
Abstract
Mitosis is a highly dynamic process during which the genetic material is equally distributed between two daughter cells. During mitosis, the sister chromatids of replicated chromosomes interact with dynamic microtubules and such interactions lead to stereotypical chromosome movements that eventually result in chromosome segregation and successful cell division. Approaches that allow quantification of microtubule dynamics and chromosome movements are of utmost importance for a mechanistic understanding of mitosis. In this chapter, we describe methods based on activation of photoactivatable green fluorescent protein (PA-GFP) that can be used for quantitative studies of microtubule dynamics and chromosome segregation.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Accurate segregation of chromosomes during mitosis is critical for ensuring equal partitioning of the genetic material between the two daughter cells. Errors in mitotic chromosome segregation can lead to the formation of daughter cells with abnormal chromosome numbers, a condition known as aneuploidy, which is a leading cause of miscarriage and genetic disorders [1] and is ubiquitously found in cancer [2–4]. Mitotic cell division is a highly dynamic process resulting from the concerted dynamic behavior of different mitotic apparatus components. The microtubules of the mitotic spindle and the chromosomes, aided by numerous microtubule- and chromosome-associated molecular motors and non-motor proteins, are the major players in this process. A key event in chromosome segregation is the establishment of microtubule–chromosome interactions, with the major point of interaction between microtubules and chromosome being the kinetochore. During prometaphase, establishment of kinetochore–microtubule attachment leads to chromosome congression to the spindle equator. In a mature, metaphase mitotic spindle, the chromosomes are lined up at the spindle equator, forming the so-called metaphase plate. The two sister chromatids are bound to microtubules, with each sister kinetochore bound to microtubules from one pole (i.e., sister kinetochores bound to opposite poles). The bound microtubules remain dynamic, undergoing poleward flux and dynamic instability at the plus end (facing the kinetochore). These microtubule dynamics produce chromosome movement, so that in many cell types the aligned chromosomes oscillate back and forth about the metaphase plate. Moreover, kinetochore-bound microtubules turn over while maintaining stable kinetochore attachment. In other words, metaphase kinetochores are stably bound to bundles of microtubules (k-fibers), but individual microtubules within such k-fibers can detach from kinetochores (and disassemble) with certain probabilities, and this results in measurable turnover rates.
A number of perturbations can cause changes in microtubule dynamics, and when such changes are specific to kinetochore microtubules, they can result in kinetochore–microtubule attachment errors and chromosome mis-segregation. Thus, methods to quantify chromosome and microtubule dynamics in mitosis represent powerful tools for the identification of specific chromosome segregation errors and the identification of the responsible mechanisms.
Here, we specifically focus on the use of photoactivatable GFP to study microtubule dynamics, including poleward flux and turnover, and chromosome segregation. We describe experimental methods as well as data acquisition and analysis, and briefly discuss some of the caveats and precautions related to these methods.
2 Materials
2.1 Cells
-
1.
PA-GFP-tub PtK1; PtK1 cells stably expressing photoactivatable-GFP-tubulin (see Note 1 ).
-
2.
H2B-PA-GFP PtK1; PtK1 cells stably expressing photoactivatable-GFP-tagged H2B (see Note 2 ).
2.2 Media, Coverslips, Dishes, Chambers, Taxol
-
1.
Ham’s F-12 medium complemented with 10 % fetal bovine serum, antibiotics, and antimycotic. Occasionally, cells (particularly the PA-GFP-tub PtK1 cells) may be cultured for a few days in the presence of antibiotics (1 mg/ml G418 for PA-GFP-tub PtK1 cells and 0.1 μg/ml puromycin for H2B-PA-GFP PtK1 cells).
-
2.
Acid-washed 22 mm × 22 mm, # 1.5 glass coverslips (see Note 3 ).
-
3.
Sterile 35 mm polystyrene petri dishes or 6-well plate.
-
4.
Modified Rose chambers, including top and bottom plates, silicone gaskets, and screws (Fig. 1; see Note 4 ).
Fig. 1 
Modified Rose chamber. (a) Image of disassembled Rose chamber with bottom plate (left ), silicone gasket and screws (center), and top plate (right). (b) Assembled Rose chamber. The top plate shown in this picture is cut off to allow for easier access of microinjection needles or for viewing/imaging with short working distance condenser. For most applications, a cutoff top plate is not required
-
5.
21 Gauge (21G) needles and 5 ml syringes.
-
6.
Glass-bottom 35 mm petri dishes.
-
7.
Imaging medium: Leibovitz’s L-15 with 1.822 g/l HEPES, and 4.5 g/l glucose. Adjust pH to 7.2 with NaOH, and complement with 10 % fetal bovine serum and antibiotics/antimycotic solution.
-
8.
Taxol, 5 mM stock solution in DMSO.

3 Methods
3.1 Microtubule Photoactivation
3.1.1 Background
Different populations of microtubules coexist within the bipolar mitotic spindle. The two major classes of microtubules relevant to this procedure are the kinetochore microtubules and the non-kinetochore microtubules present in the region between the two spindle poles. These two classes of microtubules display different dynamic behavior [5]. Indeed, attachment to the kinetochore results in microtubule stabilization, which causes kinetochore microtubules to be less dynamic than non-kinetochore microtubules. Importantly, a number of perturbations associated with mitotic defects cause changes in kinetochore microtubule dynamics, and abnormal kinetochore microtubule dynamics are associated with chromosomal instability in cancer cells [6–8]. Early studies measured microtubule poleward flux and/or microtubule turnover through a variety of methods, including fluorescence activation of injected caged-fluorescein tubulin [5, 9], a combination of microtubule depolymerizing drugs and electron microscopy [10], and fluorescent speckle microscopy [11]. In recent years, the emergence of photoactivatable and photoswitchable fluorescent proteins [12, 13] has allowed the generation of cell lines that express tubulin fused to a fluorescent protein that can be “switched on” at any given time and at any given location within the cell. Here, we describe the method used to measure rates of kinetochore microtubule poleward flux and spindle microtubule turnover in PtK1 cells expressing tubulin fused to photactivatable green fluorescent protein (PA-GFP-tub PtK1).
3.1.2 Sample Preparation
-
1.
If using glass-bottom dishes, plate PA-GFP-tub-PtK1 cells in the dishes and replace culture medium with pre-warmed imaging medium prior to the experiment.
-
2.
If planning to use modified Rose chambers, place sterile coverslips in 35 mm petri dishes or 6-well plate. To do so, use clean forceps to remove one acid-washed coverslip (see Note 3 ) from the ethanol-containing jar, drain the excess ethanol on the edge of the jar, and flame the coverslip prior to depositing it in a petri dish/well. As a cautionary measure, keep the flame source and the flaming coverslip away from the ethanol-containing jar.
-
3.
Repeat the procedure to prepare as many coverslip-containing petri dishes/wells as needed for the experiment. Then, plate the cells on the sterile coverslips inside the petri dishes/wells. Perform the whole procedure under sterile conditions (i.e., biosafety cabinet).
-
4.
Prior to the experiment, remove the coverslip and assemble a modified Rose chamber (described in [14]) with a top coverslip. Briefly, deposit the coverslip on the Rose chamber bottom plate, cell-side up, making sure to center it on the round hole; place the silicon gasket on top of the coverslip; place a top coverslip (again making sure to center it on the round hole); place the top plate; secure the chamber with screws (do not tighten too much at this stage).
-
5.
To fill the chamber with imaging media, prepare a syringe with pre-warmed imaging media and a 21G needle. Then, insert a 21G needle (exhaust) into the silicone gasket on one side of the chamber and insert the needle of the syringe on the opposite side. Hold the chamber vertically, with the syringe at the bottom and the exhaust needle at the top and fill the chamber with imaging media making sure that as the media goes in, all the air bubbles are expelled out of the chamber through the exhaust needle. When the chamber is filled, further tighten the screws and clean the bottom coverslip with a Kimwipes moist with deionized water and then with a Kimwipes moist with 70 % ethanol.
-
6.
Once ready, place the chamber or the glass-bottom dish on the stage of an inverted microscope equipped with a system for temperature control (see Note 5 ) and appropriate photoactivation (see Note 6 ) and imaging light sources.
3.1.3 Data Acquisition
-
1.
After placing your sample on the microscope stage, use transmitted light and a DIC or phase-contrast 60× objective to locate a mitotic cell (typically in prometaphase or metaphase) and acquire pre-activation GFP fluorescence (480/40 nm illumination in our setup) and transmitted light images. Using the phase-contrast image on the screen, draw a ~0.5 μm-wide line perpendicular to the spindle long axis and located on one side of the metaphase plate. Using the appropriate software command, activate the fluorescence (1–2 s with our setup) within the selected region and immediately acquire post-activation fluorescence and phase-contrast images.
-
2.
Acquire images (see Note 7 ) again after 10–15 s and then every 15–20 s thereafter for 5–10 min (or until the fluorescence signal disappears at the spindle pole due to microtubule poleward flux; Fig. 2, top, last frame).
Fig. 2 
Microtubule photoactivation. The images (stills from a time-lapse movie) show photoactivation of PA-GFP-tubulin (left column) in a prometaphase PtK1 cell (chromosomes are visible in the phase-contrast images on the right column). A narrow rectangular region across the left half spindle was activated and fluorescence and phase-contrast images were acquired nearly simultaneously immediately after activation and every 15 s thereafter. Elapsed time is shown in min:sec. The graph at the bottom reports the change in fluorescence intensity over time within the activated region (see text for details on how to quantify the fluorescence intensity)
-
3.
Acquire data sets under each experimental condition as well as in samples pretreated for ~1 h with 10 μM of the microtubule-stabilizing drug [15] taxol (in growth medium) and maintained in 10 μM taxol (in imaging medium) during imaging. The taxol data will be used to calibrate for photobleaching (see Note 8 ).
-
4.
Given that each cell is typically imaged for no longer than 10 min, multiple cells can be activated and imaged on the same coverslip one after the other both in taxol-treated and experimental samples.

3.1.4 Quantification of Microtubule Turnover
-
1.
For each photoactivated cell (both experimental samples and taxol-treated samples), fluorescence dissipation must be quantified after background subtraction. To do this, draw a rectangular region around the area of activation and an equivalent rectangular region on the opposite, non-activated, side of the spindle. Measure the total integrated fluorescence within those two regions at each time point using any image analysis software (we use either NIS elements image analysis package or the open source program Fiji/ImageJ). For cells displaying normal microtubule dynamics, the fluorescent mark will move poleward over time. Consequently, the rectangular regions (for fluorescence and background measurements) must be moved at each time frame to follow the poleward movement of the activated mark. For taxol-treated cells, the mark is expected to be static, and therefore, quantification of background fluorescence and fluorescence intensity of the activated region can be performed without moving the rectangular quantification regions.
-
2.
Fluorescence intensity at each time point will be obtained by subtracting the background value from the value of the fluorescent mark. Quantification of fluorescence dissipation over time in taxol-treated cells will represent the loss of fluorescence due to photobleaching.
-
3.
To calculate the average photobleaching-dependent fluorescence loss, collect data from 10 or more taxol-treated cells and average the fraction of fluorescence loss over time. The fluorescence intensity will be normalized to 1 at t 0 (time point immediately after photoactivation) and the fluorescence intensity at subsequent time points will be a fraction of the fluorescence intensity at t 0, and we will refer to them as photobleaching values. The data from taxol-treated cells are expected to display a linear, but very slow (how slow will depend on the imaging system, exposure times, etc., which will affect how much photobleaching occurs at each time point), decay.
-
4.
After quantifying the fluorescence intensity for the experimental data (fluorescence intensity of activated mark minus background), divide the values obtained by the “ photobleaching values.” These final values will represent the fluorescence intensity values after background subtraction and photobleaching correction and should be represented as percent fluorescence intensities, where the t 0 value is set to 100 % (see graph in Fig. 2).
-
5.
Data from multiple cells are typically averaged [16, 17] for representation and for quantitative analysis (see details below).
-
6.
To obtain quantitative information about microtubule dynamics, the data should be subject to nonlinear regression analysis. The best fit for this type of data is a double exponential curve [5], with equation y = A 1 × exp(−k 1 × t) + A 2 × exp(−k 2 × t), where A 1 and A 2 are the percentages of the total fluorescence contributed by, and hence the percentages of, non-kinetochore and kinetochore microtubules, respectively; k 1 and k 2 are the rate constants of turnover/fluorescence dissipation for non-kinetochore and kinetochore microtubules, respectively; and t is time after photoactivation. Microtubule half-lives (t 1/2) can be calculated as follows: t 1/2 = ln 2/k 1 for non-kinetochore microtubule; t 1/2 = ln 2/k 2 for kinetochore microtubules. Thus, this method allows quantification of both the percentage of microtubules incorporated into k-fibers vs. unbound microtubules and turnover rates of these two populations of microtubules.
3.1.5 Quantification of Microtubule Poleward Flux
Due to the bundling of microtubules within k-fibers, the fluorescence marks after activation look particularly well-defined along k-fibers. Quantifying microtubule poleward flux can easily be performed by tracking the poleward movement of such marks on the k-fibers. In PtK1 cells, a phase-contrast image of good quality allows visualization of the spindle pole. Alternatively, at the end of the experiment, the whole spindle can be activated and imaged, and this image can be used as a reference for distances from the spindle poles. Poleward flux can be quantified by simply measuring the mark-to-pole distance at subsequent time points (either manually or with simple MatLab scripts) and then calculating the rate of flux as d/t, where d is the distance (in μm) traveled by the mark over a given period of time, t (in minutes), which will give a rate in μm/min.
3.2 Chromosome Photoactivation
3.2.1 Background
Accurate chromosome segregation is an absolute requirement for maintenance of cell and organismal function. Indeed, chromosome segregation errors result in aneuploidy, which is a leading cause of miscarriage and genetic disorders in humans [1] and has been long recognized as a hallmark of cancer [2–4]. In addition to being aneuploid, most cancer cells also display high rates of chromosome mis-segregation [7, 18–21], a phenotype typically referred to as chromosomal instability, or CIN [21]. Given the dire consequences of chromosome segregation errors, many methods have been developed over the years to examine chromosome segregation during mitosis. These methods, including both fixed and live-cell approaches, allow labeling of one or a few specific chromosomes or labeling of all chromosomes. Fixed-cell analysis of anaphase cells, possibly combined with immunostaining of kinetochore proteins [22, 23], or live imaging of cells expressing histone-GFP [24, 25] are common methods for visualization of all chromosomes. These methods, however, preclude observation of certain chromosome segregation errors, such as that occurring when two sister chromatids segregate to the same spindle pole. To obviate this limitation, fluorescence in situ hybridization with chromosome specific probes has been performed on ana-telophase cells [24, 26, 27], but this approach can be very laborious and is unlikely to provide insight into the mechanism responsible for the mis-segregation events. Alternatively, individual chromosomes have been visualized in live cells using the LacO-LacI system, which relies on the integration of a Lac operator array at a specific locus and expression of a fluorescent protein-tagged Lac repressor [28]. This approach has been extensively used in yeast and occasionally to study chromosome segregation in vertebrate cells [29, 30]. However, with this method the site of integration along vertebrate chromosomes cannot be controlled. Recent efforts have attempted to overcome this limitation through the development of CRISPR-based methods [31, 32]. Photoactivation of H2B-PA-GFP is a relatively simple method that offers many advantages over other currently used approaches. Indeed, any chromosome can be marked and different chromosomes can be marked in different mitotic cells. Moreover, several chromosomes within the same cell can be activated if necessary and the mark can be created in any position along the chromosome. Most importantly, the decision of which chromosome or which region of a chromosome to mark and follow can be made at the time of imaging rather than at the time of experiment set up or establishment of the cell line.
3.2.2 Sample Preparation
For sample preparation, plate H2B-PA-GFP PtK1 cells following the instructions provided in Subheading 3.1.2.
3.2.3 Data Acquisition
-
1.
After placing your sample on the microscope stage, use transmitted light and a DIC or phase-contrast 60× objective to locate a mitotic cell (typically in early prometaphase) and acquire a transmitted light image.
-
2.
Using the phase-contrast image on the screen, select a region (see Note 9 ) on one or two chromosomes of choice for photoactivation (see Note 6 ). Using the appropriate software command, activate the fluorescence within the selected region and acquire post-activation fluorescence and phase-contrast images immediately after activation and thereafter at regular time intervals.
-
3.
The activation time varies depending on the goal of the experiment (as well as expression level of PA-GFP in the cell), and it will be determined accordingly (see Note 10 ).
-
4.
The time intervals for imaging of photoactivated chromosomes should be chosen in such a way that will allow imaging through mitosis without causing excessive bleaching of the activated signal (see Note 11 ). One way to keep the cell healthy and the fluorescence visible long enough is to monitor or image the cell by phase-contrast and only periodically acquire a fluorescence image, until the cell reaches the phase of interest (e.g., metaphase-anaphase transition if the goal is to examine chromosome segregation). At that point, GFP and phase images can be acquired simultaneously and at shorter time intervals. With our setup we can follow cells through anaphase (~20 min) imaging at 30 s intervals. However, the total imaging time and time intervals can vary widely depending on the purpose of the experiment and the type of microscope setup.
3.2.4 Data Analysis
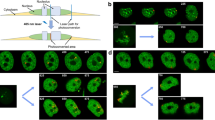
The type of analysis will depend on the goal of the experiment, and we will describe here three different types of experiments and their respective data analysis. A small region on one or two chromosomes can be marked in early prometaphase cells. Alternatively, we have marked a single region on the last unaligned chromosome in prometaphase cells. In both cases, analysis of the time-lapse video will provide information on the segregation of the marked chromosome(s). In addition to this qualitative information, the mark on individual chromosomes could also be used to determine the dynamics of the chromosome. Particularly, if the mark is located in proximity of the centromere, tracking its movement in metaphase will allow quantification of directional instability [33]. A MatLab script [34], commercial image analysis software, or open source programs, such as Fiji/ImageJ, can be used for tracking. If the mark is located away from the centromere on a chromosome arm (Fig. 3), the arm dynamics can also be quantified. By marking two spots on a single chromosome arm (Fig. 3), we were able to quantify the changes in orientation of the chromosome arm with respect to the spindle axis. Interestingly, this type of experiment revealed that in PtK1 cells the arms of peripheral metaphase chromosomes undergo extensive oscillations (Fig. 3), even though their centromeres/kinetochores were previously shown to not oscillate [34–37]. This method could be used to investigate arm dynamics for chromosomes at various spindle locations, which would, in turn, reveal possible variations in the forces exerted on chromosomes in different regions of the mitotic spindle. Finally, activation of a whole chromosome-containing micronucleus in late prophase makes it possible to distinguish that given chromosome from all the others upon nuclear envelope breakdown [38]. Analysis of the time-lapse movie enables studies of the detailed behavior, through mitosis, of the micronucleus chromosome in terms of segregation and position relative to other chromosomes or the spindle [38].

Chromosome photoactivation. The images (stills from a time-lapse movie) show photoactivation of two points (arrows in 0 min image) on the same chromosome arm of a PtK1 cell. The chromosomes can be viewed in the top row of phase-contrast images, whereas the photoactivated GFP signals are shown in the bottom row of images. The cell was imaged from metaphase into anaphase, with anaphase onset occurring at ~13 min, as indicated in the graph. During image analysis, the movement of the two activated points was tracked (using a MatLab script) in order to describe the behavior of the chromosome arm. Using these data, we calculated the angle between the chromosome arm and the spindle long axis. The change of this angle over time is shown in the graph at the bottom right. From the data reported in the graph it is evident that the orientation of the chromosome arm displays an oscillatory pattern resulting from spindle-dependent forces acting on the chromosome. After anaphase onset, the marks are only visible on one sister chromatid (due to activation at a single focal plane), and as this chromatid starts its poleward movement, the angle approaches 0°, as expected as a result of the typical V-shaped orientation of segregating chromosomes, in which individual chromosome arms become nearly parallel to the spindle axis
4 Notes
-
1.
In principle, the methods described here could be applied to any adherent cell line. However, we described methods that were specifically optimized for variants of the PtK1 cell line, a stable epithelial cell line derived from the kidney of a normal adult female rat kangaroo (Potorous tridactylus). PtK1 cells are easily maintained in culture and have been used as a model of choice for many studies of mitosis, particularly those based on imaging approaches. Two key features, remaining flat during mitosis and possessing a small number of large chromosomes, make these cells particularly suited for microscopy-based studies and allow for easy identification and/or tracking of individual chromosomes/kinetochores and individual k-fibers. Two minor drawbacks with this cell line are that the Potorous tridactylus genome has not been sequenced and that PtK1 cells do not transfect well with standard lipid-based methods or electroporation. Nevertheless, several groups have been able to sequence a number of PtK1 genes or to infer sufficient sequence information to design efficient siRNAs ([39, 40]; Shah and Berns, personal communication). Moreover, both lipid-based transfection methods and retroviral gene transduction have been successfully employed to generate PtK1 or PtK2 (equivalent cell line derived from male Potorous tridactylus) cell lines stably expressing a variety of gene products [24, 36, 41, 42]. The large size of these cells and the fact that they remain flat during mitosis has also enabled the use of microinjection in mitotic cells as a method to inhibit specific target proteins, introduce dominant-negative mutant proteins, or introduce fluorescently labeled proteins [17, 37, 43–47]. The PA-GFP-tub PtK1 cell line was initially generated by Alexey Khodjakov [36] using lipid-based transfection and has been previously used in several studies to quantify microtubule poleward flux and turnover rates [16, 17, 36].
-
2.
The H2B-PA-GFP PtK1 cell line was generated by Daniela Cimini (while still at UNC, Chapel Hill) using retrovirus-mediated gene transduction. These cells are currently being used to uncover novel mechanisms of chromosome mis-segregation and to investigate the mitotic behavior of micronucleus-derived chromosomes (He and Cimini, unpublished).
-
3.
Acid washed coverslips are prepared as follows. First, separate coverslips (from 2 to 4 packages) from one another and place them into a glass beaker containing the amount of water necessary to make a 1 M HCl solution that will cover the coverslips. Add the required amount of 37 % HCl to make a 1 M solution. Loosely cover the glass beaker with a watch glass and heat the 1 M HCl solution at 50–60 °C for 4–16 h. Cool to room temperature, pour the HCl solution out, and rinse out the 1 M HCl with deionized water. Fill container with deionized water and sonicate in ultrasonic cleaner for 30 min. Pour the water out, fill the beaker with fresh deionized water, and sonicate in ultrasonic cleaner for 30 min. Repeat this last step one more time. Pour the water out, fill the beaker with 50 % ethanol, and sonicate in ultrasonic cleaner for 30 min. Pour the 50 % ethanol out, fill the beaker with 70 % ethanol, and sonicate in ultrasonic cleaner for 30 min. Pour the 70 % ethanol out, fill the beaker with 95 % ethanol, and sonicate in ultrasonic cleaner for 30 min. Pour the 95 % ethanol out, transfer the coverslips to a glass jar with screw cap, fill with 95 % ethanol, and store for later use.
-
4.
Rose chambers (Fig. 1) are typically custom-made by in-house machine shops. Note that the plates should not be painted/varnished (the paint/varnish will peel off), but anodized. The modified Rose chambers should be disassembled immediately after use and silicone gaskets, top and bottom plates should be immersed in deionized water to prevent the media from drying out on the plates and causing corrosion. Top and bottom plates and silicone gaskets can be washed in bulk and this should be done manually using an appropriate detergent, such as Sparkleen 1 (Fisher Scientific). To wash, immerse the plates and gaskets in a large container (e.g., a dish pan) with a solution of deionized water and Sparkleen 1, scrub each piece with a glassware brush and transfer it into a secondary large container filled with deionized water. Rinse one more time and conclude by rinsing under running deionized water. Let the parts dry before use.
-
5.
We typically perform our experiments at ~35–36 °C either on a microscope stage heated with an air stream incubator (Nevtek) or in a Tokai Hit stage top incubator, and we make sure that the system has reached the desired temperature prior to starting the experiment. We also wait 5–10 min after placing the dish/Rose chamber on the stage, to make sure the sample has reached the correct temperature prior to the beginning of the experiment. Different groups use slightly different temperatures [5, 9, 16, 36], but temperatures slightly below 37 °C appear to protect cells from photodamage [48]. Importantly, because temperature is known to affect the rates of tubulin polymerization/depolymerization, it is important to maintain the temperature constant for all experimental conditions.
-
6.
We perform our photoactivation experiments on an epifluorescence microscope (Nikon Ti-E) equipped with a Mosaic Photoactivation System (Photonic Instruments/Andor) consisting of digital diaphragm optical head with micromirror array and optics with transmission spectra <370–700 nm. For activation, we use a 100 W Olympus mercury lamp and a dichroic mirror that transmits light at 365–435 nm and reflects above 435 nm (a 405 nm laser can also be used for activation of PA-GFP). The Mosaic system allows for activation of user-defined regions of interest of any shape, size, and complexity.
Other methods, besides the Mosaic digital diaphragm, can be used for fluorescence photoactivation. In earlier studies, microtubule fluorescence activation was achieved by focusing the activation light through a narrow slit introduced into the light path of a standard epifluorescence microscope [9, 49, 50]. In other more recent studies, photoactivation of a pseudo-Gaussian line region was achieved with a 408 nm laser focused by a cylindrical lens [16, 17, 36]. The problem with these methods is that the activation line/bar will have a fixed orientation, which requires either exclusive selection of cells whose spindle has the right orientation (i.e., spindle long axis perpendicular to the activation line/bar) or the use of a rotating stage [9]. Finally, another recent commercial system that has been used by some groups to study kinetochore microtubule dynamics [6, 51] is the Micropoint System (Photonic Instruments). Whereas this system shares some of the advantages offered by the Mosaic System, the area of activation is limited to a spot instead of a user-selected region. Thus, this system could be easily employed to activate small regions along chromosome arms, but would limit microtubule activation to a small region of the spindle rather than a region spanning the spindle width (i.e., all the k-fibers on a given focal plane). In fact, a bar could be created by activating multiple aligned spots, but this may compromise the accuracy of the quantification, given that different points along the line would be activated at slightly different time points.
-
7.
When choosing image acquisition settings for photoactivated tubulin, the first point that should be considered is the level of PA-GFP-tubulin expression in the cell of interest that should be high enough to allow generation of a mark that fluoresces significantly above the background. To generate a mark with good signal-to-noise ratio, however, the exposure time for post-activation image acquisition must also be carefully selected. Therefore, we suggest performing pilot experiments in which the variability in levels of PA-GFP-tubulin expression, as well as the optimal exposure times are evaluated and the experimental settings are optimized.
-
8.
Due to imaging of the fluorescently labeled tubulin, some of the sample fluorescence will photobleach, and this photobleaching should be accounted for. To this end, it is important to collect a set of data under experimental conditions in which fluorescence dissipation is only due to photobleaching and not to microtubule turnover, which is achieved by performing tubulin photoactivation in taxol-treated cells, as described in Subheading 3.1.3.
-
9.
For chromosome photoactivation, one important point to consider is the size and position of the activation region, which will vary depending on the goal of the experiment. We have successfully used photoactivation to mark chromosomes in several different ways. For instance, we labeled whole chromosome-containing micronuclei to study the segregation of such chromosomes at the mitosis following micronucleus formation [38]. In other experiments, we activated a small chromosome region close to the centromere in early prometaphase chromosomes (He and Cimini, unpublished). This results in labeling of both sister chromatids and allows for analysis of segregation of the two sisters, including segregation of two sisters to the same pole. We also performed a number of control experiments in which we labeled either entire nuclei or a portion of a nucleus and found that photoactivation per se does not affect chromosome segregation [38]. Finally, in some trial experiments, we generated two small marks on the same chromosome arm, which allowed us to measure chromosome arm movement (Fig. 3).
-
10.
For chromosome photoactivation, the activation time will vary depending on the goal of the experiment (as well as expression level of PA-GFP in the cell). For example, when the goal is to mark a small region on a prometaphase chromosome, one second illumination results in good signal, although in some cases one of the two sisters may not be marked as well as the other. Depending on the mitotic stage and how condensed the chromosomes are, activation may be performed on more than one focal plane to make sure the whole chromosome is activated. However, this should be avoided when possible, because it may result in unwanted activation of other chromosomes. On the other hand, for activation of the bulk of chromatin within an entire micronucleus, we routinely use multiple 100 μs illumination pulses over five focal planes (spaced 0.6 μm of each other), including two focal planes above and two below the center focal plane.
-
11.
One problem we encounter with activated H2B-PA-GFP is a general low-level fluorescence activation on all the chromosomes over time. This appears to be caused by slight photoactivation resulting from repetitive exposure to the GFP excitation wavelength (480/40 nm for our filter set) during imaging. This, combined with bleaching of the activated mark, leads to a reduction of the signal-to-noise ratio over time, thus limiting the time and frequency of imaging, despite PA-GFP initially being shown to remain stable for days [52]. Therefore, although we believe that labeling of chromosomes/chromosome regions by photoactivation has great potential in the study of chromosome segregation, the imaging set up must be carefully evaluated. Pilot experiments should be performed to determine exposure and imaging times compatible with collection of data that will address the desired experimental question(s). Moreover, as described in the methods Subheading 3.2.2, the fluorescent mark can be preserved by restricting GFP imaging in time. This can be achieved if the cell of interest is imaged by phase-contrast microscopy until it approaches the time window of interest, at which point imaging of the fluorescent signal is added.
References
Hassold T, Hunt P (2001) To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet 2(4):280–291
Cimini D (2008) Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim Biophys Acta 1786(1):32–40
Mitelman F, Johansson B, Mertens F (2014) Mitelman database of chromosome aberrations and gene fusions in cancer. http://cgap.nci.nih.gov/Chromosomes/Mitelman
Weaver BA, Cleveland DW (2006) Does aneuploidy cause cancer? Curr Opin Cell Biol 18(6):658–667
Zhai Y, Kronebusch PJ, Borisy GG (1995) Kinetochore microtubule dynamics and the metaphase-anaphase transition. J Cell Biol 131(3):721–734
Bakhoum SF, Genovese G, Compton DA (2009) Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol 19(22):1937–1942
Bakhoum SF et al (2014) The mitotic origin of chromosomal instability. Curr Biol 24(4):R148–R149
Bakhoum SF et al (2009) Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol 11(1):27–35
Mitchison TJ (1989) Polewards microtubule flux in the mitotic spindle: evidence from photoactivation of fluorescence. J Cell Biol 109(2):637–652
Cassimeris L et al (1990) Stability of microtubule attachment to metaphase kinetochores in PtK1 cells. J Cell Sci 96(Pt 1):9–15
Waterman-Storer CM et al (1998) Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Curr Biol 8(22):1227–1230
Lippincott-Schwartz J, Patterson GH (2008) Fluorescent proteins for photoactivation experiments. Methods Cell Biol 85:45–61
Zhou XX, Lin MZ (2013) Photoswitchable fluorescent proteins: ten years of colorful chemistry and exciting applications. Curr Opin Chem Biol 17(4):682–690
Rieder CL, Hard R (1990) Newt lung epithelial cells: cultivation, use, and advantages for biomedical research. Int Rev Cytol 122:153–220
Schiff PB, Horwitz SB (1980) Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci U S A 77(3):1561–1565
Cimini D et al (2006) Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr Biol 16(17):1711–1718
DeLuca JG et al (2006) Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127(5):969–982
Nicholson JM, Cimini D (2011) How mitotic errors contribute to karyotypic diversity in cancer. Adv Cancer Res 112:43–75
Nicholson JM, Cimini D (2013) Cancer karyotypes: survival of the fittest. Front Oncol 3:148
Thompson SL, Compton DA (2008) Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol 180(4):665–672
Lengauer C, Kinzler KW, Vogelstein B (1997) Genetic instability in colorectal cancers. Nature 386(6625):623–627
Cimini D et al (2003) Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol Biol Cell 14(9):3821–3833
Cimini D et al (2001) Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol 153(3):517–527
Cimini D et al (2002) Merotelic kinetochore orientation versus chromosome mono-orientation in the origin of lagging chromosomes in human primary cells. J Cell Sci 115(Pt 3):507–515
Kanda T, Sullivan KF, Wahl GM (1998) Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 8(7):377–385
Cimini D, Tanzarella C, Degrassi F (1999) Differences in malsegregation rates obtained by scoring ana-telophases or binucleate cells. Mutagenesis 14(6):563–568
Torosantucci L et al (2009) Aneuploidy in mitosis of PtK1 cells is generated by random loss and nondisjunction of individual chromosomes. J Cell Sci 122(Pt 19):3455–3461
Robinett CC et al (1996) In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol 135(6 Pt 2):1685–1700
Thompson SL, Compton DA (2010) Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 188(3):369–381
Thompson SL, Compton DA (2011) Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc Natl Acad Sci U S A 108(44):17974–17978
Chen B et al (2013) Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155(7):1479–1491
Ma H et al (2015) Multicolor CRISPR labeling of chromosomal loci in human cells. Proc Natl Acad Sci U S A 112(10):3002–3007
Skibbens RV, Skeen VP, Salmon ED (1993) Directional instability of kinetochore motility during chromosome congression and segregation in mitotic newt lung cells: a push-pull mechanism. J Cell Biol 122(4):859–875
Wan X et al (2012) The coupling between sister kinetochore directional instability and oscillations in centromere stretch in metaphase PtK1 cells. Mol Biol Cell 23(6):1035–1046
Civelekoglu-Scholey G et al (2013) Dynamic bonds and polar ejection force distribution explain kinetochore oscillations in PtK1 cells. J Cell Biol 201(4):577–593
Cameron LA et al (2006) Kinesin 5-independent poleward flux of kinetochore microtubules in PtK1 cells. J Cell Biol 173(2):173–179
Cimini D, Cameron LA, Salmon ED (2004) Anaphase spindle mechanics prevent mis-segregation of merotelically oriented chromosomes. Curr Biol 14(23):2149–2155
He B et al (2014) Chromosomes mis-segregated into micronuclei cause chromosomal instability by further mis-segregating at subsequent mitoses. Mol Biol Cell 25:P1839
Guimaraes GJ et al (2008) Kinetochore-microtubule attachment relies on the disordered N-terminal tail domain of Hec1. Curr Biol 18(22):1778–1784
Stout JR et al (2006) Deciphering protein function during mitosis in PtK cells using RNAi. BMC Cell Biol 7:26
Dumont S, Salmon ED, Mitchison TJ (2012) Deformations within moving kinetochores reveal different sites of active and passive force generation. Science 337(6092):355–358
Shah JV et al (2004) Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr Biol 14(11):942–952
Canman J, Salmon E, Fang G (2002) Inducing precocious anaphase in cultured mammalian cells. Cell Motil Cyto 52:61–65
Canman JC, Hoffman DB, Salmon ED (2000) The role of pre- and post-anaphase microtubules in the cytokinesis phase of the cell cycle. Curr Biol 10(10):611–614
Howell BJ et al (2000) Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J Cell Biol 150(6):1233–1250
Shannon KB, Canman JC, Salmon ED (2002) Mad2 and BubR1 function in a single checkpoint pathway that responds to a loss of tension. Mol Biol Cell 13(10):3706–3719
De Antoni A et al (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol 15(3):214–225
Schulze E, Kirschner M (1988) New features of microtubule behaviour observed in vivo. Nature 334(6180):356–359
Mitchison TJ, Salmon ED (1992) Poleward kinetochore fiber movement occurs during both metaphase and anaphase-A in newt lung cell mitosis. J Cell Biol 119(3):569–582
Salmon ED et al (2007) A high-resolution multimode digital microscope system. Methods Cell Biol 81:187–218
Maffini S et al (2009) Motor-independent targeting of CLASPs to kinetochores by CENP-E promotes microtubule turnover and poleward flux. Curr Biol 19(18):1566–1572
Patterson GH, Lippincott-Schwartz J (2002) A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297(5588):1873–1877
Acknowledgements
We would like to thank Lisa Cameron (Dana Farber Cancer Institute) for critical reading of the manuscript. Funding in the Cimini lab provided by NSF grants MCB-0842551 and MCB-1517506 and HFSP grant RGY0069/2010.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
He, B., Cimini, D. (2016). Using Photoactivatable GFP to Study Microtubule Dynamics and Chromosome Segregation. In: Chang, P., Ohi, R. (eds) The Mitotic Spindle. Methods in Molecular Biology, vol 1413. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3542-0_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3542-0_2
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3540-6
Online ISBN: 978-1-4939-3542-0
eBook Packages: Springer Protocols