
Abstract
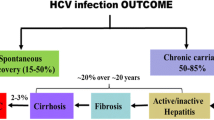
The hepatitis C virus (HCV) is a major cause of severe liver disease worldwide. It is estimated that around 130–170 million individuals are chronic carriers of the infection and they are over time at an increased risk of developing severe liver disease. HCV is often referred to as a silent epidemic because the majority of infected individuals do not develop any symptoms. Hence, many individuals are diagnosed at a late stage and thus in need of immediate treatment. Today we have very effective direct-acting antivirals (DAAs), which cure more than 90–95 % of all treated patients. However, this treatment is associated with high-costs and the use is limited to the patients with most advanced liver disease in high-income countries. Notably, a majority of the chronic HCV carriers live in resource-poor countries and do not have access to the new effective DAAs. We therefore need to develop alternative treatments for chronic HCV infection such as therapeutic vaccines. The idea with therapeutic vaccines is to reactivate the infected patient’s own immune system. It is well known that patients with chronic HCV infection have dysfunctional immune responses to the virus. Hence, the vaccine should activate HCV-specific T cells that will home to the liver and eradicate the HCV infected hepatocytes. Importantly, one should also consider the combination of a therapeutic vaccine and DAAs as a treatment strategy to equip the resolving patients with post-cure HCV-specific immune responses. This would provide patients with a better protection against reinfection. Numerous genetic vaccine candidates for HCV have been developed and tested in clinical trials with limited effects on viral load and in general inefficient activation of HCV-specific immune responses. In this chapter we describe the rational of developing highly immunogenic vaccines for HCV. Different strategies to improve vaccine immunogenicity and methods to evaluate vaccine efficacy are described. Detailed description of vaccine delivery by intramuscular immunization in combination with in vivo electroporation/electrotransfer (EP/ET) is covered, as well as immunological analysis of primed immune responses by determination of interferon-γ (IFN-γ) production by ELISpot assay and direct ex vivo quantification of HCV NS3/4A-specific CD8+ T cells by pentamer staining. To analyze the in vivo functionality of primed NS3/4A-specific T cells we utilized the in vivo bioluminescence imaging technology. In conclusion, this chapter describes a method to design HCV vaccines and also a protocol to assess their efficacy.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
1.1 Characteristics of the Hepatitis C Virus Infection and Treatment Options
The hepatitis C virus (HCV) is one of the major causative agents responsible for development of severe liver disease and cancer worldwide. It is estimated that 130–170 million individuals are chronically infected with HCV [1, 2]. Due to the fact that the majority of the infected individuals do not develop symptoms explains why so many are diagnosed late in their infection. Hence, patients with late stage chronic HCV infection are in need of effective treatment to avoid severe liver complications. The currently used highly efficient direct-acting antivirals (DAAs) can cure more than 90–95 % of all chronic HCV patients [3, 4]. The only limitation is the high-cost for the treatment, which only allow patients with severe liver disease to be treated. Hence, alternative treatment options are therefore needed. To understand how to develop alternative treatments for HCV a brief introduction of the virus will follow. HCV has a positive sense single stranded (ss) RNA molecule of approximately 9.6 kb that encodes for at least ten structural and nonstructural proteins. Each of these proteins may be a target for antiviral drugs and vaccines. The currently approved DAAs target the viral replication machinery by inhibiting the protease (e.g., NS3/4A), the polymerase (e.g., NS5B), and the NS5A protein, which is important for replication and assembly [5]. Notably is that the DAAs does not protect against reinfection [6], which highlights the importance of post-cure HCV-specific T cell responses. Priming of post-cure HCV-specific immune response s may preferable be achieved by vaccinating the HCV infected and/or resolving patients with a specific and potent vaccine . There are several strategies to develop HCV vaccines: such as inactivated, attenuated, subunit, and experimental (e.g., genetic) vaccines. However, the use of some of these vaccines is not possible due to safety concerns (e.g., inactivated and attenuated vaccines). In addition, inactivated, attenuated and subunit vaccines preferentially prime humoral immune responses, which is not suitable for therapeutic vaccines that require activation of T cell responses. We here focus on the development of genetic vaccines, which may be used for both prophylactic and therapeutic purposes. Numerous genetic vaccines have been developed and tested for efficacy in clinical trials [7–13], however none have so far cured HCV infection. To develop a successful vaccine for chronic HCV infection it is expected that multiple parameters need to be optimized such as the vaccine antigen , inclusion of adjuvants, dosing, tissue targeting, delivery, and possibly use of a prime-boost approach. In this chapter we describe some of these important parameters and how they affect immune priming and effector functions.
1.2 Vaccine Design
1.2.1 Antigen Optimization
Firstly, when designing a vaccine for HCV one should select the antigen or antigens carefully. The antigen/s should preferentially be highly immunogenic, advantageously be a relative large protein to maximize the number of targeted epitopes, to include epitopes that correlate with clearance or resolution of infection, and the antigen should demonstrate a low genetic variability. One such antigen is the HCV non-structural 3/4A (NS3/4A) protein, which encodes the viral protease and helicase. Herein, we show examples of strategies to improve the immunogenicity of HCV NS3/4A expressed as a DNA vaccine (e.g., naked plasmid DNA). Other genetic vaccine types include recombinant proteins, peptides, and viral vectors. Secondly, the in vivo expression level of the DNA vaccine is commonly correlated with the potency of the primed immune response . One strategy to increase the protein-expression levels of a DNA vaccine is to optimize the gene sequence based on codon usage, GC content, CpG motifs, mRNA secondary structures, RNA instability motifs, and repeat sequences. Additional sequences that preferentially should be included are Kozak sequence for efficient translation initiation and the TGA stop codon for most efficient translational termination. All mentioned parameters might affect how efficiently a gene is expressed in vivo. We have shown that antigen optimization significantly enhances the HCV NS3/4A immunogenicity [14].
1.2.2 Molecular Adjuvants
Another way to improve the immunogenicity of a selected vaccine -antigen is to add heterologous gene-sequences and/or co-expression with immune stimulatory or inhibitory molecules. When adding heterologous gene-sequences to the vaccine-antigen one should carefully consider the choice of gene-sequence/s. Ideally one should include a gene-sequence encoding a highly immunogenic antigen that will recruit healthy heterologous T cells to the site of immune priming. This is utmost important in the therapeutic vaccination setting, where the infected patient´s immune system often has encountered the viral antigen and its T cells may be dysfunctional. Here, heterologous T cells may help in reactivating the dysfunctional T cells or aid in priming new potent T cell responses targeted to HCV. We have shown that fusion constructs of HCV NS3/4A and human or stork hepatitis B core antigen (HBcAg) significantly enhance the immunogenicity of NS3/4A in a mouse model with dysfunctional T cells to HCV [15,16]. One concern with heterologous gene-sequences is whether they cause any unwanted effects when expressed in vivo. This needs to be investigated for every combination of heterologous gene-sequences and viral antigen/s.
The use of cytokine, chemokine, and co-stimulatory genes as genetic adjuvants has been tested extensively. Some of the most commonly used cytokine genes are IL-2, IL-12, and IL-15 in respect of priming antiviral T cell responses [17, 18]. Also, TLR agonists [19] have been utilized as well as nuclear localization signals to target genetic material to the nucleus [20]. Our results show that co-expression of NS3/4A DNA and IL-12 is superior NS3/4A alone in priming potent T cell responses (Levander et al., submitted for publication). Similar beneficial effects have been shown for a DNA vaccine targeting hepatitis B virus [17].
1.2.3 Vaccine Delivery
It is well known that delivery of DNA vaccine s to small animals and humans differs significantly in uptake and transfection efficiency [21]. Early results showed that DNA vaccines delivered with a regular needle injection primed potent immune response s in mice [22]. However, when researchers performed early DNA vaccine trials in humans the results were disappointing [23, 24]. The main reason thought to be the inefficient uptake of the injected plasmid DNA in muscle and/or skin. This was the starting point for development of delivery devices, which should facilitate the uptake of plasmid DNA in animal and human tissues. Numerous delivery devices have been developed and tested in preclinical and clinical trials. The most commonly used devices are: (1) apparatus for electroporation (EP)/electro transfer (ET), (2) biolistic delivery device (e.g., gene gun, gg), and (3) needle-free high-pressure injection device (e.g., Biojector). Early results showed that intramuscular delivery of DNA could be improved by EP/ET [25]. We have shown that intramuscular NS3/4A DNA immunization combined with in vivo EP/ET significantly improved the NS3/4A-specific T cell activation in mice [26]. The same NS3/4A DNA vaccine was later delivered as a therapeutic DNA vaccine to patients with chronic HCV infection. Results revealed that the vaccine was considered safe, induced HCV-specific T cell responses, and had a transient effect on the viral load [9] but none of the patients were cured. Hence, additional efforts are needed to improve vaccine immunogenicity . One strategy to further improve the uptake of DNA vaccines through EP/ET based delivery is to manipulate the pulse parameters and voltages [27]. Our experience is that a short high-voltage pulse followed by a longer low-voltage pulse is most efficient for in vivo transfection [21]. Herein we present results obtained using the Cliniporator2 EP/ET device (Fig. 1, IGEA, Carpi, Italy). This EP/ET device allows customized pulse parameters, voltages, and the choice of using two to eight electrodes for intramuscular EP/ET and/or two plates for skin EP/ET. Our results show that intramuscular administration of NS3/4A DNA can be significantly improved when applying in vivo EP/ET (Fig. 2). In line with these results, frequencies of NS3-specific CD8+ T cells were significantly higher in EP/ET treated mice (Fig. 3). Hence, it is evident that NS3/4A DNA immunization benefits from in vivo EP/ET. In addition, we found that NS3/4A DNA immunized mice challenged with a hydrodynamic injection [28] of NS3/4A and firefly luciferase DNA, had significantly less hepatic NS3/4A-firefly luciferase compared to non-immunized mice (Fig. 4a, b, p < 0.05). This highlights that NS3/4A DNA immunization prime T cell responses that home to the liver and eradicate NS3/4A-expressing hepatocytes. The major companies developing EP/ET devices are IGEA (Carpi, Italy), Inovio Pharmaceuticals, Inc. (Plymouth Meeting, PA), and Ichor Medical Systems Inc. (San Diego, CA).

Illustrative pictures of the Cliniporator2 EP/ET device. (a) Picture of the complete Cliniporator2 device. Dimensions are 43 cm × 52 cm × 147 cm (width × length × height). Weight is 49 kg. (b) Picture of the Cliniporator2 handle connected with a device electrode. The two needles have a length of 10 mm and the distance between needles are 4 mm

In vivo EP/ET enhances the priming of HCV NS3/4A-specific T cell responses in C57BL/6 mice. Groups of five wild-type C57BL/6 mice were immunized once with 5 μg coNS3/4A-pVAX1 intramuscularly (i.m.) with or without in vivo EP/ET. One group of mice was left untreated. Two weeks after immunization the mice were sacrificed and splenocytes harvested for determination of T cell responses. A comparison of the number of IFN-γ spot forming cells (SFCs) by ELISpot assay after stimulation with indicated antigens was done in immunized and non-immunized groups of mice. Results are given as the mean SFCs/106 (+SD) splenocytes with a cutoff set at 50 SFCs/106 splenocytes. The statistical difference shown, indicate a statistical difference between groups with and without in vivo EP/ET (***p < 0.001, by AUC and ANOVA)

In vivo EP/ET enhances the frequency of HCV NS3-specific CD8+ T cells after immunization. Groups of ten to thirty wild-type C57BL/6 mice were immunized once with 5 μg coNS3/4A-pVAX1 intramuscularly (i.m.) with or without in vivo EP/ET. One group of mice was left untreated (n = 5). Two weeks after immunization the mice were sacrificed and splenocytes harvested for determination of the frequency of NS3-specific CD8+ T cells. (a) Gating scheme for identification of NS3-specific CD8+ T cells. (b) Representative dot plots from each group are shown. (c) Expansion of NS3-specific CD8+ T cells was determined using direct ex vivo pentamer staining . GAVQNEVTL epitope -specific CD8+ T cells are shown as the percentage of NS3-pentamer positive CD8+ T cells where each filled black circle represent an individual mouse. The black horizontal line indicates the mean of the group. The statistical difference between the groups is indicated as p < 0.05 determined by the Mann–Whitney U test

In vivo clearance of HCV NS3/4A-expressing hepatocytes. (a) Biodistribution of NS3/4A and firefly luciferase determined by using in vivo imaging (Caliper Life Science) is shown in immunized (2 weeks post priming) and non-immunized C57BL/6 mice (four mice/group) 12 h after NS3/4A transfection of hepatocytes. (b) Statistical difference has been indicated ( p < 0.05) using area under the curve (AUC) and analysis of variance (ANOVA) at 12–60 h after NS3/4A transfection of hepatocytes
1.2.3.1 Prime-Boost Approaches
One way to enhance immune activation of genetic vaccines is to employ the prime-boost approach. The idea is to utilize one antigen /vector for immune priming, followed by a booster immunization using another antigen/vector to enhance the previous immune activation. A few different prime-boost approaches have been evaluated in animal models for HCV [29–31]. So far only two HCV prime/boost studies have reached clinical trials, notably in healthy human volunteers and not in chronic HCV patients. The first study utilized human adenovirus 6 (Ad6) to prime, and chimpanzee adenovirus 3 (Ad3) to boost [7]. The second study utilized chimpanzee adenovirus 3 (Ad3) to prime, and modified vaccinia Ankara (MVA) to boost [8]. Both studies showed evidence of potent activation of HCV-specific T cell responses. However, efficacy of these vaccines to protect or clear HCV infection is so far unknown.
2 Materials
2.1 Delivery of DNA Vaccines to Mice Using Intra Muscular Immunization and In Vivo Electro Transfer
-
1.
Ethical permission for the proposed animal experimentation.
-
2.
Required documentation that allows you to work with laboratory mice.
-
3.
Laboratory mice (strain of your choice, we have used H-2b (C57BL/6)).
-
4.
Ear puncher.
-
5.
Hypnorm: 0.315 mg/mL fentanyl citrate, 10 mg/mL fluanisone (VetaPharma Ltd) or other neuroleptanalgesic drug.
-
6.
Ethanol (70 %).
-
7.
Purified plasmid DNA (1 mg/mL) resuspended in PBS. We have used a codon-optimized HCV NS3/4A gene inserted into the pVAX1 vector [14].
-
8.
1 mL syringe.
-
9.
27G, 19 mm needle.
-
10.
Cliniporator2 (IGEA, Carpi, Italy).
-
11.
Cliniporator2 handle (IGEA, Carpi, Italy).
-
12.
Electrode tips (REF N-10-4B) using two electrodes (IGEA, Carpi, Italy).
2.2 Media Preparation and Preparation of Spleen Cells
-
1.
Incomplete medium: RPMI 1640 plus 100 U/mL penicillin, 100 μg/mL streptomycin.
-
2.
Complete medium: RPMI 1640 medium plus 10 % fetal bovine serum (FBS, prior to use the FBS is heat inactivated for 30 min at +56 °C), 100 U/mL penicillin, 100 μg/mL streptomycin, 10 mM HEPES buffer, 2 mM L-glutamine, 1 mM sodium pyruvate, 1 mM nonessential amino acids, 50 μM 2-mercaptoethanol.
-
3.
Marking pen.
-
4.
Lab timer.
-
5.
Scissor.
-
6.
Forceps.
-
7.
Biological safety cabinet.
-
8.
Cell strainer, 70 μm pore size.
-
9.
Petri dish, 10 mm.
-
10.
1 mL syringe.
-
11.
Centrifuge (+4 °C).
-
12.
15 mL Falcon tubes with screw-cap.
-
13.
50 mL Falcon tubes with screw-cap.
-
14.
Red Blood Cell Lysing buffer (Sigma-Aldrich)
-
15.
1 × PBS.
-
16.
Adjustable pipettors and tips (0.5–1000 μL) and multi-pipettors (50–200 μL).
-
17.
Trypan blue stain 0.4 %.
-
18.
Bürkner chamber.
-
19.
Microscope.
2.3 ELISpot Assay
-
1.
Marking pen.
-
2.
Lab timer.
-
3.
Incubator 37 °C, 5 % CO2.
-
4.
Adjustable pipettors and tips (0.5–1000 μL) and multi-pipettors (50–200 μL).
-
5.
Absolute ethanol (>99.5 %). Diluted to 70 % in dH2O.
-
6.
96-well PVDF plates.
-
7.
BCIP/NBT-plus substrate (Mabtech, Nacka Strand, Sweden).
-
8.
Primary IFNγ-antibody, AN18 (Mabtech, Nacka Strand, Sweden).
-
9.
Secondary IFNγ-antibody, R4-6A2-biotin (Mabtech, Nacka Strand, Sweden).
-
10.
Streptavidin-ALP (Mabtech, Nacka Strand, Sweden).
-
11.
10 × PBS.
-
12.
1 × PBS.
-
13.
dH2O.
-
14.
Sterile syringe filters 0.22 μm.
-
15.
Sterile syringe filters 0.45 μm.
-
16.
15 mL Falcon tubes with screw-cap.
-
17.
50 mL Falcon tubes with screw-cap.
-
18.
NS3-CTL peptide, amino acid sequence: GAVQNEITL (NS3 CTL).
-
19.
NS3-Th, peptide, amino acid sequence: EIPFYGKAIPLEAIK (E13K).
-
20.
Ovalbumin-CTL peptide, amino acid sequence: SIINFEKL (OVA CTL).
-
21.
Ovalbumin-Th peptide, amino acid sequence: ISQAVHAAHAEINEAGR (OVA Th).
-
22.
Concanavalin-A (Con A).
-
23.
Dissection microscope.
-
24.
Automated ELISpot reader.
2.4 Quantification of CD8+ T Cell Responses
-
1.
Marking pen.
-
2.
Lab timer.
-
3.
Adjustable pipettors and tips (0.5–1000 μL) and multi-pipettors (50–200 μL).
-
4.
MHC pentamer-R-PE (stored at +4 °C in dark, ProImmune) specific for the mouse H-2Db (GAVQNEVTL) peptide.
-
5.
Anti-mouse CD19 (APC-label).
-
6.
Rat anti-mouse CD8 (FITC-label, monoclonal: KT15).
-
7.
Rat anti-mouse CD3 (PE-label).
-
8.
Purified anti-mouse CD16/CD32 (FCγ III/II, 2.4G2) “Fc block.”
-
9.
Wash buffer/staining buffer (1 % fetal bovine serum (FBS) in PBS). Prepare fresh every time!
-
10.
Fix solution (stock solution: 4 % paraformaldehyde (PFA) in PBS (can be stored at +4 °C for up to one month), working solution: 2 % PFA diluted in wash buffer/staining buffer).
-
11.
V-bottom 96-well plate.
-
12.
FACS tubes (5 mL, polystyrene round-bottom tube).
-
13.
FACSVerse (BD Biosciences, San Jose, CA) or other brand of flow cytometer.
-
14.
FlowJo 9.2 software (Tree Star, Ashland, OR) or other brand of analysis software.
2.5 Detection of Liver Specific Protein Expression by In Vivo Imaging
-
1.
Transiently transgenic mice with intrahepatic expression of the gene of interest and a reporter gene (e.g., Firefly luciferase) for measurements of bioluminescence signals. The transiently transgenic mouse model has been described previously [28].
-
2.
IVIS Spectrum in vivo imaging system (PerkinElmer, Waltham, MA) or similar system for in vivo bioluminescence detection.
-
3.
Living Image Software version 4.2 (PerkinElmer, Waltham, MA).
-
4.
XGI-8 Gas Anesthesia System (PerkinElmer, Waltham, MA) or similar system for anesthesia.
-
5.
Isoflurane (IsoFlo®, Abbott Laboratories Ltd, Berkshire, UK) or similar inhalational anesthesia.
-
6.
Oxygen supply.
-
7.
Luciferin (15 mg/mL, d-luciferin, K+ salt, PerkinElmer, Waltham, MA).
-
8.
1 mL syringe.
-
9.
27G, 19 mm needle.
-
10.
Shaver (for laboratory mice).
3 Methods
The methodology section outlines the following procedures: (1) preparation and intramuscular delivery of DNA vaccine s to laboratory mice in combination with in vivo electro transfer, (2) monitoring the primed T cell responses using (a) ELISpot assay, (b) pentamer staining , and (c) in vivo imaging .
3.1 Delivery of DNA Vaccines to Laboratory Mice Using In Vivo Electro Transfer
The following protocol describes the procedure for intramuscular immunization of laboratory mice using a regular needle and syringe in combination with in vivo electro transfer. The procedure requires laboratory work involving one person. The time needed depends on the number of laboratory mice to be immunized. Roughly, the procedure takes 1 h for immunization and in vivo electro transfer of 20 laboratory mice.
-
1.
Prepare 5 μg DNA in 50 μL PBS (for each mouse to immunized) (see Note 1 ).
-
2.
Switch on the Cliniporator2 device (see Fig. 1a), connect the handle and attach the electrode tip (e.g., two electrodes, see Fig. 1b). Select your electro transfer program of choice (see Note 2 ). We used an optimized program utilizing a short high-voltage pulse followed by a long low-voltage pulse for efficient uptake of the plasmid DNA.
-
3.
Restrain the mouse in your hand, earmark, and inject the mouse intraperitoneally with Hypnorm using a 1 mL syringe and a 27G needle 5–10 min prior to the immunization. This will anesthetize the mouse during the procedure (see Note 3 ).
-
4.
Restrain the mouse in your hand with the abdominal area facing up and keep the right tibialis anterior/cranialis muscle stretched (see Note 4 ).
-
5.
Wet the muscle with 70 % ethanol (see Note 5 ).
-
6.
Inject the mouse with 50 μL PBS containing 5 μg DNA using a regular 1 mL syringe and a 27G needle (see Notes 6 and 8 ). Figure 5 visualizes a successful immunization of the tibialis anterior/cranialis muscle using a tissue marking dye instead of DNA.
Fig. 5 
Illustrative picture of a tibialis anterior/cranialis muscle injected with a tissue marking dye to visualize the injected area. The red arrow indicates site of injection and the direction of the needle insertion
-
7.
Immediately after the DNA injection, treat the same area of the muscle with in vivo electro transfer by penetrating the muscle with the two electrodes and thereafter delivering the electrical pulses (see Notes 7 and 8 ).
-
8.
Place the mouse in a cage and check the status of the mouse until fully recovered from anesthesia.

3.2 Media Preparation
-
1.
Prepare the media in sterile conditions working in a biological safety cabinet (BSC).
-
2.
Discard the volume media that will be replaced by the addition of supplements (discard 70 mL for complete media and 5 mL for incomplete media).
-
3.
Add supplements and store the media in +4 °C until use.
3.3 Preparation of Spleen Cells (for Detection of ELISpot and Quantification of CD8+ T Cell Responses)
The following protocol describes preparation of spleen cells to be used in ELISpot and pentamer staining /quantification of CD8+ T cells. The procedure requires laboratory work involving one person. The time needed depends on the number of laboratory mice to be sacrificed and spleens to prepare. Roughly, the procedure takes 3 h for 20 laboratory mice.
-
1.
Sacrifice the mouse through cervical dislocation and excise the spleen using scissors and forceps.
-
2.
Immediately put the spleen in a 15 mL Falcon tube containing 2 mL of incomplete medium (see Subheading 2.2, item 1).
-
3.
The following procedures should be performed under sterile conditions in a biological safety cabinet.
-
4.
Make single cell suspension by teasing the spleen with the plunger of a 1 mL syringe in the cell strainer placed in a petri dish.
-
5.
Transfer the cell suspension to a new 15 mL Falcon tube, wash the cell strainer with 5 mL of incomplete medium and collect the wash in the same tube.
-
6.
Centrifuge for 5 min at 450 × g, +4 °C.
-
7.
Discard the supernatant and resuspend the pellet in the remaining medium.
-
8.
Add 1 mL of Red Blood Cell Lysing buffer and incubate at room temperature for 1 min (see Note 9 ).
-
9.
Add 12 mL of PBS to the Falcon tube to inactivate the Red Blood Cell Lysing buffer and proceed immediately with centrifugation for 5 min at 450 × g, +4 °C.
-
10.
Discard the supernatant and resuspend the cell pellet in 2 mL of complete medium (see Subheading 2.2, item 2).
-
11.
Prepare cells for counting by mixing 10 μL cell suspension + 90 μL trypan blue stain 0.4 %.
-
12.
Count cells in a Bürkner chamber.
-
13.
13a. Prepare a cell suspension (from individual or pools of spleen cells) at a concentration of 2 × 106 cells/mL for the ELISpot assay (see Subheading 3.4).
-
14.
13b. Calculate and transfer a cell suspension containing 1 × 106 cells (from individual mice) to a V-bottom plate for quantification of HCV-specific CD8+ T cells (see Subheading 3.5).
3.4 Determination of Immune Responses after DNA Immunization by ELISpot Assay
The following section describes the procedure to determine in vitro T cell responses in spleen cells from DNA immunized mice by measuring the number of IFN-γ spot forming cells (SFCs) after stimulation with HCV-derived antigens (Fig. 2). The procedure requires laboratory work involving one person for approximately 1 h day 1, 5 h day 2, and 6 h day 4.
Day 1. Preparation of ELISpot plates (under sterile conditions). Coating of plates:
-
1.
Dilute the coating antibody (IFN-γ, AN18) to 10 μg/mL in sterile PBS, pH 7.4.
-
2.
Pre-wet each well with 50 μL of 70 % ethanol for maximum 1 min (step 2–4, one plate at a time) (see Note 10 ).
-
3.
Wash plates 4 times with 200 μL sterile water (see Note 11 ). Immediately move to step 3 without allowing the membrane to dry (see Note 12 ).
-
4.
Coat ELIIP plates with 100 μL anti-IFN-γ (AN18) antibody in sterile PBS.
-
5.
Incubate overnight at 4–8 °C (in a refrigerator).
Day 2. Incubation of cells in plate (under sterile conditions). Block membrane:
-
6.
Decant primary antibody solution by flicking the plate over a waste bin.
-
7.
Wash off unbound antibody with 200 μL PBS/well as described in Note 11 , incubate for 5 min.
-
8.
Decant washing buffer and repeat item 7, three times.
-
9.
Block with 200 μL complete medium (see Subheading 2.2, item 2) for at least 2 h at 37 °C.
Preparation of spleen cells and antigens used for in vitro stimulation:
-
10.
Prepare cells according to Subheading 3.3 and dilute cells to 2 × 106/mL.
-
11.
Remove blocking media and gently add 100 μL/well (200,000 cells/well) of isolated spleen cells.
-
12.
Prepare dilutions of your antigens of interest. In this chapter we have used different antigens from HCV NS3 (e.g., NS3-CTL peptide and NS3-Th peptide) and control antigens (Ovalbumin-CTL peptide and Ovalbumin-Th peptide). As positive control ConA was used and as negative control medium was used (see Note 13 ).
-
13.
Sterile filter all antigen -solutions through a 0.22 μm filter before use.
-
14.
Add 100 μL/well of antigen -solution according to protocol.
-
15.
Incubate for 24–48 h in 37 °C, 5 % CO2.
Day 4. Detection of spot-forming cells (SFCs). Secondary antibody:
-
16.
Inspect all plates/wells and mark wells with leakage before decanting the cells (see Note 14 ).
-
17.
Wash plate 5 times with PBS (see Note 15 ).
-
18.
Dilute the biotinylated anti-IFN-γ antibody (R4-6A2-biotin) to 2 μg/mL in PBS + 0.5 % FBS.
-
19.
Add 100 μL/well. Incubate for 2 h at room temperature.
Development of spot-forming cells (SFCs):
-
20.
Wash wells 5 times with PBS as described in item 16 and see Note 15 .
-
21.
Dilute the streptavidin-ALP 1:1000 in PBS containing 0.5 % FBS
-
22.
Add 100 μL/well. Incubate for 1 h at room temperature.
-
23.
Wash wells 5 times with PBS as described in item 16 and see Note 15 .
-
24.
Filtrate the ready-to-use substrate solution (BCIP/NBT-plus) through a 0.45 μm sterile filter.
-
25.
Add 100 μL/well of the substrate solution (see Note 16 ).
-
26.
Develop plates for 12 min (IFNγ) or until distinct spot-forming cells emerge (see Note 17 ).
-
27.
Stop color development by washing the plate extensively in tap water for 10 min. Wash both sides of the plate (see Note 18 ).
-
28.
Forcefully decant the remaining water and wipe off excess of water using paper tissues.
-
29.
Let the plates dry for at least 2 h before analysis.
-
30.
When the plates are completely dry, inspect and count spot-forming cells in a dissection microscope (×40) or using an automated ELISpot reader. Herein we used the AID iSpot (EliSpot/FluoroSpot) reader system (Autoimmun Diagnostica GmbH, Strassberg, Germany).
-
31.
The automated ELISpot reader will count the number of spots-forming cells (cytokine producing cells) at each concentration of the included antigens and the results given as the number of IFN-γ producing cells per 106 cells. A mean number of cytokine producing cells of less than 50 per 106 cells are considered as negative.
-
32.
Store plates at room temperature in the dark (see Note 19 ).
3.5 Detection of HCV-Specific CD8+ T Cells by Pentamer Staining
The following section describes the procedure to direct ex vivo quantify the number of HCV-specific CD8+ T cells in spleen cells from DNA immunized mice (Fig. 3). The procedure requires laboratory work involving one person for approximately 10 h.
-
1.
Centrifuge the pentamer to pellet any protein aggregates present in the solution (12,000 × g, 10 min, +4 °C). Thereafter, directly dilute pentamer using the supernatant after centrifugation (see Note 20 ).
-
2.
Dilute 10 μL of pentamer in 40 μL of wash buffer/staining buffer (per sample). The total volume is 50 μL per sample to be stained. Store the diluted pentamer at room temperature in the dark until use.
-
3.
Prepare a single spleen cell suspension according to Subheading 3.3.
-
4.
Calculate the volume of cell suspension corresponding to 1 × 106 cells.
-
5.
Add 1 × 106 spleen cells to the 96-well V-bottom plate. Individual samples should be added to single wells. In addition to the individual samples, the following controls should be included:
-
(a)
Unstained cells (from both immunized and non-immunized mice)
-
(b)
Single stained cells (from immunized mice).
-
(c)
Double stained cells (from immunized mice).
-
(d)
Cells stained with pentamer only (from both immunized and non-immunized mice)
-
(a)
-
6.
Centrifuge for 3 min at 450 × g, +4 °C.
-
7.
Forcefully discard the supernatant by hand.
-
8.
Wash cells once with 200 μL wash buffer/staining buffer. Pipette the solution up and down thrice in the well to loosen the cell pellets.
-
9.
Centrifuge for 3 min at 450 × g, +4 °C.
-
10.
Forcefully discard the supernatant by hand.
-
11.
Carefully resuspend the pellet in 50 μL of pre-diluted pentamer (item 2). Control wells without pentamer should instead be filled up with 50 μL wash buffer/staining buffer and resuspended.
-
12.
Incubate the plate at room temperature for 15 min in the dark (see Note 21 ).
-
13.
Centrifuge for 3 min at 450 × g, +4 °C.
-
14.
Forcefully discard the supernatant by hand.
-
15.
Wash cells once with 200 μL wash buffer/staining buffer. Pipette the solution up and down thrice in the well to loosen the cell pellets.
-
16.
Centrifuge for 3 min at 450 × g, +4 °C.
-
17.
Forcefully discard the supernatant by hand.
-
18.
Carefully resuspend the pellet in 50 μL Fc block (purified anti-mouse CD16/CD32 (FCγ III/II)) diluted at 1:50 in wash buffer/staining buffer (see Note 22 ).
-
19.
Incubate the plate on ice for 15 min in the dark.
-
20.
Centrifuge for 3 min at 450 × g, +4 °C.
-
21.
Forcefully discard the supernatant by hand.
-
22.
Wash cells once with 200 μL wash buffer/staining buffer. Pipette the solution up and down thrice in the well to loosen the cell pellets.
-
23.
Centrifuge for 3 min at 450 × g, +4 °C.
-
24.
Forcefully discard the supernatant by hand.
-
25.
Add optimal amounts of anti-CD8, anti-CD19, and anti-CD3 antibodies in a total volume of 50 μL. Dilute antibodies in wash buffer/staining buffer. Mix by pipetting the solution up and down thrice (see Notes 23 and 24 ).
-
26.
Incubate the plate on ice for 20 min in the dark.
-
27.
Centrifuge for 3 min at 450 × g, +4 °C.
-
28.
Forcefully discard the supernatant by hand.
-
29.
Wash cells once with 200 μL wash buffer/staining buffer. Pipette the solution up and down thrice in the well to loosen the cell pellets. Repeat item 27–29 once.
-
30.
Fix cells by adding 150 μL fixing solution to each well (see Note 25 ).
-
31.
Transfer the fixed cell suspension to FACS vials. Add another 150 μL fixing solution to each well and transfer the remaining cells to the same FACS vials.
-
32.
Store the fixed cell suspension at +4 °C in the dark. Perform data acquisition within 2 days of staining (see Note 26 ).
Flow cytometric analysis:
-
33.
To view the pentamer-positive cells we used the following gating strategy (see Fig. 3a). Live lymphocytes were gated, and from this gate the CD19+ cells were excluded, and further gating on CD8+ and Pentamer+ cells were performed to determine the frequency of HCV-specific CD8+ T cells (see Fig. 3b, c).
3.6 Detection of Liver-Specific Protein Expression by In Vivo Imaging
The following section describes the procedure to determine the presence of intrahepatic HCV protein expression in DNA immunized and non-immunized mice (Fig. 4). Bioluminescent imaging allows longitudinal studies and comparison of data sets. The procedure requires laboratory work involving one person for approximately 2 h.
Pre-imaging procedure:
-
1.
The IVIS Spectrum in vivo imaging system should be turned on at all time.
-
2.
Log into the computer and start the Living Image Software.
-
3.
Initiate the system (see Note 27 ).
-
4.
Check the charcoal filters on top of the anesthesia station (XGI-8 Gas Anesthesia System) by weighing (see Note 28 ).
-
5.
Turn on the evacuation pump.
-
6.
Turn on the oxygen supply switch.
-
7.
Turn on the gas switch for the imaging chamber.
-
8.
Set the vaporizer at 0 % isoflurane position. Thereafter turn on the imaging chamber toggle valve and set at 0.25 L/min.
-
9.
Turn on the induction chamber toggle valve and set at 1.0 L/min.
-
10.
Confirm the flow rate, and then turn off the two toggles.
-
11.
Set the vaporizer to 2.0 % isoflurane (for mice) (see Note 29 ).
-
12.
How to generate mice with transient intrahepatic expression of HCV NS3 and firefly luciferase has been described previously [28] (see Subheading 2.5). Procedure for intramuscular immunization of mice using a regular needle and syringe in combination with in vivo electro transfer has been described in Subheading 3.1.
-
13.
Prior to anesthetizing mice the area of interest for bioluminescent measurement should be shaved. We measure the bioluminescent signal in the liver and therefore carefully shave the abdominal area of the mouse (see Note 30 ).
-
14.
Weigh all mice to be analyzed (see Note 31 ).
-
15.
Thaw aliquots of luciferin on ice (see Note 32 ).
-
16.
Inject 10 μL luciferin (0.15 mg)/g of body weight intraperitoneally (i.p.) (see Notes 33 and 34 ).
-
17.
Wait 4 min and thereafter put the animals (maximum 5 mice) in the induction chamber and turn on the induction chamber toggle valve. Keep the mice in the induction chamber for 4 min.
-
18.
Turn on the imaging chamber toggle valve.
-
19.
When all mice are anesthetized, transfer them from the induction chamber to the imaging chamber. Put the mice on the back with the nose in the nose cone. The shaved mouse abdomens should be exposed (see Note 35 ).
-
20.
Turn off the induction chamber toggle.
-
21.
Acquire a luminescent image using predefined protocols. The protocol is set up in the Living Image software. Enable the auto-save function. You can select auto exposure, manual exposure, and sequence acquisition. We routinely perform both auto exposure and sequence acquisition. Using the auto exposure setting the software automatically determines the binning and F/Stop settings. Using the manual exposure setting you select the exposure time, binning, and F/Stop. Using the sequence acquisition you can select several segments with different exposure time (see Note 36 ).
-
22.
Select the bioluminescent imaging mode, check mark photograph and chose an appropriate Field of View (FOV) (see Note 37 ).
-
23.
Capture image/s by clicking the acquire button.
-
24.
Enter relevant image label information.
Post-imaging procedure:
-
25.
Carefully return mice to their cage. Continuously observe mice during recovery from the gas anesthesia (see Note 38 ).
Turn off the anesthesia station:
-
26.
Turn off the vaporizer and keep the oxygen supply on for approximately 5 min to clear the isoflurane in the anesthesia system before switching off the oxygen tank.
-
27.
Turn on the induction and imaging chamber toggle valves until the pressure drop to zero.
-
28.
Turn off the toggle valves.
-
29.
Turn off oxygen valve.
-
30.
Turn off the evacuation pump.
-
31.
Securely copy your imaging data.
-
32.
Exit the imaging software and log out from the computer.
-
33.
Leave the IVIS Spectrum in vivo imaging system on at all time.
Image analysis and data presentation:
-
34.
Analyze your optical images using the Living Image Software. Within the software you can perform different analyses. You may organize your images, view image information, adjust image appearance, perform background subtraction to the image, view intensity data, make measurements, overlay multiple images, export and print images.
-
35.
Your optical imaging results may be presented in various ways. We commonly show representative photographs of mice from different groups/time-points, supported with the light emitted/radiance within a region of interest (ROI) and statistical comparison (Fig. 4a, b). We present the results as light emitted/radiance (photons/second/cm2/steradian, e.g., p/s/cm2/sr) + standard deviation (SD).
4 Notes
-
1.
Mix the DNA and PBS to a homogeneous solution. Measure the DNA concentration to ensure correct concentration. This is especially important when higher concentrations of DNA are prepared.
-
2.
The electro transfer program need to be evaluated prior to use. In general, a short high-voltage pulse increases the permeability of the cellular membrane. The long low-voltage pulse is believed to facilitate the transport of the DNA into the cell. Thus, both the high- and low-voltage pulses enhance the uptake of DNA into the cell.
-
3.
The volume of Hypnorm to be used has to be adjusted depending on mouse strain and weight of the animals. The Hypnorm will anesthetize the laboratory mice for approximately 30–60 min depending on the dose.
-
4.
Stabilize your hand by placing it against the bench. It is important that the mouse is completely restrained during the immunization and in vivo electro transfer procedure. Movements may affect the efficiency of the primed immune response . Figure 5 visualizes a tibialis anterior/cranialis muscle successfully injected with a tissue marking dye.
-
5.
Wipe the muscle with ethanol prior to immunization to make it more visible.
-
6.
The 27G needle tip should be injected approximately 2–3 mm into the muscle for optimal delivery of the DNA. If injected to shallow the DNA solution may leak out (e.g., subcutaneous delivery) and if injected to deep you may pass through the tibialis anterior/cranialis muscle.
-
7.
The electrode tips should be injected approximately 3–4 mm into the muscle for an optimal in vivo electro transfer.
-
8.
Wait a few seconds and thereafter carefully withdraw the needle/electrodes from the muscle to avoid leakage of the DNA solution from the needle/electrode hole/s.
-
9.
Shorter incubation time will result in insufficient lysis of the red blood cells. Longer incubation time will affect the condition of the splenocytes and may results in low cell yield.
-
10.
If the well is pre-wetted with ethanol for longer time than 1 min, the well may dry out, which often causes leakage in the PVDF membrane.
-
11.
Decant the wash solution by flicking the plate over a waste bin in the BSC. Remove excess of water on the plate using sterile paper tissue.
-
12.
Leaving the plate for a longer time in water or if the membrane dries out may result in leaking wells.
-
13.
All antigens should be prepared at a 2× final concentration since antigens will be diluted in an equal volume of cell suspension.
-
14.
Wells with leakage will give a false signal and should be excluded. It is recommended to use triplicates for all samples and dilutions.
-
15.
Decant cell suspension by emptying the plate in a waste bin for biological waste. Wash the plate by completely immersing it in 1 × PBS and thereafter decant the PBS in the sink. Repeat the procedure at least 5 times. After the last wash, forcefully decant the remaining PBS and wipe off excess of PBS using paper tissues before proceeding to next step.
-
16.
If bubbles appear in some wells after pipetting the substrate solution, carefully remove them by using a pipette tip. If bubbles remain on the bottom of the plate it may cause an incomplete development and thus lack of detectible spot-forming cells.
-
17.
The plate development time varies and needs to be determined for each new protocol. Commonly one is utilizing the dilutions of the positive control (ConA) to set the time for development. Too long development time will cause a high background due to dark wells.
-
18.
When spot-forming cells are visible in the positive control, the development should be stopped by immersing the plate in water. The spot-forming cells will be clearly visible when the plates have dried completely.
-
19.
Plates may be stored for several years without substantial loss of SFC intensity.
-
20.
The protein aggregates in the pentamer solution may cause nonspecific staining.
-
21.
Prolonged incubation time may increase the sensitivity but also the nonspecific staining.
-
22.
Addition of Fc block is important to block nonspecific antibody binding to Fc receptors on immune cells.
-
23.
We diluted the antibodies as follows: anti-CD8 (FITC-label) 1:33, anti-CD19 (APC-label) 1:40, and anti-CD3 (PE-label) 1:33.
-
24.
We included the following staining controls: unstained cells, single stained cells (anti-CD19, anti-CD8, anti-CD3, pentamer), double stained cells (anti-CD8 + anti-CD19, anti-CD8 + pentamer, anti-CD19 + pentamer), and triple stained cells (anti-CD8 + anti-CD8 + penatmer).
-
25.
The working solution should be prepared fresh every time for optimal fixation.
-
26.
You should aim at performing the data acquisition as soon as possible after staining. The longer you wait for data acquisition, the weaker fluorescent signal you will detect. This is very important if you are detecting low frequencies of antigen -specific CD8+ T cells.
-
27.
Allow several minutes for controlling the system and for the camera to cool down to −90 °C.
-
28.
A filter that is weighting >50 g than the original weight should be replaced. This is to ensure a good absorption of the gas anesthesia.
-
29.
The % isoflurane needed may vary between mouse strains or if rats are used. Hence, the optimal % isoflurane needs to be predetermined.
-
30.
The fur will significantly reduce the bioluminescent signal and should therefore be removed prior to imaging.
-
31.
This is important to calculate the accurate dose of luciferin.
-
32.
The luciferin solution should be sterile filtered through a 0.2 μm filter. Avoid freeze-thaw ing of luciferin aliquots.
-
33.
Mice should be injected with 150 mg luciferin/kg body weight. A 20 g mouse should be injected with 200 μL = 3 mg of luciferin. This is of utmost importance since luciferin (the substrate) is the limiting factor for the luminescent signal. Thus, weight-based dosing of luciferin is important to be able to compare luminescent signals between animals. Otherwise, differences in the luminescent signal may depend on the amount of substrate administered to the animals.
-
34.
Luciferin should be injected 11 min prior to luminescent imaging.
-
35.
Check that all mice have their noses completely inside the nose cone. Check that all mice breathe rhythmically. If the mice are not completely anesthetized increase the percentage of isoflurane by 0.25–0.5 % and wait a few minutes before analysis. Caution: If the mice wake up during the imaging procedure they may move around inside the imaging chamber and cause damage to themselves and the instrument.
-
36.
Exposure time: the length of the time that the shutter is open during image acquisition. Binning: Controls the pixel size on the CCD camera. Hence, increasing the binning will result in increased pixel size and sensitivity. This will reduce the spatial resolution. F/Stop: defines the size of the camera lens aperture. A small aperture size results in lower sensitivity since less light is collected for the image. Small apertures produce sharp imagines whereas large apertures maximize the sensitivity.
-
37.
The choice of FOV depends on how many mice you will image. FOV is the size of the stage area to be imaged. FOV A (part of 1 mouse), B (1 mouse), C (up to 3 mice), and D (up to 5 mice).
-
38.
Mice usually recover within 1–5 min.
References
Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST (2013) Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342
Thomas DL (2013) Global control of hepatitis C: where challenge meets opportunity. Nature Med 19:850–858
Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR et al (2014) Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. New Engl J Med 370:211–221
Feld JJ, Kowdley KV, Coakley E, Sigal S et al (2014) Treatment of HCV with ABT-450/r–ombitasvir and dasabuvir with ribavirin. New Engl J Med 370:1594–1603
Ahlen G, Frelin L, Brenndorfer ED, Brass A et al (2013) Containing “The Great Houdini” of viruses: combining direct acting antivirals with the host immune response for the treatment of chronic hepatitis C. Drug Resist Updat 16:60–67
Callendret B, Eccleston HB, Hall S, Satterfield W et al (2014) T-cell immunity and hepatitis C virus reinfection after cure of chronic hepatitis C with an interferon-free antiviral regimen in a chimpanzee. Hepatology 60:1531–1540
Barnes E, Folgori A, Capone S, Swadling L et al (2012) Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med 4:115ra111
Swadling L, Capone S, Antrobus RD, Brown A et al (2014) A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci Transl Med 6:261ra153
Weiland O, Ahlen G, Diepolder H, Jung MC et al (2013) Therapeutic DNA vaccination using in vivo electroporation followed by standard of care therapy in patients with genotype 1 chronic hepatitis C. Mol Therapy 21:1796–1805
Di Bisceglie AM, Janczweska-Kazek E, Habersetzer F et al (2014) Efficacy of immunotherapy with TG4040, peg-interferon, and ribavirin in a Phase 2 study of patients with chronic HCV infection. Gastroenterology 147:119–131
Habersetzer F, Honnet G, Bain C, Maynard-Muet M et al (2011) A poxvirus vaccine is safe, induces T-cell responses, and decreases viral load in patients with chronic hepatitis C. Gastroenterology 141:890–899
Klade CS, Schuller E, Boehm T, von Gabain A, Manns MP (2012) Sustained viral load reduction in treatment-naive HCV genotype 1 infected patients after therapeutic peptide vaccination. Vaccine 30:2943–2950
Wedemeyer H, Schuller E, Schlaphoff V, Stauber RE et al (2009) Therapeutic vaccine IC41 as late add-on to standard treatment in patients with chronic hepatitis C. Vaccine 27:5142–5151
Frelin L, Ahlen G, Alheim M, Weiland O et al (2004) Codon optimization and mRNA amplification effectively enhances the immunogenicity of the hepatitis C virus nonstructural 3/4A gene. Gene Ther 11:522–533
Chen A, Ahlen G, Brenndorfer ED, Brass A et al (2011) Heterologous T cells can help restore function in dysfunctional hepatitis C virus nonstructural 3/4A-specific T cells during therapeutic vaccination. J Immunol 186:5107–5118
Levander S, Sällberg M, Ahlén G, Frelin L. A non-human hepadnaviral adjuvant for hepatitis C virus-based DNA vaccines. Submitted for publication.
Brass A, Frelin L, Milich DR, Sallberg M, Ahlen G (2015) Functional aspects of intrahepatic Hepatitis B Virus-specific T cells induced by therapeutic DNA vaccination. Mol Therapy 23:578–590
Tovey MG, Lallemand C (2010) Adjuvant activity of cytokines. Methods Mol Biol 626:287–309
Applequist SE, Rollman E, Wareing MD, Liden M et al (2005) Activation of innate immunity, inflammation, and potentiation of DNA vaccination through mammalian expression of the TLR5 agonist flagellin. J Immunol 175:3882–3891
Branden LJ, Mohamed AJ, Smith CI (1999) A peptide nucleic acid-nuclear localization signal fusion that mediates nuclear transport of DNA. Nature Biotechnol 17:784–787
Sallberg M, Frelin L, Ahlen G, Sallberg-Chen M (2015) Electroporation for therapeutic DNA vaccination in patients. Medical Microbiol Immunol 204:131–135
Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH et al (1993) Heterologous protection against influenza by injection of DNA encoding a viral protein. Science 259:1745–1749
MacGregor RR, Boyer JD, Ugen KE, Lacy KE et al (1998) First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: safety and host response. J Infect Dis 178:92–100
Calarota S, Bratt G, Nordlund S, Hinkula J et al (1998) Cellular cytotoxic response induced by DNA vaccination in HIV-1-infected patients. Lancet 351:1320–1325
Aihara H, Miyazaki J (1998) Gene transfer into muscle by electroporation in vivo. Nature Biotechnol 16:867–870
Ahlen G, Soderholm J, Tjelle T, Kjeken R, Frelin L et al (2007) In vivo electroporation enhances the immunogenicity of hepatitis C virus nonstructural 3/4A DNA by increased local DNA uptake, protein expression, inflammation, and infiltration of CD3+ T cells. J Immunol 179:4741–4753
Gothelf A, Gehl J (2012) What you always needed to know about electroporation based DNA vaccines. Human Vaccines Immunother 8:1694–1702
Ahlen G, Sallberg M, Frelin L (2013) Methods for monitoring gene gun-induced HBV- and HCV-specific immune responses in mouse models. Methods Mol Biol 940:239–267
Fournillier A, FrelinL JE, Ahlen G et al (2013) A heterologous prime/boost vaccination strategy enhances the immunogenicity of therapeutic vaccines for hepatitis C virus. J Infect Dis 208:1008–1019
Lin Y, Kwon T, Polo J, Zhu YF et al (2008) Induction of broad CD4+ and CD8+ T-cell responses and cross-neutralizing antibodies against hepatitis C virus by vaccination with Th1-adjuvanted polypeptides followed by defective alphaviral particles expressing envelope glycoproteins gpE1 and gpE2 and nonstructural proteins 3, 4, and 5. J Virol 82:7492–7503
Pancholi P, Perkus M, Tricoche N, Liu Q, Prince AM (2003) DNA immunization with hepatitis C virus (HCV) polycistronic genes or immunization by HCV DNA priming-recombinant canarypox virus boosting induces immune responses and protection from recombinant HCV-vaccinia virus infection in HLA-A2.1-transgenic mice. J Virol 77:382–390
Acknowledgements
The following work was supported by grants from the Swedish Research Council (K2012-99X-22017-01-3), the Swedish Society of Medicine, Goljes Memorial Fund, the Åke Wiberg Foundation, the Ruth and Richard Juhlin Foundation, and from Karolinska Institutet.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Ahlén, G., Frelin, L. (2016). Methods to Evaluate Novel Hepatitis C Virus Vaccines. In: Thomas, S. (eds) Vaccine Design. Methods in Molecular Biology, vol 1403. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3387-7_11
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3387-7_11
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3385-3
Online ISBN: 978-1-4939-3387-7
eBook Packages: Springer Protocols