
Abstract
Mobile genetic elements (MGEs) are of critical importance in genomics and developmental biology. Polymorphic and somatic MGE insertions have the potential to impact the phenotype of an individual, depending on their genomic locations and functional consequences. However, the identification of polymorphic and somatic insertions among the plethora of copies residing in the genome presents a formidable technical challenge. Whole genome sequencing has the potential to address this problem; however, its efficacy depends on the abundance of cells carrying the new insertion. Robust detection of somatic insertions present in only a subset of cells within a given sample can also be prohibitively expensive due to a requirement for high sequencing depth. Here, we describe retrotransposon capture sequencing (RC-seq), a sequence capture approach in which Illumina libraries are enriched for fragments containing the 5′ and 3′ termini of specific MGEs. RC-seq allows the detection of known polymorphic insertions present in an individual, as well as the identification of rare or private germline insertions not previously described. Furthermore, RC-seq can be used to detect and characterize somatic insertions, providing a valuable tool to elucidate the extent and characteristics of MGE activity in healthy tissues and in various disease states.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
- Retrotransposition
- Somatic mosaicism
- Whole genome sequencing (WGS)
- LINE-1
- Alu
- Mobile genetic element
- Neurogenesis
- Oncogenesis
1 Introduction
The degree to which somatic cell genomes within a single individual differ from one another is an active area of research. Somatic genome alterations can be generated by aberrant mutational processes, but in some instances are the products of precise, programmed genomic rearrangements. Somatic mutations vary in their physiological impact, depending on the nature of the alteration, as well as the type and number of cells affected. Abnormalities arising during cell division and DNA replication [1, 2] range from point mutations to large genomic alterations that affect chromosome content and structure. By contrast, as an example of controlled genomic mosaicism, V(D)J recombination and somatic hypermutation are confined to the lymphocyte antigen receptor loci and generate variability necessary for adaptive immunity [3, 4].
Recent work has led to an increased appreciation of insertional mutagenesis generated by retrotransposons, a class of replicative MGE, as an additional source of somatic genome mosaicism in mammals. Their activity is frequently associated with deletion or duplication of genomic DNA, but can also facilitate mobilization of other cellular transcripts [5, 6]. Studies involving engineered retrotransposon reporter constructs and copy number variation (CNV) assays have demonstrated that retrotransposons are active in embryonic stem cells and also in adult neuronal progenitor cell s [7–9]. Retrotransposon activity in embryonic stem cell s can be attributed to the impetus for a “selfish” element to reach the germline and generate a heritable insertion. In contrast, retrotransposition in adult neural progenitor cells has no obvious evolutionary benefit for the retrotransposon. Such activity may therefore represent the “domestication” of retrotransposons into a functional role in the brain , although the physiological impact, if any, of neuronal retrotransposition remains to be elucidated. Well-characterized examples of transposon domestication include the Transib transposon-derived V(D)J recombinase mentioned above [10], as well as the Drosophila retroelements HeT-A, TART and Tahre which function to support telomere stability [11].
RC-seq was first published in 2011 [12]. At this time, L1 had been established as the only active autonomous retrotransposon in humans, and was also known to be responsible for the mobilization of other non-autonomous elements including Alu and SVA [13–15]. The canonical mechanism of L1 mobilization is Target-Primed Reverse Transcription (TPRT ) [16, 17], which occurs at an L1-encoded endonuclease cleavage motif (5′-TT/AAAA-3′), and is characterized by the generation of short direct target site duplication s (TSDs) flanking the new insertion (Fig. 1). There is also evidence that L1 can integrate at sites of existing DNA damage, which generally results in insertions lacking TSDs [18, 19].

Schematic representation of a pre-integration site and an inserted new L1 copy with TPRT signatures. The TPRT insertion depends on two single-strand cleavages of the target DNA. The single asterisk indicates the first cleavage that occurs in the EN consensus site, while the double asterisk indicates the second strand cleavage. The regions between the two cleavages becomes a target site duplication (TSD ) flanking the new insertion. The 5′ TSD contains the EN consensus site in the 5′ end (blue), while the 3′ TSD starts just after the 3′ poly(A) tract at the end of the inserted copy (green). The L1 copy is represented by thick-outlined white box and the two open reading frames (ORF) are depicted by two inside boxes encoding nucleic acid chaperone (NAC), endonuclease (EN), and reverse transcriptase (RT) activities
Nearly one-third of the human genome is comprised of L1 or L1-dependent MGEs [20]. In 1988, a de novo L1 insertion into the X-linked Factor VIII gene was isolated as the cause of a case of hemophilia A in a male patient. This discovery constituted unequivocal evidence that L1s were actively retrotransposing in the human population [21]. Indeed, ~100 cases of human disease have been demonstrated to arise from L1-mediated mutagenesis, and current estimates suggest that there are hundreds of polymorphic L1 insertions segregating in the human population [22–26]. However, the discovery of L1 promoter activity during neurogenesis [8, 27], as well as age- and region-dependent L1 transcription and CNV in the healthy human brain [27–30], and the mobilization of engineered L1s in vitro and in transgenic animals [8, 29] supported the existence of L1-driven somatic mosaicism in the human brain. Nonetheless, prior to the development of RC-seq, evidence for endogenous L1 retrotransposition in the brain lacked critical proof, namely genomic mapping and sequence characterization of the resulting L1 insertions. Indeed, such evidence is essential to elucidate a putative physiological impact of L1 activity during neurogenesis through some particular insertional pattern, perhaps mediated by differential euchromatinization [31]. RC-seq allowed the large-scale identification and genomic localization of somatic MGE insertions present in one or a few cells within a tissue, revealing the surprising extent of somatic mosaicism induced by L1 in the brain [12, 32].
RC-seq is typically performed on genomic DNA extracted from a tissue of interest and a matched control tissue from the same individual (i.e., brain and liver, Fig. 2a), in order to distinguish somatic L1 insertions from unannotated polymorphic insertions. Genomic DNA is sheared by sonication , and the sheared DNA serves as the substrate for Illumina sequencing library preparation (Fig. 2b). The library is then hybridized to two biotin-labeled locked nucleic acid (LNA) probes targeting the 5′ and 3′ termini of the L1-Ta consensus (Fig. 2c). Then, streptavidin-based pull down enriches the library for fragments containing the junctions between L1 sequence and flanking genomic DNA . The subsequent amplification and sequencing of the post-hybridization library by a high-throughput platform produces a collection of sequencing reads enriched for L1-genome junctions (Fig. 2d). The data are then analyzed in silico to identify previously annotated fixed or polymorphic insertions, as well as find any previously unknown polymorphic insertions present in the individual (Fig. 2e). The remaining L1-genome junctions represent putative somatic insertions unique to the tissue of interest.

RC-seq workflow. (A) Genomic DNA is extracted from the biological tissue by standard phenol-chloroform extraction. (B) Library preparation is performed using Illumina technology. (C) Hybridization and pull down of the subset of fragments containing the boundaries of L1 element using a L1-specific biotin-LNA probe and Dynabeads M-270 Streptavidin. (D) The captured library is sequenced using an Illumina platform. (E) Computational analysis maps new somatic insertions in the annotated genome, and also can provide information about the particular genotype concerning to the polymorphic insertions described in the population and even detect new unknown ones. Note: the red sections within the library fragments represent L1 sequences; the grey sections represent other genomic regions
The location of the L1-specific RC-seq probes at the extreme 5′ and 3′ termini of the L1-Ta consensus sequence lead to RC-seq generally only capturing the 5′ junctions of full-length or extremely 5′ truncated L1 insertions. Indeed, most insertions are only detected at their 3′ L1-genome junction, as the vast majority of L1 insertions are variably 5′ truncated but retain their 3′ end. For those insertions detected at only a 3′ L1-genome junction, PCR amplification and capillary sequencing of the matched 5′ L1-genome junction is the gold standard validation to confirm bona fide L1 insertions. In this scenario, a collection of primers tiled along, and oriented antisense to, the L1-Ta consensus are combined with a primer in the presumed 5′ genomic flank and sense oriented relative to the L1. Capillary sequencing the resulting amplicon(s) allows characterization of the 5′ L1-genome junction, typically revealing TPRT hallmark TSDs and an L1 EN motif and thereby distinguishing true insertions from putative artifacts.
RC-seq was first applied to elucidate L1-driven somatic mosaicism in the brain , but it has also been successfully applied to tumor samples, identifying insertional mutations that trigger oncogenic pathways [33]. In addition, RC-seq has the potential to profile the unique subset of annotated polymorphic insertions in a single individual. Thus it can be used as a genotyping tool to match different tissues or samples from the same donor, and also constitutes a source of potential traceable genetic markers applicable, for example, in genome wide association studies.
The most significant advantage of the sequence capture approach used in RC-seq is that the number of PCR cycles can be kept to a minimum. In contrast to other methods for identifying endogenous L1 insertions [23], minimization of PCR cycles preserves the integrity of library content by reducing PCR artifact generation and amplification, giving greater resolution in the detection of rare L1 variants in tissue samples. Note that RC-Seq was originally performed with custom sequence capture arrays consisting of a pool of DNA probes covering 1 and 0.2 Kb at either end of the L1-Ta consensus sequence in the first (V1) [12] and second (V2) [33] RC-seq designs. However, the technique described here, the third (V3) RC-seq design, utilizes two locked nucleic acid (LNA) probes targeting suitable regions at the 5′ and 3′ termini of the L1-Ta consensus sequence, and achieves more than 15-fold improvement in enrichment over previous iterations of RC-seq [34].
2 Materials
Solutions should be prepared with molecular grade water. Examples are water purified by filtration and deionization to achieve a resistivity of 18.2 MΩ cm at 25 °C (such as Mili-Q water produced by Millipore Corporation water filtration stations) or water distilled and filtered by 0.1 μm membrane filters (such as Ultrapure Water provided by Invitrogen—Life Technologies).
2.1 DNA Extraction
-
1.
Benchtop centrifuge for 1.5 ml tubes.
-
2.
Two thermoblocks.
-
3.
Nanodrop spectrophotometer.
-
4.
Scalpel blades, blade holder, forceps and spatula.
-
5.
Disposable plastic Petri dishes.
-
6.
1.5 ml tubes.
-
7.
Phenol (equilibrated with 10 mM TRIS pH 8).
-
8.
Phenol:Chloroform:Isoamyl alcohol (12:12:1, TRIS saturated).
-
9.
Chloroform:Isoamyl alcohol (24:1, TRIS saturated).
-
10.
Isopropanol (molecular grade).
-
11.
Absolute Ethanol (molecular grade).
-
12.
TE buffer: 10 mM Tris–HCl, 1 mM EDTA, pH 8.
-
13.
Lysis Buffer: TE, 2 % SDS, 100 μg/ml Proteinase K , see Note 1 ).
-
14.
3 M Sodium Acetate.
-
15.
10 mg/ml RNase A.
-
16.
Dry ice (if the tissue sample is frozen).
2.2 DNA Shearing
-
1.
Covaris M220 Focused-ultrasonicator electronically controlled by Sonolab 7 software.
-
2.
Covaris MicroTube AFA Snap-Cap, 130 μl sample.
-
3.
Buffer TE: 10 mM Tris–HCl, 1 mM EDTA, pH 8.
-
4.
Molecular grade water.
-
5.
Low lint paper wipes (such as Kimtech Science Kimwipes).
2.3 Library Preparation
-
1.
Two thermocyclers.
-
2.
Qubit® Fluorometric Quantification technology (Life Technologies) or similar. This includes Qubit® dsDNA HS Assay kit, Qubit® Assay Tubes and Qubit® Fluorometer.
-
3.
Illumina® TruSeq® Nano DNA Sample Prep Kit. This kit provides End Repair Mix, A-Tailing Mix, Ligation Mix, Stop Ligation Mix, Resuspension Buffer and a set of Illumina Barcoded Library Adapters that will be used in the following protocol.
-
4.
DynaMag™-2 Side (Life Technologies) magnetic rack (see Note 2 ).
-
5.
0.2 and 1.5 ml tubes. 0.2 ml tubes must be PCR-grade.
-
6.
Agentcourt® AMPure® XP beads (Beckman Coulter).
-
7.
Absolute Ethanol (molecular grade).
2.4 Agarose Gel-Size Selection and PCR Enrichment of the DNA Library
-
1.
Thermocycler.
-
2.
Safe Blue Light Imager or UV trans-illuminator (Safe Blue Light source is preferable).
-
3.
Gel tray, gel combs, electrophoresis tank and power pad.
-
4.
Agilent Bioanalyzer technology or similar. This includes Agilent 2100 Bioanalyzer instrument, Agilent DNA 1000 Reagents and DNA Chips (Agilent Technologies).
-
5.
UV source.
-
6.
MiniElute® Gel Extraction Kit (Qiagen). This includes Buffer QG, Buffer PE, Buffer EB and MiniElute® Spin Columns.
-
7.
Scalpel blades.
-
8.
0.2 and 1.5 ml tubes. 0.2 ml tubes must be PCR-grade.
-
9.
UV or Safe Blue Light-transparent cling film.
-
10.
High Resolution Agarose (Sigma-Aldrich).
-
11.
Molecular grade water.
-
12.
SYBR® Gold Nucleic Acid Gel Stain (Life Technologies).
-
13.
TAE Buffer: 40 mM Tris–HCl, 20 mM Acetic acid, 1 mM EDTA.
-
14.
Gel Loading Buffer: 20 % glycerol, 0.04 % Orange G stain.
-
15.
Phusion® High-Fidelity PCR Master Mix 2× (New England Biolabs).
-
16.
DNA ladder. Recommended: a ladder with several bands on the 200–400 bp rage, like GenRuler 1 Kb Plus DNA ladder 0.5 μg/μl (Thermo Scientific).
-
17.
Isopropanol (molecular grade).
-
18.
100 μM each LM-PCR primers: TS-F Primer (5′AATGATACGGCGACCACCGAGA3′) and TS-F Primer (5′CAAGCAGAAGACGGCATACGAG3′).
2.5 Hybridization
-
1.
Thermoblock with two tube removable holders, one of them able to host 1.5 ml tubes.
-
2.
Two thermocyclers. One of them must be connected to a power source capable of holding uninterrupted supply for 3 days.
-
3.
Roche NimbleGen Sequence Capture Kit. Components used in the hybridization step are 2× Hybridization Buffer and Hybridization Component A.
-
4.
Roche Diagnostics Sequence Capture Developer Reagent.
-
5.
0.2 ml and 1.5 tubes. 0.2 ml tubes must be PCR-grade.
-
6.
100 μM Universal Blocking Oligo. Sequence: 5′AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC*/3ddC/3′.
-
7.
100 μM Index-specific Blocking Oligos. Sequence: 5′CAAGCAGAAGACGGCATACGAGATN8GACTGGAGTTCAGACGTGTGCTCTTCCGATCT/3ddC/3′. N8 fragment is an Illumina index-specific sequence indicated in Note 3 .
-
8.
10 μM each Capture probes: LNA-5′ (/5Biosg/CTCCGGT+C+T+ACAGCTC+C+C+AGC) and LNA-3′ (/5Biosg/AG+A+TGAC+A+C+ATTAGTGGGTGC+A+GCG). Note that /5Biosg/ denotes the presence of a biotin moiety in the 5′ end and + denotes the LNA positions within each probe.
-
9.
ToughTag stickers.
2.6 Capture Recovery and Amplification
-
1.
Thermoblock.
-
2.
Thermocycler (the same used for the Subheading 2.5).
-
3.
Agilent Bioanalyzer technology or similar. This includes Agilent 2100 Bioanalyzer instrument, Agilent DNA 1000 Reagents and DNA Chips (Agilent Technologies).
-
4.
Qubit® Fluorometric Quantification technology (Life Technologies) or similar. This includes Qubit® dsDNA HS Assay kit, Qubit® Assay Tubes and Qubit® Fluorometer.
-
5.
Roche NimbleGen Capture Wash Kit. Components used in the capture recovery step are 10× Stringent Wash Buffer, 10× Wash Buffer I, II and III, and 2.5× Bead Wash Buffer.
-
6.
MiniElute® Gel Extraction kit (Qiagen).
-
7.
DynaMag™-2 Magnet rack (Life Technologies, see Note 2 ).
-
8.
DynaMag™-96 Side Magnet plate (Life Technologies).
-
9.
Agentcourt® AMPure® XP beads (Beckman Coulter).
-
10.
Dynabeads ® M-270 Streptavidin (Life Technologies).
-
11.
Molecular grade water.
-
12.
Ethanol.
-
13.
Phusion® High-Fidelity PCR Master Mix 2× (New England Biolabs).
-
14.
100 μM each LM-PCR primers: TS-F Primer (5′AATGATACGGCGACCACCGAGA3′) and TS-F Primer (5′CAAGCAGAAGACGGCATACGAG3′).
2.7 Sequencing
RC-seq libraries can, theoretically, be sequenced on any Illumina platform able to sequence WGS libraries. Thus far, RC-seq has been performed on Illumina HiSeq2000, HiSeq2500 and MiSeq platforms. This choice is dependent upon how many libraries are to be sequenced, the desired depth of sequencing, and the maximum insert size that can be spanned by paired-end reads generated on a given platform.
2.8 Bioinformatic Analysis
The computational resources required for RC-seq library analysis scale with project size. A laptop or PC with 4 GB RAM, a Linux operating system, and 100 GB of hard disk space would likely be sufficient to run a single library over the course of 72 h, depending on library size and L1 enrichment. Most projects would involve multiple RC-seq libraries and therefore would require a larger server. For example, the Translational Research Institute server used by the Faulkner laboratory has 2200 CPUs, 8 TB RAM, and 3 PB hard disk space. This enables parallelization and accelerated analysis.
2.9 PCR Validation
The PCR validation is a highly variable procedure that requires researcher expertise in molecular biology, particularly in PCR amplification, cloning, and sequencing. The materials listed here provide a recommended starting point for the validation process. Additionally, more complex approaches might be necessary according to the inherent difficulty of amplification of some insertions.
For primer design:
-
1.
Primer3 software.
For amplification:
-
2.
Roche Expand Long Range dNTPack.
-
3.
Platinum® Taq DNA Polymerase High Fidelity.
For DNA imaging and purification:
-
4.
Agarose electrophoresis material and Safe Blue Light Imager or UV trans-illuminator (Safe Blue Light source is preferable).
-
5.
Material for agarose gel-purification of DNA.
For cloning:
-
6.
Promega pGEM®-T vector system.
-
7.
Life Technologies TOPO® PCR cloning system.
-
8.
Material for molecular cloning, bacteria transformation, culture, and DNA extraction.
3 Methods
As described in the Introduction, to effectively discern between previously unannotated polymorphic insertions and somatic insertions within a tissue of interest of a single donor, it is necessary to analyze a control tissue from the same donor. Those insertions not previously described as fixed or polymorphic insertions, but found in both tissues, very likely represent a germline insertion rather than a somatic one. Insertions present in only one of the tissues are putative somatic insertions. In the example described here, we describe the use of brain and liver tissues, with liver as the control tissue.
Due to limitations in the number of elongation cycles possible during sequencing, it is important to choose an appropriate fragment size for the library. The sequencing reaction described here is designed for a 300-cycle Illumina paired-end sequencing kit (150 cycles from each end). To produce a minimum overlap of the paired-reads initiated at both ends of a single molecule sufficient to reconstitute the fragment sequence, the preferable sequenced fragment size is 250 bp.
Unless otherwise indicated, it is recommended to bring thermocycles to reach the indicated block and lid temperatures before preparing the reaction. To do that, start the run of the thermocycler and pause it when it reaches the first step of the cycling protocol.
3.1 DNA Extraction
3.1.1 Preparation
-
1.
Take an aliquot of TE 2%SDS with the appropriate volume necessary for the extraction (10 μl of Lysis Buffer per 1 mg of tissue) and add the Proteinase K to a final concentration of 100 μg/ml (see Note 1 ).
-
2.
Place the forceps, spatula, one scalpel blade, one 1.5 ml tube and one Petri dish per sample on dry ice.
-
3.
Set up one thermoblock at 37 °C and the other at 65 °C.
-
4.
Prepare 500 μl of 70–80 % ethanol per sample with Ultrapure Water.
3.1.2 Procedure
-
1.
Working on a bed of dry ice, place the tissue sample in a pre-cooled Petri dish using the forceps and shave it using a pre-cooled scalpel blade. The dissociation will be quicker if the shaving is thinner. Transfer the shaved sample to a pre-cooled 1.5 ml tube and keep it frozen until all the tissue samples are processed.
-
2.
Add 10 μl of Lysis Buffer per 1 mg tissue to each tube and incubate at 65 °C in thermoblock until the tissue is completely dissolved. Shake the tubes every 10 min (see Note 4 ).
-
3.
Once dissociated, allow the sample to cool briefly at room temperature and add RNase A to a final concentration of 20 μg/ml (see Note 5 ). Incubate the tubes in the thermoblock at 37 °C for 30 min.
-
4.
Add an equal volume of phenol saturated solution and mix by inversion (not vigorously) until the sample is homogeneous.
-
5.
Centrifuge at maximum speed for 10 min.
-
6.
Transfer the aqueous phase to a fresh tube. Make the pipette tip end wider by cutting it. Repeat steps 4–6 once if the sample is from a tissue highly rich in organic compounds (for example, liver. See Note 6 ).
-
7.
Add an equal volume of Phenol:Chloroform:Isoamyl alcohol (12:12:1) and repeat steps 5–6. Repeat steps 7 and 5–6 if the sample is from a tissue highly rich in organic compounds (for example, liver).
-
8.
Add an equal volume of Chloroform:Isoamyl alcohol (24:1) and repeat steps 5–6.
-
9.
Add 0.1 volume of 3 M Sodium Acetate and 2 volumes of isopropanol and invert the tube several times to precipitate DNA until all DNA is dehydrated (when the viscous transparent goo turns white).
-
10.
Spool DNA with a pipette tip and transfer to a new tube (see Note 7 ). Rinse DNA with 500 μl 70–80 % ethanol and briefly air dry. Stop drying samples before they turn clear. Overdrying can result in extremely difficult resuspension of the pellet.
-
11.
Resuspend samples in 100–200 μl TE and incubate samples at 4 °C overnight for complete resuspension (see Note 8 ).
-
12.
Quantify DNA concentration by Nanodrop.
SAFETY STOPPING POINT. You can store the samples at 4 °C for short periods, or freeze at −20/−80 °C for longer periods.
3.2 DNA Shearing
3.2.1 Preparation
-
1.
Switch on the Covaris M220 Focused-Ultrasonicator (see Note 9 ) and the computer that manages the ultrasonicator. Open SonoLab 7.1 software and prepare the instrument for sonication . Ensure that the water temperature and the water level in the “Instrument Status” window are correct.
-
2.
Select the appropriate sonication method. For a library with an ideal fragment size of 250 bp, the sonication method is: Peak power 50, Duty factor 20, Cycles per burst 200 and Timer 120 s (see Note 9 ).
3.2.2 Procedure
-
1.
Dilute 1–5 μg of genomic DNA from each tissue sample into 130 μl of buffer TE (see Note 10 ). Do not use the whole amount of DNA for the sonication , keep an aliquot and dilute it to 5 ng/μl to use for PCR validation reactions (see Subheading 3.9).
-
2.
Transfer each DNA dilution to a MicroTube AFA Snap-Cap and sonicate the sample following manufacturer’s instructions.
-
3.
Wipe out any water remaining in the edge of the Cap of the MicroTube AFA Snap-Cap using a low lint paper wipe and transfer the sheared DNA solution to a fresh 1.5 ml tube.
-
4.
Repeat the sonication with each sample. Check the water level every time (see Note 11 ).
SAFETY STOPPING POINT. You can store the samples at 4 °C for short periods, or freeze at −20/−80 °C for longer periods.
3.3 Library Preparation
3.3.1 Preparation
-
1.
Take an aliquot of AMPure® XP beads, resuspend well by vortex and allow it to reach room temperature (~30 min).
-
2.
Prepare 2–4 ml of 80 % ethanol per sample (see Note 2 ).
-
3.
Set up a thermocycler with a 30 °C block, 40 °C lid (absolutely critical).
-
4.
Set up a thermocycler with A-tailing program: 37 °C for 30 min, 70 °C for 5 min, 4 °C hold; lid at 80 °C.
3.3.2 Procedure
To concentrate DNA.
-
1.
Add 1.1 volumes of Ampure® XP beads to each tube, mix by pipetting ten times and incubate at room temperature for 15 min.
-
2.
Place the tubes on a magnetic rack for 2 min. Aspirate and discard the supernatant.
-
3.
Add 200–400 μl of 80 % ethanol to each tube without disturbing the beads and incubate at room temperature for 30 s (see Note 2 ).
-
4.
Remove the ethanol and repeat step 3.
-
5.
Remove the ethanol and leave the tubes air dry on the magnetic rack for 15 min.
-
6.
Add 52 μl of resuspension buffer to each tube. Remove the tubes from the magnetic rack and flick it until beads are completely resuspended.
-
7.
Incubate at room temperature for 2 min and place the tubes back in the magnetic rack. Incubate at room temperature for 2 min (the liquid must appear clear).
-
8.
Transfer 50 μl of supernatant to a new fresh tube.
-
9.
Quantify DNA concentration using Qubit® Fluorometric Technology. To check the efficiency of the sonication setting, use Agilent Bioanalyzer technology to check the DNA size distribution (Fig. 3a).
Fig. 3 
Gel-size selection for library sequencing. (a) The figure shows an Agilent® 2100 Bioanalyzer distribution (graph and electropherogram) of a genomic DNA sample sheared by sonication after step 9 of the Subheading 3.3.2. The post-shearing Agencourt® AMPure® beads size selection performed to concentrate DNA and remove fragments under 100 bp (steps 1–8 in same Subheading). The arrowhead indicates the 100 bp cut-off in the size distribution. The arrow in the electropherogram indicates the electrophoresis direction. (b) A typical gel electrophoresis distribution of a pre-hybridization library is shown in the central lane. GenRuler 1 Kb Plus DNA Ladder is used as Marker at both sides. Note that there is an empty well at each side of the library to prevent cross-contamination . White numbers indicate the size (bp) of the corresponding fragment of the ladder. (c) An example of size selection by gel-cutting. White boxes represent a recreation of the gel-cuts performed on the gel shown in b, aiming for sizes between 290 and 400 bp. The electropherogram corresponding to four consecutive gel-cuts produced by the Agilent® 2100 Bioanalyzer instrument shows an effective size selection. (d) Size distribution corresponding to the electropherograms shown in c. The gel-cut shown in green has an approximate distribution between 330–400 bp and an average size of 365 bp

SAFETY STOPPING POINT. You can store the samples at 4 °C until the next day.
To prepare TruSeq Libraries:
-
10.
Aliquot 1 μg of sheared DNA per sample into a 0.2 ml tube and bring it to a final volume of 60 μl with resuspension buffer. Add 40 μl of End Repair Mix, mix by pipetting ten times, and place in the thermocycler at 30 °C for 30 min.
-
11.
Transfer the whole volume of each tube to a 1.5 ml tube and add 100 μl of well resuspended room temperature AMPure® XP beads (ratio 1:1) and proceed as in steps 1–5.
-
12.
Resuspend the beads in 19 μl of resuspension buffer by flicking and incubate at room temperature for 2 min.
-
13.
Place the tube back in the magnetic rack and incubate at room temperature for 2 min. Transfer 17.5 μl of supernatant to a new 0.2 ml tube.
-
14.
Add 12.5 μl of A-tailing mix to each sample and mix by pipetting ten times (do not vortex). Place the tubes in the thermocycler with the A-tailing program (37 °C for 30 min, 70 °C for 5 min, 4 °C hold) and resume the run.
-
15.
Take the 0.2 ml tubes with the samples and add 2.5 μl of resuspension buffer, 2.5 μl of Ligase Mix and 2.5 μl of each adapter specific for each sample, and mix by pipetting up and down ten times (do not vortex, see Note 12 ).
-
16.
Place the tubes in a thermocycler at 30 °C for 10 min.
-
17.
Add 5 μl of Stop Ligase Mix and mix by pipetting ten times.
-
18.
Transfer the whole volume (42.5 μl) to a 1.5 ml tube and add 42.5 μl of AMPure® XP beads. Mix by pipetting ten times and incubate at room temperature for 15 min.
-
19.
Repeat steps 2–8 (two ethanol washes and resuspension of the sample in 50 μl). Pay attention to avoid confusing the elution step with a washing step (see Note 13 ).
-
20.
Add 50 μl of AMPure® XP beads to each tube. Mix by pipetting ten times and incubate at room temperature for 15 min.
-
21.
Repeat steps 2–5.
-
22.
Add 21 μl of resuspension buffer to each tube, resuspend by flicking and incubate at room temperature for 2 min.
-
23.
Place the tubes in the magnetic rack and incubate them at room temperature for 2 min (the liquid must appear clear).
-
24.
Transfer 20 μl of supernatant to a new fresh tube (avoid carry-over of beads).
SAFETY STOPPING POINT. You can store the samples at 4 °C until the next day.
3.4 Agarose Gel-Size Selection
3.4.1 Preparation
-
1.
Wash the gel tray and comb with 70 % ethanol and wipe out. UV-irradiate the gel tray and comb to eliminate contaminant DNA.
-
2.
Prepare a 2 % high-resolution agarose gel using the following proportions: 5 g Agarose in 250 ml of final volume 1× TAE buffer. Partially cool the molten gel in a 65° water bath for ~20 to 30 min. Add 25 μl of SYBR® Gold Stain to the melted agarose solution. Leave the gel sit in a 4 °C room until it solidify (the gel can be prepared the previous day).
-
3.
Set the agarose gel in the gel tank. Use TAE buffer 1× as electrophoresis buffer.
-
4.
Set up a thermocycler with the LM-PCR cycling protocol: 98 °C 45 s; 98 °C 15 s, 60 °C 30 s and 72 °C 30 s for six times (see Note 14 ); 72 °C 5 min and 4 °C hold. Do not bring the thermocycler to the first step, wait until the reaction tubes are inside.
-
5.
Wrap the trans-illuminator with cling film to protect the gel sample from cross-contamination during the gel cutting (this can be done during the step 3 of the Subheading 3.4.2).
3.4.2 Procedure
-
1.
Add 4 μl of Gel Loading Buffer 6× to each tube and load the 24 μl samples in the gel. Leave an empty space between each sample to prevent cross-contamination . Load also 5 μl of DNA ladder every two or three samples and make sure you have a DNA ladder line in both ends of the gel.
-
2.
Run the gel electrophoresis at 120 mA until the separation of the fragmented DNA is enough to comfortably purify bands of ∆30–50 nt size fragments in the frame 250–400 bp (a typical run time for a 15 cm length gel is 3 h).
-
3.
Place the gel at 4 °C for 5–20 min.
-
4.
Perform cut out of bands ranging from 290–310 bp, 310–350 bp, 350–380 bp and 380–410 bp (see Note 15 and Fig. 3b). Place in labeled tubes. Wrap the remaining pieces of gel in cling film and keep them as a backup at 4 °C in case of the need to do new gel cuts.
-
5.
Proceed with at least the fragment of 350–380 bp size to DNA gel-purification using the MiniElute® Gel Extraction Kit by adding 6 μl of Buffer QG per 1 mg of gel band to each sample tube.
-
6.
Dissolve the agarose at room temperature (avoid incubation at 50 °C as indicated in the manufacturer instructions, which can result in a GC-bias in the sequencing data).
-
7.
Add 2 μl of isopropanol per 1 mg of gel band to each sample tube and mix by inverting.
-
8.
Place the MiniElute® Spin Columns in provided 2 ml collection tubes and apply each sample to a single column. Centrifuge at maximum speed for 1 min.
-
9.
Discard the flow-through and add 500 μl of Buffer QG. Centrifuge at maximum speed for 1 min.
-
10.
Discard the flow-through and add 750 μl of Buffer PE. Incubate at room temperature for 2–5 min and centrifuge at maximum speed for 1 min.
-
11.
Discard the flow-through and centrifuge again at maximum speed for 1 min.
-
12.
Transfer the column to a fresh 1.5 ml tube and add 16 μl of Buffer EB pre-heated to 60 °C. Incubate at room temperature for 1 min and centrifuge at maximum speed for 1 min.
-
13.
Repeat step 12 in the same 1.5 ml tube, ending with slightly >30 μl final volume. Discard the column.
-
14.
Transfer 30 μl of each sample to a 0.2 ml tube and add: 50 μl of Phusion® High-Fidelity PCR MasterMix (2×), 18 μl of Ultrapure Water, 1 μl of TS-F Primer (100 μM) and 1 μl of TS-R Primer (100 μM). Mix my pipetting ten times.
-
15.
Put the tubes in the thermocycler with the LM-PCR program and start the run.
-
16.
Transfer the whole volume (100 μl) to a 1.5 ml tube and add 110 μl of AMPure® XP beads to each tube. Mix by pipetting ten times and incubate at room temperature for 15 min.
-
17.
Place the tubes on a magnetic rack for 2 min. Aspirate and discard the supernatant.
-
18.
Add 200–400 μl of 80 % ethanol to each tube without disturbing the beads and incubate at room temperature for 30 s (see Note 2 ).
-
19.
Remove the ethanol and repeat step 3.
-
20.
Remove the ethanol and leave the tubes air dry on the magnetic rack for 15 min.
-
21.
Add 32 μl of molecular grade water (do not use Resuspension Buffer, see Note 16 ), remove the tubes from the magnetic rack and flick them until completely resuspend the beads.
-
22.
Incubate at room temperature for 2 min and place the tubes back in the magnetic rack. Incubate at room temperature for 2 min (the liquid must appear clear).
-
23.
Transfer 30 μl of supernatant to a new fresh tube.
-
24.
Quantify the concentration and size distribution of the DNA library by analyzing 1 μl of each sample in an Agilent DNA 1000 chip. The preferable fragment size distribution should be between 340 and 410 bp, with a median peak of 370 bp (see Fig. 3c, d, and Note 17 ).
SAFETY STOPPING POINT. You can store the samples at 4 °C until the next day.
3.5 Hybridization
3.5.1 Preparation
-
1.
Pre-heat Speed-Vac at 70 °C.
-
2.
Prepare two 0.2 ml tubes with 4.5 μl of each LNA capture probe (10 μM): LNA-5′ and LNA-3′.
-
3.
Set up a thermocycler with 47 °C block, 57 °C lid (absolutely critical). This one will host an incubation for 3 days.
-
4.
Set up a thermocycler with 95 °C block, 105 °C lid.
-
5.
Set up a thermoblock at 95 °C.
3.5.2 Procedure
-
1.
Pool the same amount of brain and liver libraries in the same tube in a ratio 1:1 by molecular mass to reach 1 μg of amplified DNA (see Note 18 about pooling several libraries). This will be used for both 5′ end and 3′ end captures. You can proceed with smaller amount of DNA (up to 100 ng) but keep the ratio 1:1 for the different libraries.
-
2.
Add 10 μl of Sequence Capture Developer Reagent and 10 μl of Universal Blocking Oligo per 1 μg of total DNA. Add also Index-specific Blocker Oligos according to the libraries’ indices at the same ratio (10 μl of Blocker Oligo per 1 μg of each specific library within the pool).
-
3.
Mix the sample by pipetting up and down, spin down and take half of the sample to a fresh tube (each tube will be used for capturing the 5′ and the 3′ end, respectively).
-
4.
Make a hole in the lid of the tubes using a needle.
-
5.
Dry the whole sample by heat and vacuum in the Speed-Vac for 30–60 min.
-
6.
When the sample is absolutely dry, cover the hole in the lid with a ToughTag sticker and add 7.5 μl of 2× Hybridization Buffer and 3 μl of Hybridization Component A. Be careful not to dislodge the dried sample when opening tube. Vortex the mixture and centrifuge the tubes at maximum speed for 10 s.
-
7.
Incubate the tubes on the thermoblock at 95 °C for 5 min. Cover them with a 95 °C pre-heated heat block to prevent any evaporation of the sample through condensation in the tube lid.
-
8.
Mix by flicking to ensure a complete resuspension of the sample and transfer the whole sample of each tube (10.5 μl) to the corresponding 0.2 ml tube with pre-aliquoted LNA probes.
-
9.
Place the tubes in the thermocycler at 95 °C for 3 min.
-
10.
Transfer the tubes to the thermocycler at 47 °C and incubate for 3 days (see Note 19 ).
3.6 Capture Recovery and Amplification
3.6.1 Preparation
-
1.
Prepare 1× solutions of each wash buffer in the Roche NimbleGen Capture Wash Kit. The following amounts are indicated for a single RC-seq sample involving one 5′ and one 3′ capture.
10× Stock (μl)
Water (μl)
Volume (μl)
Stringent Wash Buffer
100
900
1000
Wash Buffer I (first aliquot)
50
450
500
Wash Buffer I (second aliquot)
50
450
500
Wash Buffer II
50
450
500
Wash Buffer III
50
450
500
2.5× Stock (μl)
Water (μl)
Volume (μl)
Beads Wash Buffer
440
660
1100
-
2.
Set up a thermoblock at 47 °C and place one aliquot of 1× Wash Buffer I and the one of 1× Stringent Wash Buffer.
-
3.
Set up a thermocycler with 47 °C block, 57 °C lid (this one is the same that just held the 3 days incubation for the hybridization).
-
4.
Set vortex on constant (not pressure-sensitive), at minimum speed.
-
5.
Allow the streptavidin Dynabeads ® M-270 Streptavidin and the AMPure® XP beads to warm to room temperature 30 min before use and resuspend them well by inverting or vortex for 1 min.
-
6.
Set up a thermocycler with the LM-PCR cycling protocol: 98 °C 45 s; 98 °C 15 s, 60 °C 30 s and 72 °C 30 s for eight times (see Note 14 ); 72 °C 5 min and 4 °C hold. Do not bring the thermocycler to the first step, wait until the reaction tubes are inside.
-
7.
Prepare 2–4 ml of 80 % ethanol per each pair of 5′ and 3′ captures of the same pooled sample (see Note 2 ).
-
8.
Place an aliquot of 50 μl of MiniElute® Gel Extraction Kit buffer EB on a thermoblock at 60 °C (this thermoblock can be the same listed above at 47 °C, that must be set up after step 18 in the Subheading 3.6.2).
3.6.2 Procedure
To pull down the captured library subset:
-
1.
Dispense 200 μl of Dynabeads ® M-270 Streptavidin per each pair of 5′ and 3′ captures in one 1.5 ml tube (up to a maximum of 400 μl per tube).
-
2.
Place the tube in the magnetic rack. Aspirate and discard the supernatant once it becomes clear.
-
3.
With the tube in the magnetic rack, add two volumes of 1× Bead Wash Buffer per initial volume of Dynabeads ® M-270 Streptavidin.
-
4.
Remove the tubes from the magnetic rack and vortex well.
-
5.
Place it back in the magnetic rack until the liquid becomes clear. Aspirate and discard the supernatant.
-
6.
Repeat steps 3–5.
-
7.
Resuspend the Dynabeads ® M-270 Streptavidin in the original volume of 1× Beads Wash Buffer and aliquot 100 μl of beads in 0.2 ml tubes, one tube for the 5′ capture and one tube for the 3′ capture.
-
8.
Place the 0.2 ml tubes against the magnetic rack until the liquid becomes clear. Aspirate and discard the supernatant.
-
9.
Place the 0.2 ml tubes for 30 s in the thermocycler at 47 °C where the hybridization reaction is taking place.
-
10.
Transfer each 5′ and 3′ capture hybridization reactions to each 0.2 ml tube with Dynabeads ® M-270 Streptavidin while keeping the tubes in the thermocycler block. Close the thermocycler lid and incubate for 30 s.
-
11.
Take the tubes, secure the lid and flick them to resuspend the mixture. Spin down briefly (see Note 20 ).
-
12.
Place the tube back in the thermocycler and incubate at 47 °C for 45 min. Resuspend the samples every 15 min by pipetting up and down ten times. During this incubation, warm up the magnetic rack for 0.2 ml tubes on the thermoblock at 47 °C.
-
13.
Place the 0.2 ml tubes in the magnetic rack at 47 °C until the liquid is clear. Aspirate and discard the supernatant.
-
14.
Place the tubes in the thermocycler at 47 °C, add 100 μl of 47 °C 1× Wash Buffer I to each tube and mix by pipetting for 10 s.
-
15.
Repeat step 13.
-
16.
Place the tubes in the thermocycler at 47 °C and add 200 μl of 47 °C 1× Stringent Wash Buffer to each tube. Mix by pipetting ten times and incubate for 5 min.
-
17.
Repeat step 13.
-
18.
Repeat step 16–17.
-
19.
Remove the tubes from the magnetic rack, add 200 μl of room temperature 1× Wash Buffer I to each tube, mix by vortexing at minimum speed for 2 min.
-
20.
Bring the magnetic rack to room temperature.
-
21.
Place the tubes in the magnetic rack at room temperature until the liquid is clear. Aspirate and discard the supernatant.
-
22.
Remove the tubes from the magnetic rack, add 200 μl of room temperature 1× Wash Buffer II to each tube, mix by vortexing at minimum speed for 1 min.
-
23.
Repeat step 21.
-
24.
Remove the tubes from the magnetic rack, add 200 μl of room temperature 1× Wash Buffer III to each tube, mix by vortexing at minimum speed for 30 s.
-
25.
Repeat step 21.
-
26.
Resuspend the Dynabeads ® M-270 Streptavidin pellet of each tube in 50 μl of ultrapure water and transfer the whole mixture to a fresh 0.2 ml tube (including the beads).
SAFETY STOPPING POINT. You can store the samples at −20 °C or proceed immediately.
To amplify the post-hybridization library:
-
27.
Add, directly to the dynabeads bound to your sample, 100 μl of Phusion® High-Fidelity PCR MasterMix (2×), 46 μl of Ultrapure Water, 2 μl of TS-F Primer (100 μM) and 2 μl of TS-R Primer (100 μM). Mix by pipetting ten times and split the sample in two tubes of 0.2 ml with 100 μl each (this is to ensure that the whole reaction volume is in the tube section contained within the block).
-
28.
Place the tubes in the thermocycler with the LM-PCR program and start the run. Do not pre-heat cycler block to 95° prior to the run (see step 6 in Subheading 3.6.1).
-
29.
Combine the identical reactions (5′ + 5′, 3′ + 3′) into a 1.5 ml tube.
-
30.
Proceed to a clean up reaction using the MiniElute® Gel Extraction Kit by adding 1 ml of Binding Buffer to each tube and mix well (including the beads).
-
31.
Add 700 μl of each sample to a MiniElute® column and centrifuge at maximum speed for 1 min.
-
32.
Discard the flow-through and add the remaining sample to the corresponding column. Centrifuge at maximum speed for 1 min and discard the flow-through.
-
33.
Add 700 μl of Washing Buffer and incubate at room temperature for 2 min. Centrifuge at maximum speed for 1 min and discard the flow-through.
-
35.
Rotate each column 180° in its well of the centrifuge rotor and centrifuge at maximum speed for 30 s.
-
36.
Transfer each column to a fresh 1.5 ml tube, add 16 μl of 60 °C Elution Buffer in the center of the column and let it sit for 5 min.
-
37.
Centrifuge at maximum speed for 1 min to elute the library. Discard the column.
-
38.
Quantify each 5′ and 3′ capture analyzing 1 μl of the sample by a Qubit® dsDNA HS Assay. Usually, 3′ capture concentration is >1 ng/μl and 5′ capture concentration is >7 ng/μl (see Note 21 ).
-
39.
Pool 5′ and 3′ capture samples in a ratio 3:7 by molecular mass.
-
40.
Quantify the concentration and size distribution of the capture sample by analyzing 1 μl on an Agilent DNA 1000 Assay.
3.7 Sequencing
3.7.1 Preparation
-
1.
Prepare the Sample Sheet for sequencing on Illumina Instruments using Illumina Experiment Manager software according to the following guidelines:
-
Choose the application “FASTQ Only” for the Sample Sheet, that appears in the “Other” category for MiSeq Instrument and is listed directly as application “HiSeq FASTQ Only” for HiSeq Instruments. This is designed to generate demultiplexed FASTQ files from any type of library.
-
Select the number of Index Reads according to the Illumina adapters used for the library preparation.
-
Select “Paired End” type for sequencing run. This sets the distribution of the Illumina reads after the sequencing reaction in two files according to their forward and reverse orientation in their fragment. The paired reads originated from the same fragment cluster will appear in the same position in both documents.
-
Select 151 cycles for each end. Illumina recommends this number of cycles for a sequencing reaction by a 300 elongation cycles kit, when preparing libraries of the fragment size described in this protocol. Cycle number will vary according to the fragment size and, thus, the sequencing kit.
-
Introduce Samples IDs and Indexes for each sample pooled in the hybridization mix.
-
-
2.
Prepare an appropriate dilution of the capture library containing both 5′ and 3′ captures using molecular grade water (i.e., for MiSeq sequencing the protocol starts with a 4 nM dilution of the library).
-
3.
Ensure that you have enough storage space in the hard disk of the instrument (i.e., a MiSeq run requires 100 GB).
-
4.
Ensure that the Illumina Sequencing instrument is connected to an uninterrupted power supply or is equipped with a power supply unit able to support the instrument during the whole length of the run.
3.7.2 Procedure
-
1.
Dilute the library and denature the DNA following Illumina instructions, according to the sequencing run you are performing.
-
2.
Set up the sequencing components (flow cell, sequencing cartridge, and sequencing buffers) as described by Illumina instructions.
-
3.
Start the sequencing run.
-
4.
After the run, the Instrument performs an analysis of the primary data. Paired-end sequencing indicated here produces two different FASTQ files for each indexed library. These files contain the reads from the forward and the reverse sequencing reaction of the same fragment, which are located in the same position of each file. Check that you transfer both files to the computer designated for the analysis.
3.8 Bioinformatic Analysis
The goal of RC-seq is to elucidate true positive L1 insertions, relying primarily on L1-genome junction sequences, rather than read count, to distinguish these events from artifacts. Although this can be computationally intensive, it is preferable to stringently exclude false positives (artifacts) bioinformatically rather than having to PCR validate every L1 insertion. Similarly, the analysis must not be so strict as to exclude any L1 insertion structure that is unusual, or cause false negatives. The procedure outlined below is intended to balance these considerations when searching for rare somatic L1 insertions in human brain tissue; a user should evaluate whether the algorithms and parameters in each step are appropriate to other retrotransposons, spatiotemporal contexts or types of L1 insertions (e.g. germline polymorphisms). A user will also need to be able to write scripts to process the output of each step. Custom scripts referred to below are written in Python, similar scripts may be written in other programming languages.
3.8.1 Preparation
-
1.
Paired-end Illumina sequencing will have generated two fastq files, one for each read pair end. Prior to analyzing these files, it is recommended to establish a file name nomenclature that is consistent across projects, including the date of sequencing, the project identifier and a sample identifier. For example, 011214.GBM.1T.R1.fastq and 120114.GBM.1T.R2.fastq are two files for RC-seq sequenced on December 1, 2014 for a glioblastoma (GBM) project sample identifier 1T (individual #1 tumor). Using this system will simplify project management and data archiving prior to publication.
3.8.2 Procedure
-
1.
Write a Python script to trim reads in each fastq file from their 5′ and 3′ ends to remove any bases with Illumina quality scores <10.
-
2.
Input fastq files into FLASH [35], using default parameters, to assemble overlapping read pairs into read contigs in a single fasta file. These read contigs potentially span L1-genome junctions.
-
3.
Align read contig fasta file to the latest human reference genome build (e.g. hg19, available from UCSC Genome Browser) hg19 using SOAP2 [36] (parameters –M 4 –v 2 –r 1 –p 8). These parameters will retain only read contigs aligning to one genomic location each. SOAP2 can also output unmapped read contigs (option –u). L1 insertions not present in the reference genome will be unmapped, and are what a user is seeking.
-
4.
Align the unmapped read contigs to an active L1 consensus sequence (e.g. L1.4 from [37]) using LAST [38] (parameters –s 2 –l 11 –d 30 –q 3 –e 30).
-
5.
Write a Python script to retain read contigs aligned at >95 % identity to L1.4 and spanning ≥33 nt of one contig end and arrange read contigs with a 5′ non-retrotransposon section (≥33 nt) followed by a 3′ retrotransposon section (≥33 nt).
-
6.
Align read contigs to hg19 using LAST (s2 –l11 –d30 –q3 –e30), which excels in reporting split alignments found for translocations and, for RC-seq, where one end of the assembled read contig maps to one location on the genome and the other end maps elsewhere (Fig. 4).
Fig. 4 
Logic for the fragment size-selection by electrophoresis for a 300-cycle sequencing run. The sequencing reaction is primed by two oligonucleotides (Primer L in red and Primer R in blue), one from each end of the molecule. The 300-cycle run catalyzes 150 elongation cycles for the extension of each primer (reads and arrows in red and blue). Since 30–80 nt overlap between the paired reads is necessary for a successful alignment (double-headed black arrow) along the RC-seq pipeline, the overall size of the readable fragment is 220–290 bp. The agarose electrophoresis size-selection is performed after the ligation of Illumina sequencing adapters, increasing fragment size by 120 bp (60 bp per adapter). Therefore, for a library to be sequenced on a 300-cycle run, the preferred fragment for the size selection is 340–390 nt in length
-
7.
Write a Python script to remove any contig read with an alignment of the non-retrotransposon section plus 10 nt (potential molecular chimera or genomic rearrangement). The remaining contig reads with a uniquely mapped non-retrotransposon section indicate the nucleotide position and strand of an L1-genome junction not present in the reference genome, and the nucleotide position and end of L1.4 detected.
-
8.
Write a Python script to cluster aligned contig reads join opposing clusters separated by ≤100 nt and detecting different ends of a common L1 insertion, and annotate this list of clusters with existing databases of polymorphic L1 insertions. L1 insertions found only in one brain sample by ≥1 RC-seq reads, and absent from any matched control RC-seq library, or previous publications, can be annotated as somatic insertions. These can be further filtered by, for example, removing L1-genome junctions that indicate substantial L1 3′ truncations or requiring reported insertions to present TSDs. This process should generate a table of putative somatic L1 insertions in each sample.

3.9 PCR Validation
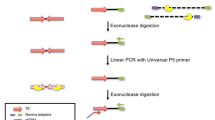
The validation process is highly dependent on the researcher’s inventiveness and ability. To validate a new somatic insertion it is required that both ends of the insertion represent a genomic continuity interrupted by the new L1 copy. The validation will be more robust if, additionally, canonical TPRT signatures like TSDs, a polyA track in the 3′ end of the element and an EN-motif at the 5′ end of the 5′ TSD are found, otherwise it will be necessary to assume that the copy has been inserted by non-canonical pathways already characterized via in vitro approaches. However, many of the insertions detected by RC-Seq are only indentified at one junction [12, 33] (Prof. Faulkner group, unpublished results), so the basis of the PCR validation resides in the specific PCR amplification of the missing junction of each insertion. To amplify that junction sequence, it is necessary to “recreate” the structure of the missing junction, deduced by combining the annotated genome sequence following the genomic section of the known junction and the consensus sequence of the L1 end opposite the one detected in the junction (Fig. 5). It will be necessary to design primers annealing within the L1 sequence and within the genomic region expected for the missing junction.

Rationale of PCR validation. (a) Validation design for empty-site PCR. (b) Validation design for 5′ junction amplification
The PCR amplification of 3′ junctions consistently yields off-target amplicons [12]. Additionally, due to the location of the LNA-5′ probe and the fact that TPRT frequently generates 5′ truncated insertions, the 3′ junction is the most frequently captured one. The PCR validation described here is therefore based exclusively on PCR amplification of 5′ junctions.
The PCR validation of a 5′ junction has as a major handicap the fact that a priori the extent of 5′ truncation is not known, so it is impossible to predict the 5′ L1 sequence constituting the junction. As a guideline, there are two consecutive approaches: empty-filled site PCR and 5′ junction PCR.
The empty-filled site PCR consists of amplification by primers annealing in the 5′ and 3′ genomic flanks of the putative insertion (Fig. 5a). For this PCR, we recommend the use of a high processive enzyme with PCR-cycling conditions able to amplify a ≤8 kb amplicon. As a starting point we suggest the Expand Long Range dNTPack (Roche) with the following conditions: 1× Buffer (with 2.5 mM MgCl2 final concentration), 5 % DMSO, 2 mM dNTPs (0.5 mM each), 2 mM primers (1 mM each), 1.75 U of polymerase and 10 ng template DNA in a final volume of 25 μl with molecular grade water; and the suggested cycling conditions are: 92 °C 3 min; 92 °C 30 s, 60 °C 30 s and 68 °C 7.5 min for ten times; 92 °C 30 s, 58 °C 30 s and 68 °C 7 min plus ∆20 s increase per cycle for 30 times; 68 °C 10 min and 10 °C hold. The expected empty site is usually favored during the amplification and competes with the amplification of the filled site. It is important to screen faint bands in an electrophoresis gel in the size frame around 7 Kb. In any case, any band above the empty site amplicon is a candidate to be purified, cloned and sequenced in order to identify the 5′ junction. We recommend designing the genomic primers within 200 nt from the breaking point. Primer design can be initially attempted using a software like Primer3; if Primer3 fails to find good candidate primers then primers must be chosen by hand.
In the second approach, the 5′ junction amplification will require a primer annealing in the genomic section of the missing junction (designed as described above) and a primer annealing in the L1 sequence in reverse sense. If the empty-site PCR has provided positive results about the location of the junction (estimating by amplicon size or directly by sequencing), a specific primer can be designed in the known L1 region next to the truncation point and this PCR will confirm the result obtained with the empty-filled PCR. Otherwise, a set of L1 primers located along the whole L1 sequence in reverse sense can be used for multiple PCR reactions aiming to capture the 5′ L1-genome junction (Fig. 5b. See Note 22 for a list of L1 primers to start with). Note that the length of full length insertions together with the small size of the empty site amplicon will strongly oppose the filled site amplification, so if the empty-filled PCR is not successful, proceed to the 5′ junction amplification using the battery of reverse L1 primers.
For this PCR reaction a high fidelity PCR enzyme is preferable, to avoid the formation of chimeras during the amplification due to the repetition of L1 sequences in the genome. The suggested enzyme is the Platinum® Taq DNA Polymerase High Fidelity. The starting conditions are: 1× High Fidelity PCR buffer, 2 mM MgSO4, 0.8 mM dNTPs (0.2 mM each), 0.4 μM primers (0.2 μM each), 7.5 U polymerase, and 10 ng template DNA. The recommended cycling conditions are 94 °C 3 min; 94 °C 30 s, 57 °C 30 s and 68 °C 30 s for 35 cycles; 68 °C 10 min and 10 °C hold.
Sanger sequencing of the PCR products directly of via cloning will be required to fully characterize the paired junction of the insertion. According to the features of the amplification enzyme utilized, we recommend AT cloning by pGEM®-T vector system or AT or blunt-ends cloning by TOPO® PCR system (Life Technologies) for regular size and large amplicons respectively. Due to the low yield of some amplification products such as full-length insertions, agarose gel-purification and re-amplification may be attempted to obtain enough amplified fragment for cloning. In some cases, phenol extraction from agarose and ethanol precipitation may increase the yield for faint bands of large size DNA fragments.
4 Notes
-
1.
Prepare the Lysis Buffer without adding Proteinase K and store it at room temperature. Take an aliquot for each extraction and add Proteinase K to the aliquot at final concentration of 100 μg/ml just before use. If using Ambion® 20 mg/ml Proteinase K Solution (Life Technologies), add 5 μl of Proteinase K to each 995 μl of Lysis Buffer.
-
2.
The whole protocol can be adapted to 0.2 ml tube-strips for multichannel adaptation. In this case, a DynaMag™-96 Side (Life Technologies) magnetic rack must be used and, consequently, the 80 % ethanol volume used for the washes must be 200 μl.
-
3.
N8 segment sequence of Illumina Indices-specific Blocking Oligos: Index 1, CGTGATGT; Index 2, ACATCGGT; Index 3, GCCTAAGT; Index 4, TGGTCAGT; Index 5, CACTGTGT; Index 6, ATTGGCGT; Index 7, GATCTGGT; Index 8, TCAAGTGT; Index 9, CTGATCGT; Index 10, AAGCTAGT; Index 11, GTAGCCGT; Index 12, TACAAGGT; Index 13, TGTTGACT; Index 14, ACGGAACT; Index 15, TCTGACAT; Index 16, CGGGACGG; Index 18, GTGCGGAC; Index 19, CGTTTCAC; Index 20, AAGGCCAC; Index 21, TCCGAAAC; Index 22, TACGTACG; Index 23, ATCCACTC; Index 25, ATATCAGT; Index 27, AAAGGAAT.
-
4.
Additional Proteinase K can be added to the dissociation reaction if the tissues are not dissolving at a timely rate.
-
5.
If using 10 mg/ml DNase-free, protease-free RNase A (Thermo Scientific), add 1 μl per 500 μl of sample.
-
6.
The quality of the starting DNA solution is really important for the library preparation. It is worthwhile to take extra time to duplicate the phenol and phenol:chlorophorm:isoamyl alcohol extraction steps in order to ensure the success of the subsequent steps.
-
7.
Alternatively the sample can be centrifuged at >12,000g in a benchtop centrifuge and supernatant thoroughly removed.
-
8.
Short incubations (~30 min) at 65 °C can be used to aid resuspension, but high temperatures should be avoided where possible.
-
9.
Alternatively, Covaris S220 Focused-Ultrasonicator can be used with the following parameters: Duty Cycle 10 %, Intensity 5, Pulses per Burst 200 and Duration 120 s.
-
10.
DNA quantification by Nanodrop usually overestimates genomic DNA concentration. After shearing the genomic DNA, the overestimation is reduced but is still higher than results produced by analysis by Agilent Bioanalyzer Technology. In step 10 from the Subheading 3.3.2, up to 1 μg of DNA is required, so if only a single library is being prepared, we recommend starting the whole process with 5 μg of genomic DNA.
-
11.
If the sonication is interrupted by a sudden drop of the water level, immediately proceed to add more water with the Wash Bottle through the water sense aperture in the Tube Holder and click “resume” in the warning panel.
-
12.
For two different libraries combine indices 6 and 12; or 5 and 19. For three libraries combine indices 2, 7 and 19; 5, 6 and 15; or any combination for two libraries plus any other index. For four different libraries combine indices 5, 6, 12 and 19; 2, 4, 7 and 16; or any combination for three libraries plus any other index. For 5–11 different libraries, use the combination for 4 different libraries plus any other adapter (for more information please visit http://supportres.illumina.com/documents/documentation/chemistry_documentation/samplepreps_truseq/truseqsampleprep/truseq_sample_prep_pooling_guide_15042173_a.pdf).
-
13.
Pay special attention to these two consecutive AMPure XP beads purification steps. They are necessary to completely remove the adapter excess from the samples and avoid any interference in the following amplification steps. The procedure is highly repetitive and it is very easy to trash the library by mistaking the elution step for an ethanol washing step.
-
14.
The number of cycles of PCR must be kept to a minimum. The amplification of certain repeated sequences is relatively unfavored, diminishing their relative abundance in the pool with increasing amplification cycles. In both the pre- and post-hybridization LM-PCR steps, start with the indicated number of cycles. The hybridization mix before completely drying the sample has no restrictions concerning sample volume, so it is preferable to include more pre-hybridization library input in case of low concentration instead of running more PCR cycles.
-
15.
We recommend taking one or two initial cuts below and above the preferred one to increase the chances of obtaining a library with right size distribution. The gel cuts can be stored at 4 °C or can be processed together. Consider the fact that each Agilent DNA chip has 12 wells, so it can be convenient to proceed with more than one gel cut depending on the number of libraries to maximize the chip yield.
-
16.
Salt in the hybridization mix can dramatically affect to the reaction, so for this step is essential to use molecular grade water instead of resuspension buffer. This library will be substrate for the immediate hybridization and it is necessary to reduce the salt content to minimum.
-
17.
After this analysis, two possible problems can come up: low amount of library (<20 ng/μl) or an inappropriate size. If the amount is low, one solution is to perform additional cycles of LM-PCR. To do that, go to step 14 and proceed with a first attempt of 2–3 cycles depending on the concentration of the input library. If the fragment size is not appropriate, because it is too big or too small, then go to the backup gel-cuts of step 4 and proceed with the immediately smaller or bigger one respectively. If two consecutive gel-cuts are obtained, one of them bigger and the other one smaller that the preferred size, it is possible to combine the two libraries and reanalyze the mixture. Usually the resulting peak will be the preferred size.
-
18.
If analyzing tissues A and B from the same donor, pool 500 ng of each sample library; if analyzing tissues A, B and C, pool 333 ng of each sample library; if analyzing tissues A and B tissues from donor X and Y, then pool 250 ng of each library; and so.
-
19.
It is very important the temperature of the incubation not to go below 47 °C. LNA probes have a high ability for hybridization and the reversibility is strongly disabled. The unspecific binding occurring below 47 °C will not dissociate upon bringing the temperature back to 47 °C. This is why it is very important that the thermocycler has an uninterrupted power supply during the 3 days the incubation requires. Any drop in the incubation temperature will result in a reduction in the library enrichment in L1-specific sequences.
-
20.
Try to avoid holding the tubes out of the thermocycler more than 10 s. It is better to put them back in the thermocycler and do several resuspension re-attempts.
-
21.
In case one or both libraries end with low concentration insufficient for sequencing, proceed as in Note 17 . Briefly, bring the sample to 30 μl final volume with molecular grade water and go to step 14–23 from the Subheading 3.4.2. Similarly, do a first attempt of reamplification by LM-PCR with 2–3 cycles. In step 21, add only 16.5 μl of resuspension buffer and in step 23 recover 15 μl to a new tube.
-
22.
This is a guide of suggested reverse primer mapping in the L1 consensus sequence with their approximate location in brackets. L1-1R(554) 5′CCAGAGGTGGAGCCTACAGA3′; L1-2R (1085) 5′ATGTCCTCCCGTAGCTCAGA3′; L1-3R (1426) 5′TGGTTCCATTCTCCACATCA3′; L1-4R (2080) 5′TCCAACTTGCCAGTCTGTGT3′; L1-5R (2591) 5′TAGGTGTGGTGTGGTGCTGA3′; L1-6R (3085) 5′ACCAGCTCCTGGATTCATTG3′; L1-7R (3550) 5′CCGGCTTTGGTATCAGAATG3′; L1-8R (4041) 5′TTCCTTCTCCTGCCTGATTG3′; L1-9R (4263) 5′TGGGAGTTCACCCATGATTT3′; L1-10R (5084) 5′TGCCTGTTCACTCTGATGGT3′; L1-11R (5627) 5′CATTTGGGTTGGTTCCAAGT3′; L1-12R (5799) 5′TGAGAATATGCGGTGTTTGG3′.
References
Taylor TH, Gitlin SA, Patrick JL et al (2014) The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum Reprod Update 20:571–581
Stern C (1936) Somatic crossing over and segregation in Drosophila melanogaster. Genetics 21:625–730
Hozumi N, Tonegawa S (1976) Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions. Proc Natl Acad Sci U S A 73:3628–3632
Muramatsu M, Kinoshita K, Fagarasan S et al (2000) Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102:553–563
Gilbert N, Lutz S, Morrish TA et al (2005) Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol 25:7780–7795
Garcia-Perez JL, Doucet AJ, Bucheton A et al (2007) Distinct mechanisms for trans-mediated mobilization of cellular RNAs by the LINE-1 reverse transcriptase. Genome Res 17:602–611
Kano H, Godoy I, Courtney C et al (2009) L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev 23:1303–1312
Muotri AR, Chu VT, Marchetto MC et al (2005) Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435:903–910
McConnell MJ, Lindberg MR, Brennand KJ et al (2013) Mosaic copy number variation in human neurons. Science 342:632–637
Kapitonov VV, Jurka J (2005) RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol 3, e181
Levis RW, Ganesan R, Houtchens K et al (1993) Transposons in place of telomeric repeats at a Drosophila telomere. Cell 75:1083–1093
Baillie JK, Barnett MW, Upton KR et al (2011) Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479:534–537
Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH Jr (1996) High frequency retrotransposition in cultured mammalian cells. Cell 87:917–927
Dewannieux M, Esnault C, Heidmann T (2003) LINE-mediated retrotransposition of marked Alu sequences. Nat Genet 35:41–48
Raiz J, Damert A, Chira S et al (2012) The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res 40:1666–1683
Jurka J (1997) Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci U S A 94:1872–1877
Luan DD, Korman MH, Jakubczak JL et al (1993) Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell 72:595–605
Ostertag EM, Kazazian HH Jr (2001) Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res 11:2059–2065
Morrish TA, Gilbert N, Myers JS et al (2002) DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet 31:159–165
Lander ES, Linton LM, Birren B et al (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
Kazazian HH Jr, Wong C, Youssoufian H et al (1988) Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 332:164–166
Huang CR, Schneider AM, Lu Y et al (2010) Mobile interspersed repeats are major structural variants in the human genome. Cell 141:1171–1182
Ewing AD, Kazazian HH Jr (2010) High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res 20:1262–1270
Stewart C, Kural D, Stromberg MP et al (2011) A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet 7, e1002236
Faulkner GJ (2011) Retrotransposons: mobile and mutagenic from conception to death. FEBS Lett 585:1589–1594
Hancks DC, Kazazian HH Jr (2013) Active human retrotransposons: variation and disease. Curr Opin Genet Dev 22:191–203
Muotri AR, Marchetto MC, Coufal NG et al (2010) L1 retrotransposition in neurons is modulated by MeCP2. Nature 468:443–446
Faulkner GJ, Kimura Y, Daub CO et al (2009) The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41:563–571
Coufal NG, Garcia-Perez JL, Peng GE et al (2009) L1 retrotransposition in human neural progenitor cells. Nature 460:1127–1131
Belancio VP, Roy-Engel AM, Pochampally RR et al (2010) Somatic expression of LINE-1 elements in human tissues. Nucleic Acids Res 38:3909–3922
Cost GJ, Golding A, Schlissel MS et al (2001) Target DNA chromatinization modulates nicking by L1 endonuclease. Nucleic Acids Res 29:573–577
Richardson SR, Morell S, Faulkner GJ (2014) L1 retrotransposons and somatic mosaicism in the brain. Annu Rev Genet 48:1–27
Shukla R, Upton KR, Munoz-Lopez M et al (2013) Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 153:101–111
Upton KR, Gerhardt DJ, Jesuadian JS et al (2015) Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161:228-239
Magoc T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics (Oxford) 27:2957–2963
Li R, Yu C, Li Y et al (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics (Oxford) 25:1966–1967
Dombroski BA, Scott AF, Kazazian HH Jr (1993) Two additional potential retrotransposons isolated from a human L1 subfamily that contains an active retrotransposable element. Proc Natl Acad Sci U S A 90:6513–6517
Kielbasa SM, Wan R, Sato K et al (2011) Adaptive seeds tame genomic sequence comparison. Genome Res 21:487–493
Acknowledgments
G.J.F. acknowledges the support of an NHMRC Career Development Fellowship (GNT1045237). F.J.S-L. was supported by a postdoctoral fellowship from the Alfonso Martín Escudero Foundation (Spain). Work in the Faulkner laboratory was funded by Australian NHMRC Project grants GNT1042449, GNT1045991, GNT1067983 and GNT1068789, as well as the European Union’s Seventh Framework Programme (FP7/2007–2013) under grant agreement No. 259743 underpinning the MODHEP consortium.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Sanchez-Luque, F.J., Richardson, S.R., Faulkner, G.J. (2016). Retrotransposon Capture Sequencing (RC-Seq): A Targeted, High-Throughput Approach to Resolve Somatic L1 Retrotransposition in Humans. In: Garcia-Pérez, J. (eds) Transposons and Retrotransposons. Methods in Molecular Biology, vol 1400. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3372-3_4
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3372-3_4
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3370-9
Online ISBN: 978-1-4939-3372-3
eBook Packages: Springer Protocols