
Abstract
Most apoptotic cell death events in the body occur via engagement of the mitochondrial pathway of apoptosis. This signaling pathway involves the regulated release, by members of the BCL2 protein family, of mitochondrial proteins following mitochondrial outer membrane permeabilization (MOMP). This in turn activates caspase proteases, generally leading to rapid destruction of the cell. However, recent reports have demonstrated that MOMP in the absence of full-blown caspase activation can have unexpected and detrimental effects. Here, we consider the mitochondrial pathway of apoptosis, how it came to be, and its place in the cell’s innate immune and cell death pathways. We then discuss two unexpected consequences of MOMP in the absence of cell death: activation of innate immune pathways via cytosolic DNA sensing and induction of DNA damage and cellular transformation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Mitochondrial outer membrane permeabilization (MOMP)
- PAMPs
- Cytochrome c
- Caspase-independent cell death (CICD)
11.1 Introduction
11.1.1 Engagement of MOMP
Apoptotic cell death is a highly regulated pathway of cell suicide that is fundamentally required for development, tissue homeostasis, immunity, and tumor suppression (Green 2011). Tens of billions of cells are deleted from our bodies by apoptosis each day, and nearly all of these die following engagement of the mitochondrial pathway of apoptosis. This cell death pathway has been described in great detail, and other chapters of this book discuss its intricacies; here, we present only a general overview.
The pathways of apoptosis are generally broadly divided into “intrinsic” and “extrinsic” pathways ; these names refer to the origin of the apoptosis-initiating signals. Intrinsic apoptosis is initiated by signals from within the dying cell, such as DNA damage, aberrant proliferation or cell cycle dysregulation, ER stress, or lack of sufficient trophic support. These signals converge on members of the BCL2 protein family. This group of proteins comprises both pro- and anti-apoptotic members, and their complex interactions determine whether mitochondrial permeabilization will occur. Upon activation, the BCL2 proteins BAX and BAK form pores in the mitochondrial outer membrane; these proteins are activated by BH3-only proteins such as BIM or BID, and this activation is opposed by the anti-apoptotic family members BCL2, BCLXL, and MCL1. These activation and inhibition interactions are influenced by other family members, and have been exhaustively described elsewhere (Chipuk et al. 2010; Czabotar et al. 2014). For our purposes, it is sufficient to understand that BAX and BAK-mediated pore formation in the outer mitochondrial membrane is the key event that triggers mitochondrial outer membrane permeabilization (or MOMP).
Extrinsic apoptosis is triggered by signaling events that originate outside the dying cell, mediated by engagement of a subset of the TNF receptor superfamily termed the “death receptors.” These include TNFR1, the TRAIL receptors DR4 and DR5, and Fas (also called CD95) (Dickens et al. 2012). Engagement of these receptors by their cognate ligands can lead to the activation of caspase 8. While in some conditions caspase 8 can directly engage a downstream caspase cascade (Jost et al. 2009), in most cells the apoptotic signal must be amplified via the mitochondrial pathway. To accomplish this, caspase 8 cleaves the activating BCL2 protein BID; this cleavage event triggers BID-mediated activation of BAX and/or BAK, and thereby engages MOMP.
11.1.2 Consequences of MOMP
Once activated, BAX and/or BAK insert into the mitochondrial outer membrane, oligomerize, and allow proteins sequestered within the intermembrane space to escape. Notably, this event generally occurs in a rapid, coordinated and complete manner, meaning that all mitochondria in a cell become permeabilized within a few minutes; however, as discussed below this is not always the case (Goldstein et al. 2000). Once mitochondria are permeabilized, intermembrane space proteins gain access to the cytosol. These include cytochrome c, an iron-containing, essential component of the respiratory machinery, as well as the proteins SMAC (also called DIABLO) and OMI (also called HTRA2) (Tait and Green 2010). Importantly, the inner mitochondrial membrane, which is responsible for maintaining the ion gradient that drives oxidative phosphorylation, remains intact following MOMP (Cookson and Silverstone 1976).
Once cytochrome c, SMAC and OMI gain access to the cytosol, they coordinate the activation of the caspase proteolytic cascade. This occurs by binding of the adapter APAF1 to cytochrome c, which in turn recruits caspase 9 to form a heptameric complex called the “apoptosome.” Recruitment of caspase 9 to the apoptosome triggers its activation through dimerization. Once activated, caspase 9 cleaves and activates the downstream executioner caspases 3 and 7. These potent enzymes cleave hundreds of targets within the cell, leading to the ordered destruction, condensation, and phagocytic elimination of the dying cell (McIlwain et al. 2013). SMAC and OMI contribute to this process by binding to and inactivating the IAPs, a family of E3 ubiquitin ligases that keep caspases in check in healthy cells. MOMP thereby simultaneously activates the executioners (caspases) and inactivates their inhibitors (IAPs), leading to rapid and coordinated destruction of the cell.
11.1.3 Why MOMP at All?
The description above is cursory, largely because MOMP is such a well-described phenomenon. However, our familiarity with MOMP as a part of the apoptotic pathway obscures what a fundamentally weird process it is: That cells posses proteins whose sole function is to permeabilize and destroy an essential organelle—the mitochondria—is itself quite striking. That the key effect of this event is the release of an otherwise unexceptional member of the metabolic machinery, cytochrome c, and that this of all things is the precipitating event in the initiation of the apoptotic cascade is truly remarkable. Why would the signaling be wired this way?
Hints to the answer to this question can be gleaned by examining the proteins that orchestrate MOMP, and their homologues. Strikingly, the cytochrome c binding adapter APAF1 bears significant homology to members of the NOD-like receptor (NLR) family of innate immune sensors. These proteins bind to pathogen-associated molecular patterns (PAMPs) such as bacterial flagellin or toxin molecules, and in so doing, alert cells to the presence of these invaders, initiating immune responses (Kanneganti 2010; Man and Kanneganti 2015). Many of the NLRs signal by forming circular protein complexes termed inflammasomes, which recruit the inflammatory protease caspase 1. This in turn can cause cell death via a non-apoptotic program termed “pyroptosis,” and can also promote the processing and maturation of the cytokines IL-1β and IL-18.
The similarities between PAMP-mediated activation of NLRs and cytochrome c mediated activation of APAF1 are notable: the structures of the apoptosome and the inflammasome are similar, both recruit caspases via interactions mediated by CARD domains, and both can trigger cell death. But why would cytochrome c be standing in for bacterial PAMPs in these two parallel pathways?
Of course, mitochondria entered eukaryotic cells as prokaryotic symbionts about 1.5 billion years ago, bringing with them a unique set of metabolic tools that allowed host cells to more efficiently extract energy from carbohydrate bonds. One of the key tricks that bacteria brought to the cell was the ability to orchestrate complex redox chemistry through proteins that coordinated inorganic molecules; the ancestral cytochrome c was such a protein. Thus, prior to establishment of mitochondria as integral symbionts, cytochrome c would have been an excellent PAMP, a molecule that would signal the presence of a prokaryotic invader. This observation leads to the idea that the mitochondrial pathway of apoptosis may represent a regulated form of an ancient pathway of pathogen-induced cell death. Further bolstering this idea is the observation that BAX and BAK, the BCL2 family members responsible for permeabilizing the outer mitochondrial membrane, bear structural homology to bacterial pore-forming toxins called colicins (Lazebnik 2001). But how do we make sense of these observations?
11.1.4 A Long Time Ago, in an Ocean not So Very Far Away…
Here is a scenario that provides a plausible—albeit speculative—answer to the questions above. A few billion years ago, single-celled organisms lived and grew in large clonally similar populations. Being single-celled, they had no need to develop pathways of cell death for developmental or homeostatic purposes. However, altruistic suicide in response to invasion of these cells by other organisms evolved as a means to deprive the invaders of replicative niches. This provided a selective advantage to clonal groups that developed these pathways, since death of a few individuals allowed survival of the population as a whole. In order to induce cell death in response to invasion, these organisms developed sensors that would recognize key reactive molecules in the invaders, such as the iron-coordinating molecules these invaders encoded. These single-celled organisms sequestered the invaders inside vesicles within their cytoplasm, and the invaders responded by producing pore-forming toxins in an effort to escape these membranes. It was war!
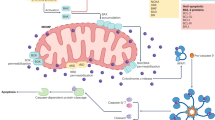
Flashing forward a billion years, cooler heads have prevailed; our single-celled protagonists have discovered that their invaders encode metabolic enzymes that allow them to use nutrients more efficiently, and they’ve harnessed this new efficiency by sequestering the invaders inside vacuoles and providing them with the building blocks they need to produce energy. These are proto-mitochondria. Genetic exchanges between the host and (former) invader take place, and the sensors that triggered cell death in response to cytochrome c-like molecules lie dormant. However, as time goes by these single-celled organisms begin to band together and to specialize, eventually forming multicellular communities. As these become more sophisticated, the death of some cells is required for the proto-organism to survive. To achieve this, evolution fashions an elegant solution: the pathways that used to trigger cell death in response to invading bacteria are dusted off and reconfigured to allow controlled engagement of these same pathways by the molecules contained within the now-symbiotic bacteria, i.e., the mitochondria. The bacterial toxins that had formerly been used to puncture the vacuoles enclosing the invaders are reconfigured to perform this task in a controlled way, becoming the BCL2 family. And the innate immune sensor that recognized the PAMP cytochrome c was reactivated, becoming APAF1; thus, the cell was able to control the emergence of a former PAMP into its cytosol, and thereby reformat a pathway of innate immune sensing and defensive suicide into a regulated process for organismal health. A summary of parallels between MOMP and bacterial sensing are presented in Fig. 11.1a–c.

Parallels between bacterial innate immunity and MOMP. (a) Modern pathways of inflammasome activation. PAMPs from invading bacterial pathogens are sensed by members of the NLR family, forming inflammasomes. These activate caspase 1 to trigger pyroptotic cell death. (b) A hypothetical ancient pathway for “defensive cell suicide” in response to detection of a cytochrome c like molecule encoded by proto-bacteria. (c) Modern pathways of MOMP-driven apoptosis, as a reconfiguration of the pathway depicted in (b) following establishment of symbiosis. This allows programmed cell death in response to controlled release of cytochrome c. (d) When caspase activation is prevented, mitochondrial DNA released during MOMP can activate the cGAS/STING pathway, leading to IFN production and establishment of an anti-viral immune state
As an aside, it is interesting to note that the mitochondrial pathway is not present in all animals, being absent in nematodes and apparently having been lost in Drosophila melanogaster (Bender et al. 2012; Oberst et al. 2008). These organisms likely represent different mechanisms by which the evolutionary process reconfigured its innate immune pathways for regulated cell death. For example, in the nematode Caenorhabditis elegans , the BCL2 homologue (CED9) binds to and sequesters the APAF1 homologue (CED4) on the mitochondria; once displaced, CED4 can activate the caspase homologue CED3 (Chinnaiyan et al. 1997; Spector et al. 1997). This configuration may represent a different PAMP-sensing pathway, one centered around the BCL2-like bacterial pore-forming toxins. It is fascinating to note, however, that despite the absence of MOMP, the C. elegans cell death pathways remain arrayed around the mitochondria.
11.1.5 Mitochondria: The Enemy Within
This long and speculative interlude should illustrate a key idea: that mitochondria can be thought of as foreign matter within our cells. Our innate immune system remains on high alert, and is capable of sensing invaders through the recognition of triggers such as cytosolic DNA or changing redox and ion status; both of these conditions can be triggered by the mitochondria. Furthermore, the apoptotic pathway itself is an altered version of an ancient pathogen-destruction mechanism, and the enzymes involved in apoptosis are evolved specifically to wreak molecular destruction on the cell. Thus, the mitochondria behave like highly reactive molecular bombs within the host cytosol, and as such, dysregulated MOMP would be expected to trigger detrimental effects. Here, we discuss two scenarios in which this has been shown to be the case.
11.2 MOMP: The Ultimate Killer
11.2.1 MOMP Triggers Caspase-Dependent and -Independent Cell Death
As we have discussed, the intricate molecular mechanisms linking MOMP to caspase protease activity and apoptotic cell death are well defined. Nevertheless, we have also known for a long time that once MOMP has occurred, the cell is destined to die irrespective of caspase activity (Tait et al. 2014). The ability of MOMP to trigger caspase-dependent apoptosis or, in its absence, caspase-independent cell death (CICD) directly relates to the extent of mitochondrial permeabilization. Following a cell death trigger, MOMP is widespread and rapid leading to robust caspase activity (Fig. 11.2a). Because of its extensive, and likely irreversible nature, MOMP ensures cell death even in the absence of caspase activity, by triggering massive mitochondrial dysfunction and metabolic catastrophe (Lartigue et al. 2009). In vivo studies have shown that CICD often represents an effective cell death modality; for example, mice deficient in MOMP-dependent caspase activity (Apaf1 or Casp9 knockout) display grossly normal development, contrasting to the much severe developmental defects observed in MOMP-deficient mice (BAX/BAK knockout) (Cecconi et al. 1998; Kuida et al. 1998; Lindsten et al. 2000). These findings argue that CICD can effectively substitute for apoptosis under caspase-deficient conditions, however, whether CICD is engaged under pathophysiological conditions remains an open question. Addressing this, comparative morphological analysis suggests that up to 10 % of cells dying during development may engage CICD, although definitive assessment necessitates combined single-cell analysis of MOMP and caspase activity in vivo (Chautan et al. 1999).

Three degrees of MOMP drive lethal and nonlethal functions. (a) During apoptosis, MOMP often occurs in all mitochondria triggering massive caspase activation and rapid cell death. (b) Following an apoptotic stress, MOMP can sometimes be incomplete, allowing mitochondria to remain intact. In specific settings, cells can survive dependent on these intact mitochondria, provided caspase activity is lacking. (c) Following a sublethal stress, MOMP can be engaged in a minority of mitochondria. Although minority MOMP induces caspase activity, this fails to kill the cell. Instead, caspase activity can cause DNA damage and contribute to other non-apoptotic caspase signaling functions
11.2.2 In Death, Timing Is Everything
If MOMP usually kills cells irrespective of caspase activity, then why have caspases and dedicated activation machinery? One likely reason is that caspase activity dramatically enhances the speed of cell death . Following MOMP, caspases typically kill the cell within a matter of minutes, whereas, in the absence of caspase activity, cells can remain viable for hours or even days. In some contexts, the kinetics of cell death are crucial; as discussed, while many aspects of embryonic development in Apaf1 or Casp9-deficient mice are grossly normal, mice lacking these proteins often display massive forebrain outgrowth. This was recently shown to be due to the prolonged survival of morphogen (FGF-8) producing cells, that otherwise would have died rapidly via apoptosis (Nonomura et al. 2013). This extended window of FGF-8 production wreaks havoc on the developing brain, contributing to forebrain outgrowth. Besides enhancing the kinetics of cell death, as we will now discuss, a s econd reason as to why caspases are activated following MOMP is distinct from their role in cellular execution.
11.3 MOMP STINGs Cells into an Antiviral State
The innate immune system recognizes patterns associated with pathogen invasion—PAMPs—and responds by initiating defensive mechanisms. These include altered transcriptional responses leading to cytokine production and immune cell activation. Nucleic acids are a major source of PAMPs, and multiple, interlocking PAMP sensors have evolved to sense unusual or mislocalized nucleic acids as signatures of pathogen infection. These include sensors that recognize nucleic acids that do not occur naturally in cells; examples of this include recognition of viral signatures such as 5′ triphosphates or double-stranded RNA by RIG-I and PKR, respectively (Fitzgerald et al. 2014).
Cytosolic DNA is another key nucleic acid PAMP, but of course the host cell nucleus also contains DNA; thus, innate immune DNA sensors can’t rely on molecular differences between pathogen and host DNA to distinguish between the two. While the precise mechanisms by which innate immune sensors differentiate self from nonself DNA remain incompletely understood, one key mechanism relates to localization: nuclear DNA is thought to be ignored, while DNA in the cytosol is regarded as evidence of invasion of this compartment by a pathogen (Stetson and Medzhitov 2006).
Cytosolic DNA is sensed by at least three mechanisms: (1) AIM2, which can trigger the formation of inflammasomes that activate caspase 1 (Hornung et al. 2009); (2) DAI, which can trigger NF-κB transcriptional responses or necroptosis via interaction with the RIP kinases (Rebsamen et al. 2009); or (3) the cGAS/STING pathway, which triggers interferon responses via activation of IRF3/7 signaling (Cai et al. 2014; Zhang et al. 2014; Li et al. 2013). Notably, while the AIM2 and STING pathways were initially described as mediators of antiviral responses, recent work has indicated that they also play key roles in responding to bacterial infection (Meunier et al. 2015; Karki et al. 2015; Belhocine and Monack 2012; Watson et al. 2012). In this latter context, it is thought that release of DNA from damaged bacteria leads to their activation. Indeed, recent studies have shown that cells possess specific mechanisms to ensure that bacterial PAMPs gain access to the cytosol, presumably to promote their sensing by these and other mechanisms. Many species of bacteria attempt to “hide” in vacuolar compartments, and it appears that cells have countered this strategy by evolving mechanisms to disrupt bacteria-containing vacuoles. One such strategy involves a family of cellular GTPases called the guanylate binding proteins (GBPs), which disrupt bacterial vacuoles to induce release and recognition of bacterial DNA and other PAMPs (Meunier et al. 2015). Another involves recognition of secreted bacterial DNA by the STING pathway, which in turn unleashes the autophagy machinery to promote bacterial clearance (Watson et al. 2012).
The purposeful release of bacteria and their contents from vacuoles by cellular factors may sound familiar; as we’ve already discussed, mitochondria can be thought of as ancient bacteria enclosed within cellular membranes. MOMP is, in essence, the disruption of the vacuole (the mitochondrial outer membrane) surrounding the proto-bacterium (the mitochondrial inner membrane.) And of course, like bacterial invaders, mitochondria have their own genomes.
These observations set the stage for recent reports that MOMP can lead to release of mitochondrial genomic DNA (mtDNA), which is sensed by the STING pathway to trigger production of type-I interferon (Rongvaux et al. 2014; White et al. 2014). This rather interesting observation can be put into context with our earlier discussion of cytochrome c as a vestigial PAMP: mitochondria do not contain other bacterium-specific PAMPs, such as LPS or peptidoglycans, possibly because these were lost during the establishment of symbiosis. But because DNA is an irreducible and irreplaceable molecule rather than a bacterium-specific “pattern,” and because mitochondria continue to contain their own vestigial genomes, mtDNA remains able to aberrantly trigger the cell’s DNA sensing machinery (Fig. 11.1d).
These recent studies have demonstrated that when downstream caspase 9-mediated apoptosis is prevented (either by knockout of Casp9, double-knockout of Casp3 and Casp7, or chemical inhibition of all caspases), MOMP induction triggers interferon responses that depend on the cGAS/STING pathway (Rongvaux et al. 2014; White et al. 2014). This observation explains the unexpected finding that mice in which caspase-9 is deleted in the endothelial lineage are resistant to viral infection; they have an elevated basal interferon signature mediated by sensing of cytosolic DNA released from the mitochondria.
These reports provide clear evidence for mtDNA as a PAMP, but they also raise questions. One obvious issue is with the release of mtDNA itself; mtDNA resides in the mitochondrial matrix, inside the impermeable inner membrane. MOMP itself does not lead to permeabilization of this membrane, so how DNA escapes—and if this process is regulated in some way—remains unclear. A broader question is: When might this happen under physiological conditions? Normally MOMP leads to rapid cell death, and these interferon responses only manifest when this is prevented by ablation or inhibition of the caspases. So, is sensing of mitochondrial DNA a biologically relevant driver of the interferon response? We can speculate that minority MOMP (discussed further below) may be able to release enough DNA to promote IFN responses, without sufficient cytochrome c release to drive full-blown apoptosis.
Another interesting recent observation that is likely to prove relevant is the finding that knockout of a mtDNA packaging protein, TFAM, promotes escape of mtDNA into the cytosol and also triggers a STING-dependent antiviral response (West et al. 2015). This finding indicates that mtDNA stress, which is a feature of several human diseases, can lead to mislocalization of mtDNA and thus to elevated IFN signaling. It may therefore be the case that limited MOMP could similarly act to a mitochondrial stressor.
While these effects have been explored in the context of potentiated antiviral signaling, they may also have detrimental effects. Several human mutations have been described that lead to aberrant sensing of cytosolic DNA derived from genomic retro-elements (Rice et al. 2009; Crow et al. 2006a, b; Volkman and Stetson 2014; Stetson et al. 2008). This in turn leads to STING-dependent IFN responses that cause lupus-like autoimmune diseases collectively called Aicardi–Goutieres syndrome. It is therefore possible that mitochondrial DNA could also contribute to such deleterious IFN responses, either due to incomplete MOMP or other forms of mitochondrial stress. It is also unclear whether DNA released from the mitochondria may trigger other cytosolic DNA sensing pathways, such as AIM2-mediated inflammasome activation and concurrent IL-1β maturation, which could likewise contribute to autoimmune and inflammatory responses.
11.4 Cell Survival Following MOMP
11.4.1 Survival: A Fate Worse Than Death?
Seminal live-cell imaging studies initially revealed MOMP as a binary all or nothing event (Goldstein et al. 2000). However, a more nuanced view of MOMP has since emerged. Specifically, during apoptosis some mitochondria have been found to evade MOMP, a process called incomplete MOMP or iMOMP (Tait et al. 2010) (Fig. 11.2b). The ability of mitochondria to avoid permeabilization becomes important under caspase-deficient settings. While widespread MOMP normally triggers cell death regardless of caspase activity, the presence of intact mitochondria can serve to repopulate the cell, restoring mitochondrial function and allowing cell survival. Survival under these conditions may be important in various pathophysiological situations, for example by enabling tumor cells to evade therapy-induced cell death or by allowing the survival of post-mitotic cells such as neurons that are required lifelong. More recently, it has emerged that sublethal levels of apoptotic stress can engage MOMP in a minority of mitochondria—so-called minority MOMP (Fig. 11.2c). Importantly, minority MOMP still engages caspase activity but at levels which are insufficient to kill the cell (Ichim et al. 2015). Rather than killing, sublethal caspase activity causes DNA damage and genomic instability that can actually promote oncogenic transformation and tumorigenesis (Ichim et al. 2015; Liu et al. 2015). Why might this be important? To consider this we must first take a step back. Apoptosis is widely considered a potent anticancer mechanism—tumor suppressors prevent cancer by inducing apoptosis and anticancer therapies often kill cancer cells by apoptosis (Czabotar et al. 2014). These recent findings demonstrate that the same core apoptotic machinery that prevents cancer may also have the potential to promote cancer, if cell death is not executed. The ability of minority MOMP-induced DNA damage and genomic instability may, in part, provide a mechanistic basis for recurrent reports demonstrating an oncogenic role for apoptotic signaling (Labi and Erlacher 2015). Developing this idea further, an unfortunate consequence of some anticancer therapies is that they increase the likelihood of developing leukemia in later life. The chromosomal translocations that underlie these leukemias have been associated with sublethal caspase activity, which potentially may be initiated via minority MOMP (Hars et al. 2006). From these findings, a desirable therapeutic goal will be to enhance minority MOMP-induced tumor cell killing while preventing unwanted DNA damage in healthy tissue.
11.4.2 MOMP in Nonlethal Signaling Functions
By triggering limited caspase activity, minority MOMP may also play various physiological signaling functions. Various non-apoptotic functions for ca spase protease activity have been described in processes that range from cell cycle to migration (Connolly et al. 2014). Perhaps their non-apoptotic role is best supported in neurons (Hyman and Yuan 2012). Modification of neuronal synaptic plasticity is important for both the development of the central nervous system as well as information storage. Non-apoptotic caspase activity modulates synaptic plasticity in different ways, for example by causing retraction of neuronal dendrites that form synapses (so-called pruning) (Cusack et al. 2013; Wang et al. 2014; Nikolaev et al. 2009). Additionally, non-apoptotic caspase activity has been shown to regulate the endocytosis of receptors that control synaptic plasticity (Li et al. 2010). In both settings, MOMP-dependent caspase activity has been implicated. The ability of neurons to activate caspases at non-apoptotic levels is likely due to two reasons. First, due to their large size, the spatial separation between permeabilized, caspase-activating mitochondria at the synapse and the rest of the cell likely restrains propagation of a pro-apoptotic signal. Secondly, neurons are remarkably resistant to cell death following MOMP, such that they must develop a competency to die (Deshmukh et al. 2000; Martinou et al. 1999).
11.5 Concluding Remarks
MOMP was discovered as an integral part of the apoptotic program, and a defining characteristic of MOMP was its rapid, all-or-nothing character. However, further analysis has demonstrated that MOMP is not always the end of a cell’s life, and that MOMP-related phenomena can trigger unforeseen and detrimental cellular processes independent of cell death. These findings make more sense when viewed in the context of the evolution of the mitochondria themselves, and of the mitochondrial pathway of cell death as a “special case” of an ancient pathogen-sensing response. Future studies will no doubt shed additional light on the regulation of these fascinating processes.
References
Belhocine K, Monack DM (2012) Francisella infection triggers activation of the AIM2 inflammasome in murine dendritic cells. Cell Microbiol 14:71–80. doi:10.1111/j.1462-5822.2011.01700.x
Bender CE et al (2012) Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc Natl Acad Sci U S A 109:4904–4909. doi:10.1073/pnas.1120680109
Cai X, Chiu YH, Chen ZJ (2014) The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54:289–296. doi:10.1016/j.molcel.2014.03.040
Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94:727–737
Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P (1999) Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr Biol 9:967–970
Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM (1997) Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science 275:1122–1126
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR (2010) The BCL-2 family reunion. Mol Cell 37:299–310. doi:10.1016/j.molcel.2010.01.025
Connolly PF, Jager R, Fearnhead HO (2014) New roles for old enzymes: killer caspases as the engine of cell behavior changes. Front Physiol 5:149. doi:10.3389/fphys.2014.00149
Cookson J, Silverstone T (1976) 5-Hydroxytryptamine and dopamine pathways in mania: a pilot study of fenfluramine and pimozide. Br J Clin Pharmacol 3:942–943
Crow YJ et al (2006a) Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 38:910–916. doi:10.1038/ng1842
Crow YJ et al (2006b) Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38:917–920. doi:10.1038/ng1845
Cusack CL, Swahari V, Hampton Henley W, Michael Ramsey J, Deshmukh M (2013) Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat Commun 4:1876. doi:10.1038/ncomms2910
Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15:49–63. doi:10.1038/nrm3722
Deshmukh M, Kuida K, Johnson EM Jr (2000) Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol 150:131–143
Dickens LS, Powley IR, Hughes MA, MacFarlane M (2012) The ‘complexities’ of life and death: death receptor signalling platforms. Exp Cell Res 318:1269–1277. doi:10.1016/j.yexcr.2012.04.005
Fitzgerald ME, Rawling DC, Vela A, Pyle AM (2014) An evolving arsenal: viral RNA detection by RIG-I-like receptors. Curr Opin Microbiol 20:76–81. doi:10.1016/j.mib.2014.05.004
Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2:156–162. doi:10.1038/35004029
Green DR (2011) Means to an end: apoptosis and other cell death mechanisms. Cold Spring Harbor, New York, NY
Hars ES, Lyu YL, Lin CP, Liu LF (2006) Role of apoptotic nuclease caspase-activated DNase in etoposide-induced treatment-related acute myelogenous leukemia. Cancer Res 66:8975–8979. doi:10.1158/0008-5472.CAN-06-1724
Hornung V et al (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. doi:10.1038/nature07725
Hyman BT, Yuan J (2012) Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci 13:395–406. doi:10.1038/nrn3228
Ichim G et al (2015) Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 57:860–872. doi:10.1016/j.molcel.2015.01.018
Jost PJ et al (2009) XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 460:1035–1039. doi:10.1038/nature08229, nature08229 [pii]
Kanneganti TD (2010) Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol 10:688–698. doi:10.1038/nri2851
Karki R et al (2015) Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe 17:357–368. doi:10.1016/j.chom.2015.01.006
Kuida K et al (1998) Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94:325–337
Labi V, Erlacher M (2015) How cell death shapes cancer. Cell Death Dis 6:e1675. doi:10.1038/cddis.2015.20
Lartigue L et al (2009) Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell 20:4871–4884. doi:10.1091/mbc.E09-07-0649
Lazebnik Y (2001) Why do regulators of apoptosis look like bacterial toxins? Curr Biol 11:R767–R768
Li Z et al (2010) Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141:859–871. doi:10.1016/j.cell.2010.03.053
Li XD et al (2013) Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. doi:10.1126/science.1244040
Lindsten T et al (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 6:1389–1399
Liu X et al (2015) Caspase-3 promotes genetic instability and carcinogenesis. Mol Cell 58:284–296. doi:10.1016/j.molcel.2015.03.003
Man SM, Kanneganti TD (2015) Regulation of inflammasome activation. Immunol Rev 265:6–21. doi:10.1111/imr.12296
Martinou I et al (1999) The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol 144:883–889
McIlwain DR, Berger T, Mak TW (2013) Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5:a008656. doi:10.1101/cshperspect.a008656
Meunier E et al (2015) Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol 16:476–484. doi:10.1038/ni.3119
Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457:981–989. doi:10.1038/nature07767
Nonomura K et al (2013) Local apoptosis modulates early mammalian brain development through the elimination of morphogen-producing cells. Dev Cell 27:621–634. doi:10.1016/j.devcel.2013.11.015
Oberst A, Bender C, Green DR (2008) Living with death: the evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ 15:1139–1146. doi:10.1038/cdd.2008.65
Rebsamen M et al (2009) DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep 10:916–922. doi:10.1038/embor.2009.109
Rice GI et al (2009) Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41:829–832. doi:10.1038/ng.373
Rongvaux A et al (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159:1563–1577. doi:10.1016/j.cell.2014.11.037
Spector MS, Desnoyers S, Hoeppner DJ, Hengartner MO (1997) Interaction between the C. elegans cell-death regulators CED-9 and CED-4. Nature 385:653–656. doi:10.1038/385653a0
Stetson DB, Medzhitov R (2006) Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24:93–103. doi:10.1016/j.immuni.2005.12.003
Stetson DB, Ko JS, Heidmann T, Medzhitov R (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134:587–598. doi:10.1016/j.cell.2008.06.032
Tait SW, Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11:621–632. doi:10.1038/nrm2952
Tait SW et al (2010) Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell 18:802–813. doi:10.1016/j.devcel.2010.03.014
Tait SW, Ichim G, Green DR (2014) Die another way--non-apoptotic mechanisms of cell death. J Cell Sci 127:2135–2144. doi:10.1242/jcs.093575
Volkman HE, Stetson DB (2014) The enemy within: endogenous retroelements and autoimmune disease. Nat Immunol 15:415–422. doi:10.1038/ni.2872
Wang JY et al (2014) Caspase-3 cleavage of dishevelled induces elimination of postsynaptic structures. Dev Cell 28:670–684. doi:10.1016/j.devcel.2014.02.009
Watson RO, Manzanillo PS, Cox JS (2012) Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150:803–815. doi:10.1016/j.cell.2012.06.040
West AP et al (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520:553–557. doi:10.1038/nature14156
White MJ et al (2014) Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159:1549–1562. doi:10.1016/j.cell.2014.11.036
Zhang X et al (2014) The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep 6:421–430. doi:10.1016/j.celrep.2014.01.003
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this chapter
Cite this chapter
Oberst, A., Ichim, G., Tait, S.W.G. (2016). Mitochondrial Permeabilization: From Lethality to Vitality. In: Hockenbery, D. (eds) Mitochondria and Cell Death. Cell Death in Biology and Diseases. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3612-0_11
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3612-0_11
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3610-6
Online ISBN: 978-1-4939-3612-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)