
Abstract
Accumulated lines of evidence reveal that a large number of circular RNAs are produced in transcriptomes from fruit fly to mouse and human. Unlike linear RNAs shaped with 5′ cap and 3′ tail, circular RNAs are characterized by covalently closed loop structures without open terminals, thus requiring specific treatments for their identification and validation. Here, we describe a detailed pipeline for the characterization of circular RNAs. It has been successfully applied to the study of circular intronic RNAs derived from intron lariats (ciRNAs) and circular RNAs produced from back spliced exons (circRNAs) in human.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Single-stranded circular RNA molecules were firstly observed by electron microscopy in plant viroids [1], yeast mitochondrial RNAs [2], and hepatitis δ virus [3]. Later, a handful of circular RNAs processed from splicing were identified by Northern blots and/or RT-PCRs in higher eukaryotes, including human and mouse [4–6]. Due to their low abundance and scrambled order of exon–exon joining, these circular RNAs had long been considered as by-products of aberrant splicing, thus unlikely with important biological functions [6]. Nevertheless, circular RNAs produced from human INK4a/ARF or CDR1 locus were reported to affect human atherosclerosis risk [7] or regulate gene expression [8–10], suggesting that some circular RNAs can be functional.
The application of next generation sequencing in transcriptome analyses (mRNA-seq) has provided unprecedented insight and quantitative measurements of gene expression , alternative splicing , etc. [11]. However, as formed covalently closed structures, circular RNAs lack canonical 3′ polyadenylation, thus falling below the radar of most canonical polyadenylated (linear) transcriptome profiling . Until recently, non-polyadenylated RNAs could be enriched from total RNAs by depleting both polyadenylated RNAs and ribosomal RNAs for high throughput sequencing analyses [12], allowing the identification of new RNA transcripts. Non-polyadenylated RNA-seq signals were frequently identified in specific intron and exon regions [12], many of which were further proven to be circular RNAs produced from either introns [13] or exons [14, 15].
So far, two major groups of circular RNAs have been described [16]. One is produced from intronic regions (circular intronic RNAs , ciRNAs) [13] by escaping from debranching after splicing. The other group of circular RNAs (circRNAs ) is generated from reversely back-spliced exons [10, 15, 17, 18]. Importantly, these two groups of circular RNAs have been successfully recapitulated with specific expression vectors [13, 15]. Currently, ten thousands of circular RNAs have been widely identified in metazoans from fruit fly to mouse and human [10, 15, 17–20], greatly expanding the complexity of transcriptomes and the diversity of noncoding RNAs [16]. To be noticed, special attention is required for circular RNAs enrichment and detection due to their intrinsic circular structures.
In this chapter, we describe an integrated pipeline for circular RNA characterization (Fig. 1), from the fractionation of non-polyadenylated RNAs from total RNAs, to the enrichment of circular RNAs with the RNase R digestion, and then to the validation of their existence by Northern blots and divergent PCRs. This pipeline provides a standard lab protocol to characterize circular RNAs , and has been successfully applied to the study of ciRNAs [13] and circRNAs [15] in human.

A diagram of circular RNA fractionation , enrichment, and validation. See text for details
2 Materials
All solutions/recipes are prepared from analytical grade chemicals with deionized DEPC-treated water. All reagents are RNase-free. Sterilized reagents are aliquoted and stored at room temperature for immediate usage or −20 °C for long term storage. 1.5 ml RNase-free microcentrifuge tubes are purchased from Crystalgen (catalog number L-2507). 15 ml RNase-free centrifuge tubes are from Nest (catalog number 601052). 50 ml RNase-free centrifuge tubes are from Corning (catalog number 430829). Equipment required for polyacrylamide gel electrophoresis and electrophoretic transfer are from BIO-RED (catalog numbers 165-8001 and 170-3930, respectively).
2.1 RNA Fractionations
All reagents and recipes needed in this step have been described previously in great details [21].
2.2 RNase R Digestion
-
1.
Ribonuclease R (RNase R): Epicentre, catalog number RNR07250, lot number RNR40620.
-
2.
Phenol–chloroform–isoamyl alcohol (25:24:1, v/v): Life Technologies, catalog number 15593-031.
-
3.
4 M LiCl solution: weigh 3.3912 g LiCl (Sigma-Aldrich, catalog number L9650-500 G) and transfer to a 15 ml RNase-free centrifuge tube, add DEPC-treated water to 10 ml. Mix thoroughly and filter through a 0.22 μm Millex-GP Syringe Filter Unit (Millipore, catalog number SLGP05010). Split into small aliquots and store at −20 °C.
-
4.
Glycogen, RNA grade: Thermo SCIENTIFIC, catalog number R0551.
2.3 Validation of Circular RNAs
2.3.1 RNA Deep-Sequencing and Genome-Wide Analysis of Circular RNAs
-
1.
75 % ethanol (v/v): transfer 30 ml absolute ethanol to a 50 ml RNase-free centrifuge tubes and add 10 ml DEPC-treated water. Mix well and store at −20 °C.
2.3.2 Denaturing Urea Polyacrylamide Gel Electrophoresis (Urea PAGE)
-
1.
Urea: aMRESCO, catalog number 037-1 KG.
-
2.
30 % acrylamide: Sangon Biotech, catalog number SD6017.
-
3.
10× TBE Electrophoresis Buffer: weigh 108 g Tris base (Sigma-Aldrich, catalog number 15456-3), 55 g boric acid (Sigma-Aldrich, catalog number B6768-500 G), transfer to a beaker with 40 ml 0.5 M EDTA (pH 8.0), and add DEPC-treated water to 1 l. Filter through a 0.22 μm Millex-GP Syringe Filter Unit, and store at room temperature. 1× TBE buffer is diluted from 10× TBE buffer with DEPC-treated water.
-
4.
0.5 M EDTA, pH 8.0: weigh 186.1 g Na2EDTA · 2H2O (Sigma-Aldrich, catalog number E5134-250 G), transfer to a beaker with 500 ml DEPC-treated water, adjust pH to 8.0 with 10 M NaOH, and add DEPC-treated water to 1 l. Filter through a 0.22 μm Millex-GP Syringe Filter Unit, and store at room temperature.
-
5.
10 % (w/v) ammonium persulfate (APS): dissolute 0.1 g APS (Sigma-Aldrich, catalog number A3678-25 G) with DEPC-treated water to 1 ml. Store at 4 °C for few weeks.
-
6.
TEMED ((N,N,N′,N′-tetramethylethylenediamine): BIO-RED, catalog number 161-0801.
-
7.
2× urea loading buffer: 8 M urea, 90 % formamide (Sigma-Aldrich, catalog number F9037-100 mL), 20 mM EDTA, 0.1 % (w/v) xylene cyanol, bromophenol blue. Store at 4 °C.
-
8.
50× Transfer Buffer: weigh 30.285 g Tris base, 17.02 g sodium acetate trihydrate (Sigma-Aldrich catalog number 236500-500 G), and 9.306 g EDTA, transfer to a beaker with 10 ml acetate, and add DEPC-treated water to 500 ml. Filter through a 0.22 μm Millex-GP Syringe Filter Unit, and store at room temperature. 1× transfer buffer is diluted from 50× transfer buffer with DEPC-treated water.
-
9.
RiboMAX™ Large Scale RNA Production Systems: Promega, catalog number P1280 (SP6), P1300 (T7) for prepare RNA probe.
-
10.
Dig RNA Labeling Mix: Roche, catalog number 11277073910.
-
11.
DNA-free™ Kit, DNase Treatment and Removal Reagents: Ambion, catalog number AM1906.
-
12.
Dig Easy Hyb Granules: Roche, catalog number 11796895001.
-
13.
20× SSC: weigh 175 g NaCl (Sigma-Aldrich, catalog number S9625-1 KG) and 88 g sodium citrate dihydrate (Sigma-Aldrich, catalog number W302600), transfer to a beaker with 500 ml DEPC-treated water, adjust pH to 7.0 with 1 M HCl, and add DEPC-treated water to 1 l. Filter through a 0.22 μm Millex-GP Syringe Filter Unit, and store at room temperature. 2× SSC and 0.2× SSC are diluted from 20× SSC with DEPC-treated water.
-
14.
10 % (w/v) SDS: dissolute 100 g SDS (Sigma-Aldrich, catalog number L3771-1KG) with DEPC-treated water to 1 l, filter through a 0.22 μm Millex-GP Syringe Filter Unit, store at room temperature. 0.1 % SDS dilute from 10 % SDS.
-
15.
DIG Wash and Block Buffer Set: Roche, catalog number 11585762001.
-
16.
Anti-Digoxigenin-AP, Fab fragments: Roche, catalog number 11093274910.
-
17.
CDP-Star, ready-to-use solution: Roche, catalog number 12041677001.
2.3.3 Divergent RT-PCR
-
1.
SuperScript™ III Reverse Transcriptase: Invitrogen, catalog number 18080.
-
2.
Random hexamers: TaKaRa, catalog number RR037A.
-
3.
dNTP mixture: TaKaRa, catalog number T4030.
-
4.
Recombinant RNasin® Ribonuclease Inhibitor: Promega, catalog number N2511.
3 Methods
3.1 Fractionation of Non-polyadenylated RNAs from Mammalian Cells
Ribo-minus RNAs (ribo− RNAs) is enriched from DNase I treated total RNAs by depleting most redundant ribosomal RNAs with RiboMinus™ Human/Mouse Transcriptome Isolation Kit (Invitrogen™, catalog number K1550-01) as previously described [21]. In addition, fractionation of non-polyadenylated and ribo-minus RNAs (poly(A)−/ribo− RNAs, poly(A)− RNAs for short) from total RNAs by removal of poly(A) + RNA transcripts and ribosomal RNAs [21] shows cleaner background for circular RNA analysis (see Note 1 ).
3.2 RNase R Digestion (See Note 2 )
-
1.
Dissolve fractionated ribo− RNAs (or poly(A)− RNAs) from 20 μg total RNAs from Subheading 3.1 with 52 μl DEPC-treated water. Mix well.
-
2.
Split RNAs to two aliquots in new 1.5 ml RNase-free microcentrifuge tubes: one for RNase R digestion and another for control with digestion buffer only.
-
3.
For RNase R digestion, add 3 μl 10× RNase R Reaction Buffer and 1 μl RNase R (20 U/μl); for control, add 3 μl 10× RNase R Reaction Buffer and 1 μl DEPC-treated water. Mix well and quick spin the tubes for a few second. Incubate the samples at 37 °C for 1–2 h according to manufacturer’s instructions (see Note 3 ).
-
4.
After incubation, add 30 μl of phenol–chloroform–isoamyl alcohol to stop the exonuclease digestion. Vertex the tubes vigorously, and spin the tubes in a microcentrifuge at 13,000 × g at 4 °C for 5 min.
-
5.
Carefully remove the upper aqueous layers and transfer them to new 1.5 ml RNase-free microcentrifuge tubes, add 6 μl 4 M LiCl, 1 μl glycogen, 90 μl prechilled absolute ethanol (−20 °C). Mix well by inverting the tubes several times, and incubate the tubes at −80 °C for 1 h to precipitate RNAs. These RNA samples (RNase R treated or untreated) can be stored at −80 °C until use. This is a good stopping point in the process (see Note 4 ).
-
6.
Precipitate RNAs by spinning at 13,000 × g at 4 °C for 20 min. Remove the supernatants and wash the RNA pellet twice with 700 μl prechilled 75 % (v/v) ethanol and air-dry.
-
7.
Resuspend RNA pellets with 20 μl DEPC-treated water.
3.3 Validation of Circular RNAs
3.3.1 High-Throughput Sequencing Analysis of Circular RNAs
-
1.
Isolated ribo− RNAs (or poly(A)− RNAs) and/or their RNase R treatment can be directly used for RNA-seq library preparation according to the manufacturer’s instructions, and then subjected to deep sequencing [21].
-
2.
Specific pipelines can be used for genome-wide identification of ciRNAs [13] and/or circRNAs [10, 15, 17] by retrieving junction reads for circular RNAs.
-
3.
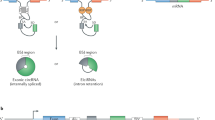
Circular RNAs can be individually visualized at genome browser with uploaded track files, as indicated in Fig. 2 (Fig. 2(a) ci-ankrd52 and (b) circCAMSAP1).
Fig. 2 
Visualization of two types of circular RNAs with RNA-seq . (a) An example of ciRNAs (circular intronic RNAs), ci-ankrd52, from the second intron of ANKRD52 gene. Deep sequencing signal from poly(A)+ (black), poly(A)− (red), poly(A)−/RNase R (purple) are shown. The predicted ci-ankrd52 is indicated in pink.(b) An example of circRNAs from back spliced exons, circCAMSAP1, from the CAMSAP1 loci. Deep sequencing signal from poly(A)+ (black), poly(A)− (red), poly(A)−/RNase R (purple) are shown. The predicted circCAMSAP1 is indicated in blue

3.3.2 Northern Blots with Denaturing Polyacrylamide Gel Electrophoresis (PAGE)
3.3.2.1 Prepare the Gel
-
1.
Assemble the gel plates according to the manufacturer’s instructions, and fix the gel plates in the gel-casting apparatus.
-
2.
Prepare the appropriate denaturing polyacrylamide gel solution by mixing 8 M urea, 10× TBE and 30 % acrylamide with DEPC-treated water (see Note 5 ). 10 ml gel solution is enough for a denaturing polyacrylamide gel of 10 cm × 7.5 cm × 1 mm. Dissolve the mixture by rotation, and filter the solution through 0.22 μl Millex-GP Syringe Filter Unit. Sequentially add 100 μl 10 % APS and 8 μl TEMED, and mix thoroughly. Immediately pour the gel solution between the gel plates and insert the comb. Wait for about 30 min to let the gel polymerize.
3.3.2.2 Prepare the Samples
-
3.
Meanwhile, prepare RNA samples. Total RNAs (Subheading 3.1) were digested with RNase R (Subheading 3.2), cleaned up with phenol, precipitated by ethanol, and resuspended by DEPC-treated water. The final RNA concentration/amount was determined by measure UV absorption with a spectrophotometer. RNA concentration is determined by the OD reading at 260 nm. To get better signals for circular RNAs, similar amounts of RNAs with or without RNase R treatment were used for Northern blots (see Note 6 ).
-
4.
Add 2× urea loading buffer to RNA samples (10 μl total volume is recommended to get sharp bands), for example RNAs with or without RNase R treatment as Subheading 3.2. Boil the RNA samples at 100 °C for 5 min, and chill on ice immediately.
3.3.2.3 Load the Samples and Run the Gel
-
5.
Dismount the gel from the casting chamber, and assemble the gel electrophoresis apparatus according to the manufacturer’s instructions.
-
6.
Remove the comb, and fill the chamber with 1× TBE electrophoresis buffer (see Note 7 ).
-
7.
Load denatured RNA samples and run the gel at 120 V for 3 h (see Note 8 ).
3.3.2.4 RNA Transfer and Fixation
-
8.
Dissemble the gel electrophoresis apparatus, and assemble the gel transfer “sandwich” in a transfer blot. Put the bottom of the gel face to the nylon membrane, remove bubbles between the gel and nylon membrane. Transfer the RNA from gel to membrane in 1× transfer buffer with 100 V for 90 min.
-
9.
Dissemble the gel transfer apparatus. Immobilize RNAs with UV cross-linking by an Ultravoliet Crosslinker (UVP, CL-1000, 254-nm wavelength) for the appropriate length of time (usually 180 mJ/cm2 is used) (see Note 9 ).
3.3.2.5 Prepare RNA Probe
-
10.
Prepare Dig (Digoxigenin)-labeled RNA probes according to the manufacturer’s instructions (RiboMAX™ Large Scale RNA Production Systems, Promega). Briefly, mix DNA template (with T7 or SP6 promoter), 10× Dig labeling mixture, 5× transcription buffer (T7 or SP6) and enzyme mixture (T7 or SP6) with DEPC-treated water. Mix gently by pipetting, and incubate reaction at 37 °C for 2–3 h.
-
11.
Add 1 μl DNase I and mix well. Incubate at 37 °C for 15 min to remove the DNA template.
-
12.
To precipitate the RNA probe, sequentially add 4 μl 4 M LiCl, 100 μl DEPC-treated water and 300 μl prechilled absolute ethanol (−20 °C), mix well by inverting the tubes several times, and incubate the tubes at −20 °C for 1 h.
-
13.
Spin the tubes at 13,000 × g at 4 °C for 15 min. Remove the supernatants and wash the RNA pellet twice with 700 μl 75 % (v/v) cold ethanol (−20 °C) and air-dry.
-
14.
Dissolve the RNA probes by adding 40 μl DEPC-treated water (usually 8–10 μg RNA probes can be obtained).
3.3.2.6 Hybridization
-
15.
Pre-hybridize membrane with DIG Easy Hyb (3 ml/100 cm2) for 30 min at 68 °C with gentle rotation.
-
16.
Denature DIG-labeled RNA probe by heating at 100 °C for 5 min, and immediately place the tube on ice.
-
17.
Discard prehybridization buffer, and add new prewarmed DIG Easy Hyb with denatured DIG-labeled RNA probe (100 ng/ml). Incubate overnight with gentle rotation.
3.3.2.7 Immunological Detection
The following steps followed protocol for DIG Wash and Block Buffer Set, and all performed at room temperature (unless indicated otherwise).
-
18.
Wash the membrane twice with 2× SSC, 0.1 % SDS for 5 min with gentle rotation.
-
19.
Wash the membrane twice with 0.2× SSC, 0.1 % SDS at 68 °C for 30 min with gentle rotation.
-
20.
After stringency washes, briefly rinse the membrane in 1× Washing buffer.
-
21.
Incubate the membrane in 1× Blocking solution for 30 min in an appropriate container with gentle agitation.
-
22.
Incubate the membrane in Antibody Solution for 30 min with gentle agitation.
-
23.
Wash the membrane three times with 1× Washing buffer for 20 min.
-
24.
Incubate in Detection buffer for 5 min.
-
25.
Place the membrane (RNA side-up) on a development folder, add CDP-Star ready-to-use solution (1 ml for 100 cm2 membrane), and incubate for 2–5 min.
-
26.
Expose to X-ray film for an appropriate time. Multiple exposures can be taken to achieve appropriate signals. Examples of Northern blots of circular RNAs migration in denaturing PAGE are shown in Fig. 3 (Fig. 3(a) ci-ankrd52, (b) circCAMSAP1).
Fig. 3 
Validation of circular RNAs by Northern blots . (a) Northern blot of ci-ankrd52. RNase R treated or untreated total RNAs from H9 cells are loaded on 5 % denaturing urea PAGE for Northern blot with a DIG-labeled antisense RNA probe (pink bar) as previously reported [13]. Note that ci-ankrd52 remains stable after RNase R treatment and runs slowly on the denaturing urea PAGE. (b) Northern blot of circCAMSAP1. RNase R treated or untreated total RNAs from H9 cells are loaded on 5 % denaturing urea PAGE for Northern blot with a DIG-labeled antisense RNA probe (blue bar) as previously reported [15]. Note that circCAMSAP1 remains stable after RNase R treatment and runs slowly on the denaturing urea PAGE. Asterisk, linear mRNAs

3.3.3 RT-PCR with Divergent Primers
3.3.3.1 First Strand cDNA Synthesis
-
1.
Before the first strand cDNA synthesis, ribo− RNAs (Subheading 3.1) were digested with RNase R (Subheading 3.2), cleaned up with phenol, precipitated by ethanol, and resuspended by DEPC-treated water. The final RNA concentration/amount was determined by measure UV absorption with a spectrophotometer. RNA concentration is determined by the OD reading at 260 nm. To get better signals for circular RNAs, similar amounts of ribo− RNAs with or without RNase R treatment were used for RT-PCR validation (see Note 6 ). Prepare the first strand cDNA according to the manufacturer’s instructions (SuperScript™ III Reverse Transcriptase, Invitrogen). Briefly, mix RNAs samples, random hexamers (100 μM), dNTP (2.5 mM each) with DEPC-treated water. Heat at 65 °C for 5 min, and immediately chill on ice for at least 1 min.
-
2.
Briefly spin the tubes, add 5× First-Strand Buffer, 0.1 M DTT, RNasin, and SuperScript™ III RT. Pipette gently and incubate reaction at 25 °C for 5 min.
-
3.
Incubate at 50 °C for 60 min, and inactivate the reaction by heating at 70 °C for 15 min.
3.3.3.2 Circular RNAs Divergent Primer PCR
-
1.
Unlike linear RNA, circular RNAs can be amplified by divergent primers from both ribo− RNAs with or without RNase R treatment. Examples of circular RNAs semi-quantitative RT-PCR results are shown in Fig. 4 (Fig. 4(a) ci-ankrd52, (b) circCAMSAP1).
Fig. 4 
Validation of circular RNAs by divergent PCRs. (a) PCR validation of ci-ankrd52 with a divergent primer set (pink arrows) as previously reported [13]. Primers for linear mRNA are indicated as black arrows. Note that ci-ankrd52 remains stable after RNase R treatment, while the linear mRNA is largely degraded with RNase R treatment. (b) PCR validation of circCAMSAP1 with a divergent primer set (blue arrows) as previously reported [15]. Primers for linear mRNAs are indicated as black arrows. Note that circCAMSAP1 remains stable after RNase R treatment, while the linear mRNA is largely degraded with RNase R treatment

4 Notes
-
1.
Ribo-minus RNAs (ribo− RNAs) is enriched from total RNAs by depleting ribosomal RNAs, which contain both poly(A) + and poly(A)− RNAs. However, the fractionation of poly(A)− /ribo− RNAs (poly(A)− RNAs for short) removes both poly(A) + RNAs and ribosomal RNAs, which shows a much cleaner background for circular RNA analysis. We thus prefer to use poly(A)− RNAs (with or without further RNase R digestion) for RNA-seq analyses.
-
2.
RNase R is a magnesium-dependent 3′ → 5′ exribonuclease that digests linear RNAs and Y-structure RNAs, while preserving circular RNAs [22]. Besides RNase R, tobacco acid phosphatase and terminator exonuclease can also efficiently degrade linear RNAs, while leave circular RNAs intact [9].
-
3.
RNase R activity may vary from batch to batch. Thus, check the amount of enzyme and the incubation time for your particular experiment is highly recommended before carrying out further sequencing . For example, we have observed that circular RNAs can also be degraded with the RNase R incubation (Fig. 5). In this case, some RNase R-sensitive circular RNAs could be lost with long time incubation with RNase R . From the time course of the RNase R treatment (Fig. 5), we recommend that one-hour incubation of this batch of RNase R is good enough for circular RNA validation by Northern blots . Meanwhile, it is worthy to note that the short time course of RNase R digestion can lead to only partial digestion of linear RNAs, which may in turn result in unwanted linear RNA signals when such samples are applied to RNA-seq analyses (data not shown).
Fig. 5 
Time course of the RNase R treatment. Ribo− RNAs from 10 μg total RNAs in either wild type (a) or transfected (b) cells are incubated with 1 μl RNase R at 37 °C for 0, 5, 15, 30, 60, 120 min, respectively, and then applied for Northern blots on either native agarose gel for endogenous circCAMSAP1 (a) or denaturing PAGE for over-expressed circPOLR2A (b). The probes are the same ones as previously reported [15]. Note that linear RNAs (asterisks) are largely degraded with the short time of RNase R incubation, and circular RNAs are also reduced during the prolonged incubation
-
4.
RNA samples are recommended to be used immediately or stored at −80 °C for later usage. RNA precipitation in ethanol can be stored at −80 °C for years.
-
5.
Although we have applied the denaturing urea PAGE to detect some long circular RNAs [13], such PAGE is usually used to analyze RNAs less than 500 nt. In addition, due to the special covalently closed loop structure, circular RNAs migrate much more slowly than linear RNAs with the same molecular weight [13, 15]. On the contrast, the migration of circular RNAs on native agarose gels is similar with linear RNAs with similar molecular weights [13].
-
6.
During the preparation of the RNase R treated RNA samples or RNA samples subjected to other treatments, RNAs will be lost due to different treatments and the subsequent RNA recovery. Thus, we generally use similar amounts of RNAs prior to any treatment (total RNA) and after the treatment of interest for Northern blots . For instance, about 20 μg total RNAs were first digested with RNase R (about 5–10 μg RNAs could be retrieved) and then applied for Northern blot ; correspondingly, about 5–10 μg total RNAs were directly applied for Northern blot without RNase R digestion (Fig. 3). A similar strategy has been applied to RT-PCR analyses (Fig. 4).
-
7.
Before loading the samples, rinse gel wells several times to remove dissolved urea, and then load the samples immediately.
-
8.
The time of electrophorese depends on the molecular weights of interested circular RNAs. Usually, it requires about 3 h at 120 V to sufficiently resolve 400–nt circular RNAs in 5 % denaturing PAGE.
-
9.
The cross-linked membrane can be immediately used for prehybridization or store at −80 °C for weeks after drying up.

References
Sanger HL et al (1976) Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A 73:3852–3856
Arnberg AC et al (1980) Some yeast mitochondrial RNAs are circular. Cell 19:313–319
Kos A et al (1986) The hepatitis delta (delta) virus possesses a circular RNA. Nature 323:558–560
Nigro JM et al (1991) Scrambled exons. Cell 64:607–613
Cocquerelle C et al (1992) Splicing with inverted order of exons occurs proximal to large introns. EMBO J 11:1095–1098
Capel B et al (1993) Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 73:1019–1030
Burd CE et al (2010) Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet 6:e1001233
Hansen TB et al (2011) miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 30:4414–4422
Hansen TB et al (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495:384–388
Memczak S et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495:333–338
Graveley BR et al (2008) Molecular biology: power sequencing. Nature 453:1197–1198
Yang L et al (2011) Genomewide characterization of non-polyadenylated RNAs. Genome Biol 12:R16
Zhang Y et al (2013) Circular intronic long noncoding RNAs. Mol Cell 51:792–806
Salzman J et al (2012) Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7:e30733
Zhang XO et al (2014) Complementary sequence-mediated exon circularization. Cell 159:134–147
Zhang Y, Yang L, Chen LL (2014) Life without A tail: new formats of long noncoding RNAs. Int J Biochem Cell Biol 54:338–349
Jeck WR et al (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19:141–157
Salzman J et al (2013) Cell-type specific features of circular RNA expression. PLoS Genet 9:e1003777
Wang PL et al (2014) Circular RNA is expressed across the eukaryotic tree of life. PLoS One 9:e90859
Westholm JO et al (2014) Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 9:1966–1980
Yin QF, Chen LL, Yang L (2015) Fractionation of non-polyadenylated and ribosomal-free RNAs from mammalian cells. Methods Mol Biol 1206:69–80
Suzuki H et al (2006) Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 34:e63
Acknowledgements
This work was supported by 31271390, 31322018, 91440202 from NSFC to L.Y. and L.L.C.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Zhang, Y., Yang, L., Chen, LL. (2016). Characterization of Circular RNAs. In: Feng, Y., Zhang, L. (eds) Long Non-Coding RNAs. Methods in Molecular Biology, vol 1402. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3378-5_17
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3378-5_17
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3376-1
Online ISBN: 978-1-4939-3378-5
eBook Packages: Springer Protocols