
Abstract
Virus discovery based on cDNA-AFLP (amplified fragment length polymorphism) (VIDISCA) is a novel approach that provides a fast and effective tool for amplification of unknown genomes, e.g., of human pathogenic viruses. The VIDISCA method is based on double restriction enzyme processing of a target sequence and ligation of oligonucleotide adaptors that subsequently serve as priming sites for amplification. As the method is based on the common presence of restriction sites, it results in the generation of reproducible, species-specific amplification patterns. The method allows amplification and identification of viral RNA/DNA, with a lower cutoff value of 105 copies/ml for DNA viruses and 106 copies/ml for the RNA viruses. Previously, we described the identification of a novel human coronavirus, HCoV-NL63, with the use of the VIDISCA method.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key Words
1 Introduction
To date, there is still a variety of human diseases of unknown etiology, including several chronic diseases such as amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS), but also acute infections such as Kawasaki disease and multiple respiratory diseases (1,2). A viral origin has been suggested for many of these diseases, emphasizing the importance of a continuous search for new viruses. Identification of previously unrecognized viral agents in patient samples is of great medical interest, but remains a major technical challenge. Identification of novel viral pathogens is difficult with the virus discovery tools known to date. Several problems are encountered when searching for new viruses. First, most of the unidentified viruses do not replicate in vitro, at least not in the cells that are commonly used in viral diagnostics. Second, the molecular biology techniques previously employed to identify unknown viruses have their specific drawbacks. Several techniques are in use for virus discovery, e.g., universal primer PCR, random priming based PCR, and representational difference analysis (RDA). Although every technique has proven to be useful for virus discovery in certain circumstances, they all have serious limitations and restrictions.
Universal PCR primers should amplify new members of an already known virus family, but this method has two major drawbacks. First, a choice for a specific virus family has to be made. This limits the possibility of identifying a member of an unsuspected family or the founding member of a totally new one. Second, the universal primers may simply not match the genome sequence of novel members of a virus family. This is illustrated by the lack of success of universal coronavirus primers that were designed before the new members—SARS-CoV, HCoV-NL63, and HCoV-HKU1—were identified. None of the studies that used such primers was able to detect a novel human coronavirus (3,4). Obviously, such primers gradually improve once more family members are known.
Another technique uses nonspecific amplification of viral sequences in a random priming PCR at low annealing temperatures. However, most ingredients of this assay contain contaminating DNA. For instance, the enzymes used may contain trace amounts of DNA from the bacteria in which they are produced. This contaminating DNA is also amplified and it is therefore not possible to determine at an early stage whether amplification products represent a new virus or contaminating nucleic acids. This can be resolved only after excessive cloning and sequencing. Therefore, high throughput screening of many clinical samples is impractical. Moreover, this technique has only been successful with viruses that replicate in vitro, in which case cell culture supernatant was used as input for the assay (5).
Representational difference analysis (RDA) is a subtractive hybridization technique that enriches for nucleic acid sequences that are present in one tissue but absent or present at lower concentration in an otherwise identical tissue sample. RDA utilizes PCR to generate sets of nucleic acids in a target and a (negative control) tester sample. After subtractive hybridization, there is selective amplification of target-enriched sequences. The method was developed for tissue material and not for nontissue samples such as serum/plasma or virus culture supernatants (6). The fact that these liquid samples have low concentrations of DNA and RNA in the tester sample may restrain the selective amplification of an unknown viral target. A disadvantage of this technique is that it requires a negative control tissue from the same person from whom the diseased tissue was obtained.
We recently developed a general, simple, and easy to use new virus discovery method that allows large-scale screening for any RNA or DNA virus in samples such as serum/plasma or virus culture supernatant (7). The method is based on the cDNA-AFLP technique (8) (Virus discovery cDNA-AFLP: VIDISCA). The main feature of VIDISCA is that prior knowledge of the genome sequence is not required as the presence of restriction enzyme sites is sufficient to guarantee PCR amplification.
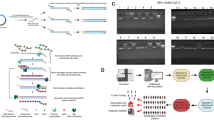
VIDISCA begins with a treatment to selectively enrich for viral nucleic acid, which includes a centrifugation step to remove residual cells and mitochondria (Fig. 1). In addition, a DNase treatment is used to remove interfering chromosomal DNA and mitochondrial DNA from degraded cells, whereas RNases in the sample will degrade RNA. During this step, the viral nucleic acid is specifically protected within the viral particle. Next, DNase/Rnases are inactivated and the viral nucleic acids are subsequently extracted from the particles, RNA is reverse transcribed into cDNA, and second-strand synthesis is performed to make dsDNA (from a viral RNA or DNA genome). The dsDNA is digested with frequently cutting restriction enzymes that are likely to be present in every viral target (HinP1-I and Mse-I), and HinP1-I- and Mse-I-anchors are ligated to the digested DNA. Essential to the method is that the restriction enzymes remain active during the ligase reaction, thus preventing concatamerization of digested fragments. The anchors themselves are not removed because they are designed in such a way that the restriction site is lost.

Overview of the VIDISCA method.
The target is subsequently PCR-amplified with primers that anneal to the anchor sequences, followed by a round of selective amplification with primers that are extended with one nucleotide (G, A, T, or C). Thus, 16 primer combinations are used and each sample is compared to a representative negative control (negative serum, plasma, or supernatant from an uninfected culture). The PCR fragments that are specific to the “infected” clinical sample can then be cloned and sequenced. Because amplification is based on the presence of restriction sites, the PCR is reproducible (in duplicate samples the same fragments are amplified) and these PCR products can be distinguished from background amplification. The assay is relatively high-throughput as multiple samples (about ten) can be tested per cycle of VIDISCA.
We were able to amplify viral nucleic acids from EDTA-plasma of a person with hepatitis B virus infection and a person with an acute parvovirus B19 infection. Using urine, we could detect adenoviral DNA and influenza B RNA in two patients. The technique can also detect HIV-1 and picornaviruses in cell culture. These results illustrate that the VIDISCA technique has the capacity to identify both RNA and DNA viruses directly from patient material or from cell cultures. In fact, it was the first experiment with a suspected virus culture that led to identification of a novel human coronavirus [HCoV-NL63 (9)]. Only three human coronaviruses were known at that time: HCoV-229E, HCoV-OC43, and SARS-CoV (10,11), and HCoV-NL63 represents the fourth species. The rapid identification of this novel coronavirus demonstrates the power of our virus discovery tool, which can now be used to test large sample sets suspected of containing viral pathogens.
2 Materials
2.1 Pretreatment of the Sample
-
1.
DNase I, RNase-free at concentration 2 U/μl (Ambion)
-
2.
DNase buffer: 10X concentrated (Ambion)
-
3.
Sterile HPLC pure water (Baker)
2.2 Nucleic Acid Isolation
-
1.
L2 buffer: 0.1 M Tris-HCl pH 6.4. Prepare by mixing 12.1 g Tris, 9.4 ml of 32% HCl, and adjust with sterile water (Baker) to 1 liter (1 2).
-
2.
L2 solution: prepare by mixing 480 g guanidine thiocyanate (SIGMA) and 400 ml of L2 buffer (1 2).
-
3.
L6 solution: prepare by mixing 480 g guanidine thiocyanate (SIGMA), 88 ml of 0.2 M EDTA, 10.4 g Triton X-100 (Merck), and 400 ml of L2 buffer (1 2).
-
4.
Silica. Prepare 60 g of silicon dioxide (Sigma) in a 500-ml glass graduated cylinder and adjust the volume to 500 ml with sterile water (Baker). Resuspend the silica with vortexing and incubate at room temperature for 25 h. Remove 430 ml of top liquid. Adjust the volume with sterile water (Baker) to 500 ml and resuspend. Incubate at room temperature for 5 h and remove 440 ml of water. Resuspend the silica and stir adding 600 μl of 32% HCl. Aliquot and autoclave. Store at room temperature (1 2).
-
5.
70% ethanol (Merck)
-
6.
100% acetone (Merck)
-
7.
Sterile HPLC pure water (Baker)
2.3 Reverse Transcription, Digestion, Ligation, and PCR Amplification
-
1.
Sequenase 2.0, T7 DNA polymerase at concentration 13 U/μl (Amersham Biosciences).
-
2.
Random primers (hexamers; Amersham Biosciences). Working solution 1 μg/μl.
-
3.
RNase H, 5 U/μl (Amersham Biosciences).
-
4.
MMLV-RT (Moloney murine leukemia virus reverse transcriptase enzyme; 200 U/μl; Invitrogen)
-
5.
CMB buffer (10X): 100 mM Tris-HCl pH 8.3, 500 mM KCl, 1% Triton-X100. Prepare by mixing 1 ml of 2 M Tris-HCl pH 8.3, 5 ml of 2 M KCl, 2 ml of 10% Triton X-100, and 12 ml of sterile water (Baker). Store at –20°C in 250 μl portions.
-
6.
SEQII buffer (10X): 350 mM Tris-HCl pH 7.5, 250 mM NaCl, 175 mM MgCl2. Prepare by mixing 2.25 ml sterile water (Baker), 3.5 ml of 1 M Tris-HCl pH 7.5, 2.5 ml of 1 M NaCl, 1.75 ml of 1 M MgCl2. Store at –20°C in 100 μl portions.
-
7.
Magnesium chloride (100 mM).
-
8.
dNTP’s (25 mM of each; Amersham Biosciences).
-
9.
PCR buffer (10X): 100 mM Tris-HCl pH 8.3, 500 mM KCl, 100 mg BSA. Prepare by mixing 10 ml of 2 M Tris-HCl pH 8.3, 25 ml of 2 M KCl, 60 ml of sterile water (Baker), and 5 ml of BSA (Bovine serum albumin, 20 mg/ml; Roche). Store at –20°C in 250 μl portions.
-
10.
First PCR primer set: HinP1-I standard primer 5′-GAC GAT GAG TCC TGA CCG C-3′ and Mse-I standard primer 5′-CTC GTA GAC TGC GTA CCT AA-3′.
-
11.
Nested PCR primer set: HinP1-I-X Selective primers 5′-GAC GAT GAG TCC TGA CCG CA-3′; 5′-GAC GAT GAG TCC TGA CCG CT-3′; 5′-GAC GAT GAG TCC TGA CCG CC-3′; and 5′-GAC GAT GAG TCC TGA CCG CG-3′.
-
12.
Nested PCR primer set: Mse-I-X Selective primers. Mse-I-A: 5′-CTC GTA GAC TGC GTA CCT AAA-3′; Mse-I-T: 5′-CTC GTA GAC TGC GTA CCT AAT-3′; Mse-I-C: 5′-CTC GTA GAC TGC GTA CCT AAC-3′; Mse-I-G: 5′-CTC GTA GAC TGC GTA CCT AAG-3′.
-
13.
HinP1-I anchors. Top strand: 5′-GAC GAT GAG TCC TGA C-3′; Bottom strand: 5′-CGG TCA GGA CTC AT- 3′. Oligonucleotides should be diluted to the 10 μM concentration.
-
14.
Mse-I anchor: Top strand: 5′-CTC GTA GAC TGC GTA CC-3′; Bottom strand: 5′-TAG GTA CGC AGT C-3′. Oligonucleotides should be diluted to the 10 μM concentration.
-
15.
Mse-I restriction enzyme, 10 U/μl (New England Biolabs). BSA and NEB-2 buffer are included.
-
16.
HinP1-I restriction enzyme. 10 U/μl (New England Biolabs).
-
17.
Ligase, 5 U/μl (Invitrogen).
-
18.
Ligase buffer (Invitrogen).
-
19.
UltraPureTM Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v) (Invitrogen).
-
20.
3 M sodium acetate (pH 5.2).
-
21.
100% ethanol.
-
22.
70% ethanol.
-
23.
AmpliTaq® DNA Polymerase polymerase (5 U/μl; Applied Biosystems).
-
24.
Sterile HPLC pure water (Baker).
2.4 Gel Electrophoresis and Gel Extraction
-
1.
MetaPhor agarose (Cambrex).
-
2.
Agarose MP (Roche).
-
3.
Ethidium bromide (BioRad).
-
4.
Tris-Borate-EDTA buffer (Sigma).
-
5.
A 25-bp DNA ladder (Invitrogen).
-
6.
Smart ladder DNA size marker (Eurogentec).
-
7.
Sterile razor blades.
-
8.
QIAquick gel extraction kit.
-
9.
Sterile HPLC pure water (Baker).
-
10.
Agarose gel loading buffer: 0.1% orange G, 30% glycerol in 0.5X TBE.
2.5 Cloning and Sequencing
-
1.
TOP10 E. coli chemically competent bacteria (Invitrogen).
-
2.
TOPO TA dual promoter or TOPO 2.1 TA cloning kit (Invitrogen).
-
3.
Luria-Broth (LB, Gibco) agar plates supplemented with ampicilline.
-
4.
BigDye terminator kit (Applied Biosystems).
-
5.
M13 reverse primer and T7 primer (10 μM and 1 μM) (Eurogentec).
3 Methods
3.1 Sample Purification
-
1.
Upon receipt samples are stored without thawing at –80°C to preserve the nucleic acids. On the day of the assay, the sample is thawed, vortexed, and 110 μl is immediately centrifuged at room temperature at 13,500 rpm (in a microfuge) for 10 min, in order to remove the cells, cell debris, and insoluble particles such as mucus. Every analyzed sample is tested in duplicate and in every experiment an appropriate negative control is included. The negative control can be a sample of the same type derived from a healthy person or virus-negative cell culture of the same cell type if the pathogen was cultured.
-
2.
Immediately after centrifugation, 100 μl of sample is transferred into a fresh tube. Care should be taken that pelleted material is not transferred. If the sample is exceptionally full of cells/insoluble material, the primal volume may be increased as needed.
-
3.
DNase treatment. DNase I solution is prepared in the nucleic acid free environment by mixing 15 μl of DNase I enzyme, 15 μl of DNase I buffer, and 20 μl of sterile water per 100 μl of the original sample. Subsequently, the DNase I solution is added to the sample material and incubated at 37°C for 45 min.
3.2 Nucleic Acid Isolation Using the Boom Method (see Note 1)
-
1.
Immediately after the DNase I treatment, 900 μl of L6 solution is added to the sample to lyse the material (see Notes 2–5). The lysis is done at room temperature for 10 min. Sample should by thoroughly mixed by inverting and vortexing.
-
2.
40 μl of silica is added and the sample is incubated at room temperature with gentle shaking for 10 min.
-
3.
Sample is centrifuged (13,200 rpm) for 10 sec to pellet the silica particles, and the L6 supernatant is discarded.
-
4.
The pelleted silica is washed twice with 900 μl of L2. After addition of L2 solution, the sample is vortexed thoroughly until no pellets or large particles are visible and centrifuged for 10 sec at 13,200 rpm. Washing with L2 is necessary to remove all traces of Triton-X100 and EDTA that may inhibit the following enzymatic reactions.
-
5.
The sample is washed twice with room temperature 70% ethanol and once with 100% acetone, in the same manner as described above for L2. Ethanol is added to wash out the guandine thiocyanate and residual traces of detergent and EDTA, whereas the acetone washing is needed primarily to speed up the drying process.
-
6.
After the removal of the acetone, silica is dried for 5 min at 56°C with the lid open.
-
7.
To elute bound nucleic acids, 50 μl of sterile water is added and the sample is vortexed until all silica particles are in suspension and incubated at 56°C for 10 min with shaking (500 rpm). After the elution, the sample is centrifuged for 2 min at 13,200 rpm. About 30 μl of the liquid fraction is transferred into a fresh tube. Samples should be stored at –80°C until needed.
3.3 Reverse Transcription and Second-Strand Synthesis
3.3.1 RT Reaction
The reverse transcription (RT) reaction mixture is assembled under nucleic acid and nuclease free conditions and consists of a two-step reaction.
-
1.
Two RT solutions (I and II) are prepared according to the formula:
-
a.
RT solution I (10 μl per sample)
-
i.
2.5 μl of random primers
-
ii.
3.0 μl of 10X concentrated CMB buffer
-
iii.
2.4 μl of MgCl2
-
iv.
2.1 μl of sterile water
-
i.
-
b.
RT solution II (20 μl per sample)
-
i.
2.0 μl of 10X concentrated CMB buffer
-
ii.
1.0 μl of MMLV-RT enzyme
-
iii.
0.8 μl of dNTPs
-
iv.
16.2 μl of sterile water
-
i.
-
a.
-
2.
The nucleic acids isolated by the Boom method are centrifuged (1 min, 13,200 rpm) in order to remove the residual silica particles.
-
3.
After centrifugation, 20 μl of the supernatant is mixed with 10 μl of RT solution I and incubated at room temperature for 2 min in order to support primer-template annealing.
-
4.
After 2 min of incubation, 20 μl of RT solution II is added and samples are incubated for 90 min at 37°C to allow efficient reverse transcription. The RT reaction is followed by 5 min 95°C to deactivate the enzyme.
3.3.2 Second-Strand Synthesis
-
1.
After the RT reaction, the resulting single-stranded cDNA cannot be used as a template for restriction enzyme cleavage or adaptor ligation. Therefore, second-strand synthesis is performed using RNase H to digest any residual RNA and Sequenase enzyme to synthesize the second strand DNA.
-
2.
Second-strand reaction mixture (100 μl per sample):
-
a.
10 μl of 10 ×concentrated SEQII buffer
-
b.
2.0 μl of Sequenase 2.0
-
c.
1.5 μl of RNase H
-
d.
1.0 μl of dNTPs
-
e.
85.5 μl of water
-
a.
The mixture is added to the 50.0 μl RT reaction product
-
3.
Incubate for 90 min at 37°C.
3.3.3 Phenol/Chloroform Extraction
-
1.
After the second-strand synthesis, the sample (150 μl) is mixed with an equal volume of UltraPureTM phenol: chloroform: isoamyl alcohol and vortexed vigorously until the two phases are completely mixed.
-
2.
The separation of phases is done by centrifugation (2 min, 13,200 rpm, room temperature) and the water phase containing ds-DNA is collected into a new tube. The usual volume recovery rate is about 93%.
-
3.
Subsequently, the recovered ds-DNA is precipitated with ethanol. Water phase is mixed with 350 μl of 100% ethanol and 14 μl of 3 M sodium acetate and incubated for 14 h at –20°C.
-
4.
The ds-DNA is pelleted by 25 min centrifugation (15,000 rpm) at 4°C, supernatant is discarded and the pellet is washed with 200 μl of freshly prepared 70% ethanol (centrifugation for 25 min at 15,000 rpm at 4°C).
-
5.
The ethanol is discarded, and the pellet is air dried for 15 min at room temperature.
-
6.
The pellet is dissolved in 30 μl of sterile water by incubation at room temperature.
3.4 Construction of the Adaptors
-
1.
Adaptors are ds-DNA oligonucleotides with its 3′-ends designed to anneal to the cleavage site in the target DNA molecule. Introduction of the single mutation in the region recognized by the restriction enzyme prevents the cleavage of ligated adaptor-target DNA molecule (Fig. 2). Adaptors are homemade, using single-stranded oligonucleotides.
Fig. 2. 
Ligation of adaptors to the target sequence: (A) HinP1-I and Mse-I adaptors. (B) the restriction sites specific for HinP1-I and Mse-I enzymatic cleavage. (C) The product of ligation of the adaptors to the target sequence. Incorporation of a single nucleotide in the region of cleavage result in a nonfunctional cleavage site.
-
2.
To prepare the functional adaptor, the bottom and top oligonucleotides are annealed by adding:
-
a.
20.0 μl of top adaptor (Mse-I or HinP1-I)
-
b.
20.0 μl of bottom adaptor (Mse-I or HinP1-I)
-
c.
5.0 μl of ligase buffer
-
d.
49.0 μl of sterile water
-
a.

-
3.
Premixes for each adaptor should be prepared independently.
-
4.
Solution should be heated up to 65°C for 5 min and cooled down slowly to the room temperature.
-
5.
Prepared adaptors are stored at –20°C.
3.5 Digestion of Target Sequence and Ligation of Adaptors
-
1.
Digestion of the ds-cDNA is performed with restriction enzymes. In the protocol described here we included only digestion with HinP1-I and Mse-I enzymes, but this combination may be altered (1 3). The use of two different restriction enzymes is essential, as we observed that fragments that are cleaved on both sides by the same enzyme have less of a chance to be amplified by PCR. If one is planning to change the enzyme combination, care should be taken that the combination of restriction enzymes generates restriction fragments from virtually all templates.
-
2.
For digestion of ds-cDNA material, the premix is prepared, containing per sample (10 μl per sample):
-
a.
4.0 μl of NEB-2 buffer
-
b.
4.0 μl of 1:10 diluted BSA
-
c.
1.0 μl of Hinp1-I restriction enzyme
-
d.
1.0 μl of Mse-I restriction enzyme
-
a.
-
3.
10 μl of the digestion premix is added to 30 μl of purified ds-cDNA and incubated for 2 h at 37°C.
-
4.
The ligation mix should be prepared before the digestion reaction is finished. The ligation premix contains per sample (15 μl per sample):
-
a.
1.0 μl of HinP1-I adaptor
-
b.
1.0 μl of Mse-I adaptor
-
c.
2.0 μl of 5X concentrated ligase buffer
-
d.
1.0 μl of ligase
-
e.
10.0 μl of sterile water
-
a.
-
5.
15 μl of the ligation premix is added to the digested sample (40 μl). There is no inactivation step between the digestion and ligation, as restriction activity prevents generation of the concatameric forms of the target templates. Fragments that are properly ligated with adaptors will not be cleaved, because of the point mutation introduced (Fig. 2). The ligation should be performed for 2 h at 37°C.
3.6 PCR Reactions
The main part of the VIDISCA method is amplification of the genetic material without prior knowledge of the sequence. The preprocessed ds-cDNA with adaptors can be now amplified using the primers specific for the adaptors. During development of the method it was determined that a single PCR round does not provide sufficient specificity and sensitivity, so a second “nested” PCR is included in the protocol. This PCR uses primers that are similar to the primers used in the first PCR, but with one nucleotide added to the 3′-end of the primers.
-
1.
The first PCR reaction is done with the standard PCR thermocycling program (Table 1) and is optimized for 50 μl reaction.
Table 1 First PCR Thermocycling Profile -
2.
10 μl of ligated sample is mixed with 40 μl of PCR mix. The PCR mix is prepared as described below (the volumes are calculated per sample):
-
a.
31.25 μl of sterile water
-
b.
0.75 μl of MgCl2
-
c.
5.0 μl of 10X concentrated PCR buffer
-
d.
0.5 μl of dNTPs
-
e.
1.0 μl of Hinp1-I standard primer (10 μM)
-
f.
1.0 μl of Mse-I standard primer (10 μM)
-
g.
0.5 μl of AmpliTaq® DNA polymerase
-
a.
-
3.
The PCR reaction thermocycling is performed according to the scheme presented in Table 1 (see Note 6). After successful thermocycling the sample can be store at –20°C until needed.
-
4.
Second PCR—selective amplifications: The second, nested PCR reaction is necessary to provide high specificity and sensitivity. This selective PCR is performed with primers with sequence identical as the standard primers, but with an additional nucleotide on its 3′ part. This additional nucleotide is outside the adaptor sequence and thus belongs to the unknown material (Fig. 2). Use of an additional nucleotide allows separation of the reactions in 16 different primer combinations and enables better analysis of the sample. To have a selectivity that is required when one wants to amplify only those fragments with a 100% match, the thermocycling profile is designed to increase the specificity of reaction by using the starting annealing temperature of 65°C, which gradually decreases during first ten cycles to 56°C.
-
5.
The PCR mix is prepared as described below (47.5 μl per sample). The HinpI-X and MseI-X primers denote primers with an additional 3′-nucleotide.
-
a.
40.3 μl of sterile water
-
b.
0.75 μl of MgCl2
-
c.
5.0 μl of 10X concentrated PCR buffer
-
d.
0.2 μl of dNTPs
-
e.
0.5 μl of HinpI-X primer (10 μM)
-
f.
0.5 μl Mse-I-X primer (10 μM)
-
g.
0.25 μl AmpliTaq® DNA polymerase
-
a.
-
6.
16 PCR premixes are prepared with different primer combinations (Hinp1-I - G,C,A,T; Mse-I - G,C,A,T) and 47.5 μl per sample of each premix is combined with 2.5 μl of the first PCR product. The second PCR thermocycling profile is presented in Table 2 (see Note 6). The PCR product may be analyzed immediately or stored at –20°C until needed.
Table 2 Touch Down PCR profile
3.7 Gel Analysis of the PCR Product and Purification of the Amplified DNA
-
1.
The second PCR product is analyzed on agarose gel. Most of generated fragments are less than 300 bp in size. Owing to the need for high-quality separation and small differences in fragment sizes, the MetaPhor agarose is being used (it allows differentiation among fragments varying 1 bp in size). Additionally, the MetaPhor agarose provides an easy setup and high-throughput processing for gel analysis and purification, compared to the polyacrylamide gels. The MetaPhor agarose gel is prepared as described below.
-
2.
150 ml of 0.5X concentrated TBE buffer is poured into an Erlenmeyer flask and stirred with a magnetic stirrer. 4 g of MetaPhor agarose is weighted and gently poured into the Erlenmeyer flask while mixing. Addition of all agarose powder at once will result in clumping of the agarose. The solution is stirred for another 10 min to soak the agarose grains and heated in the microwave for 60 sec with low power. After the primary heating, the agarose is stirred and heated in 30-sec cycles (low power) with extensive stirring in between. All the agarose is solubilized during a final heating step for 60 sec with medium power. The agarose is cooled down to ∼65°C and 10 μl of ethidium bromide (10 mg/ml) is added. The agarose is poured into the electrophoresis tray and combs are inserted. It is crucial to remove all air bubbles from the gel (e.g., with a pipette tip). The agarose is solidified at room temperature and further incubated at 4°C for at least 20 min (the incubation at 4°C improves the gel resolution). The gel is positioned in the electrophoresis box filled with 0.5X TBE buffer
-
3.
15 μl of the second PCR product is mixed with 5 μl of the loading buffer and the samples are layered on the prepared agarose gel. 5 μl of the 25-bp ladder is used as a DNA size marker. Electrophoretic separation is performed at 150 V for about 1 h.
-
4.
Immediately after the electrophoresis is completed, the gel is analyzed on the UV transilluminator. A picture is taken for analysis and the gel is stored at 4°C, wrapped in plastic (Saran Wrap). The picture of the gel is used to search for fragments that are present in the sample of interest and not in the control sample All the fragments that are present exclusively in the sample of interest are marked on the picture (Fig. 3). If the bands appear very faint on the gel, the PCR products can be concentrated by vacuum centrifuge and reanalyzed on a MetaPhor gel. After fragment selection, the gel is again positioned on the UV transilluminator and the selected bands are excised with sterile razors (about 100 mg per slice) and stored in coded 1.5-ml Eppendorf tubes at 4°C (see Note 7). After excision of all bands, a second picture of the gel should be taken to document the proper excision.
Fig. 3. 
Representative VIDISCA fragments on the MetaPhor agarose gel. Samples with ‘+’ were supernatants of LLC-MK2 cells infected with HCoV-NL63 and samples ‘–’ were supernatants of control LLC-MK2 cells.
-
5.
The DNA fragments from the gel are extracted with the QIAquick gel extraction kit following the manufacturer’s protocol. The gel slices are solubilized in 600 μl of QG buffer and 100 μl of isopropanol is added. After extraction, resulting DNA is dissolved in 30 μl of EB buffer. Alternatively, any other gel extraction method may be used.

3.8 Cloning, Selection of Plasmids, and Sequencing
-
1.
Purified DNA is subsequently cloned into a vector and transformed into bacteria. The usual procedure is to use the TOPO cloning kit from Invitrogen (TOPO TA Cloning® Kit Dual Promoter). Cloning is done with 0.5 μl of vector, 0.5 μl of salt solution, and 2 μl of gel purified DNA. Chemically competent TOP10 E. coli (Invitrogen) bacteria are transformed using the TOPO reaction sample (10 μl of bacteria per reaction). The E. coli are plated on LB agar plates supplemented with ampicilline. The growth on the LB plates is carried on for 16 h at 37°C. Eight colonies per plate are collected with a pipette tip into 50 μl of the BHI medium supplemented with ampicilline on the 96-well PCR plate. The suspended bacteria are subjected directly to a colony-PCR procedure, described below.
-
2.
Colony PCR. The PCR mix is prepared by mixing (45 μl per sample):
-
a.
0.5 μl of M13 reverse primer (10 μM)
-
b.
0.5 μl of T7 primer (10 μM)
-
c.
5.0 μl of 10X concentrated PCR buffer
-
d.
0.5 μl of dNTPs
-
e.
0.75 μl of MgCl2
-
f.
0.2 μl of AmpliTaq® DNA Polymerase
-
g.
37.55 μl of sterile water
-
a.
-
3.
5 μl of suspended E. coli bacteria in BHI medium is added to the PCR mixture.
-
4.
The thermocycling is performed as described in Table 3. After the PCR is completed, 10 μl of the PCR product is mixed with gel loading dye and analyzed on 0.8% agarose MP gel with a Smart ladder DNA size marker. A representative picture of such a gel is shown in Fig. 4.
Table 3 Colony PCR Thermocycling Scheme Fig. 4. 
Gel analysis of colony PCR of VIDISCA fragments cloned into the TOPO 2.1 vector.
-
5.
The lanes that seem to contain the plasmid with proper insert are selected, and corresponding PCR products are subjected to sequencing reactions.
-
6.
Sequencing reactions are performed on the colony-PCR product with the BigDye chemistry, using the M13 reverse and T7 primer, according to the manufacturers’ instructions (Applied Biosystems).

3.9 Data Analysis
The sequence data obtained in the survey is analyzed with the BLAST server (http://www.ncbi.nlm.nih.gov/BLAST/). The raw sequence is edited to remove the sequence derived from the vector and the adaptors. This procedure can be done manually or using designated program, e.g., CodonCode (http://www.codoncode.com/). After the cleanup, the sequences are analyzed for their quality and only those that show a clear, single signal are exported in FASTA format for further analysis. Once imported to the BioEdit program (http://www.mbio.ncsu.edu/BioEdit/bioedit.html), the sequences are subjected to batch BLAST analysis with default settings. This batch analysis allows preselection of the sequences of interest, as mRNA and rRNA fragments are frequently found as background. All results that indicate the presence of a virus, or an unknown sequence should be selected and reanalyzed with the BLAST server (nblast) with the expectation number of 1000 against all databases. If the results are still not clear the following steps might be taken:
-
1.
Analysis against translated database (tblastx)
-
2.
Search the conserved domain database (rpsblast)
-
3.
Analysis against virus database (nblast)
The sequences in tblastx and rpsblast that display similarity to viral sequences should be considered as possibly unknown pathogens. If the sequence is analyzed against a viral database, care should be taken with each hit, because virtually all fragments show some similarity to viral sequences. In that case, the pathogen might be considered identified only if the results from different fragments from one sample show similarity to the same virus family.
In all cases, it is essential to design a diagnostic primer set and retest the original material for the presence of the pathogen. It is only when the pathogen can be detected by the diagnostic (RT)-PCR in the original sample that efforts to sequence the entire genome can be undertaken.
4 Notes
-
1.
For the nucleic acid isolation, any highly efficient method may be used. It is not advisable to use TRIzol isolation, as that is intended for isolation of nucleic acids from cells and tissues.
-
2.
The L6 buffer lysis is sufficient to inactivate the virus. After a 10-min incubation it is safe to process the sample in a normal biochemistry laboratory.
-
3.
The L6 and L2 buffer contain concentrated guanidine thiocyanate (GTC) and thus should be considered as highly toxic. Remember to store the GTC waste separately with addition of one-tenth volume of 1 N sodium hydroxide to prevent GTC degradation.
-
4.
All RNA and cDNA handling before the first PCR should be performed in a nucleic-acid-free environment. The sequence independent amplification will result in overamplification of contaminating DNA.
-
5.
The use of chlorine as a decontaminant should be limited as it may decrease the viability of reverse transcription enzyme.
-
6.
If the thermocycling is performed in a PCR machine that does not include heating of the cover, two drops of paraffin oil should be layered on top of the PCR solution to prevent evaporation during the PCR reaction.
-
7.
It is advised to use a fresh razor for each band during excision. The exposure of the gel to UV light should be limited, as such exposure results in DNA degradation.
References
Burgner, D., and Harnden, A. (2005) Kawasaki disease: what is the epidemiology telling us about the etiology? Int. J. Infect. Dis. 9, 185–194.
Fujinami, R. S., von Herrath, M. G., Christen, U., and Whitton, J. L. (2006) Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin. Microbiol. Rev. 19, 80–94.
Stewart, J., Talbot, P., and Mounir, S. (1995) Detection of coronaviruses by the polymerase chain reaction. In: Becker, Y., and Darai, G. (eds.) Diagnosis of Human Viruses by Polymerase Chain Reaction Technology, Springer-Verlag, New York, pp. 316–327.
Stephensen, C. B., Casebolt, D. B., and Gangopadhyay, N. N. (1999) Phylogenetic analysis of a highly conserved region of the polymerase gene from 11 coronaviruses and development of a consensus polymerase chain reaction assay. Virus Res. 60, 181–189.
Drosten, C., Gunther, S., Preiser, W., van der Werf, S., Brodt, H. R., Becker, S., Rabenau, H., Panning, M., Kolesnikova, L., Fouchier, R. A., Berger, A., Burguiere, A. M., Cinatl, J., Eickmann, M., Escriou, N., Grywna, K., Kramme, S., Manuguerra, J. C., Muller, S., Rickerts, V., Sturmer, M., Vieth, S., Klenk, H. D., Osterhaus, A. D., Schmitz, H., and Doerr, H. W. (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976.
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M., and Moore, P. S. (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266, 1865–1869.
van der Hoek L., Pyrc, K., Jebbink, M. F., Vermeulen-Oost, W., Berkhout, R. J., Wolthers, K. C., Wertheim-van Dillen, P. M., Kaandorp, J., Spaargaren, J., and Berkhout, B. (2004) Identification of a new human coronavirus. Nature Med. 10, 368–373.
Bachem, C. W., van der Hoeven, R. S., de Bruijn, S. M., Vreugdenhil, D., Zabeau, M., and Visser, R. G. (1996) Visualization of differential gene expression using a novel method of RNA fingerprinting based on AFLP: analysis of gene expression during potato tuber development. Plant J. 9, 745–753.
van der Hoek L., Pyrc, K., Jebbink, M. F., Vermeulen-Oost, W., Berkhout, R. J., Wolthers, K. C., Wertheim-van Dillen, P. M., Kaandorp, J., Spaargaren, J., and Berkhout, B. (2004) Identification of a new human coronavirus. Nature Med. 10, 368–373.
Holmes, K. V., and Lai, M. M. C. (1996) Coronaviridae: The viruses and their replication. In: Fields, B. N., Knipe, D. M., Howley, P. M., et al. (eds.) Fields Virology. Lippincott-Raven, Philadelphia, pp. 1075–1093.
Drosten, C., Gunther, S., Preiser, W., van der Werf, S., Brodt, H. R., Becker, S., Rabenau, H., Panning, M., Kolesnikova, L., Fouchier, R. A., Berger, A., Burguiere, A. M., Cinatl, J., Eickmann, M., Escriou, N., Grywna, K., Kramme, S., Manuguerra, J. C., Muller, S., Rickerts, V., Sturmer, M., Vieth, S., Klenk, H. D., Osterhaus, A. D., Schmitz, H., and Doerr, H. W. (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976.
Boom, R., Sol, C. J., Salimans, M. M., Jansen, C. L., Wertheim-van Dillen, P. M., and van der Noordaa, J. (1990) Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 28, 495–503.
Bachem, C. W., van der Hoeven, R. S., de Bruijn, S. M., Vreugdenhil, D., Zabeau, M., and Visser, R. G. (1996) Visualization of differential gene expression using a novel method of RNA fingerprinting based on AFLP: analysis of gene expression during potato tuber development. Plant J. 9, 745–753.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2008 Humana Press, a part of Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Pyrc, K., Jebbink, M.F., Berkhout, B., van der Hoek, L. (2008). Detection of New Viruses by VIDISCA. In: Cavanagh, D. (eds) SARS- and Other Coronaviruses. Methods in Molecular Biology, vol 454. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-59745-181-9_7
Download citation
DOI: https://doi.org/10.1007/978-1-59745-181-9_7
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-58829-867-6
Online ISBN: 978-1-59745-181-9
eBook Packages: Springer Protocols