
Abstract
Well-defined as signaling hormones for the programming of cell type-specific and context-dependent gene expression signatures, glucocorticoids control experience-driven allostasis. One unifying model is that glucocorticoids help maintaining the integrity and plasticity of cellular networks in changing environments through the mobilization of cellular energy stores, profiling of gene expression, and changes in the electrical and morphological properties of cells. The nucleus is their primary site of action, yet recent discoveries point to additional gene transcription-independent functions at the plasma membrane of neuronal synapses. Glucocorticoids are secreted factors that reflect intrinsically the changes coming from the external world, temporally and regionally, during development and adulthood. In this review, we will enumerate the properties and signaling attributes of glucocorticoids and their receptors that characterize them as allostatic modulators. The molecular mechanisms used to support their role at the synapse will be highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Glucocorticoid hormones are chemical messengers that signal via alterations of gene expression in cells expressing cognate receptors. Nuclear receptors are an evolutionarily conserved class of transcription factors that regulate gene expression in a cell type-specific and context-dependent manner. Importantly, ligand-activated nuclear receptors are able to integrate multiple signals giving rise to distinct patterns of gene expression, and rapid non-genomic signaling outcomes yet to be discovered. Converging signaling pathways act in part by altering receptor phosphorylation that fosters the recruitment of interacting co-regulatory molecules in the mitochondria, cytoplasm, synapse and nucleus. This chapter explores how glucocorticoid-mediated allostasis employs rapid non-genomic and slow genomic mechanisms together with ongoing neuronal activity, thus providing a molecular framework for understanding normal and pathological glucocorticoid functions. In particular, the mechanisms underlying the slow appearance of glucocorticoid resistance in numerous human disease-states are not understood although several molecular correlates have been identified. One hypothesis that shows great promise is that the survival/ growth MAPK pathway modulates glucocorticoid-mediated allostasis, in part, through the phosphorylation of the glucocorticoid receptors. This chapter synthesizes our current understanding of glucocorticoid signaling with an emphasis on cellular networks of the brain.
Ligands
Sources
Glucocorticoids are cholesterol-derived steroid hormones that prepare the organism to adapt to changing environments during development and adulthood. Metabolism, immunity, cognition, circadian learning and allostatic response (defined as the physiological processes engaged to return to homeostasis [1]) to physical or psychological threats are well-characterized physiological responses that engage glucocorticoid hormone signaling. Cortisol, the major endogenous glucocorticoid in humans (corticosterone in rodents), is secreted by the adrenal cortex where biosynthetic enzymes are produced [2]. The classical view is that cortisol reaching the brain comes from the adrenal gland. Yet, the expression of some of these enzymes like 11βhydroxysteroid dehydrogenase (11βHSD1) exists in other tissues like the brain, which supports the possibility of local production sites for glucocorticoids and related hormones called neurosteroids [3]. Therefore, adrenal glands are the major source of glucocorticoids but the presence of biosynthetic or degradation enzymes modulate the availability of these hormones at target organs. Future investigations shall clarify the impact of the presence of 11βHSD1 in the brain. Interestingly, it is the brain that processes sensory information and activates the hypothalamic-pituitary-adrenal (HPA) axis that triggers the secretion of cortisol. Hypothalamic paraventricular neurons secrete corticotropin releasing hormone (CRH) and arginine-vasopressin (AVP) into the portal vessels to reach the anterior lobe of the pituitary gland and stimulate adrenocorticotropic hormone (ACTH) secretion. Circulating ACTH binds to cognate receptor in the cortex of the adrenal gland, which stimulates the (i) biosynthesis and (ii) release of cortisol in the bloodstream. Through the vasculature, crossing the blood–brain-barrier, cortisol can readily access every organ to prepare a coordinated cellular allostatic response.
Thanks to robust anti-inflammatory and immunosuppressive effects, glucocorticoids are a mainstay of treatment for numerous inflammatory and immune diseases, and against trauma despite a wide range of side effects in multiple organs (brain, bone, liver, lung, eye, muscle). Side effects of glucocorticoid therapy that involve the brain include manifestations of emotional liability, psychosis, gain or loss of appetite, insomnia, and memory impairments.
Secretion Modes
Pulsatile
Glucocorticoids are secreted in synchrony with circadian rhythms and in response to stress. At rest, the HPA axis displays a circadian pattern of activity, which produces a cortisol secretion peak during the active period of the day and a trough during the inactive phase (Fig. 2.1). A recent study demonstrates that glucocorticoid peaks and troughs are critical determinants of circadian learning [4]. That is, mice learning at glucocorticoid peaks (evening) acquire a learned motor coordination task better than mice trained at glucocorticoid trough (morning). If learning is sensitive to glucocorticoid circadian rhythms, recall of the learned motor task is not sensitive to glucocorticoid peaks and troughs. This study illustrates how naturally occurring glucocorticoid oscillations impacts distinct phases required for behavioral adaptation to novelty. Circadian activation of the HPA axis is controlled by the suprachiasmatic nucleus, which functions as a light–dark oscillator with direct output projections to the hypothalamic PVN. Thus, neurons of the suprachiasmatic nucleus order CRH-producing neurons to activate the HPA axis, at the time period that precedes the awakening and active phase [5–7].

Typical superimposed ultradian (fine line) and circadian (thick line) circulating corticosterone plasma level. Due to a very high affinity for corticosterone, MR are constantly activated contrary to GR only activated at secretion peaks (adapted from [6])
Superimposed to the circadian rhythms are the ultradian oscillations. The frequency of ultradian pulsatory release of glucocorticoid is much more rapid than that of circadian rhythms. An ultradian oscillation usually resolves within a 2 h period [8–10]. The coincidence of ultradian and circadian peaks and troughs produces the highest amplitudes of glucocorticoid oscillations [6] (Fig. 2.1). Counter-intuitively, glucocorticoid secretion at ultradian trough during the circadian peak may be lower than that resulting from ultradian peak at circadian trough. One question that arises from this notion is whether learning at the ascending ultradian peak is more effective than learning at the ultradian trough. According to proposed mathematical models, this ultradian oscillation is the consequence of the slightly delayed feedforward control of pituitary ACTH on glucocorticoid release and the feedback control of adrenal glucocorticoids on ACTH release that depends on an underlying CRH drive but does not necessitate CRH pulsatility [11]. This model contrast with the best described endocrine system controlled by ultradian oscillations in which the oscillating secretion of gonadotropin-releasing hormone from the hypothalamus directly controls pulses of LH and FSH in the pituitary [12, 13]. Disruption of circadian glucocorticoid rhythms in animal models is sufficient to impair learning and memory and to produce symptoms of depression [4, 14]. Consistently, perturbation of glucocorticoid oscillations have been associated with numerous diseases of the central nervous system [6].
In Response to Stress
Circulating levels of glucocorticoids not only vary as a result of intrinsic rhythms, they are also secreted in response to stress. Polysynaptic limbic circuits converge on the hypothalamic paraventricular nucleus (PVN) to activate CRH-producing neurons [15]. These input circuits involve multiple brain regions including the hippocampus, prefrontal cortex, amygdala, bed nucleus of the stria terminalis (BNST), and locus coeruleus (LC). The intricacy of these circuits allow for a diversity of stressor specific responses like physical stress or emotionally arousing experiences, leading to the activation of the HPA axis. Deactivation of the HPA axis is critical to prevent escalation of glucocorticoid levels beyond physiological range. A remarkable feature of this pathway is the ability of glucocorticoid to exert feedback inhibition on CRH, AVP, ACTH and glucocorticoid levels, resulting in the deactivation of the HPA axis. The dexamethasone-suppression test allows for the determination of the strength of the negative feedback in humans and animal models. About 50 % of depressed individuals in multiple cohorts present with diminished negative feedback [16]. Such signs of glucocorticoid resistance gave rise to the glucocorticoid hypothesis of depression [17]. Therefore, maintenance of the HPA axis involves homeostatic equilibrium between the activation pathway and inhibitory feedback.
Availability
Bioavailability of glucocorticoids to target tissues is not only regulated by its secretion patterns but also by carrier proteins in the blood. Corticosteroid binding globulin (CBG) a high affinity plasma protein and albumin bind up to 95 % of plasma glucocorticoids. Only the remaining unbound fraction is free to diffuse across plasma membranes and bind receptors, be degraded by the liver or processed by converting enzymes [18]. CBG is saturated at glucocorticoid concentrations corresponding to the peak concentration of each pulse (400–500 nmol cortisol for humans), permitting an increase of free cortisol only at these time points [6, 19]. CBG-glucocorticoid coupling regulation, allows for spatial and temporal modulation of glucocorticoid availability. Minute temperature rise leads to a reversible decrease in CBG affinity for glucocorticoids, permitting in case of fever or inflammation an increment of glucocorticoid availability [19]. Inflammation sites are rich in proteinases, like elastase that cleaves CBG such that steroid binding is irreversibly lost [20, 21]. Glucocorticoid availability in target tissues is also regulated by locally expressed 11βHSD enzymes (see Chap. 16 for more discussion on this topic). These exist in two isoforms that convert corticosteroid precursors into cortisol and vice-versa [22]. Type 2 (11βHSD-2) catalyzes inactivation by converting cortisol in inactive cortisone. Type 1 (11βHSD-1) catalyzes activation by converting inactive keto-forms like cortisone in active cortisol, locally within the cells. 11βHSD-1 is abundant in the liver, adipose tissue and brain, where it plays a role in ageing related cognitive decline, as demonstrated by stable learning ability of 11βHSD-1 KO mice [23, 24]. Finally, to access target tissues like the brain, glucocorticoids need to cross the blood–brain barrier, a specialized layer of endothelial cells. Despite their lipophilic nature permitting diffusion through plasma membranes, transporters pump glucocorticoids out of the intracellular space. Among these transporters, multidrug resistance-p-glycoproteins 1a and b (Mdr1) are expressed by endothelial cells of the blood–brain barrier and cortical and hippocampal neurons [25]. Interestingly, Mdr1a expression in neurons is upregulated following seizures [25, 26].
Receptors
Early postnatal development is characterized by a glucocorticoid hypo-responsive period during which the organism is highly vulnerable to extreme environmental changes. Extensive research using early life stress paradigms indicates that glucocorticoid signaling is not fully mature during embryogenesis and the first weeks of life. In consequence, pups are unable to cope with extreme stress and easily develop defects that persist during adulthood. For example, early life stress (or glucocorticoid treatment during the same period) impairs neuronal growth and differentiation that could result in neural circuit wiring defects responsible for the development of cognitive and neuropsychiatric illnesses during adulthood [27]. It is believed that such critical period of glucocorticoid responsiveness depends on receptor expression patterning that is temporally and spatially regulated.
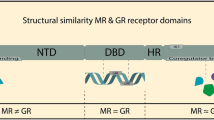
To date, glucocorticoids have been shown to signal trough two nuclear receptors, the mineralocorticoid receptor (MR), and the glucocorticoid receptor (GR), denominations that correspond to their peripheral activity, as MR regulates electrolyte balance, and GR is involved in gluconeogenesis. Both of these receptors belong to the nuclear hormone receptor family. Nuclear hormone receptors are comprised of multiple, independent functional domains [28]. These include (i) a variable amino-terminal domain (NTD or A/B region), containing an activation function (AF)-1 with ligand-independent transactivation activity; (ii) a conserved DNA binding domain (DBD or C region) involved in the recognition of specific DNA ligand sequences; (iii) a variable hinge region (D) connecting the DBD to the ligand binding domain (LBD); (iv) and a conserved LBD (E) containing an additional region for transactivation activity named AF-2. Some receptors contain an additional highly variable carboxyl-terminal region of unknown function (Fig. 2.2). Of these functional domains, the NTD is the most variable among nuclear receptors in terms of length and sequence similarities and is the major target for ligand-dependent phosphorylation at multiple serine residues [29, 30].

A schematic diagram of the functional domains and best characterized phosphorylation sites of human GR. Sites in red are BDNF-dependent sites. NTD amino terminal domain, AF-1 activation function-1, DBD DNA-binding domain, HR hinge region, LBD ligand-binding domain, AF-2 activation function-2, S serine, and P proline
The Cloned Receptor Subtypes
Mineralocorticoid Receptor
MR is encoded by the NR3C2 gene. The human gene is composed of ten exons and eight introns. The first two exons, 1β and 1α undergo alternative splicing, giving rise to two different mRNA forms [31]. This alternative transcription is under control of two distinct promoters whose activities are tissue specific and regulated during development. The alternative spliced exons encode the 5′ untranslated region of the transcript and translation starts at exon 2. Both transcript variants result in the same protein of 984 amino acids [31, 32]. In the adult brain, MR is mostly expressed in the dorsolateral septum and hippocampus. The highest levels of MR receptors are detected in the pyramidal layer of the CA1 and CA2 region of the hippocampus and the granular layer of the dentate gyrus, with lower expression on the pyramidal layer of the CA3 region of the hippocampus [33, 34]. Outside the brain, the main ligand for MR is aldosterone, a corticoid hormone also secreted by the adrenal gland at a concentration 100-fold less than glucocorticoids. Having a similar affinity for glucocorticoids and mineralocorticoids, the coexpression of MR with 11βHSD-2 in peripheral organs like the kidney provides deterministic signaling properties. Indeed, the degradation of intra-kidney cortisol by 11βHSD-2 allows MR to bind to the less abundant aldosterone [22]. Because 11βHSD-2 is mostly absent from the adult brain, the preferred MR ligand within the brain is cortisol in humans and corticosterone in rodents. Corticosterone binding affinity to MR K d of 0.1–0.3 nM is very high compared to that of GR K d of 2–5 nM [35, 36]. One consequence of this very high affinity is the relatively high occupation of MR with endogenous corticosterone that can reach 80 % at a low corticosterone circadian trough level. This translates into a constant activation of MR signaling regardless of circadian and ultradian rhythms (Fig. 2.1). What could be the function of a hippocampal glucocorticoid receptor that would be constantly activated? The hippocampus plays an important role in the HPA axis regulation as demonstrated by lesion studies [37]. Using different pharmacological antagonists, it was demonstrated that blocking MR receptor activates the HPA axis, suggesting a role of MR on the tonic inhibition of the HPA axis [36, 38, 39]. Additionally, dentate granular neurons death resulting from adrenalectomy can be rescued by hormonal replacement with aldosterone, a specific MR receptor agonist [40]. Aldosterone treatment also enhances the proliferation and survival of newly-born granule cells of naïve mice [41]. Given that GR activation inhibits granule cell proliferation in the hippocampus, it is believed that a balance of GR/MR activation along the glucocorticoid circadian and ultradian oscillations controls the electrical activity and number of newly-born neurons in the adult hippocampus.
Glucocorticoid Receptor
GR is encoded by the NR3C1 gene, comprising 9 exons and 11 introns, and the protein is coded from exons 2 to 9. Exons 1 and 9 undergo alternative splicing. Alternative splicing of exon 9 gives rise to two isoforms: the most prevalent GRα and, and GRβ form that has a shorter C terminus transactivation region [42, 43]. GRα is the majoritarian form and classically shuttles between the cytoplasm and nucleus depending on signaling. GRβ resides permanently in the nucleus and acts as a dominant negative inhibitor of the GRα isoform, but can also directly regulate genes that are not regulated by the α isoform [43, 44]. Additional isoforms of GR are generated by alternative translation via different initiation sites giving rise to eight variants with truncated N termini. The resulting isoforms all bind glucocorticoids with similar affinities but have different transcriptional activity depending on the presence or not of the AF1 domain [43]. The expression of GR is ubiquitous and several isoforms can be co-expressed in many tissues adding to the complexity of GR signaling. Consequently, GR signaling depends on tissue-specific isoforms, expression level and glucocorticoid availability (time at exposure, duration and dose). Given that mice lacking NR3C1 die of lung maturation defects [45, 46], further tissue specific gene inactivation studies provided valuable information regarding the role of limbic circuits in the control of the HPA axis and allostatic responses to drugs of abuse and fear [47–49].
In stark contrast with the MR, it is unlikely that GR is activated at circadian and ultradian glucocorticoid troughs due to its low affinity for endogenous corticoids. This is one important feature of glucocorticoid signaling that alternates between cycles of MR followed by a MR/GR co-activation both of which are critical for cellular networks of the limbic system known to co-express both receptor subtypes (Fig. 2.1). For instance, glucocorticoid circadian peaks are as important as glucocorticoid troughs for learning and memory. If training at glucocorticoid peaks facilitates the acquisition of a motor coordination task, the glucocorticoid trough is paramount for its retention. In vivo imaging studies indicate that glucocorticoid peaks via GR enhance the formation of dendritic spines, the post-synaptic entity of excitatory synapses whereas the following troughs via MR permits the elimination of pre-existing old spines [4]. Overall, the coincidence of glucocorticoid naturally occurring circadian oscillations with procedural learning may favor the patterning of dendritic spines needed to adjust neural circuit wiring with behavioral demands.
Sub-cellular Distribution
The classic view is that both MR and GR reside in the cytoplasm of cells and shifts to their nucleus upon ligand binding for transcriptional regulation. Yet, unliganded GR can be found in the nucleus unbound to chromatin and yet, readily accessible to cortisol [50–52]. Sub-cellular distribution is actually controlled by the rates of import and export through the nuclear pores, the directionality being determined by glucocorticoid binding and recruitment of specific co-factors like importins, HSP90, FKBP52 and FKBP51. For instance, cytoplasmic location is favored in the absence of ligand, when GR is bound to a HSP90-FKBP51-based chaperone complex. This chaperone complex stabilizes the receptor, represses its regulatory activities, and favors a conformation that facilitates ligand binding [53]. Upon glucocorticoid binding, the receptor undergoes a conformational change and is released from the cytoplasmic chaperone-complex. Structural rearrangements elicited upon ligand binding notably expose nuclear localization signal sequences facilitating the binding to import proteins and active transport through the nuclear pores. Once in the nucleus, the interaction of MR and GR with DNA ligands and transcription factors specify the transcriptional targets involved in the glucocorticoid genomic signaling. The nucleus is not the only organelle where DNA ligands for the GR have been characterized [54]. Such discovery opens new avenue for understanding cell autonomous metabolic effects of glucocorticoids through the mitochondria [55] (Fig. 2.3). Since then, mitochondrial GR has been purified in brain extracts from mice subjected to chronic stress [56]. The functional role of mitochondrial GR is still in its infancy but experiments that forced GR localization to the mitochondria suggests it is highly toxic [57]. Beyond the dogmatic genomic effects, which are slow at onset (minutes to hours from the transcription to the bioactivity of regulated genes) exists rapid non-genomic effects that could rely upon the subcellular distribution of the receptors. One example is illustrated by the effects of glucocorticoids on neuronal excitability that cannot be accounted for genomic effects [58, 59], because they are not only faster than expected from a transcriptional response, but also resistant to protein synthesis inhibitors [58, 60].

Subcellular distribution of GR associates with distinct signaling outcomes. Neuronal GR is abundant in the nucleus, somatic cytoplasm and less abundant but detectable in the mitochondria and synaptic terminals like post-synaptic dendritic spines. (1) Ligand activated nuclear GR binds to DNA to induce or repress gene transcription, which target Y may also function as transcription factor to trigger a second wave of glucocorticoid response involving the gene Z. GR phosphorylation is tightly control by kinases and phosphatases that regulates the binding of biased signaling co-factors. (2) Ligand activated GR can translocate to the mitochondria to regulate the production of ATP and the release of cytochrome C known to endanger the survival prognosis of targeted cells. (3) Ligand-activated GR signaling from a membrane origin rapidly activates the LIMK1-Cofilin pathway that impinges on the turnover of the actin cytoskeleton. As a result, glucocorticoids rapidly enhance the formation of post-synaptic dendritic spines. (4) The postsynaptic membrane bound GR first facilitates neurotransmission by enhancing the transport of AMPA receptor subunits to the active zone of the synapse, and diminish neurotransmission through a pre-synaptic mechanism that requires the release of a retrograde messenger (endocannabinoids) that employs a G-protein coupled receptor (CB1) to suppress the release of the excitatory neurotransmitter glutamate
Other Receptors
Rapid glucocorticoid effects on neuronal excitability can be reproduced with a functional synthetic glucocorticoid conjugated with BSA that cannot penetrate the plasma membrane nor access the cytoplasmic and nuclear receptors. Such glucocorticoid signaling of membrane-bound origin has been replicated in numerous paradigms and frequently associated with a MR type of pharmacology because it can be abolished by a MR antagonist and does not occur in mice lacking MR [61]. Yet, in some tissues, the membrane-bound glucocorticoid receptor is also sensitive to GR antagonists and GR knockdown at least on dendritic spine formation [4]. Localization of MR and GR in close proximity to the plasma membrane notably at the synapse was evidenced by biochemical purification and electron microscopy [60, 62–65]. The existence of membrane-bound MR and GR has been functionally proved in other models [66, 67] (Fig. 2.3). Despite the evidence, the pharmacology of membrane-bound cortisol receptors supports the possibility that several transmembrane proteins coupled to small heterotrimeric G proteins and sensitive to MR and or GR could signal rapidly.
Associated Protein Complexes
Ligand-Independent Partners
Chaperone Complex
The unliganded MR and GR are permanently scanning the cellular environment thanks to a large complex of chaperone proteins that control the architecture of the glucocorticoid ligand binding domain. Numerous GR and MR binding partners have been characterized. Here, we present only a few examples. Invariably, HSP90 serves as scaffold for the ‘foldosome’ complex (HSP70, HSP40 and Hop) that molds the ligand-binding pocket of GR, in an ATP-dependent manner [53, 68, 69]. Binding of p23 by this heterocomplex, further stabilizes the conformational change [69]. The release of Hop from the complex leads to the dynamic and competitive addition of other co-chaperones like the immunophilins FKBP51, FKBP52 and cyp 40 and the immunophilin-like phosphatase PP5 that mediate changes in receptor mobility, ligand affinity and signaling capacity [70, 71]. For example, FKBP52 facilitates GR signaling activity by bringing together GR and motor proteins onto the microtubules for active nuclear transport [70, 72]. In contrast, binding of FKBP51 to the chaperone supercomplex attenuates glucocorticoid signaling by facilitating the nuclear export of GR. The robust induction of FKBP51 expression by glucocorticoid, unlike FKBP52, suggests that glucocorticoid signaling feedback can also be cell autonomous [73].
Ligand Dependent Partners
Numerous proteins have been functionally associated with the liganded MR and GR. By no means is it the goal of this chapter to enumerate them. Only a small selection of relevant partners will be emphasized, as it is dependent on post-translational modifications and subcellular distribution.
Post-translational Modifications
With low basal phosphorylation at rest, MR and GR get hyperphosphorylated upon ligand binding offering binding sites for the 14-3-3 family of proteins. Despite multiple phosphorylated serine or threonine residues identified in the N-terminal domain of GR [74], only a select number of sites have been functionally characterized. For instance, glucocorticoid-dependent phosphorylation at conserved serines 203 (S203), 211 (S211), 226 (S226), and 404 (S404) in the human GR numbering scheme impacts on transcriptional capabilities at specific target gene. Three of these sites demonstrate some interdependency upon one another [75]. All four sites exhibit low basal phosphorylation in the absence of cortisol as long as the protein phosphatase PP5 remains associated with GR and the HSP90 chaperone complex [76]. S203 phosphorylation, however, is characteristically higher without hormone compared to S211 and S226 sites. Upon glucocorticoid binding, the S203 phosphorylated form of the receptor appears perinuclear, suggesting that GR phosphorylated at S203 does not participate in DNA-bound GR transcriptional regulation [75]. Consistent with this idea, S203 phosphorylated GR does not occupy select GREs whereas S211 and S226 phospho-isoforms can bind to DNA ligands in the genome [77]. Classically, phosphorylation of GR at S211 serves as surrogate marker of ligand-activated GR because basal phosphorylation is null in absence of glucocorticoids and ligand-induced S211 phosphorylation correlates with the magnitude of GR transcriptional activity [77]. In mitotic cells, S211 phosphorylation depends on CDK2 whereas CDK5 or ERK may phosphorylate GR in postmitotic neurons [78, 79]. In contrast, phosphorylation at S226 by JNK serve as docking site for specific cofactors that increases nuclear export and reduces GR transcriptional activation [77, 80, 81]. S404, a substrate of GSK3β reduces GR transactivation by increasing the turnover of the liganded GR and by hindering GR-mediated NF-κB repression [82].
Other post-translational modifications are dependent on the phosphorylation code of GR. For example, JNK-dependent phosphorylation of S246 enhances sumoylation at K293 and K277, which are implicated in the control of GR transactivation at multiple GREs [83, 84]. Lastly, ligand-mediated GR degradation by the proteasome requires ubiquitination of K419, which is also dependent upon phosphorylation, as a phospho-defective GR mutant is resistant to ligand-induced degradation [84, 85]. Functionally, GR phosphorylation at rat serine 232 (analogous to S211 in humans), responds to neurotoxic insults to induce specifically the ectopic expression of hdac2 that correlates with the occurrence of cognitive defects [86]. GR phosphorylation at serine 232 also increases in the rodent brain after exposure to glucocorticoids or stress [87, 88]. The MR is also phosphorylated at rest and upon ligand binding, yet the functional characterization of individual sites is far less advanced [89]. Although the kinases have more or less been characterized for most sites, it is unclear how glucocorticoid signaling can activate kinases.
Nuclear Trafficking
It was long believed that nuclear translocation of GR necessitated the release from the chaperone complex [90]. The emerging picture is that the entire complex moves along the microtubule cytoskeleton depending on the recruitment of subtype specific immunophilins, importins and exportins [91, 92]. GR has two nuclear localization signals, NL1 and NL2. NL1 supports rapid and hormone independent translocation [93], whereas NL2 facilitates slower hormone dependent nuclear import [84, 94]. Several importins interact with GR NL regions and mediate its nuclear translocation across the nuclear pore complex. Importins 7 and 8 bind NL1 and NL2, whereas importin α/β bind exclusively to NL1 [94]. Evidence of importins and nuclear pore proteins binding with components of the chaperone complex shed light into the molecular mechanism of nuclear import. Importin β and nuclear pore glycoprotein Nup62 bind GR and HSP90, p23, and FKBP52 to accelerate nuclear import rate [92, 95]. Nuclear export is dependent on exportin/CRM1 as pharmacological blockade abolishes GR translocation to the cytosol [52]. Nuclear calcium levels also impacts upon GR nuclear residency via a direct interaction between the NES sequences and the calcium-sensing protein calreticulin and other related co-factors such as SRC-1 and 14-3-3σ [79, 84, 96–98].
DNA Binding
Despite a high degree of homology between MR and GR, common protein complexes, and similar post-translational modifications such as ubiquitination, sumoylation and phosphorylation, MR and GR affect distinct gene targets [99]. Yet, GR and MR can form heterodimers [100, 101], which may modulate transcription of a few target genes in a unique way that differ from either GR or MR homodimers. One described example is the serotonin receptor 5HT1A gene which transcription is repressed by GR and MR alone but GR/MR heterodimers exert an even stronger inhibition [102].
Zinc fingers of the DNA binding domain mediate of GR-DNA interaction that usually requires the receptor to dimerize with itself or other transcription factors. Yet, the GRdim mutant that cannot homodimerize is still capable of robust transcriptional regulation at multiple but not all targets-genes [43]. The DNA sequence also acts as an allosteric ligand that influences GR structure and activity [99]. Classically, GREs have been grouped into three classes: (i) simple GREs;(ii) composite GREs; (iii) and tethering elements [103]. Each class requires GR binding in different conformations and orientations as follows. Simple GREs are most often inverted, repeat hexameric sequences separated by three nucleotides, supporting the homodimerization model of GR. A few negative GREs (nGREs) that mediate GR-dependent repression have been well characterized [104]. The inverted repeat IR nGREs found in numerous glucocorticoid-repressed genes [105, 106] support a model whereby direct binding of GR as monomer is not always required but may instead involve the recruitment of specific co-factors like nuclear receptor corepressors 1 and 2 (N-CoR) and (SMRT) [105, 107]. Composite GREs contain non-GR binding sequences surrounding a core GR-binding site important for gene-specific synergistic or antagonistic regulation. GR may interact physically and/or functionally with the transcription factors associated with those motifs. An increasing number of mechanistic and genome-wide studies suggest that most functional transactivating GREs are composite elements, composed of binding sites for GR as well as additional DNA-binding regulatory factors that act in synergy with GR in the particular context of each GRE [108, 109]. Such composite elements might explain some of the cell-type specific regulation by GR and is discussed in additional detail in the following Chapter.
Transcription Cofactors
The typical inverted U-shape responses to increasing dose of glucocorticoids could be in part explained by “squelching” or titration of a limiting co-factor required for full response by the activated GR [110–112]. At high concentrations of activated GR, a factor becomes limiting for full GR activity and as such, transcriptional activation is reduced. Thus, the squelching model emphasizes the availability of transcription co-factors. In the case of tethering GREs, it is the DNA bound co-factor of MR and GR that specifies the glucocorticoid-targets genes. Such factors are AP-1, CREB, NF-κB, or STAT5, C/EBP, SP-1, Egr-1 and others less characterized. Most tethering GREs do not contain canonical sequence motifs for GR, but rather contain motifs for the interacting transcription factor that may also mediate induction of target genes in the absence of glucocorticoids. For example, tethering of GR to AP-1 or NF-κB alters the assembly of coactivator complexes and recruits the corepressor glucocorticoid receptor interacting protein 1 (GRIP1) [113, 114]. In the case of direct GR-DNA binding, the transcriptional output depends on recruitment of a distinct class of co-factors that include P300/CBP, HDAC2, p160, MED1,14, and SWI/SNF complex. These factors are thought to affect chromatin structure or the stability of transcriptional machinery at the transcription start site. Coactivator complexes are assembled through interactions with GR’s AF-1 or more commonly the AF-2 domain [115]. The p160 family of coactivators include SRC-1, SRC-2/GRIP1/transcriptional intermediary factor 2 (TIF2), and SRC-3 (also known as pCIP/ACTR/AIB1/RAC3). P160 proteins often increase GR transcriptional activity, but GRIP1 was shown to act both as a coactivator and corepressor via intrinsic activation and repression domains [113, 116]. GRIP1 specifically utilizes its repression domain at AP-1 and NF-κB tethering GREs, whereas SRC-1 and SRC-2 lack this domain and fail to assist repression at AP-1 [82]. Many p160 proteins facilitate transcriptional activation by recruiting p300/CBP. P300/CBP contains potent acetyltransferase activity that target histones and other proteins. GR has also been shown to interact with a number of histone deacetylases (HDACs) including: HDAC 1 and 6 as coactivators [117–119] and HDAC2 as both a coactivator and corepressor, and its recruitment depend on S211 phosphorylation [82, 86, 119, 120]. Through its AF1 and AF2 domains GR interacts with MED14 and MED1 two components of the mediator complex [121]. The mediator complex is a large multiunit complex that bridges transcription factors and RNA pol II, affecting transcription initiation [122] and elongation [123]. The assembly of select mediator subunits alters promoter responsiveness to various transcription factors, so distinct subunits and confirmations are important for GR transcriptional activity. In cell-based reporter assays, overexpression of MED14 enhances GR transcriptional activity, while MED1 only enhanced GR activity in the context of MED14 [124].
Signaling
A large number of well-documented rapid and slow cellular responses to glucocorticoids contribute to glucocorticoid-mediated allostasis. It is remarkable that slow effects can either oppose or reinforce the rapid effects. In this section, we will describe a few examples illustrating how pertinent the spatial and temporal resolution of glucocorticoid signaling is to adjust neuronal networks to changing environments.
Minutes
Rapid glucocorticoid signaling has been particularly studied in the context of neuronal excitability within the range of seconds to minutes. For instance, the excitability of dorsal hippocampal CA1 neurons is sensitive to glucocorticoids within minutes of exposure as demonstrated by an increase in the frequency of spontaneous excitatory neurotransmission (Fig. 2.3). Pharmacological and genetic proof of concept studies revealed the contribution of the membrane-bound MR [125, 126]. Pharmacological characterization of downstream signaling revealed the requirement of the ERK1/2 pathway [126]. An increase in frequency of spontaneous excitatory neurotransmission usually results from an increased quantal release of the neurotransmitter glutamate, and extracellular glutamate levels are augmented following acute glucocorticoid treatment. As much as glucocorticoids affect presynaptic neurotransmitters release, rapid glucocorticoid effects on the post-synaptic neuronal terminal were also characterized. Indeed, glucocorticoids through the membrane-bound MR rapidly (few minutes) increase the mobility and dwell time at the post-synaptic density of glutamate receptors GluR2-AMPAR [127] (Fig. 2.3). Such effects are usually interpreted as enhancing neurotransmission. Similar observations were reported in the basoloateral amygdala (BLA) that yet, expresses much lower MR levels than the hippocampus [66]. Interestingly, the rapid increase of neuronal excitability by the membrane-bound MR is context dependent as it varied with a history of glucocorticoid exposure or stress [66]. That is, a second pulse stimulation of brain slice preparations with corticosterone after washout of a first pulse decreased the excitability of BLA neurons that a single pulse could increase [21, 66]. This suggests that time at exposure with glucocorticoids also determines the signaling outcome. This picture is not the rule because glucocorticoids cause a rapid (few minutes) suppression of excitatory synaptic inputs in hypothalamic CRH- and AVP-releasing neurons of the paraventricular nucleus (PVN) [58, 128]. Physiologically, this effect contributes to one of the rapid components of the negative feedback that deactivates the HPA axis. In this paradigm, a membrane-bound GR pathway was highlighted using a synthetic membrane impermeant GR agonist that curiously cannot be abolished by a specific GR antagonist, suggesting the involvement of a membrane bound GR with non-canonical pharmacological features [58]. Suppression of PVN neuronal excitation employs endocannabinoids released from the post-synaptic membrane and signaling through the CB1 receptor at the pre-synaptic terminals to suppress glutamate release [58, 129, 130]. Given this signaling pathway involves small heterotrimeric G protein, this finding reconciles previous data indicating that glucocorticoid signaling of membrane origin is sensitive to antagonists of MR, GR and G proteins [58, 129]. Finally, glucocorticoids enhance dendritic spine formation in the living cortex within minutes of exposure via GR-mediated activation of a LIMK-cofilin pathway that impinges on the dynamics of the post-synaptic actin cytoskeleton [4] (Fig. 2.3).
Hours
While the rapid glucocorticoid signaling serves immediate purposes like the deactivation of the HPA axis, slow glucocorticoid signaling is classically viewed as an adaptive response of cellular networks to changing environments like the suppression of CRH transcription to maintain HPA axis homeostasis [131]. Both the rapid and slow signaling components often studied separately shall be considered as integrated response overtime. For instance, it is intriguing that only slow effects of GR and MR phosphorylation on transcriptional activity have been studied given that phosphorylation occurs within minutes of stimulation with cortisol. In the hippocampus, GR mediated genomic effects (transcription and RNA decay) follow the rapid increase of hippocampal excitability mediated by MR in order to reduce neuronal excitability by changing the expression levels of signaling molecules and ion channels like the L type calcium channel [21, 132]. Such a short-term increase of hippocampal neurons excitability is viewed as means to consolidate stress-related memory whereas the following decrease in excitability is viewed as means to protect hippocampal cell networks from noise information [21]. This picture is not the rule because glucocorticoids increase excitability in the BLA that persists after drug washout and is sensitive to protein synthesis inhibitors, GR inhibitors, and absent in GR knockout mice, pointing towards a genomic GR-dependent mechanism [66, 133].
Cell-type specific GR-dependent genomic effects are well-illustrated by the transcription of crh, the major molecular trigger of the HPA axis that is increased in the amygdala but decreased in the hypothalamic PVN as a function of elevated levels of glucocorticoids [134]. In the cortex, learning-dependent weaving of neural networks relies on the formation and elimination of synaptic connectivity as a function of glucocorticoid levels. At glucocorticoid circadian trough when only MR is activated, elimination of dendritic spines compensate for the increased GR-dependent spine formation that occurred during the glucocorticoid circadian peak [4]. This result is interpreted as if learning associated new spines are offset by the elimination of pre-existing old spines within distinct temporal domains to shape neural circuits as a function of novelty.
Context-Dependent Glucocorticoid Signaling
Glucocorticoid-mediated allostasis employs genomic and non-genomic mechanisms together with ongoing, experience-driven neural activity mediated by excitatory amino acids neurotransmitters, neurotrophic factors such as BDNF, neuropeptides such as CRH and cell adhesion molecules (NCAM) [135, 136]. Therefore, it is particularly interesting to discuss how glucocorticoid activity superimposes with other signaling pathways. For instance, the enhancement of emotionally arousing memories by glucocorticoids requires co-incident norepinephrine signaling in the amygdala [137]. Norepinephrine is capable of potentiating ligand-induced GR transcriptional capabilities and DNA binding in a PI3K-dependent manner [138]. Adrenergic receptor agonists, generally used in the treatment of asthma, synergistically enhance GR-dependent transcription in a cAMP/PKA-dependent manner. Thus, maximal GR transcriptional capabilities are achieved approximately at tenfold lower glucocorticoid concentrations in the presence of cAMP-elevating drugs [139]. Another set of factors are proinflammatory cytokines, such as interleukin IL1, IL-2, IL-6, TNF-a, and interferon (IFN)-a, signaling pathways that alter GR signaling and neuroendocrine function [140, 141]. Co-incident growth factor signaling may also intersect with GR actions. For instance, bFGF and IGF1 can enhance ligand-induced GR transcriptional capabilities in a PI3K-dependent manner [138]. The neurotrophin BDNF through its receptor TrkB can also specify the transcription of select GR-mediated genes [142] that may be relevant for glucocorticoid-induced memory consolidation of fear and inhibitory avoidance [143]. Mechanistically, BDNF signaling results in phosphorylation of rat GR at serines 134 and 267, which fosters cofactor recruitment to promote a novel gene expression signature. Thus, BDNF utilizes GR as a transcription factor to alter glucocorticoid-regulated transcription in neurons [142] (Fig. 2.4). Similarly, GR phosphorylation at rat serine 232 responds to neurotoxic insults to induce specifically the ectopic expression of hdac2 that correlates with the occurrence of cognitive defects [86]. Pharmacological blockade of HDAC2 activity resolved cognitive impairments produced by the prolonged activation of GR in a model of Alzheimer’s disease featuring chronic high glucocorticoid levels known to worsen neuropathological features [86, 144, 145]. In contrast, the expression of HDAC2 is downregulated in glucocorticoid-resistant cases of severe asthma and pharmacological activation of HDAC2 with theophiline and antioxidants present therapeutic value [146, 147]. Glucocorticoid activity may also diverge as a function of estrogen signaling, which increases the expression of the GR phosphatase-PP5 [148]. The opposing effect of cortisol on crh expression in the hypothalamus and the amygdala is another good example of context-dependent glucocorticoid signaling [134]. It is the CREB co-activator CRTC2 that determines the ability of neurotrophins and glucocorticoids to activate or suppress hypothalamic CRH expression, respectively [112, 149] (Fig. 2.5).

Glucocorticoid transcriptional effects diverge as a function of BDNF signaling. On top of glucocorticoid-mediated GR phosphorylation exists a parallel converging pathway that allows for site-specific GR phosphorylation by the neurotrophic factor BDNF and its receptor TrkB. The resulting hyperphosphorylated GR can activate or repress new select target genes enriched with the indicated transcription factors binding sites, as well as potentiate the expression of GR-sensitive genes. Thus, BDNF-induced GR phosphorylation rewrites the GR transcriptome toward a cellular network signature by fostering the recruitment of phospho-specific cofactors

Glucocorticoids employ GR-dependent mechanisms to enhance the formation and the elimination of post-synaptic dendritic spines within distinct temporal domains. Rapid transcription-independent GR signaling facilitates the formation of dendritic spines associated with learning capabilities whereas the slow genomic GR/MR signaling accounts for the stabilization as well as the elimination of pre-existing old spines, a process that is critical for memory retention. The patterning of dendritic spines through the processes of formation and elimination is critical to adjust neural connectivity networks in changing environments
Conclusions
One paramount feature of the body’s allostasis resides in the flexibility of the naturally occurring glucocorticoid rhythms and signaling to changing environments. For instance, glucocorticoid-mediated allostasis is critical for behavioral adaptation to novelty, stress coping, learning and memory. Disruption of glucocorticoid circadian and ultradian rhythms is a hallmark of numerous diseases notably neuropsychiatric. In contrast, administration of glucocorticoids, which are mainstay of treatment for numerous disorders, can produce side effects related to brain functions, like psychosis, depression and memory loss. One hypothesis to account for these effects is that glucocorticoid resistance whether innate or acquired increases the vulnerability to neurotoxic insults that slowly contribute to the development of numerous disorders of the nervous and immune systems. Two examples: glucocorticoids do not cause but worsen the neuropathological feature of Alzheimer’s disease and glucocorticoid resistance that interferes with inflammation, increases the sensitivity to develop a common cold. Several studies suggest that the growth/survival MAPK pathway regulates glucocorticoid signaling because diseases featuring glucocorticoid resistance also exhibit disrupted MAPK activity like asthma and depression. Evolution may have selected the MAPK pathway to cope with the allostatic overload of stress and form new memories. One putative mechanism that shows great promises is that MAPKs modulate glucocorticoid activities in part through the phosphorylation of the glucocorticoid receptors. Indeed, MAPK-mediated modulation of GR function appears to be a central player in the development of glucocorticoid resistance. Future functional characterization of the GR and MR phosphorylation codes could help comprehend how glucocorticoid actions can be changed from harmful to protective.
References
McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. 2003;43(1):2–15.
de Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269–301.
Compagnone NA, Mellon SH. Neurosteroids: biosynthesis and function of these novel neuromodulators. Front Neuroendocrinol. 2000;21(1):1–56.
Liston C, Cichon JM, Jeanneteau F, Jia Z, Chao MV, Gan WB. Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat Neurosci. 2013;16(6):698–705.
Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5(1):1–24.
Lightman SL, Conway-Campbell BL. The crucial role of pulsatile activity of the HPA axis for continuous dynamic equilibration. Nat Rev Neurosci. 2010;11(10):710–8.
Kalsbeek A, Buijs RM. Output pathways of the mammalian suprachiasmatic nucleus: coding circadian time by transmitter selection and specific targeting. Cell Tissue Res. 2002;309(1):109–18.
de Kloet ER, Sarabdjitsingh RA. Everything has rhythm: focus on glucocorticoid pulsatility. Endocrinology. 2008;149(7):3241–3.
Cook CJ. Measuring of extracellular cortisol and corticotropin-releasing hormone in the amygdala using immunosensor coupled microdialysis. J Neurosci Methods. 2001;110(1–2):95–101.
Jasper MS, Engeland WC. Synchronous ultradian rhythms in adrenocortical secretion detected by microdialysis in awake rats. Am J Physiol. 1991;261(5 Pt 2):R1257–68.
Walker JJ, Terry JR, Lightman SL. Origin of ultradian pulsatility in the hypothalamic-pituitary-adrenal axis. Proc Biol Sci. 2010;277(1688):1627–33.
Papavasiliou SS, Zmeili S, Khoury S, Landefeld TD, Chin WW, Marshall JC. Gonadotropin-releasing hormone differentially regulates expression of the genes for luteinizing hormone alpha and beta subunits in male rats. Proc Natl Acad Sci U S A. 1986;83(11):4026–9.
Wildt L, Hausler A, Marshall G, et al. Frequency and amplitude of gonadotropin-releasing hormone stimulation and gonadotropin secretion in the rhesus monkey. Endocrinology. 1981;109(2):376–85.
Gregus A, Wintink AJ, Davis AC, Kalynchuk LE. Effect of repeated corticosterone injections and restraint stress on anxiety and depression-like behavior in male rats. Behav Brain Res. 2005;156(1):105–14.
Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10(6):397–409.
Holsboer F, Ising M. Stress hormone regulation: biological role and translation into therapy. Annu Rev Psychol. 2010;61:81–109.
Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49(5):391–404.
Rosner W. The functions of corticosteroid-binding globulin and sex hormone-binding globulin: recent advances. Endocr Rev. 1990;11(1):80–91.
Cameron A, Henley D, Carrell R, Zhou A, Clarke A, Lightman S. Temperature-responsive release of cortisol from its binding globulin: a protein thermocouple. J Clin Endocrinol Metab. 2010;95(10):4689–95.
Klieber MA, Underhill C, Hammond GL, Muller YA. Corticosteroid-binding globulin, a structural basis for steroid transport and proteinase-triggered release. J Biol Chem. 2007;282(40):29594–603.
Joels M, Sarabdjitsingh RA, Karst H. Unraveling the time domains of corticosteroid hormone influences on brain activity: rapid, slow, and chronic modes. Pharmacol Rev. 2012;64(4):901–38.
Wyrwoll CS, Holmes MC, Seckl JR. 11beta-hydroxysteroid dehydrogenases and the brain: from zero to hero, a decade of progress. Front Neuroendocrinol. 2011;32(3):265–86.
Yau JL, Noble J, Kenyon CJ, et al. Lack of tissue glucocorticoid reactivation in 11beta-hydroxysteroid dehydrogenase type 1 knockout mice ameliorates age-related learning impairments. Proc Natl Acad Sci U S A. 2001;98(8):4716–21.
Seckl JR, Walker BR. 11beta-hydroxysteroid dehydrogenase type 1 as a modulator of glucocorticoid action: from metabolism to memory. Trends Endocrinol Metab. 2004;15(9):418–24.
Pariante CM. The role of multi-drug resistance p-glycoprotein in glucocorticoid function: studies in animals and relevance in humans. Eur J Pharmacol. 2008;583(2–3):263–71.
Volk HA, Burkhardt K, Potschka H, Chen J, Becker A, Loscher W. Neuronal expression of the drug efflux transporter P-glycoprotein in the rat hippocampus after limbic seizures. Neuroscience. 2004;123(3):751–9.
Akers KG, Nakazawa M, Romeo RD, Connor JA, McEwen BS, Tang AC. Early life modulators and predictors of adult synaptic plasticity. Eur J Neurosci. 2006;24(2):547–54.
Kumar R, Thompson EB. The structure of the nuclear hormone receptors. Steroids. 1999;64(5):310–9.
Bodwell JE, Webster JC, Jewell CM, Cidlowski JA, Hu JM, Munck A. Glucocorticoid receptor phosphorylation: overview, function and cell cycle-dependence. J Steroid Biochem Mol Biol. 1998;65(1–6):91–9.
Ismaili N, Garabedian MJ. Modulation of glucocorticoid receptor function via phosphorylation. Ann N Y Acad Sci. 2004;1024:86–101.
Zennaro MC, Keightley MC, Kotelevtsev Y, Conway GS, Soubrier F, Fuller PJ. Human mineralocorticoid receptor genomic structure and identification of expressed isoforms. J Biol Chem. 1995;270(36):21016–20.
Martinerie L, Munier M, Le Menuet D, Meduri G, Viengchareun S, Lombes M. The mineralocorticoid signaling pathway throughout development: expression, regulation and pathophysiological implications. Biochimie. 2013;95(2):148–57.
Van Eekelen JA, Jiang W, De Kloet ER, Bohn MC. Distribution of the mineralocorticoid and the glucocorticoid receptor mRNAs in the rat hippocampus. J Neurosci Res. 1988;21(1):88–94.
Gesing A, Bilang-Bleuel A, Droste SK, Linthorst AC, Holsboer F, Reul JM. Psychological stress increases hippocampal mineralocorticoid receptor levels: involvement of corticotropin-releasing hormone. J Neurosci. 2001;21(13):4822–9.
Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117(6):2505–11.
Reul JM, Gesing A, Droste S, et al. The brain mineralocorticoid receptor: greedy for ligand, mysterious in function. Eur J Pharmacol. 2000;405(1-3):235–49.
Jacobson L, Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev. 1991;12(2):118–34.
Ratka A, Sutanto W, Bloemers M, de Kloet ER. On the role of brain mineralocorticoid (type I) and glucocorticoid (type II) receptors in neuroendocrine regulation. Neuroendocrinology. 1989;50(2):117–23.
Cole MA, Kalman BA, Pace TW, Topczewski F, Lowrey MJ, Spencer RL. Selective blockade of the mineralocorticoid receptor impairs hypothalamic-pituitary-adrenal axis expression of habituation. J Neuroendocrinol. 2000;12(10):1034–42.
Woolley CS, Gould E, Sakai RR, Spencer RL, McEwen BS. Effects of aldosterone or RU28362 treatment on adrenalectomy-induced cell death in the dentate gyrus of the adult rat. Brain Res. 1991;554(1–2):312–5.
Fischer AK, von Rosenstiel P, Fuchs E, Goula D, Almeida OF, Czeh B. The prototypic mineralocorticoid receptor agonist aldosterone influences neurogenesis in the dentate gyrus of the adrenalectomized rat. Brain Res. 2002;947(2):290–3.
Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol. 2006;102(1–5):11–21.
Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34(9):518–30.
Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. Human glucocorticoid receptor beta binds RU-486 and is transcriptionally active. Mol Cell Biol. 2007;27(6):2266–82.
Kellendonk C, Eiden S, Kretz O, et al. Inactivation of the GR in the nervous system affects energy accumulation. Endocrinology. 2002;143(6):2333–40.
Tronche F, Kellendonk C, Kretz O, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23(1):99–103.
Howell MP, Muglia LJ. Effects of genetically altered brain glucocorticoid receptor action on behavior and adrenal axis regulation in mice. Front Neuroendocrinol. 2006;27(3):275–84.
Kolber BJ, Roberts MS, Howell MP, Wozniak DF, Sands MS, Muglia LJ. Central amygdala glucocorticoid receptor action promotes fear-associated CRH activation and conditioning. Proc Natl Acad Sci U S A. 2008;105(33):12004–9.
Schmidt MV, Sterlemann V, Wagner K, et al. Postnatal glucocorticoid excess due to pituitary glucocorticoid receptor deficiency: differential short- and long-term consequences. Endocrinology. 2009;150(6):2709–16.
Nishi M, Ogawa H, Ito T, Matsuda KI, Kawata M. Dynamic changes in subcellular localization of mineralocorticoid receptor in living cells: in comparison with glucocorticoid receptor using dual-color labeling with green fluorescent protein spectral variants. Mol Endocrinol. 2001;15(7):1077–92.
Piwien Pilipuk G, Vinson GP, Sanchez CG, Galigniana MD. Evidence for NL1-independent nuclear translocation of the mineralocorticoid receptor. Biochemistry. 2007;46(5):1389–97.
Hache RJ, Tse R, Reich T, Savory JG, Lefebvre YA. Nucleocytoplasmic trafficking of steroid-free glucocorticoid receptor. J Biol Chem. 1999;274(3):1432–9.
Dittmar KD, Banach M, Galigniana MD, Pratt WB. The role of DnaJ-like proteins in glucocorticoid receptor.hsp90 heterocomplex assembly by the reconstituted hsp90.p60.hsp70 foldosome complex. J Biol Chem. 1998;273(13):7358–66.
Lee SR, Kim HK, Song IS, et al. Glucocorticoids and their receptors: insights into specific roles in mitochondria. Prog Biophys Mol Biol. 2013;112(1–2):44–54.
Du J, Wang Y, Hunter R, et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci U S A. 2009;106(9):3543–8.
Adzic M, Lukic I, Mitic M, et al. Brain region- and sex-specific modulation of mitochondrial glucocorticoid receptor phosphorylation in fluoxetine treated stressed rats: effects on energy metabolism. Psychoneuroendocrinology. 2013;38(12):2914–24.
Sionov RV, Cohen O, Kfir S, Zilberman Y, Yefenof E. Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. J Exp Med. 2006;203(1):189–201.
Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23(12):4850–7.
Di S, Maxson MM, Franco A, Tasker JG. Glucocorticoids regulate glutamate and GABA synapse-specific retrograde transmission via divergent nongenomic signaling pathways. J Neurosci. 2009;29(2):393–401.
Groeneweg FL, Karst H, de Kloet ER, Joels M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol. 2012;350(2):299–309.
Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102(52):19204–7.
Qiu S, Champagne DL, Peters M, et al. Loss of limbic system-associated membrane protein leads to reduced hippocampal mineralocorticoid receptor expression, impaired synaptic plasticity, and spatial memory deficit. Biol Psychiatry. 2010;68(2):197–204.
Prager EM, Brielmaier J, Bergstrom HC, McGuire J, Johnson LR. Localization of mineralocorticoid receptors at mammalian synapses. PLoS One. 2010;5(12), e14344.
Wang CC, Wang SJ. Modulation of presynaptic glucocorticoid receptors on glutamate release from rat hippocampal nerve terminals. Synapse. 2009;63(9):745–51.
Johnson LR, Farb C, Morrison JH, McEwen BS, LeDoux JE. Localization of glucocorticoid receptors at postsynaptic membranes in the lateral amygdala. Neuroscience. 2005;136(1):289–99.
Karst H, Berger S, Erdmann G, Schutz G, Joels M. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci U S A. 2010;107(32):14449–54.
Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 2010;151(10):4811–9.
Dittmar KD, Demady DR, Stancato LF, Krishna P, Pratt WB. Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery. J Biol Chem. 1997;272(34):21213–20.
Stancato LF, Silverstein AM, Gitler C, Groner B, Pratt WB. Use of the thiol-specific derivatizing agent N-iodoacetyl-3-[125I]iodotyrosine to demonstrate conformational differences between the unbound and hsp90-bound glucocorticoid receptor hormone binding domain. J Biol Chem. 1996;271(15):8831–6.
Pratt WB, Silverstein AM, Galigniana MD. A model for the cytoplasmic trafficking of signalling proteins involving the hsp90-binding immunophilins and p50cdc37. Cell Signal. 1999;11(12):839–51.
Chen S, Smith DF. Hop as an adaptor in the heat shock protein 70 (Hsp70) and hsp90 chaperone machinery. J Biol Chem. 1998;273(52):35194–200.
Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18(3):306–60.
Jaaskelainen T, Makkonen H, Palvimo JJ. Steroid up-regulation of FKBP51 and its role in hormone signaling. Curr Opin Pharmacol. 2011;11(4):326–31.
Miller ML, Jensen LJ, Diella F, et al. Linear motif atlas for phosphorylation-dependent signaling. Sci Signal. 2008;1(35):ra2.
Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem. 2002;277(29):26573–80.
Wang Z, Chen W, Kono E, Dang T, Garabedian MJ. Modulation of glucocorticoid receptor phosphorylation and transcriptional activity by a C-terminal-associated protein phosphatase. Mol Endocrinol. 2007;21(3):625–34.
Chen W, Dang T, Blind RD, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22(8):1754–66.
Galliher-Beckley AJ, Cidlowski JA. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 2009;61(10):979–86.
Kino T, Ichijo T, Amin ND, et al. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol. 2007;21(7):1552–68.
Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol. 2002;16(10):2382–92.
Avenant C, Kotitschke A, Hapgood JP. Glucocorticoid receptor phosphorylation modulates transcription efficacy through GRIP-1 recruitment. Biochemistry. 2010;49(5):972–85.
Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3β-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. 2008;28(24):7309–22.
Davies L, Karthikeyan N, Lynch JT, et al. Cross talk of signaling pathways in the regulation of the glucocorticoid receptor function. Mol Endocrinol. 2008;22(6):1331–44.
Beck IM, De Bosscher K, Haegeman G. Glucocorticoid receptor mutants: man-made tools for functional research. Trends Endocrinol Metab. 2011;22(8):295–310.
Webster JC, Jewell CM, Bodwell JE, Munck A, Sar M, Cidlowski JA. Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J Biol Chem. 1997;272(14):9287–93.
Graff J, Rei D, Guan JS, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483(7388):222–6.
Adzic M, Djordjevic J, Djordjevic A, et al. Acute or chronic stress induce cell compartment-specific phosphorylation of glucocorticoid receptor and alter its transcriptional activity in Wistar rat brain. J Endocrinol. 2009;202(1):87–97.
Jeanneteau F, Garabedian MJ, Chao MV. Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc Natl Acad Sci U S A. 2008;105(12):4862–7.
Faresse N, Vitagliano JJ, Staub O. Differential ubiquitylation of the mineralocorticoid receptor is regulated by phosphorylation. FASEB J. 2012;26(10):4373–82.
Denis M, Poellinger L, Wikstom AC, Gustafsson JA. Requirement of hormone for thermal conversion of the glucocorticoid receptor to a DNA-binding state. Nature. 1988;333(6174):686–8.
Davies TH, Ning YM, Sanchez ER. A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem. 2002;277(7):4597–600.
Vandevyver S, Dejager L, Libert C. On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic. 2012;13(3):364–74.
Savory JG, Hsu B, Laquian IR, et al. Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol Cell Biol. 1999;19(2):1025–37.
Freedman ND, Yamamoto KR. Importin 7 and importin alpha/importin beta are nuclear import receptors for the glucocorticoid receptor. Mol Biol Cell. 2004;15(5):2276–86.
Echeverria PC, Mazaira G, Erlejman A, Gomez-Sanchez C, Piwien Pilipuk G, Galigniana MD. Nuclear import of the glucocorticoid receptor-hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin beta. Mol Cell Biol. 2009;29(17):4788–97.
Olkku A, Mahonen A. Calreticulin mediated glucocorticoid receptor export is involved in beta-catenin translocation and Wnt signalling inhibition in human osteoblastic cells. Bone. 2009;44(4):555–65.
Holaska JM, Black BE, Rastinejad F, Paschal BM. Ca2+-dependent nuclear export mediated by calreticulin. Mol Cell Biol. 2002;22(17):6286–97.
Amazit L, Alj Y, Tyagi RK, et al. Subcellular localization and mechanisms of nucleocytoplasmic trafficking of steroid receptor coactivator-1. J Biol Chem. 2003;278(34):32195–203.
Datson NA, van der Perk J, de Kloet ER, Vreugdenhil E. Identification of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression. Eur J Neurosci. 2001;14(4):675–89.
Liu W, Wang J, Sauter NK, Pearce D. Steroid receptor heterodimerization demonstrated in vitro and in vivo. Proc Natl Acad Sci U S A. 1995;92(26):12480–4.
Nishi M, Tanaka M, Matsuda K, Sunaguchi M, Kawata M. Visualization of glucocorticoid receptor and mineralocorticoid receptor interactions in living cells with GFP-based fluorescence resonance energy transfer. J Neurosci. 2004;24(21):4918–27.
Ou XM, Storring JM, Kushwaha N, Albert PR. Heterodimerization of mineralocorticoid and glucocorticoid receptors at a novel negative response element of the 5-HT1A receptor gene. J Biol Chem. 2001;276(17):14299–307.
Newton R, Holden NS. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol. 2007;72(4):799–809.
Sakai DD, Helms S, Carlstedt-Duke J, Gustafsson JA, Rottman FM, Yamamoto KR. Hormone-mediated repression: a negative glucocorticoid response element from the bovine prolactin gene. Genes Dev. 1988;2(9):1144–54.
Surjit M, Ganti Krishna P, Mukherji A, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145(2):224–41.
Reddy TE, Pauli F, Sprouse RO, et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19(12):2163–71.
Hudson WH, Youn C, Ortlund EA. The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol. 2013;20(1):53–8.
So AY-L, Chaivorapol C, Bolton EC, Li H, Yamamoto KR. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet. 2007;3(6), e94.
Reddy TE, Gertz J, Crawford GE, Garabedian MJ, Myers RM. The hypersensitive glucocorticoid response specifically regulates period 1 and expression of circadian genes. Mol Cell Biol. 2012;32(18):3756–67.
Wright AP, Gustafsson JA. Mechanism of synergistic transcriptional transactivation by the human glucocorticoid receptor. Proc Natl Acad Sci U S A. 1991;88(19):8283–7.
Lefstin JA, Thomas JR, Yamamoto KR. Influence of a steroid receptor DNA-binding domain on transcriptional regulatory functions. Genes Dev. 1994;8(23):2842–56.
Jeanneteau FD, Lambert WM, Ismaili N, et al. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. Proc Natl Acad Sci U S A. 2012;109(4):1305–10.
Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR. Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci. 2002;99(26):16701–6.
Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10(5):365–76.
McInerney EM, Rose DW, Flynn SE, et al. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12(21):3357–68.
Chinenov Y, Gupte R, Dobrovolna J, et al. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc Natl Acad Sci. 2012;109(29):11776–81.
Espallergues J, Teegarden SL, Veerakumar A, et al. HDAC6 regulates glucocorticoid receptor signaling in serotonin pathways with critical impact on stress resilience. J Neurosci. 2012;32(13):4400–16.
Qiu Y, Zhao Y, Becker M, et al. HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Mol Cell. 2006;22(5):669–79.
Govindan MV. Recruitment of cAMP-response element-binding protein and histone deacetylase has opposite effects on glucocorticoid receptor gene transcription. J Biol Chem. 2010;285(7):4489–510.
L-b L, Leung DYM, Martin RJ, Goleva E. Inhibition of histone deacetylase 2 expression by elevated glucocorticoid receptor β in steroid-resistant asthma. Am J Respir Crit Care Med. 2010;182(7):877–83.
Hittelman AB, Burakov D, Iniguez-Lluhi JA, Freedman LP, Garabedian MJ. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J. 1999;18(19):5380–8.
Malik S, Roeder RG. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet. 2010;11(11):761–72.
Takahashi H, Parmely TJ, Sato S, et al. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell. 2011;146(1):92–104.
Chen W, Rogatsky I, Garabedian MJ. MED14 and MED1 differentially regulate target-specific gene activation by the glucocorticoid Receptor. Mol Endocrinol. 2006;20(3):560–72.
Karst H, Joels M. Corticosterone slowly enhances miniature excitatory postsynaptic current amplitude in mice CA1 hippocampal cells. J Neurophysiol. 2005;94(5):3479–86.
Olijslagers JE, de Kloet ER, Elgersma Y, van Woerden GM, Joels M, Karst H. Rapid changes in hippocampal CA1 pyramidal cell function via pre- as well as postsynaptic membrane mineralocorticoid receptors. Eur J Neurosci. 2008;27(10):2542–50.
Groc L, Choquet D, Chaouloff F. The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nat Neurosci. 2008;11(8):868–70.
Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress. 2011;14(4):398–406.
Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, Tasker JG. Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and gamma-aminobutyric acid inputs to hypothalamic magnocellular neurons. Endocrinology. 2005;146(10):4292–301.
Malcher-Lopes R, Di S, Marcheselli VS, et al. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26(24):6643–50.
Watts AG. Glucocorticoid regulation of peptide genes in neuroendocrine CRH neurons: a complexity beyond negative feedback. Front Neuroendocrinol. 2005;26(3–4):109–30.
Karst H, Karten YJ, Reichardt HM, de Kloet ER, Schutz G, Joels M. Corticosteroid actions in hippocampus require DNA binding of glucocorticoid receptor homodimers. Nat Neurosci. 2000;3(10):977–8.
Duvarci S, Pare D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J Neurosci. 2007;27(16):4482–91.
Makino S, Gold PW, Schulkin J. Corticosterone effects on corticotropin-releasing hormone mRNA in the central nucleus of the amygdala and the parvocellular region of the paraventricular nucleus of the hypothalamus. Brain Res. 1994;640(1–2):105–12.
McEwen BS. The ever-changing brain: cellular and molecular mechanisms for the effects of stressful experiences. Dev Neurobiol. 2011;72(6):878–90.
Prager EM, Johnson LR. Stress at the synapse: signal transduction mechanisms of adrenal steroids at neuronal membranes. Sci Signal. 2009;2(86):5.
Roozendaal B, Okuda S, Van der Zee EA, McGaugh JL. Glucocorticoid enhancement of memory requires arousal-induced noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci U S A. 2006;103(17):6741–6.
Schmidt P, Holsboer F, Spengler D. Beta(2)-adrenergic receptors potentiate glucocorticoid receptor transactivation via G protein beta gamma-subunits and the phosphoinositide 3-kinase pathway. Mol Endocrinol. 2001;15(4):553–64.
Kaur M, Chivers JE, Giembycz MA, Newton R. Long-acting beta2-adrenoceptor agonists synergistically enhance glucocorticoid-dependent transcription in human airway epithelial and smooth muscle cells. Mol Pharmacol. 2008;73(1):203–14.
Miller AH, Pariante CM, Pearce BD. Effects of cytokines on glucocorticoid receptor expression and function. Glucocorticoid resistance and relevance to depression. Adv Exp Med Biol. 1999;461:107–16.
Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24–31.
Lambert WM, Xu CF, Neubert TA, Chao MV, Garabedian MJ, Jeanneteau FD. Brain-derived neurotrophic factor signaling rewrites the glucocorticoid transcriptome via glucocorticoid receptor phosphorylation. Mol Cell Biol. 2013;33(18):3700–14.
Chen DY, Bambah-Mukku D, Pollonini G, Alberini CM. Glucocorticoid receptors recruit the CaMKIIalpha-BDNF-CREB pathways to mediate memory consolidation. Nat Neurosci. 2012;15(12):1707–14.
de Quervain DJ, Poirier R, Wollmer MA, et al. Glucocorticoid-related genetic susceptibility for Alzheimer’s disease. Hum Mol Genet. 2004;13(1):47–52.
Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2006;26(35):9047–56.
Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352(19):1967–76.
Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373(9678):1905–17.
Zhang Y, Leung DY, Nordeen SK, Goleva E. Estrogen inhibits glucocorticoid action via protein phosphatase 5 (PP5)-mediated glucocorticoid receptor dephosphorylation. J Biol Chem. 2009;284(36):24542–52.
Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12(3):141–51.
Acknowledgement
We are thankful to Michael Garabedian (New York University) for support and Inserm’s AVENIR funding program.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Arango-Lievano, M., Lambert, W.M., Jeanneteau, F. (2015). Molecular Biology of Glucocorticoid Signaling. In: Wang, JC., Harris, C. (eds) Glucocorticoid Signaling. Advances in Experimental Medicine and Biology, vol 872. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2895-8_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2895-8_2
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2894-1
Online ISBN: 978-1-4939-2895-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)