly concerning neo-angiogenesis or tissue invasion and metastasis.
Leukemia/Lymphoma
Chronic Lymphocytic Leukemia
As mentioned, the first evidence of alterations of microRNA genes in human cancer came from studies of CLL. In a large study of indolent versus aggressive CLL, Calin et al. discovered a signature of 13 microRNAs capable of distinguishing between indolent and aggressive CLL [24]. Interestingly, it was found that miR-155, overexpressed in different lymphomas including the ABC type of diffuse large B-cell lymphoma, is also upregulated in aggressive CLLs (where it is induced by MYB, [25], whereas members of the miR-29 family and miR-181 were found to be underexpressed and later demonstrated to directly regulate the TCL1 oncogene, overexpressed in the aggressive form of CLL [26].
More recently, a prognostic signature has been identified in CLL patients with chromosome 17p deletions (who develop a more aggressive disease), revealing that miR-15a, miR-21, miR-34a, miR-155, and miR-181b are differentially expressed in comparison with normal 17p and normal karyotype. Moreover, miR-21 expression levels were significantly higher in patients with poor prognosis and predicted overall survival (OS), and miR-181b expression levels significantly predicted treatment-free survival [27].
Because of the “wait and watch” approach to the treatment of CLL, a signature able to distinguish between CLL with good and bad prognosis was also found. Sequencing of many microRNAs, including those in the signature, allowed the identification of germ line and somatic mutations of microRNA genes, including miR-15 and miR-16-1 and miR-29 family members. Interestingly, mutations in the miR-15/16 precursor were also identified, affecting the processing of the pri-miR into the pre-miR. In two cases, the mutant was in homozygosity in the leukemic cells, while normal cells of the two patients were heterozygous for this abnormality, indicating a loss of the normal miR-15/16 allele in the leukemic cell [24]. Thus miR-15a and miR-16-1 behave like typical tumor suppressors in CLL. Interestingly, Raveche et al. [28] mapped a gene responsible for an indolent form of CLL in the New Zealand Black (NZB) mouse strain on chromosome 14, in a region homologous to 13q14 in humans. Sequence analysis of this region showed a mutation in the precursor of miR-15/16 in the NZB mouse strain 6 nts 3′ to miR-16-1 (in the human cases, the mutation was 7 nts 3′ to miR-16-1), that also affected the processing of the miR-15/16 precursor. Thus, germline mutation of miR-15/16 can cause the indolent form of CLL both in human and mouse. By using different algorithms to identify targets of miR-15a and miR-16-1, it was found that BCL2, an oncogene protecting cells from apoptosis, was a putative target of both miR-15a and miR-16-1. Knock-down experiments showed this to be the case [29]. Thus, loss of miR-15a and miR-16-1 leads to high constitutive level of the oncogene BCL2, contributing to the development of an indolent B-cell leukemia. In follicular lymphoma, another common indolent B-cell malignancy, BCL2 gene becomes dysregulated as result of a t(14; 18) chromosome translocation, because of its juxtaposition to immunoglobulin enhancers, indicating that constitutive overexpression of BCL2 causes an indolent B-cell tumor. Moreover, it was also found that loss of miR-15a and miR-16-1 causes, although indirectly, overexpression of MCL1, another oncogene of the BCL2 family of inhibitors of apoptosis [30]. Interestingly, a recent clinical trial of CLL patients with ABT737, an inhibitor of BCL2 developed by Abbott, showed partial resistance of the leukemic cells to the drug, because ABT737 is specific for BCL2 but not for MCL1. Thus, treatment with either miR-15a or miR-16-1 may abrogate the resistance to the drug and improve the responsiveness. Additional experiments in vitro and in vivo also showed that miR-15a or miR-16-1 can be exploited to cause death of leukemic cells, suggesting the possibility of a microRNA-based therapeutic intervention [30].
To further demonstrate a causal role of miR-15a and miR-16-1 loss in the occurrence of CLL, Klein et al. [31] have applied a genetic approach generating sophisticated mouse models that have either deletion of DLEU2 (a noncoding RNA gene including the miR-15a and miR-16-1 cluster in its intron 4) together with both miRNA genes (MDR deleted) or deletion of the two miRNA genes only. After 15–18 months, about 5% of the animals displayed monoclonal B-cell lymphocytosis, which is a possible precursor to CLL. More importantly, 1/5 of the MDR-deleted and 1/8 of the miR-15a/16-1-deleted mice developed CLL or the related small cell lymphocytic leukemia. In addition, 9% of the MDR-deleted and 2% of the miR-15a/16-1-deleted animals developed a phenotype reminiscent of human diffuse large B-cell lymphoma, a disease known to progress from CLL at low frequency. Thus, the deletion of the MDR caused B-cell lymphoproliferative disorders, nicely recapitulating the spectrum of human CLL phenotypes.
More recently, Fabbri et al. [32] have shed more light into the molecular mechanisms behind the involvement of miR-15a and miR-16-1 in the biology of CLL, describing a feedback regulatory loop connecting miR-15a and miR-16-1, p53 and miR-34b/34c cluster, which critically influences the pathogenesis of CLL. The oncosuppressor p53 is indeed at the same time directly targeted by miR-15a and miR-16-1 and able to induce the expression of these microRNAs and of miR-34b/34c, which in turn directly regulate ZAP70. In this model, the loss of miR-15a/miR-16-1 expression, represented by CLLs with 13q deletions, not only shifts the balance toward higher levels of the antiapoptotic proteins BCL2 and myeloid cell leukemia sequence 1 (BCL2-related) (MCL1), as previously demonstrated, but also toward higher levels of the tumor suppressor protein TP53. Consequently, in patients with CLLs with 13q deletions, while the number of apoptotic cells may decrease because of the increased levels of antiapoptotic proteins, the TP53 tumor suppressor pathway remains intact, thus keeping the increase in tumor burden relatively low. This novel finding explains how 13q deletions are associated with the indolent form of CLL. Moreover, increased TP53 levels, as found in patients with CLLs with 13q deletions, are associated with transactivation of miR-34b/miR-34c and reduced levels of ZAP70, a tyrosine kinase relevant in the initial step of T-cell receptor-mediated signal transduction. Low expression levels of ZAP70 have been found to be positively correlated with survival in patients with CLL, further explaining the indolent course of CLL carrying 13q deletions.
Acute Myelocytic Leukemia
Acute myelocytic leukemia (AML) is a heterogeneous disease that includes several entities with different genetic abnormalities and clinical features. Garzon et al. have reported unique microRNA profiles in the main molecular and cytogenetic subgroups of AML. In addition, a subset of these microRNAs was associated with overall and disease-free survival [33]. Another study identified a microRNA-expression signature with prognostic significance in patients with AML belonging to the molecular high-risk group, including 12 microRNAs associated with event-free survival [34]. Five probes represented miR-181a and miR-181b; their increased expression was associated with a decreased risk of an event (failure to achieve CR, relapse, or death). This result was confirmed by a subsequent study showing that upregulated miR-181a predicted favorable outcome in CN-AML (AML with normal cytogenetics) [35].
Members of the miR-29 family are located in two clusters on two human chromosomes: miR-29b1/29a is located on chromosome 7q32, while miR-29b2/c is located on chromosome 1q23. Importantly, chromosome 7q is the region frequently deleted in myelodysplastic syndrome (MDS) and therapy-related AML [36]. Members of the miR-29 family have been shown to be downregulated in aggressive CLL [24], invasive breast cancer [37], lung cancer [38], and cholangiocarcinoma [39]. Transfection of miR-29b induces apoptosis in cholangiocarcinoma cell lines and reduces the tumorigenicity of lung cancer cells in nude mice. Moreover, it was shown that rhabdomyosarcoma looses miR-29 expression because of an elevation of NFkB and YY1 levels, and introduction of miR-29s into the tumor delays rhabdomyosarcoma progression in mice [40]. MiR-29s were also found to directly target MCL1 [39], an oncogene overexpressed in AMLs, and the de novo DNA methyltransferases DNMT-3A and -3B, while indirectly, through regulation of the transactivator Sp1, the maintenance DNA methyltransferase DNMT1 [35, 38]. Thus loss of miR-29 family members results in the constitutive overexpression of MCL1 and of DNMT, causing epigenetic changes characteristic of AML. These recent results suggest that loss of miR-29s may be important, perhaps critical, for the pathogenesis of a major group of MDSs and AMLs (Fig. 19.3).

Molecular alterations in CLL and AML. Deletion or downregulation of miR-15a/miR-16-1 cluster, located at chromosome 13q14.3 and directly involved in the regulation of BCL2 and MCL1 expression, represents an early event in the pathogenesis of CLL. During the evolution of malignant clones, other miRNAs (miRs) can be deleted (such as miR-29) or overexpressed (such as miR-155), contributing to the aggressiveness of B-CLL. Such abnormalities can influence the expression of other protein-coding genes (PCGs), such as the TCL1 oncogene, directly regulated by miR-29 and miR-181, or affect other noncoding RNAs (ncRNAs). The consequences of this steady accumulation of abnormalities are represented by the reduction of apoptosis and the induction of survival and proliferation of malignant B cells, leading to the evolution of more aggressive clones. Members of the miR-29 family, lost in AML and in other tumor types as lung cancer, have also been shown to directly target MCL1 and DNMT3A and B (adapted from Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27(34):5848–56. Reprinted with permission. © 2008 American Society of Clinical Oncology. All rights reserved)
Lymphoma
Early studies have shown that miR-155 is upregulated in a subgroup of Burkitt’s lymphoma, diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBL), and Hodgkin’s lymphoma [41, 42]. This microRNA is encoded by the terminal portion of the BIC (B-cell integration cluster) gene, which was originally identified as a common retroviral integration site in avian-leukosis-virus-induced B-cell lymphomas [43]. Dr. Croce’s group demonstrated that mice overexpressing miR-155 in B lymphocytes develop polyclonal preleukemic pre-B-cell proliferation followed by full-blown B-cell malignancy [44]. Moreover, two knock-out mice models have demonstrated a critical role of miR-155 in immunity by showing that BIC/miR-155−/− have defective dendritic cell functions, impaired cytokine secretion, and TH cells intrinsically biased toward TH2 differentiation [45, 46]. Moreover, miR-155 could represent the connection between inflammation, immunity, and cancer since its expression can be induced by mediators of inflammation and is involved in response to endotoxic shock [47].
He et al. [48] reported that miR-17-92 polycistron was upregulated in 65% of B-cell lymphoma patients and demonstrated in a mouse model that this miR cluster cooperates with the oncogene MYC in accelerating tumor development. More recently, a different group observed that the overexpression of miR-17-92 in lymphocytes caused a lymphoproliferative disease, autoimmunity, and premature death [49]. The enhanced proliferation of the transgenic lymphocytes was mediated by direct regulation of proapoptotic PTEN and Bim. O’Donnell et al. [50] investigated the regulation of miR-17-92 in lymphoma, demonstrating that the expression of this cluster is directly activated by the oncogene c-Myc. Moreover, miR-17-92 cluster, as well as its paralog, miR-106b-25 [51], establishes with the transcription factor E2F1, a downstream target of c-Myc, a negative feedback loop: E2F1 represents indeed a direct target of the two microRNA clusters, but it also induces their expression. Thus, MYC simultaneously activates E2F1 transcription and limits its expression, allowing a tightly controlled proliferative signal.
Multiple Myeloma
Few recent reports have linked microRNAs to this plasma cell malignancy, as the aberrant expression of miR-335, miR-342-3p, and miR-561 in comparison to normal plasma cells [52] or the Stat3-mediated activation of the oncogenic miR-21 in response to IL-6 [53]. Mir-15a and miR-16-1 have been described as oncosuppressor microRNAs also in this tumor subtype [54–56]. Pichiorri et al. [57] described a microRNA signature characteristic of this neoplasia. They evaluated by both microarray analysis and real-time PCR the expression of microRNAs in MM-derived cell lines, CD138+ bone marrow PCs from subjects with MM or monoclonal gammopathy of undetermined significance (MGUS), and normal donors, identifying the oncogenic miR-21 and miR-181 among the microRNAs aberrantly expressed. Two miRNAs, miR-19a and 19b, part of the miR-17-92 cluster, were also shown to downregulate expression of SOCS-1, a gene frequently silenced in MM that plays a critical role inhibiting IL-6 growth signaling. Moreover, xenograft studies using human MM cell lines treated with miR-19a and b precursors or miR-181a and b antagonists resulted in significant suppression of tumor growth in nude mice, confirming the involvement of these microRNAs in the development of multiple myeloma (MM). More recently, the same group [58] have demonstrated that miR-192, 194, and 215, which are downregulated in a subset of newly diagnosed MMs, can be transcriptionally activated by p53 and modulate MDM2 expression. In addition, miR-192 and 215 target the IGF pathway, preventing enhanced migration of plasma cells into bone marrow.
MicroRNAs in Solid Malignancies
MicroRNAs in Breast Cancer
One of the first solid tumors to be profiled for microRNAs expression was, in 2005, breast cancer. Iorio et al. [37] described indeed the first microRNA signature characteristic of breast carcinoma, identifying 13 microRNAs able to discriminate tumors and normal tissues with an accuracy of 100% (Fig. 19.4). Among the most significant microRNAs differentially expressed, some were extensively studied since their initial discovery and revealed an important role on the biology of breast cancer: miR-21, overexpressed in breast carcinoma, has been demonstrated to mediate cell survival and proliferation directly targeting the oncosuppressor genes PTEN, PDCD4, and TPM1, and it has been associated with advanced clinical stage, lymph node metastasis, and patient poor prognosis [59, 60] also in PABC (pregnancy-associated breast cancer) [61]. miR-21 has been also detected as circulating microRNA, freely present in the circulation [62, 63] or in exosomes, as described in ovarian cancer [64]. Very recently, Ota et al. [65] have interestingly demonstrated that increased expression of miR-21 can be found in bone marrow of breast cancer patients, and that the level of this microRNA and its target PDCD4 have a prognostic value.

Cluster analysis and PAM prediction in breast cancer and normal breast tissues. (a) Tree generated by a cluster analysis showing the separation of breast cancer from normal tissues on the basis of miRNA differentially expressed (P < 0.05) between breast cancer and normal tissue. The bar at the bottom indicates the group of cancer samples (red) or the group of normal breast tissues (yellow). (b) PAM analysis displaying the graphical representation of the probabilities (0.0–1.0) of each sample for being a cancerous or a normal tissue (adapted from Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70)
Moreover, miR-21, one of the first cancer microRNAs described, has been found overexpressed in a variety of other malignancies: glioblastoma [66, 67], ovary [68], lung [69, 70], and more [71]. In colorectal cancer and pancreas endocrine and exocrine tumor, miR-21 overexpression is also associated with poor survival and poor therapeutic outcome [72–74].
Conversely, downregulated microRNAs, as miR-125a and b and miR-205, regulate oncogenes as tyrosine kinase receptors HER2 and HER3, respectively [75, 76].
Let-7, tumor suppressor miR initially discovered in C. elegans, where it induces cell cycle exit and terminal differentiation, has been described as a new regulator of self-renewal and tumorigenicity of breast cancer cells [77], targeting molecules originally described in lung cancer: RAS [78] as well as the oncogene HMGA2 [79], and even MYC itself [80]. Overexpression of let-7 miRNA family can suppress tumor development in mouse models of breast and lung cancer [77, 81].
In the signature published in 2005, we could also identify miRNAs differentially expressed according to specific biopathological features, such as grade and stage of the disease, vascular invasion, proliferation index, and expression of hormone receptors [37]. In particular, we could identify a panel of miRNAs differentially expressed in estrogen receptor (ER)+ versus ER−breast carcinoma patients, being miR-191 and miR-26, the most significantly overexpressed, and miR-206, the most significantly downmodulated. miR-206 has been lately demonstrated by another group to directly target ERα [82]. Moreover, Foekens et al. described a subset of miRNAs significantly associated with ER + luminal signature, identifying in particular four miRNAs associated with breast cancer aggressiveness [83]. Among them, miR-128a has also been implicated in the resistance to AI (aromatase inhibitor) letrozole [84]. In a recent study performed in Dr. Croce’s laboratory, we demonstrated [85] that miR-221 and miR-222 are involved in a negative feedback regulation with ERalpha, been able to directly target the receptor (as demonstrated also by Zhao et al. [86], which in turn represses the transcription of the two miRNAs through direct binding to responsive elements on their promoter sequences. Moreover, other groups have demonstrated that overexpression of miR-221 and miR-222 is responsible for resistance to antiestrogenic therapies, such as tamoxifen [86, 87] and fulvestrant [88].
Our group particularly focused on the study of miR-205 involvement in breast cancer biology. Previous studies showed that miR-205 expression is significantly underexpressed in human breast cancer [89, 90] and associated with absence of vascular invasion [37], although it has also been shown to be upregulated in other tumors types, as ovarian cancer [68]. We recently demonstrated [89] that miR-205 is able to interfere with the HER receptor family-mediated survival pathway by directly targeting HER3 receptor and thus inhibiting its downstream mediator Akt. In addition, other studies indicated that miR-205 is a negative regulator of the epithelial–mesenchymal transition (EMT), an early phase of the process of metastasis, targeting the transcription factors ZEB1 and ZEB2, and that expression of miR-205 is lost in mesenchymal breast cancer cell lines [91] and triple-negative breast cancer [92]. Moreover, miR-205 also targets VEGF-A, a factor which plays a key role in the process of invasion and metastasis [90]. Finally, in a very recent study, silencing of miR-200 family and miR-205 has been associated with EMT and acquisition of stem-like properties in carcinogen-induced transformation of human lung epithelial cells [93].
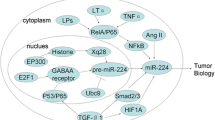
Table 19.2 summarizes the information available to date about some of the most important miRNAs involved in human breast cancer, and the cartoon reported in Fig. 19.5 illustrates the involvement of microRNAs in the complicated network of molecules regulating breast cancer biology.

miRNAs take their place in breast cancer biology. Summary of the interconnections between miRNAs and tumor suppressor genes and oncogenes in breast cancer (adapted from Iorio MV, Casalini P, Tagliabue E, et al. MicroRNA profiling as a tool to understand prognosis, therapy response and resistance in breast cancer. Eur J Cancer. 44(18):2753–9. ©2008. With permission from Elsevier)
Lung Cancer
One of the first oncosuppressor microRNAs identified is let-7a, which regulates RAS [78] as well as the oncogene HMGA2 [78, 79], and even MYC itself [80]. Overexpression of let-7 microRNA family can suppress tumor development in mouse models of breast and lung cancer [77, 81]. In the two most common forms of non-small cell lung cancers (adenocarcinomas and squamous cell carcinomas), high expression of miR-155 and low expression of oncosuppressor let-7 correlate with poor prognosis [69]. The association of let-7a with survival was also confirmed by an independent study performed by Yu et al. [94], who identified a miR signature as independent predictor of cancer relapse and survival of NSCLC (non-small cell lung cancers) patients.
As in other tumor types, also in lung cancer, microRNAs can represent accurate diagnostic markers. However, data are not always consistent: whereas in 2009, it has been described that squamous and nonsquamous NSCLCs can be distinguished according to the expression of miR-205 [95]; more recently, another group [96] underlines how, despite the relative quantification of miR-205 and miR-21 seems to be a promising diagnostic tool to discriminate adenocarcinomas (ADCs) compared with squamous cell carcinomas (SQCCs), the molecular approach is still not completely satisfactory as it may misclassify a nonnegligible percentage of cases. Therefore, the authors state that it cannot represent a substitute of accurate morphologic and immunophenotypical characterization of tumors, but it could be used as an adjunctive diagnostic criterion in selected cases.
MicroRNAs have also been found in the circulation in lung cancer patients, either free or associated with exosomes: Rabinowits et al. [97] found a similarity between the circulating exosomal miRNA and the tumor-derived miRNA patterns; in addition Hu et al. [98] found that levels of four miRNAs (i.e., miR-486, miR-30d, miR-1, and miR-499) present in the serum of lung cancer patients were significantly associated with overall survival. More recently, Sozzi’s group [20] has identified microRNA-expression signatures with strong predictive, diagnostic, and prognostic potential analyzing plasma samples of lung cancer patients collected 1–2 years before the onset of disease, thus suggesting their possible use as noninvasive biomarkers for early diagnosis.
Notably, microRNAs are also stably present in sputum and can be used as highly sensitive and specific markers for early detection of lung adenocarcinoma, in particular, a panel of four microRNAs (miR-21, miR-486, miR-375, and miR-200b) [99].
miR-200c, demonstrated to inhibit the EMT process in breast cancer [91], is lost in more aggressive and invasive NSCLC (non-small cell lung cancer) cell lines and associated with chemoresistance [100].
MiR-21, known onco-microRNA in several human tumors, seems to play an important role also in lung carcinogenesis, both in smokers and in never-smokers, being overexpressed and further enhanced by the activated EGFR signaling pathway [101]. Moreover, it has been associated with disease progression and survival [102], also in stage I lung tumors, as recently reported [103].
Among the microRNAs acting as oncogenes, Garofalo et al. [104] have shown that miR-221 and 222 are overexpressed in aggressive non-small cell lung cancer and hepatocarcinoma cells, and that they induce TRAIL resistance and enhanced cellular migration by targeting PTEN and TIMP3 tumor suppressors and activation of AKT pathway and metallopeptidases. Moreover, they demonstrated that the MET oncogene is involved in miR-221 and 222 activation through the c-Jun transcription factor.
Focusing on (c) hepatocellular carcinoma (HCC), Murakami et al. [105] reported that miR-222, miR-106a, and miR-17-92 clusters are associated with the degree of tumor differentiation, while high levels of the oncosuppressor miR-125b correlate with good survival [106]. MiR-125b has also been shown to induce growth inhibition in vitro in a model of human thyroid anaplastic carcinoma [107]. Other studies focused on the identification of molecules targeted by microRNAs deregulated in HCC: miR-122a, downmodulated in HCC, directly regulates Cyclin G1 [108], and miR-221, upregulated in this neoplasia, directly targets p27 [109], as also shown in thyroid cancer [107], and contributes to liver tumorigenesis [110], glioblastoma [111], prostate cancer [112], and melanoma [113]. One of the first evidences proving miR alteration in human melanoma is a genomic study performed by Zhang et al. [114], who reported DNA copy abnormalities in microRNA genes also in two other epithelial tumors, breast, and ovary. Interestingly, the results obtained by this genomic analysis were largely overlapping with the expression profiles on the same tumor types [37, 68].
Interestingly, the downregulation of miR-26 has been associated to poor prognosis but better response to interferon therapy [115].
Ovarian Cancer
The first report of a putative involvement of miRNAs in the biology of human ovarian cancer was the genomic study performed by Zhang et al., who used an array comparative genomic hybridization (aCGH) approach to identify miRNA loci gained/lost in ovarian cancer, breast cancer, and melanoma [114]. Many of the miRNAs resulting from this study were later confirmed to be differentially expressed in the miRNA expression profiling performed by our group in 2007 [68].
After this initial evidence, several groups have investigated the role of miRNAs in the pathogenesis of ovarian cancer, either as biomarkers, potential research tools, or targets for specific therapies. miRNA let-7i was recently found to be a tumor suppressor significantly downregulated in platinum-resistant ovarian tumors, and let-7i gain of function restored drug sensitivity of chemoresistant ovarian cancer cells, thus representing a candidate biomarker and therapeutic target [116]. An oncosuppressive role for miR-15/16 has been described also in ovarian cancer, where these two miRNAs regulate the expression of the oncogenic protein Bmi1 [117].
In another study, 27 miRNAs significantly associated with chemotherapy response, showing that (similar to DNA methylation) miRNAs represent possible prognostic and diagnostic biomarkers for ovarian cancer [118]. miR-214 has been reported to target PTEN, thus contributing to cisplatin resistance [119]. Interestingly, levels of Dicer and Drosha mRNA in ovarian cancer cells have been associated with clinical outcome [120].
Circulating microRNAs have been found in sera of ovarian cancer patients: levels of eight microRNAs (miR-21, miR-141, miR-200a, miR-200c, miR-200b, miR-203, miR-205, and miR-214), previously demonstrated as diagnostic, were compared in exosomes isolated from sera specimens of women with benign disease and various stages of ovarian cancer, and the expression profile resulted similar between tumor cells and tumor-derived exosomes in comparison with respective controls [120].
Interestingly, miR-200c, downregulated in breast cancer, where it inhibits the EMT process [121], is overexpressed in ovarian cancer [68], where the targeting of ZEB1 and 2 mediates the opposite phenomenon, the mesenchymal to epithelial transition (MET) [122].
MicroRNAs in Invasion, Angiogenesis, and Metastasis
MicroRNAs have been demonstrated to exert a crucial role not only in controlling the primary tumor growth by regulating pathways involved in cell cycle and proliferation, but also to be determinant in modulating migration, invasion, and the interaction with the microenvironment, mechanisms related to the acquisition of a more aggressive phenotype, and promoting the onset of the metastatic process: the scientific world has coined the definition “metastomiRs.”
One of the first studies reporting a prometastatic role for a miRNA was published by Ma et al. [123]. They observed that miR-10b was downmodulated in all the breast carcinomas from metastasis-free patients, as previously reported [37], but surprisingly, 50% of metastasis-positive patients had elevated miR-10b levels in their primary tumors. Induced by transcription factor Twist, miR-10b inhibits the translation of mRNA encoding homeobox D10 (HOXD10), releasing the expression of the prometastatic gene RHOC and, thus, leading to tumor cell invasion and metastasis.
The same group has later identified miR-9 as a new “metastomiR”: activated by MYC and MYCN and correlated with tumor grade and metastatic status, miR-9 directly targets CDH1, the E-cadherin-encoding messenger RNA, leading to increased cell motility and invasiveness, activation of beta-catenin signaling, and upregulation of VEGF. Moreover, overexpression of miR-9 in otherwise nonmetastatic breast tumor cells enables these cells to form pulmonary micrometastases in mice. Conversely, inhibiting miR-9 by using a “miRNA sponge” in highly malignant cells inhibits metastasis formation [124].
Through a functional screen aimed to discover miRNAs promoting cell migration in vitro, Huang et al. [125] identified miR-373 and validated its metastatic potential in tumor transplantation experiments using breast cancer cells.
MiR-34a, which is lost in several tumor types and involved into the network mediated by the well-known “genome guardian” p53 [126], inhibits migration and invasion by downregulation of MET expression in human HCC cells [127]. Oncosuppressive miR-145 inhibits not only tumor growth but also cell invasion and metastasis by direct targeting of mucin 1 [128].
EMT is thought to promote malignant tumor progression, and several groups have recently investigated whether miRNAs are involved in this process, and there are data to support this hypothesis. Indeed, members of the miR-200 family of miRNAs and miR-205 have been shown to reduce cell migration and invasiveness targeting ZEB transcription factors, known inducers of EMT [91, 129], and PKCε, as demonstrated in prostate cancer [130]. In addition ZEB1, which promotes not only tumor cell dissemination but also the tumor-initiating capacity, has been shown to repress expression of miR-200 family [131, 132] and stemness-inhibiting miR-203 [133].
The oncogenic miR-21 stimulates intravasation, extravasation, and metastasis in different tumor types, included colorectal cancer [134] and breast cancer [135], whereas oncosuppressor miR-205 has the opposite effects, reducing invasion in vitro and suppressing lung metastasis in vivo [90]. With the same aim of searching for regulators of breast cancer metastasis, Tavazoie et al. [136] identified miR-126 and miR-335 as metastasis suppressors: reduced levels of the two miRNAs are associated with poor metastasis-free survival of breast cancer patients, while their reexpression inhibits metastasis in a cell transplantation model.
Interestingly, it has been recently observed that primary tumors and metastases from the same tissue show a similar pattern of miRNAs expression [137]. Being a more accurate classifier than mRNA expression studies, miRNA profiling has thus revealed the potential to solve one of the most demanding issues in cancer diagnostics: the origin of metastasis of unknown primary tumors.
In the metastatic process, neo-angiogenesis is the crucial step allowing cells to reach and disseminate through the systemic circulation. miRNAs can control tumor progression also at this level, either promoting or inhibiting the proliferation of endothelial cells. miR-221 and miR-222 repress the proliferative and angiogenic properties of c-Kit in endothelial cells [138], and miR-221 downregulation has been recently linked to tumor progression and recurrence in a high-risk prostate cancer [116], whereas hypoxic reduction of miR-16, miR-15b, miR-20a, and miR-20b expression directly targets VEGF, supporting the angiogenic process [139]. On the other hand, VEGF levels can be indirectly increased by miR-27b, through reduction of the zinc finger protein ZBTB10 and the consequent activation of Sp transcription factor [140], and by miR-126, through repression of Sprouty-related protein SPRED1 and phosphoinositol 3-kinase regulatory subunit 2 (PIK3R2) [141]. Angiogenesis can be also promoted by miR-210, activated by hypoxia and directly represses endothelial ligand ephrin A3 [142], and by the miR-17-92 cluster, which sustains MYC angiogenic properties through repression of connective tissue growth factor (CTGF) and the antiangiogenic adhesive glycoprotein thrombospondin 1 (TSP1) [143], also targeted by miR-27b and let-7f [144].
Interestingly, Dicer expression seems to be associated with metastatic properties: a microRNA family, miR-103/107, which attenuates miRNA biosynthesis by targeting Dicer, a key component of the miRNA processing machinery, is associated with metastasis and poor outcome in human breast cancer. Functionally, miR-103/107 confers migratory capacities in vitro and empowers metastatic dissemination of otherwise nonaggressive cells in vivo. Inhibition of miR-103/107 opposes migration and metastasis of malignant cells. At the cellular level, a key event fostered by miR-103/107 is induction of epithelial-to-mesenchymal transition (EMT), attained by downregulating miR-200 levels [145]. Metastasis suppression is also mediated by TAp63, a p53 family member, which coordinately regulates Dicer and miR-130b [146].
MicroRNA Expression Regulation
General Principles of miRNA Genomic Organization
miRNAs are frequently expressed as polycistronic transcripts. To date, 1,048 human miRNA precursor sequences have been deposited in miRBase [147]. Approximately one-third (390) of these miRNAs are located in 113 clusters, each measuring ≤51 kb in the human genome (51 kb being the longest distance between miRNAs belonging to the same cluster). These miRNA clusters are coexpressed based on evidence from miRNA profiling data from a variety of tissues and cell lines [148, 149]. Presentation of miRNA profiles in the form of expression clusters provides a readily interpretable summary of expression data and stresses the importance of cistronic expression regulation; dysregulation of one member of the cluster should be accompanied by similar dysregulation of other cluster members. Since miRNA genes are frequently multicopy, determining the relative contribution of each genomic location to mature miRNA expression is challenging.
miRNA Expression Regulation: Genomic and Epigenetic Mechanisms
miRNA expression can be altered by several mechanisms in human cancer: chromosomal abnormalities, as suggested by the evidence that microRNAs are frequently located in regions of the genome involved in alterations in cancer [15], and recently confirmed by a genetic study in ovarian carcinoma, breast cancer, and melanoma [114]; mutations, as the inherited mutations in the primary transcripts of miR-15a and miR-16-1 responsible for reduced expression of the two microRNAs in vitro and in vivo in CLL [28]; polymorphisms (SNPs), as described in lung cancer [98]; defects in the miRNA biogenesis machinery, as supported by the changes in microRNA expression as a consequence of an altered Drosha or Dicer activity [120, 150–152], and epigenetic changes, as altered DNA methylation (Fig. 19.6).

Mechanisms of miRNA regulation. The deregulated miRNA expression observed in cancer can be caused by chromosomal abnormalities, mutations, polymorphisms (SNPs), transcriptional deregulation, defects in the miRNA biogenesis machinery, and epigenetic changes (adapted from Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27(34):5848–56. Reprinted with permission. ©2008 American Society of Clinical Oncology. All rights reserved)
Moreover, aberrant expression of Drosha or Dicer enzymes has been correlated with disease progression and outcome in different human tumors, even though results are still controversial: strong expression of the central microRNA biosynthesis enzyme Dicer predicts poor prognosis in patients with colorectal cancer [153] and prostate cancer, whereas in breast, lung, and ovarian cancer [120] and neuroblastoma [154], Dicer has been shown to be a marker of good prognosis. Thus further studies on the cellular functions of Dicer need to address these issues.
An extensive analysis of genomic sequences of miRNA genes have shown that approximately half of them are associated with CpG islands, suggesting that they could be subjected to this mechanism of regulation [155]. Several evidences have indeed proved that an altered methylation status can be responsible for the deregulated expression of microRNAs in cancer, as the silencing of putative tumor suppressor microRNAs: treating T24 bladder cancer cells and human fibroblasts with DNA methyltransferase (DNMT) inhibitor 5-Aza-2′-deoxycytidine, Saito et al. [156] observed a strong upregulation of miR-127, microRNA characterized by a CpG island promoter, able to target the proto-oncogene BCL-6, and silenced in several cancer cells. With the same approach of unmask epigenetically silenced microRNAs inducing chromatin remodeling by drug treatment, it has been demonstrated that miR-9-1 is hypermethylated and consequently downmodulated in breast cancer [157], as well as the clustered miR-34b and miR-34c in colon cancer [158].
Conversely, the upmodulation of putative oncogenic microRNAs in cancer can be due to DNA hypomethylation, as shown in lung adenocarcinoma for let-7a-3 [159] or in epithelial ovarian cancer for miR-21 [68].
A different approach to identify epigenetically regulated microRNAs was represented by the miR profiling of DNMT1- and DNMT3b-deficient colorectal cancer cells: among the 18 microRNAs upmodulated in comparison to WT cells, the only one resulting unmethylated in normal tissue but hypermethylated, and thus silenced, in tumor was miR-124a, embedded in a large CpG island and able to target cyclin D kinase 6, which mediates the phosphorylation of RB tumor suppressor gene [160].
Methylation is not the only epigenetic mechanism that can affect microRNAs expression: Scott et al. [161] showed that in SKBR3 breast carcinoma cells histone deacetylase inhibition is followed by the extensive and rapid alteration of microRNAs levels.
The existence of epigenetic drugs, such as DNA demethylating agents and histone deacetylase inhibitors, able to reverse an aberrant methylation or acetylation status, raises the intriguing possibility to regulate microRNA levels, for example, to restore the expression of tumor suppressor microRNAs, thus reverting a tumoral phenotype.
To complicate the scenario connecting microRNAs and epigenetics, microRNAs themselves can regulate the expression of components of the epigenetic machinery, creating a highly controlled feedback mechanism: miR-29 family directly targets the de novo DNA methyltransferases DNMT-3A and -3B, while indirectly, through regulation of the transactivator Sp1, the maintenance DNA methyltransferase DNMT1. Interestingly, introduction of miR-29s into lung cancers and AMLs results in reactivation of silenced tumor suppressors and inhibition of tumorigenesis [35, 38]. Loss of miR-290 cluster in Dicer-deficient mouse ES cells leads to the downregulation of DNMT3a, DNMT3b, and DNMT1 through upmodulation of their repressor, RBL-2, proven target of miR-290 [162, 163]; miR-1, involved in myogenesis and related diseases, directly targets HDAC4 [164].
Alterations in miRNA Transcriptional Regulation
Some autonomously expressed miRNA genes have promoter regions that allow miRNAs to be highly expressed in a cell-type-specific manner and can even drive high levels of oncogenes in cases of chromosomal translocation. The miR-142 gene, a marker of hematopoietic cells, is located on chromosome 17 and was found at the breakpoint junction of a t(8;17) translocation, which causes an aggressive B-cell leukemia due to strong upregulation of a translocated MYC gene. The translocated MYC gene, which was also truncated at the first exon, was located only four nucleotides from the 3′-end of the miR-142 precursor, placing the translocated MYC under the control of the upstream miR-142 promoter. In an animal model for HCC, a similar event placed MYC downstream the miR-122a promoter active only in hepatocytes.
Many transcription factors regulate miRNA expression in a tissue-specific and disease state-specific fashion, and some miRNAs are regulated by well-established tumor suppressor or oncogene pathways, such as TP53, MYC, and RAS. The miRNA and its transcriptional regulators can participate in complex feedback regulation loops. Examples include the TP53-regulated miR-34a [165, 166], the RAS-regulated miR-21 [167], and the MYC-regulated miR-17-92 cluster [143].
MicroRNAs/Anti-microRNAs in Cancer Treatment
An increasing body of evidence collected up to date demonstrates how microRNAs could represent valid diagnostic, prognostic, and predictive markers in cancer. Indeed, the aberrant microRNA expression correlates with specific biopathological features, disease outcome, and response to specific therapies in different tumor types. Moreover, several indications in preclinical models underline the feasibility and the efficacy of a microRNA-based therapy in cancer, using these small molecules as both targets and tools (Fig. 19.7 and Table 19.3).

miRNAs as therapeutic tools. The reintroduction by transfection of synthetic miRNAs lost during cancer development or progression or the inhibition of oncogenic miRs by using anti-miRNA oligonucleotides could help counteract tumor proliferation, extended survival, and the acquisition of a metastatic potential, thus representing potential therapeutic tools (adapted from Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27(34):5848–56. Reprinted with permission. © 2008 American Society of Clinical Oncology. All rights reserved)
Reintroduction of miR-15a/16-1, for example, induces apoptosis in leukemic MEG01 cells and inhibits tumor growth in vivo in a xenograft model [30], whereas the inhibition of oncogenic miR-21 with antisense oligonucleotides generates a proapoptotic and antiproliferative response in vitro in different cellular models and reduces tumor development and metastatic potential in vivo [168].
Moreover, microRNAs involved in specific networks, as the apoptotic, proliferation, or receptor-driven pathways, could likely influence the response to targeted therapies or to chemotherapy: inhibition of miR-21 and miR-200b enhances sensitivity to gemcitabine in cholangiocytes, probably by modulation of CLOCK, PTEN, and PTPN12 [169], whereas reintroduction of miR-205 in breast cancer cells can improve the responsiveness to tyrosine kinase inhibitors through HER3 silencing [89], and enforced expression of miR-15b or miR-16 could sensitize multidrug-resistant gastric cells to vincristine-induced apoptosis [170].
Nevertheless, effective delivery into target tissues remains a major hurdle for microRNA-based therapy, including the applications of antagomirs and synthetic miRNA duplexes.
In the case of reduction in the levels of the mature microRNA (due to deletion present in the microRNA gene or to other mechanisms as defects in the processing machinery and aberrant transcription), the therapeutic approach could be the exogenous delivery of synthetic double-stranded hairpin by complexing with lipids or delivery proteins. As reported by Tazawa et al. [171], miR-34a transiently inhibits human colon cancer tumor progression when administered subcutaneously in complexes with atelocollagen, recently shown to be a very useful system to efficiently deliver small interfering RNA molecules into tumors in vivo. Chen et al. have developed a liposome-polycation-hyaluronic acid (LPH) nanoparticle formulation modified with tumor-targeting single chain antibody fragment (scFv) for systemic delivery of miR-34a to lung metastasis of murine melanoma cells [172].
However, the vulnerability of unmodified dsRNAs to nucleases in vivo limits the use of this class of compound to privileged local environments where locally administration is feasible. Using a conditional mouse lung cancer model, in which the expression of oncogenic K-ras could be conditionally activated, Esquela-Kerscher et al. showed that the intranasal administration of an adenovirus expressing let-7a RNA hairpin reduced tumor formation in vivo [173]. In 2009, Kota et al. [174] presented a study on therapeutic microRNA delivery suppressing tumor formation in a murine liver cancer model. The authors demonstrated that systemic administration of miR-26a in a mouse model of HCC using adeno-associated virus (AVV) resulted in inhibition of cancer cell proliferation, in the induction of tumor-specific apoptosis. These results are consistent with previous findings made by that same group, which demonstrated that MYC-induced liver tumors result in concomitant downregulation of various microRNAs [175].
In short-term experiments of cardiac hypertrophy, conducted by Carè et al., [176], overexpression of miR-133 by adenovirus delivery resulted in a significant reduction in the size of left ventricular cardiac myocytes and a significant decrease in the expression of fetal genes. To achieve stable miRNA reintroduction, the expression can be enforced by a viral vector with Pol III promoter upstream an artificial short hairpin RNA (shRNA) that bypasses Drosha processing, yet is cleaved and loaded into miRISC by Dicer. Most constructs have used Pol III promoters, including U6, H1, and tRNA [177–179]; however, these promoters have no cell specificity. Moreover, exceedingly, high levels of shRNA expression increase the probability of off-target silencing and elicit nonspecific effects such as interferon response, and they can also saturate exportin 5 pathway of endogenous miRNAs with fatal consequences [180]. Alternatively, the entire pri-miRNA can be expressed from an RNA Pol II promoter, leaving open the possibility for tissue-specific or induced ectopic miRNA expression. Furthermore, most miRNAs are known to be downstream Pol II promoters, within known protein-coding genes and expressed by Pol II activity [181]. Therefore, strategies using Pol II-directed synthesis of shRNA that mimic the natural miRNA synthesis could be an efficient therapeutic approach.
To achieve miRNA loss of function, chemically modified anti-miR oligonucleotides (AMOs) have been developed [182]. The most important property of such oligonucleotides is the specificity and high binding affinity to RNA, and a number of them have been pursued in clinical trials. miRNA downregulation has been achieved by using 2-O-methyl oligonucleotides [75, 76]. miR-122 inhibition was obtained by treating mice with AMOs containing 2-O-methoxyethyl groups resulted in reduced plasma cholesterol [183]. Intravenous administration of cholesterol-conjugated AMOs against miR-16, miR-122, miR-192, and miR-194 resulted in a marked reduction of corresponding miRNA levels in liver, lung, kidney, heart, intestine, fat, skin, bone marrow, muscle, ovaries, and adrenals [182, 184].
Very interestingly, Ma et al., after the demonstration of the crucial role of miR-10b as metastomiR in breast cancer, have exploited a possible therapeutic application, reporting that systemic treatment of tumor-bearing mice with miR-10b antagomirs suppresses breast cancer metastasis [124].
Other modified AMOs are represented by locked nucleic acid (LNA) oligonucleotides, able to inhibit exogenously introduced miRNAs with high specificity [185].
The first clinical trial in human of LNA-anti-miR (a placebo-controlled, double-blind, randomized, single-dose, dose-escalating safety study of SPC3649 in a total of 64 healthy male volunteers) has been conducted by Denmark’s Santaris to study SPC3649 (LNA-anti-miR-122) (ClinicalTrials.gov Identifier: NCT00688012). miR-122 is an abundant miRNA in the liver. The hepatitis C virus (HCV) genome harbors two closely spaced miR-122 target sites in the 5′ noncoding region required for HCV replication. Kauppinen et al. showed that administration of LNA-anti-miR into mice resulted in a dose-dependent depletion of mature miR-122 [ 186]. An efficacy study was later conducted by Elmen et al. on nonhuman primates [187], where they observed a dose-dependent sequestration of mature miR-122 and a long-lasting decrease of total plasma cholesterol. The same group has very recently developed tiny (8-mer) LNAs to obtain the simultaneous inhibition of miRNAs within families sharing the same seed, with concomitant upregulation of direct targets [188].
However, the same limitations encountered with the application of synthetic miRNA duplexes are encountered in the applications of antagomirs, namely, their effective delivery into target tissues.
An interesting approach to overcome these problems is to target miRNA by saturating them with target mRNAs. Ebert et al. [189] developed miRNA inhibitory transgenes, called “microRNA sponges,” expressing an mRNA containing multiple tandem binding sites for an endogenous miRNA and thus able to stably interact with the corresponding miRNA and prevent its association with its endogenous targets. Both designed polymerase Pol II- and Pol III-driven miRNA sponges showed more efficiency for miRNA inhibition compared to standard 2′-O-Me antagomirs.
Lentiviral vectors have proven to be effective tool to ectopically express miRNAs using suitable transcriptional control units. It has been reported by Gentner et al. [179, 190] that stable miRNA-223 knockdown can be achieved in vivo by transducing bone marrow stem and progenitor cells with multiple target sequences from strong promoters and transplanting them into lethally irradiated congenic recipients. They demonstrated that overexpressing miR-targets specifically affects the targeted miRNA rather than saturating the effector pathway. However, the need for strong promoters and multiple vector integrations to obtain a high miR-target expression could increase the risk of insertional mutagenesis in target cells, potentially confounding the identification of miRNA knockdown phenotypes, and thus representing a potential limitation of this strategy.
Su et al. [191] have applied a nanotechnologic approach to the use of anti-miRNAs: systemic delivery of a chemically stabilized anti-miR-122 complexed with interfering nanoparticles (iNOPs) effectively silences the liver-expressed miR-122 in mice, thus resulting in lowering of plasma cholesterol.
Beside targeted therapies and chemotherapy, microRNAs could also alter the sensitivity to radiotherapy, as recently reported by Slack’s group [192]: a potential therapeutic use for anti-miR-34 as a radiosensitizing agent in p53-mutant breast cancer could be considered; in lung cancer cells, let-7 family can suppress the resistance to anticancer radiation therapy, probably through RAS regulation.
Evidences described up to date provide the experimental bases for the use of microRNAs as both targets and tools in anticancer therapy, but there are at least two primary issues to address to translate these fundamental research advances into medical practice: the development of engineered animal models to study cancer-associated microRNAs and the improvement of the efficiency of miRNAs/anti-miRs delivery in vivo.
Concluding Remarks and Future Perspectives
Fifteen years ago, when microRNAs seemed just a peculiar discovery in C. elegans, the scientific world did not probably even imagine that those small noncoding molecules would have a large impact on our understanding of cellular biology and gene regulation.
MicroRNAs contribute to maintain the balance among genes regulating cells’ fate, and their deregulation, a frequent hallmark in different human malignancies, can destabilize this equilibrium, thus contributing to cancer development and/or progression, from initiation to metastatic disease. However, despite the increasing and encouraging body of evidence linking microRNAs to cancer biology, many important questions remain to be addressed: in fact, although the identification and validation of microRNA targets greatly improved in the last few years, we still know very little about the cellular and molecular circuits where they are involved. The scenario is surely complicated by the ability of microRNAs to target multiple molecules, sometimes belonging to related pathways, and at the same time by the redundancy existing among microRNAs. This gives rise to a complex regulatory network where biological effects and properties of a particular microRNA do not always allow a linear explanation.
Improvement of computation programs of microRNA targets prediction and experimental methods of validation will certainly contribute to elucidate their mechanisms of action, and genetically modified murine models will likely help in determining the oncogenic and tumor suppressor potential of individual microRNAs.
Data available to date clearly support the involvement of microRNA in cancer etiology and strongly suggest a possible use of these molecules as markers of diagnosis and prognosis, and eventually, as new targets or tools for a specific therapy (Fig. 19.8): stepping from the bench to clinical applications would be the next great challenge in cancer research.

Workflow summarizing the progressive steps and the different issues considered to investigate the hypothesis of a concrete role of miRNAs in cancer biology and to support their possible clinical applications
Abbreviations
- 3′-UTR:
-
3′-Untranslated region
- AML:
-
Acute lymphocytic leukemia
- CLL:
-
Chronic lymphocytic leukemia
- EMT:
-
Epithelial–mesenchymal transition
- HCC:
-
Hepatocellular carcinoma
- MM:
-
Multiple myeloma
- Pol II:
-
Polymerase II
- Pre-miR:
-
Precursor miRNA molecule
- Pre-mRNA:
-
Precursor mRNA molecule
- Pri-miR:
-
Primary miRNA transcript
References
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75(5):843–54.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6(5):376–85.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610.
Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17(24): 3011–6.
Lingel A, Simon B, Izaurralde E, Sattler M. Structure and nucleic-acid binding of the Drosophila Argonaute 2 PAZ domain. Nature. 2003;426(6965):465–9.
Tahbaz N, Kolb FA, Zhang H, Jaronczyk K, Filipowicz W, Hobman TC. Characterization of the interactions between mammalian PAZ PIWI domain proteins and Dicer. EMBO Rep. 2004;5(2):189–94.
Mourelatos Z, Dostie J, Paushkin S, et al. miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002;16:720–8.
Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta. 2010;1803:1231–43.
Chen X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science. 2004;303(5666):2022–5.
Petersen CP, Bordeleau ME, Pelletier J, Sharp PA. Short RNAs repress translation after initiation in mammalian cells. Mol Cell. 2006;21:533–42.
Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17(4):438–42.
Kulkarni M, Ozgur S, Stoecklin G. On track with P-bodies. Biochem Soc Trans. 2010;38:242–51.
Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–9.
Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11): 857–66.
Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008; 105(30):10513–8.
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–9.
Lawrie CH, Gal S, Dunlop HM, et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br J Haematol. 2008;141(5):672–5.
Boeri M, Verri C, Conte D, et al. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc Natl Acad Sci USA. 2011;108:3713–8.
Roth C, Rack B, Muller V, Janni W, Pantel K, Schwarzenbach H. Circulating microRNAs as blood-based markers for patients with primary and metastatic breast cancer. Breast Cancer Res. 2010;12(6):R90.
Heneghan HM, Miller N, Kerin MJ. Circulating miRNA signatures: promising prognostic tools for cancer. J Clin Oncol. 2010;28(29):e573–4.
Zhao H, Shen J, Medico L, Wang D, Ambrosone CB, Liu S. A pilot study of circulating miRNAs as potential biomarkers of early stage breast cancer. PLoS One. 2010;5(10):e13735.
Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–801.
Vargova K, Curik N, Burda P, et al. MYB transcriptionally regulates the miR-155 host gene in chronic lymphocytic leukemia. Blood. 2011;117(14):3816–25.
Pekarsky Y, Santanam U, Cimmino A, et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006;66:11590–3.
Rossi S, Shimizu M, Barbarotto E, et al. microRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood. 2010;116(6):945–52.
Raveche ES, Salerno E, Scaglione BJ, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109: 5079–86.
Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–9.
Calin GA, Cimmino A, Fabbri M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci USA. 2008;105:5166–71.
Klein U, Lia M, Crespo M, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17(1):28–40.
Fabbri M, Bottoni A, Shimizu M, et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305(1):59–67.
Garzon R, Volinia S, Liu CG, et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood. 2008;111(6): 3183–9.
Marcucci G, Maharry K, Radmacher MD, et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol. 2008; 26(31):5078–87.
Garzon R, Liu S, Fabbri M, et al. MicroRNA -29b induces global DNA hypomethylation and tumor suppressor gene re-expression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–8.
Le Beau MM, Albain KS, Larson RA, et al. Clinical and cytogenetic correlations in 63 patients with therapy-related myelodysplastic syndromes and acute nonlymphocytic leukemia: further evidence for characteristic abnormalities of chromosomes no. 5 and 7. J Clin Oncol. 1986;4:325–45.
Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70.
Fabbri M, Garzon R, Cimmino A, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA. 2007;104:15805–10.
Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–40.
Wang H, Garzon R, Sun H, et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell. 2008;14:369–81.
Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A. High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer. 2004;39:167–9.
Kluiver J, Poppema S, De Jong D, et al. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol. 2005;207:243–9.
Tam W, Hughes SH, Hayward WS, Besmer P. Avian bic, a gene isolated from a common retroviral site in avian leukosis virus-induced lymphomas that encodes a noncoding RNA, cooperates with c-myc in lymphomagenesis and erythroleukemogenesis. J Virol. 2002;76:4275–86.
Costinean S, Zanesi N, Pekarsky Y, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci USA. 2006;103:7024–9.
Thai TH, Calado DP, Casola S, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–8.
Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11.
Tili E, Michaille JJ, Cimino A, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–9.
He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–33.
Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–14.
O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–43.
Petrocca F, Visone R, Onelli MR, et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13:272–86.
Ronchetti D, Lionetti M, Mosca L, et al. An integrative genomic approach reveals coordinated expression of intronic miR-335, miR-342, and miR-561 with deregulated host genes in multiple myeloma. BMC Med Genomics. 2008;1:37.
Loffler D, Brocke-Heidrich K, Pfeifer G, et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007;110:1330–3.
Roccaro AM, Sacco A, Thompson B, et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood. 2009;113(26): 6669–80.
Lerner M, Harada M, Loven J, et al. DLEU2, frequently deleted in malignancy, functions as a critical host gene of the cell cycle inhibitory microRNAs miR-15a and miR-16-1. Exp Cell Res. 2009; 315(17):2941–52.
Gatt ME, Zhao JJ, Ebert MS, et al. MicroRNAs 15a/16-1 function as tumor suppressor genes in multiple myeloma. Blood. 2010. doi:10.1182/blood-2009-11-253294.
Pichiorri F, Suh SS, Ladetto M, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci USA. 2008;105:12885–90.
Pichiorri F, Suh SS, Rocci A, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18(4):367–81.
Yan LX, Huang XF, Shao Q, et al. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA. 2008;14:2348–60.
Qian B, Katsaros D, Lu L, et al. High miR-21 expression in breast cancer associated with poor disease-free survival in early stage disease and high TGF-beta1. Breast Cancer Res Treat. 2009;117(1): 131–40.
Walter BA, Gomez-Macias G, Valera VA, Sobel M, Merino MJ. miR-21 expression in pregnancy-associated breast cancer: a possible marker of poor prognosis. J Cancer. 2011;2:67–75.
Asaga S, Kuo C, Nguyen T, Terpenning M, Giuliano AE, Hoon DS. Direct serum assay for microRNA-21 concentrations in early and advanced breast cancer. Clin Chem. 2011;57:84–91.
Wang F, Zheng Z, Guo J, Ding X. Correlation and quantitation of microRNA aberrant expression in tissues and sera from patients with breast tumor. Gynecol Oncol. 2010;119:586–93.
Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008;110(1):13–21.
Ota D, Mimori K, Yokobori T, et al. Identification of recurrence-related microRNAs in the bone marrow of breast cancer patients. Int J Oncol. 2011;38: 955–62.
Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–33.
Ciafre SA, Galardi S, Mangiola A, et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun. 2005;334:1351–8.
Iorio MV, Visone R, Di Leva G, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–707.
Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9(3):189–98.
Markou A, Tsaroucha EG, Kaklamanis L, Fotinou M, Georgoulias V, Lianidou ES. Prognostic value of mature microRNA-21 and microRNA-205 overexpression in non-small cell lung cancer by quantitative real-time RT-PCR. Clin Chem. 2008;54:1696–704.
Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103(7):2257–61.
Schetter AJ, Leung SY, Sohn JJ, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299:425–36.
Roldo C, Missiaglia E, Hagan JP, et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006;24:4677–84.
Bloomston M, Frankel WL, Petrocca F, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297:1901–8.
Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10(3): 544–50.
Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2(4):E98.
Yu F, Yao H, Zhu P, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23.
Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635–47.
Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–9.
Sampson VB, Rong NH, Han J, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67:9762–70.
Kumar MS, Erkeland SJ, Pester RE, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. 2008;105:3903–8.
Adams BD, Furneaux H, White BA. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol. 2007;21(5): 1132–47.
Foekens JA, Sieuwerts AM, Smid M, et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci USA. 2008;105: 13021–6.
Masri S, Liu Z, Phung S, Wang E, Yuan YC, Chen S. The role of microRNA-128a in regulating TGFbeta signaling in letrozole-resistant breast cancer cells. Breast Cancer Res Treat. 2010;124:89–99.
Di Leva G, Gasparini G, Piovan C, et al. A regulatory “miRcircuitry” involving miR-221&222 and ERalpha determines ERalpha status of breast cancer cells. J Natl Cancer Inst. 2010;102:706–21.
Zhao JJ, Lin J, Yang H, et al. MicroRNA-221/222 negatively regulates estrogen receptor alpha and is associated with tamoxifen resistance in breast cancer. J Biol Chem. 2008;283(45):31079–86.
Miller TE, Ghoshal K, Ramaswamy B, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem. 2008;283:29897–903.
Rao X, Di LG, Li M, et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene. 2011;30:1082–97.
Iorio MV, Casalini P, Piovan C, et al. microRNA-205 regulates HER3 in human breast cancer. Cancer Res. 2009;69:2195–200.
Wu H, Zhu S, Mo YY. Suppression of cell growth and invasion by miR-205 in breast cancer. Cell Res. 2009;19:439–48.
Gregory PA, Bracken CP, Bert AG, Goodall GJ. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle. 2008;7(20):3112–8.
Radojicic J, Zaravinos A, Vrekoussis T, Kafousi M, Spandidos DA, Stathopoulos EN. MicroRNA expression analysis in triple-negative (ER, PR and Her2/neu) breast cancer. Cell Cycle. 2011;10: 507–17.
Png KJ, Yoshida M, Zhang XH, et al. MicroRNA-335 inhibits tumor reinitiation and is silenced through genetic and epigenetic mechanisms in human breast cancer. Genes Dev. 2011;25:226–31.
Yu SL, Chen HY, Chang GC, et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell. 2008;13:48–57.
Lebanony D, Benjamin H, Gilad S, et al. Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from non-squamous non-small-cell lung carcinoma. J Clin Oncol. 2009;27: 2030–7.
Del Vescovo V, Cantaloni C, Cucino A, et al. miR-205 Expression levels in nonsmall cell lung cancer do not always distinguish adenocarcinomas from squamous cell carcinomas. Am J Surg Pathol. 2011;35(2):268–75.
Rabinowits G, Gercel-Taylor C, Day JM, Taylor DD, Kloecker GH. Exosomal microRNA: a diagnostic marker for lung cancer. Clin Lung Cancer. 2009; 10(1):42–6.
Hu Z, Chen J, Tian T, et al. Genetic variants of miRNA sequences and non-small cell lung cancer survival. J Clin Invest. 2008;118:2600–8.
Yu L, Todd NW, Xing L, et al. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int J Cancer. 2010;127(12):2870–8.
Ceppi P, Mudduluru G, Kumarswamy R, et al. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol Cancer Res. 2010;8(9):1207–16.
Seike M, Goto A, Okano T, et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc Natl Acad Sci USA. 2009;106(29):12085–90.
Markou A, Tsaroucha EG, Kaklamanis L, Fotinou M, Georgoulias V, Lianidou ES. Prognostic value of mature microRNA-21 and microRNA-205 overexpression in non-small cell lung cancer by quantitative real-time RT-PCR. Clin Chem. 2008;54(10):1696–704.
Saito M, Schetter AJ, Mollerup S, et al. The association of microRNA expression with prognosis and progression in early stage, non small cell lung adenocarcinoma: a retrospective analysis of three cohorts. Clin Cancer Res. 2011. doi:10.1158/1078-0432.CCR-10-2961.
Garofalo M, Di LG, Romano G, et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16(6):498–509.
Murakami Y, Yasuda T, Saigo K, et al. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537–45.
Li W, Xie L, He X, et al. Diagnostic and prognostic implications of microRNAs in human hepatocellular carcinoma. Int J Cancer. 2008;123:1616–22.
Visone R, Pallante P, Vecchione A, et al. Specific microRNAs are downregulated in human thyroid anaplastic carcinomas. Oncogene. 2007;26:7590–5.
Gramantieri L, Ferracin M, Fornari F, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092–9.
Fornari F, Gramantieri L, Ferracin M, et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27:5651–61.
Pineau P, Volinia S, McJunkin K, et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci USA. 2010;107(1):264–9.
Le Sage C, Nagel R, Egan DA, et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–708.
Galardi S, Mercatelli N, Giorda E, et al. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J Biol Chem. 2007;282:23716–24.
Felicetti F, Errico MC, Bottero L, et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008;68(8):2745–54.
Zhang L, Huang J, Yang N, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA. 2006;103:9136–41.
Ji J, Shi J, Budhu A, et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361(15):1437–47.
Yang N, Kaur S, Volinia S, et al. MicroRNA microarray identifies Let-7i as a novel biomarker and therapeutic target in human epithelial ovarian cancer. Cancer Res. 2008;68(24):10307–14.
Bhattacharya R, Nicoloso M, Arvizo R, et al. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 2009;69(23):9090–5.
Boren T, Xiong Y, Hakam A, et al. MicroRNAs and their target messenger RNAs associated with ovarian cancer response to chemotherapy. Gynecol Oncol. 2009;113(2):249–55.
Yang H, Kong W, He L, et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008;68(2):425–33.
Merritt WM, Lin YG, Han LY, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359(25):2641–50.
Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601.
Bendoraite A, Knouf EC, Garg KS, et al. Regulation of miR-200 family microRNAs and ZEB transcription factors in ovarian cancer: evidence supporting a mesothelial-to-epithelial transition. Gynecol Oncol. 2010;116(1):117–25.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8.
Ma L, Reinhardt F, Pan E, et al. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28(4): 341–7.
Huang Q, Gumireddy K, Schrier M, et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol. 2008;10: 202–10.
He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4.
Li N, Fu H, Tie Y, et al. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009;275:44–53.
Sachdeva M, Mo YY. MicroRNA-145 suppresses cell invasion and metastasis by directly targeting mucin 1. Cancer Res. 2010;70(1):378–87.
Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907.
Gandellini P, Folini M, Longoni N, et al. miR-205 Exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cepsilon. Cancer Res. 2009;69:2287–95.
Burk U, Schubert J, Wellner U, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9(6):582–9.
Bracken CP, Gregory PA, Kolesnikoff N, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68(19): 7846–54.
Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–95.
Asangani IA, Rasheed SA, Nikolova DA, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–36.
Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350–9.
Tavazoie SF, Alarcon C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–52.
Rosenfeld N, Aharonov R, Meiri E, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26:462–9.
Poliseno L, Tuccoli A, Mariani L, et al. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108(9):3068–71.
Hua Z, Lv Q, Ye W, et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006;1:e116.
Mertens-Talcott SU, Chintharlapalli S, Li X, Safe S. The oncogenic microRNA-27a targets genes that regulate specificity protein transcription factors and the G2-M checkpoint in MDA-MB-231 breast cancer cells. Cancer Res. 2007;67(22):11001–11.
Fish JE, Santoro MM, Morton SU, et al. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272–84.
Pulkkinen K, Malm T, Turunen M, Koistinaho J, Yla-Herttuala S. Hypoxia induces microRNA miR-210 in vitro and in vivo ephrin-A3 and neuronal pentraxin 1 are potentially regulated by miR-210. FEBS Lett. 2008;582(16):2397–401.
Dews M, Homayouni A, Yu D, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060–5.
Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res. 2007; 101(1):59–68.
Martello G, Rosato A, Ferrari F, et al. A MicroRNA targeting dicer for metastasis control. Cell. 2010; 141(7):1195–207.
Su X, Chakravarti D, Cho MS, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467(7318):986–90.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36(Database issue):D154–8.
Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005; 11(3):241–7.
Chiang HR, Schoenfeld LW, Ruby JG, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24(10):992–1009.
Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–7.
Nakamura T, Canaani E, Croce CM. Oncogenic All1 fusion proteins target Drosha-mediated microRNA processing. Proc Natl Acad Sci USA. 2007;104: 10980–5.
Karube Y, Tanaka H, Osada H, et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005;96:111–5.
Faber C, Horst D, Hlubek F, Kirchner T. Overexpression of Dicer predicts poor survival in colorectal cancer. Eur J Cancer. 2011;47(9):1414–9.
Lin RJ, Lin YC, Chen J, et al. microRNA signature and expression of Dicer and Drosha can predict prognosis and delineate risk groups in neuroblastoma. Cancer Res. 2010;70(20):7841–50.
Weber B, Stresemann C, Brueckner B, Lyko F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle. 2007;6:1001–5.
Saito Y, Liang G, Egger G, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43.
Lehmann U, Hasemeier B, Christgen M, et al. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol. 2008;214(1): 17–24.
Toyota M, Suzuki H, Sasaki Y, et al. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008;68: 4123–32.
Brueckner B, Stresemann C, Kuner R, et al. The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res. 2007;67:1419–23.
Lujambio A, Ropero S, Ballestar E, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67: 1424–9.
Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006;66(3): 1277–81.
Benetti R, Gonzalo S, Jaco I, et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15(3):268–79.
Sinkkonen L, Hugenschmidt T, Berninger P, et al. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol. 2008;15:259–67.
Chen JF, Mandel EM, Thomson JM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–33.
Chang TC, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26(5):745–52.
Raver-Shapira N, Marciano E, Meiri E, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26(5):731–43.
Frezzetti D, De MM, Zoppoli P, et al. Upregulation of miR-21 by Ras in vivo and its role in tumor growth. Oncogene. 2011;30(3):275–86.
Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene. 2007;26: 2799–803.
Meng F, Henson R, Lang M, et al. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–29.
Xia L, Zhang D, Du R, et al. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer. 2008;123(2):372–9.
Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA. 2007;104(39):15472–7.
Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther. 2010;18(9):1650–6.
Esquela-Kerscher A, Trang P, Wiggins JF, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7(6): 759–64.
Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137(6):1005–17.
Chang TC, Yu D, Lee YS, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40(1):43–50.
Care A, Catalucci D, Felicetti F, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–8.
Tiscornia G, Singer O, Ikawa M, Verma IM. A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci USA. 2003;100(4):1844–8.
Xia H, Mao Q, Eliason SL, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med. 2004; 10(8):816–20.
Kawasaki H, Taira K. Short hairpin type of dsRNAs that are controlled by tRNA(Val) promoter significantly induce RNAi-mediated gene silencing in the cytoplasm of human cells. Nucleic Acids Res. 2003;31(2):700–7.
Grimm D, Streetz KL, Jopling CL, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006; 441(7092):537–41.
Lee Y, Kim M, Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23(20):4051–60.
Weiler J, Hunziker J, Hall J. Anti-miRNA oligonucleotides (AMOs): ammunition to target miRNAs implicated in human disease? Gene Ther. 2006;13: 496–502.
Esau C, Davis S, Murray SF, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3(2):87–98.
Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9.
Orom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–41.
Elmen J, Lindow M, Silahtaroglu A, et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36:1153–62.
Elmen J, Lindow M, Schutz S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–9.
Obad S, Dos Santos CO, Petri A, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011;43(4):371–8.
Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–6.
Gentner B, Schira G, Giustacchini A, et al. Stable knockdown of microRNA in vivo by lentiviral vectors. Nat Methods. 2009;6:63–6.
Su J, Baigude H, McCarroll J, Rana TM. Silencing microRNA by interfering nanoparticles in mice. Nucleic Acids Res. 2011. doi:10.1093/nar/gkq1307.
Weidhaas JB, Babar I, Nallur SM, et al. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111–6.
Acknowledgments
This work was partially supported by Associazione Italiana per la Ricerca sul Cancro (AIRC).
Marilena V. Iorio is supported by a Start Up AIRC Grant.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Iorio, M.V., Casalini, P., Piovan, C., Braccioli, L., Tagliabue, E. (2012). Current and Future Developments in Cancer Therapy Research: miRNAs as New Promising Targets or Tools. In: Bologna, M. (eds) Biotargets of Cancer in Current Clinical Practice. Current Clinical Pathology. Humana Press. https://doi.org/10.1007/978-1-61779-615-9_19
Download citation
DOI: https://doi.org/10.1007/978-1-61779-615-9_19
Published:
Publisher Name: Humana Press
Print ISBN: 978-1-61779-614-2
Online ISBN: 978-1-61779-615-9
eBook Packages: MedicineMedicine (R0)