
Abstract
Regulation of the immune system is an important function of the gut microbiota. Increasing evidence suggests that modern living conditions cause the gut microbiota to deviate from the form it took during human evolution. Contributing factors include loss of helminth infections, encountering less microbial biodiversity, and modulation of the microbiota composition by diet and antibiotic use. Thus the gut microbiota is a major mediator of the hygiene hypothesis (or as we prefer, “Old Friends” mechanism), which describes the role of organisms with which we co-evolved, and that needed to be tolerated, as crucial inducers of immunoregulation. At least partly as a consequence of reduced exposure to immunoregulatory Old Friends, many but not all of which resided in the gut, high-income countries are undergoing large increases in a wide range of chronic inflammatory disorders including allergies, autoimmunity and inflammatory bowel diseases. Depression, anxiety and reduced stress resilience are comorbid with these conditions, or can occur in individuals with persistently raised circulating levels of biomarkers of inflammation in the absence of clinically apparent peripheral inflammatory disease. Moreover poorly regulated inflammation during pregnancy might contribute to brain developmental abnormalities that underlie some cases of autism spectrum disorders and schizophrenia. In this chapter we explain how the gut microbiota drives immunoregulation, how faulty immunoregulation and inflammation predispose to psychiatric disease, and how psychological stress drives further inflammation via pathways that involve the gut and microbiota. We also outline how this two-way relationship between the brain and inflammation implicates the microbiota, Old Friends and immunoregulation in the control of stress resilience.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Autism Spectrum Disorder
- Inflammatory Bowel Disease
- Autism Spectrum Disorder
- Quinolinic Acid
- Early Life Stress
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
This chapter concentrates on those gut-brain interactions that operate indirectly via the immune system, rather than via direct neural pathways. At least two sets of findings underlie this aspect of gut-brain interaction. First, we know that persistently raised levels of inflammatory mediators are associated with several psychiatric conditions. This will be discussed with particular reference to depression and to reduced stress resilience. The regulation of background levels of inflammation is dependent upon “learning” inputs to the immune system from appropriate microbial exposures during the prenatal and neonatal periods, and continuing diversity of input in later life. This concept, initially called the “hygiene hypothesis” is becoming renamed the “Old Friends” mechanism, which places it firmly within the field of Darwinian and evolutionary medicine. The various mammalian microbiotas, particularly the gut microbiota, are important components of the Old Friends mechanism, and have a continuing immunoregulatory role in the adult. Secondly, we know that inflammation during pregnancy can lead to abnormal development of the central nervous system (CNS). This will be illustrated by considering autism spectrum disorders (ASD) and schizophrenia, and aspects of epidemiology that suggest the importance of microbe-dependent immunoregulatory effects during pregnancy.
The expression “Hygiene hypothesis” was first published in 1989, following the observation that, when examined at 11 years old, children brought up in families with many older siblings were less likely to have developed allergic disorders. This concept was at first a narrow one, focusing on the notion that childhood infections somehow prevented subsequent allergies. In fact it had been known since the nineteenth century that the environment could modulate the likelihood of developing hay fever, which was increasing amongst wealthy townsfolk, while remaining rare amongst farmers [1]. This protective effect of the farming environment in allergic disorders has subsequently been rigorously confirmed and found to extend to juvenile-onset inflammatory bowel disease (IBD) as well [2]. Moreover to reap protection from these immune-mediated diseases it can be sufficient to expose the pregnant mother to the farming environment, rather than the infant itself [3]. The most recent observations indicate that the farm effect is mostly explained by exposure to increased microbial biodiversity, which was documented by analyzing the bacterial and fungal taxa present in the dust in children’s bedrooms [4].
Meanwhile sporadic observations in other branches of medicine have confirmed that allergic disorders are not the only chronic inflammatory conditions that have been increasing in high-income countries, particularly in urban populations. Inflammatory bowel diseases and autoimmune diseases have increased at about the same rate, and in the same places [5, 6] as allergic conditions. Subsequently large epidemiological surveys have shown that childhood infections, originally implicated as the protective mechanism behind the hygiene hypothesis, do not protect against allergic disorders, and may in some cases, such as human rhinoviruses and respiratory syncytial viruses, actually trigger allergic responses. Considered together, these observations led to a Darwinian reformulation of the hypothesis as the Old Friends mechanism, described in the next section.
Old Friends Mechanism
The Old Friends mechanism states that mammals co-evolved with various microbiotas and commensals (gut, skin, lung etc.), as well as with chronic infectious agents picked up at birth, helminths that persisted for life, and environmental organisms from animals, mud and untreated water with which we were in daily contact. Because all of these categories of organism needed to be tolerated, they took on a role of inducers of immunoregulatory circuits [7, 8]. For example, helminthic parasites need to be tolerated because, although not always harmless, once they are established in the host, efforts by the immune system to eliminate them are typically futile, and merely cause tissue damage [9].
Contact with the “Old Friends” rapidly diminished when industrialization occurred, and mankind started to inhabit a plastic and concrete environment, to consume washed food and chlorine-treated water, and to minimize our contact with mud, animals and feces. This withdrawal of the organisms that drive immunoregulatory circuits results in defective immunoregulation that, depending on the genetic background of any given individual, can manifest itself as a variety of chronic inflammatory disorders, including allergies, IBD and autoimmunity. We know that a failure of immunoregulatory mechanisms really can lead to simultaneous increases in diverse types of pathology. For example, defects in the gene encoding the immunoregulatory transcription factor Foxp3 lead to the X-linked autoimmunity-allergic dysregulation syndrome (XLAAD) that includes aspects of allergy, autoimmunity and enteropathy [10].
The underlying Darwinian principle of the Old Friends mechanism is illustrated in Fig. 15.1. The immune system at birth is analogous to a computer with hardware, some software, but very little data. The minimal data that it does have comes from T lymphocyte selection in the thymus, and probably from transfer of at least some environmental and maternal antigenic material across the placenta. After birth the immune system requires the largest possible exposure to environmental microbial biodiversity in order to build a very broad repertoire of potential effector lymphocytes. Since all life forms are ultimately constructed with similar building blocks, such diversity of “education” can even provide the system with T cells that recognize, for example, some obscure viral pathogen that might be encountered in the future [11].

The immune system requires “educational” input. The microbiota of others, tolerated organisms (such as helminths) with which we co-evolved and organisms from the natural environment are required to expand the effector and regulatory branches of the immune system. During subsequent encounters with pathogens, danger signals generated by tissue damage enhance effector mechanisms and attenuate regulatory pathways to permit an appropriate immune response. Adequate background levels of regulatory T cells and dendritic cells and other regulatory mechanisms are required to maintain suppression of responses to “forbidden targets” and to switch off inflammation completely when the danger is eliminated, so that proinflammatory mediators do not continue to circulate
However in the context of this chapter, still more important than the diverse effector repertoire is the setting up of appropriate immunoregulation. Just as exposure early in life to a wide range of microbial and parasitic organisms trains the immune system regarding what to be on guard against, it also teaches immunity what to profitably ignore because the organisms in question either confer some benefit to the host, or confer no danger or despite posing some risk are not easily eradicated by immune mechanisms once established. These immunoregulatory inputs benefit the host by teaching the immune system not to waste precious energy engaging in futile battles, by reducing the cost to the host of chronic inflammation and by reducing the risk of destruction of host tissues, either through bystander effects or via the induction of autoimmunity. Because humans in traditional environments were exposed to organisms that dampened, as well as stimulated, immune function, the Old Friends mechanism implies that inflammation should be better regulated in low-income than in high-income countries. At first sight this might seem paradoxical, because the high prevalence of infections in low-income countries might be expected to cause high levels of inflammation [12]. However recent work by McDade et al. [13, discussed in 14] has largely resolved this paradox. The results reveal that in a low-income country where there is still abundant exposure to the immunoregulation-inducing “Old Friends”, immunoregulation is efficient, and the inflammatory response is vigorous during an infection, but is terminated when no longer needed, with the result that “resting” C-reactive protein (CRP) is close to zero. These longitudinal results illuminate a previous finding by McDade et al. [15] that high levels of microbial exposure in the perinatal period and in infancy correlated with low levels of “resting” CRP in adulthood. In contrast, in the USA and other high-income countries there is often constant low-grade inflammation which tends to be stable across individuals, manifested as chronically raised CRP or interleukin (IL)-6, in the absence of any clinically apparent inflammatory stimulus. Such chronically elevated inflammation greatly increases the risk of subsequent inflammatory disease and cardiovascular problems and has been shown in some studies to predict the future development of depression [16].
Inflammation and Psychiatric Disorders
Inflammation is involved not only in chronic inflammatory disorders such as allergies, autoimmunity and IBD but also in many psychiatric disorders. We have reviewed this topic in detail elsewhere [17, 18]. Briefly, a large subset of depressed individuals has persistently raised levels of proinflammatory cytokines and other downstream inflammatory markers [19, 20], together with a relative deficit in anti-inflammatory mediators and regulatory T cells [fully referenced in 18, 21]. Interestingly, depressed individuals also show exaggerated release of inflammatory mediators in response to psychosocial stressors [22], implying altered immunoregulation (Fig. 15.2), and epidemiological studies in the United Kingdom (UK) showed that raised CRP and IL-6 predicts subsequent risk of depression assessed over a decade later [16].

Exaggerated and prolonged cytokine release in response to a psychosocial stressor in individuals with diminished immunoregulation. Populations that have poorly immunoregulatory gut microbiota and reduced exposure to immunoregulation-inducing “Old Friends” such as helminths are susceptible to excessive and prolonged cytokine release in response to psychosocial stressors, which may result in reduced stress resilience and inappropriate triggering of depressive episodes. Reprinted from Rook et al. (2013) Evolution, Medicine and Pubic Health (1): 46–64, doi: 10.1093/emph/eot004, by permission of Oxford University Press and the Foundation for Evolution, Medicine, and Public Health
The possibility that inflammatory mediators might play direct causal roles in depressive pathogenesis has been confirmed for interferon-alpha (IFN-α), interleukin 6 (IL-6), and tumor necrosis factor (TNF). When IFN-α is used therapeutically (to treat viral hepatitis or some cancers) it causes depression-like symptoms in a high percentage of patients. These symptoms have been repeatedly shown to respond to treatment with standard antidepressants, such as selective serotonin reuptake inhibitors (SSRI) [20, 23]. Similarly the cytokine antagonist infliximab, which blocks TNF actions, has been shown to have antidepressant properties, but only in depressed individuals with evidence of increased peripheral inflammation prior to treatment [24].
Finally, when IL-6 is administered to pregnant animals it causes abnormal brain development in the fetus, discussed later in relation to autism [25]. Similarly increased peripheral levels of IL-6 cause increased production of IL-6 in the CNS, and affect neurogenesis in the hippocampus [reviewed in 26]. That IL-6 is directly relevant to the changes seen is supported by the fact that these effects can be blocked by IL-6 receptor antagonists, and knockout mice with non-functional IL-6 genes have enhanced working memory compared to wild type mice [27] and are refractory to peripheral inflammation-induced impairments of spatial memory [28]. In humans raised levels of IL-6 are associated with diminished cognitive performance and reduced hippocampal gray matter [26, 29]. Mechanisms for these effects are discussed in a later section.
Immunoregulation and Stress Resilience in Developing Countries
The vicious cycle described in Fig. 15.2, considered against the background of the Old Friends mechanism, suggests that in developing countries there will be less release of inflammatory mediators in response to psychosocial stressors, and less psychiatric consequences of such stressors. Recent data support this hypothesis. In experimental animals parental deprivation is a potent inducer of long-term changes to stress responses and immunoregulation [30]. Human studies suggest similar correlations [31]. However in a recent study performed in a developing country, parental absence in childhood was a significant predictor of raised CRP in adulthood, as it would be in a rich country, but only in a subset of the cohort raised in hygienic environments [32]. However, adults who had a high level of microbial exposure in infancy were resistant to the long-term proinflammatory effects of this severe childhood stressor [32]. The same was true of perceived stress during the previous month in young adults. CRP correlated with recent perceived stress in subjects with low microbial exposure in infancy, but not in those with high microbial exposure. Again, exposure to immunoregulation-inducing “Old Friends” seemed to provide resistance to the inflammation-inducing effects of psychosocial stressors [32].
This leads to an obvious question. If psychosocial stressors cause depression at least partly by triggering the release of proinflammatory mediators (Fig. 15.2), are inhabitants of developing countries resistant to psychosocial stress-induced depression? If so the prevalence of depression should be increasing in developed countries in parallel with the chronic inflammatory disorders [33], and lower in developing countries than in developed ones. Comparative studies are difficult to do, but this is indeed what data collected by the World Health Organization indicate [34]. Moreover one study failed to find a correlation between depression and raised CRP in a developing country whereas this association is routinely found in rich ones [35].
Urbanization and Immigration to High-Income Countries
If a dysregulated immune system resulting from diminished contact with immunoregulation-inducing “Old Friends” is partly to blame for the increasing prevalence not only of chronic inflammatory disorders such as allergies, autoimmunity and IBD, but also of those psychiatric disorders that can be triggered by inflammatory mediators, then it should be useful to examine urban-rural differences in disease prevalence, and the effect of migration from low-income to rich urban environments. In each case there will be loss of exposure to Old Friends.
Urban Versus Rural
A feature shared by most of the disorders discussed here is a higher prevalence in urban communities compared to rural ones. For example a meta-analysis of high quality studies performed in high-income countries since 1985 found that the prevalence of depression in urban areas was 39 % higher than in rural areas. Similarly, the prevalence of anxiety disorders was 21 % higher in urban than in rural areas [36], though a small minority of studies fails to find this urban-rural difference [37]. Peen et al. [36] also noted an increased urban prevalence of psychiatric disorders in general (38 % more in urban communities). This agrees well with another large meta-analysis that found a significantly raised prevalence of schizophrenia in urban communities [38]. Similarly, a study of all children born in Denmark between 1 January 1984 and 31 December 1998 found that the degree of urbanization of place of birth was very significantly correlated to risk of autism [39].
The urban > rural phenomenon is also well established for chronic inflammatory disorders, where the etiology is known to involve dysregulation of the immune system. Contact with the farming environment, whether early postnatal [40] or prenatal [3, 41] protects against allergic disorders, whereas the prevalence of these conditions increases with increasing urbanization [42]. The same is true for IBD [43], and for autoimmune diseases such as multiple sclerosis (MS) [44, 45, discussed in 46].
Immigrants
Another striking parallel between chronic inflammatory diseases and psychiatric disorders concerns the effects that immigration has on these conditions. All the diseases discussed here, whether chronic inflammatory [43, 47–49] or psychiatric [50–52], tend to be more common in immigrants than in the birth population from which the immigrant was derived, at least when the migration is from a developing to a high-income country. Other relevant variables include the age of the individual at the time of immigration, and whether the prevalence increases in second generation immigrants, born in the adopted country. A study of these parameters provides some insight into whether the relevant influences, be they psychosocial or immunological, need to occur before birth, or in early childhood, or whether they can still exert their effects on adults.
Immigration and Psychiatric Disorders
Depression is particularly interesting in this respect [53, 54]. Mexicans, Cubans and African/Caribbean peoples were found to have a two to threefold increase in the prevalence of depression if immigration to the USA occurred when the individual was less than 13 years old, or was born in the USA, compared to the prevalence in those who migrated after the age of 13 [53]. But this is not likely due to psychosocial stress related to skin color, because white Eastern European immigrants show the same effect. In sharp contrast, the effect is not seen in immigrants from Western Europe, or from Puerto Rico, which is closely associated with the USA. (These last two populations already have a high prevalence of depression that is not increased by immigrating to, or being born in, the USA) [53]. These findings imply that influences important for depression occur perinatally, or in the early years of life.
The same is true for psychotic disorders [55]. A large Danish study noted that immigration into Denmark when less than 4 years old was associated with a strikingly increased risk for psychotic disorders, whereas the increased risk gradually decreased with older age at migration and disappeared in those immigrating when more than 29 years old [56]. Similarly a large meta-analysis confirmed that schizophrenia was increased amongst first generation immigrants, and further increased amongst second generation immigrants, particularly when the country of origin was a developing one [57]. Again, early events seem crucial.
Age at immigration is irrelevant to an early onset condition such as autism, but autism is strikingly (as much as tenfold) increased in second generation Caribbean or African immigrants born in the UK, compared to children of white UK-born mothers [52]. These findings implicate crucial early events in the perinatal period or early childhood as risk factors for depression, schizophrenia and autism.
Immigration and Chronic Inflammatory Disorders
Migration has clear effects on the prevalence of MS, and the crucial events that confer increased risk for the disease occur very early in life, as is true for the psychiatric disorders [reviewed and referenced in 58, 59]. Iranians who migrate to Sweden have twice the prevalence of MS seen in their birth country [49]. Interestingly, if the second (or later) generation immigrants return to their developing country of origin, they retain their increased susceptibility to MS, which remains higher than in the local population that was not born abroad [60]. A similar phenomenon was seen when people born in the UK (a high MS country) migrated to South Africa (SA: a low MS country). Migration from the UK to SA was protective when the migrant was a child, whereas adult migrants retained their high UK prevalence of MS [61]. Analysis of this and other studies suggests that the environmental factors that protect from or predispose to MS act during the first two decades of life [58, 59]. The same is true for type 1 diabetes (T1D). Here the crucial factor is to have been born in the receiving developed country, again suggesting that relevant environmental factors act very early, or even in the prenatal period [48].
The role of migration in conferring risk for allergic disorders has been intensively examined. A study of children adopted into Sweden from developing countries showed that the prevalence of asthma, hay fever and eczema were highest in those adopted when less than 2 years old [62]. Similarly, for Mexican immigrants to the USA, the prevalence of asthma was highest for those born in the USA, while in those not born in the USA, the prevalence of asthma decreased as the age at immigration increased [63]. This effect of age at the time of childhood immigration was also seen in immigrants to Israel from the former Soviet Union or Ethiopia who were assessed when 17 years old [64]. These observations suggest the importance of early environmental influences for allergy/asthma risk, a conclusion that is powerfully supported by evidence that prenatal exposure (i.e. of the pregnant mother) to the farming environment protects the infant against some allergic manifestations [3, 41]. This is discussed later in another context.
Finally, a definitive study of all first- and second-generation immigrants in Sweden between January 1, 1964, and December 31, 2007 showed that some first generation immigrants remain partially protected from both ulcerative colitis (UC) and Crohn’s disease (CD), presumably by environmental factors encountered in their countries of origin, but the diseases increased in prevalence in second generation immigrants, relative to first generation immigrants [65]. Similarly, the prevalence of UC in South Asian immigrants to Leicester in the UK was higher in second than in first generation immigrants [66]. This again implicates perinatal factors as potentially causative of this migration effect.
Thus the influence of immigration, acting via factors that occur perinatally or very early in life, is equally consistent and highly apparent for both psychiatric and chronic inflammatory disorders.
Mechanisms of Immunoregulation by Old Friends
Urbanization and immigration from low- to high-income countries cause diminished contact with Old Friends, and correlate with an increased incidence of chronic inflammatory disorders, all of which show evidence of failed immunoregulation [reviewed in 67]. Moreover adverse outcomes in animal models of all of these chronic inflammatory conditions can be prevented or treated with “Old Friends” such as helminths, certain gut commensals or probiotics that induce immunoregulation [68–70]. What are the mechanisms that enable the “Old Friends” to exert immunoregulatory effects? This is a vast topic, and here we outline some of its most studied aspects, particularly those that involve, or occur in, the gut.
Regulation of Innate Immunity
The gut microbiota has been shown to be necessary for priming of innate immunity, measured as the microbicidal activity of splenic macrophages [71]. This microbicidal activity was increased following a social disruption stressor. However, if the mice were germ-free no increase in microbicidal activity was seen [71]. Moreover depletion of microbiota with antibiotics attenuated the stressor-induced macrophage activation, and reduced stressor-induced increases in circulating bacterial cell wall peptidoglycan [71], and eliminated the increases in circulating IL-6 and monocyte chemoattractant protein-1 (MCP-1) usually seen in stressor-exposed mice [72]. These observations were in agreement with an earlier finding that systemic activation of the innate immune system by the gut microbiota involves recognition of meso-diaminopimelic acid (mesoDAP)-containing peptidoglycan found predominantly in Gram-negative bacteria, by the pattern recognition receptor nucleotide-binding, oligomerization domain-containing protein-1 (Nod1) [73]. Similarly abdominal surgery causes systemic release of Nod2-binding bacterial components and consequent rises in several inflammatory biomarkers [74].
Regulatory Macrophages
Regulatory microorganisms can also operate via macrophages. During helminth infections there is expansion of the population of alternatively-activated macrophages, activated by Th2 rather than Th1 cytokines [75]. Such macrophages secrete IL-10 and TGF-β rather than IL-12, are able to inhibit lymphocyte proliferation in a contact-dependent manner [76, 77], and may be responsible for preventing inflammation in mucosal surfaces such as the lung. However, some helminths drive types of regulatory macrophages that are distinct from alternatively activated macrophages [77]. Several species of filarial nematodes secrete cystatin, a cysteine protease inhibitor that induces macrophages to make IL-10 and IL-12 p40 through activation of intracellular signaling pathways. These can prevent allergic sensitization and airway hyperresponsiveness [78]. Similarly a colon-infiltrating macrophage population induced by Schistosoma infection was shown to prevent colitis in mice [79]. The protection was independent of T cells in general and regulatory T cells (Treg) in particular.
Regulatory B Cells
Helminths also induce regulatory B cells. S. mansoni infection prevented anaphylaxis in a mouse model, and this suppression of the effector phase of the allergic response was mediated by IL-10-secreting B cells [80]. These IL-10-secreting CD1dhiCD5+ regulatory B cells can also suppress experimental autoimmune encephalomyelitis [81], and have recently been designated B10 cells [82]. They act in part by increasing the number of pulmonary CD4+CD25+Foxp3+ regulatory T cells in the lungs [83]. IL-10+ regulatory B cells are also found in humans [84]. Depletion of human B-cells using rituximab can occasionally exacerbate Th1-mediated conditions, suggesting that the rituximab removed a B-cell-mediated regulatory mechanism [84]. IL-10 production by B cells is increased in multiple sclerosis patients developing intestinal helminth infections [85], but not in patients infected by Trypanosoma cruzi [85]. The helminths also increase circulating Treg, as discussed below.
Regulatory Dendritic Cells (DC)
A particularly important immunoregulatory function is the generation of regulatory dendritic cells (DCreg) that tend to drive regulatory rather than inflammatory responses. This is crucial because such DCreg can process gut contents, autoantigens and allergens, and so downregulate responses to the target antigens of the major groups of chronic inflammatory disease. It is likely that health requires the presence of a certain background proportion of DCreg. This could be regarded as a “Treg adjuvant” function. A mixture of several putative probiotic organisms (VSL#3; four lactobacilli, three bifidobacteria, and one streptococcal strains) was found to ameliorate recurrent Th1-mediated murine colitis by inducing IL-10 and TGF-β+Treg [86]. In vitro this preparation caused human DC to release more IL-10 and inhibited their ability to drive Th1 cells [87]. Other probiotic strains [88, 89], and a ubiquitous environmental saprophyte often present in untreated or muddy water [90] also modulate human DC function in vitro so as to induce T cell responses with a more regulatory bias. In the gut some of these DCreg express the integrin alpha chain CD103 and have unique immunoregulatory properties [91]. CD103 is involved in de novo conversion of Foxp3-CD4+ cells to Foxp3+ Treg cells [92]. Conversion of DC to this tolerogenic phenotype is driven locally by TGF-β and retinoic acid (RA). CD103+ DCs express aldh1a2, the gene encoding RALDH2. This enzyme is involved in conversion of dietary retinal to RA, which enhances development of FoxP3+ T cells rather than Th17 cells [reviewed in 93]. Some probiotic Lactobacillus strains, such as L. plantarum WCFS1 induce migration of these CD103+ DCreg as far as the spleen, and bias the response towards Treg [94].
An example of a Treg adjuvant effect that must at some stage involve DC is seen when MS patients become infected with helminths. The disease stops progressing, and circulating Treg appear in the peripheral blood [95, 96]. These Treg recognize the major epitope from myelin basic protein. Thus the immunoregulation caused by the helminths is not merely a bystander effect of IL-10 release, but rather a genuine Treg adjuvant effect that generates regulation specific for the autoantigen. Although the mechanism is not elucidated this is an exciting observation that has led to formal clinical trials [97].
Regulatory T Cells (Treg)
Ultimately many of these immunoregulatory mechanisms result in a relative increase in the numbers of Treg, whether secondary to changes in macrophages, B cells or DC. However some Old Friends release molecules that specifically expand Treg populations. The gut commensal Bacteroides fragilis releases a polysaccharide antigen that drives expansion of Treg via TLR-2 [98]. The helminth Heligmosomoides polygyrus drives Treg expansion via the TGF-β receptor [99]. Treatment with oral Lactobacillus reuteri for 9 days significantly increased the percentage and total number of CD4+CD25+Foxp3+ T cells in the spleens of experimental animals [70]. Colonization of mice by commensal Clostridium strains increased TGF−β levels and numbers of Foxp3+ Treg in the colon [100].
Gut Microbiota Diversity and Regulation of Inflammation
Interestingly the diversity of the gut microbiota appears to have consequences for immunoregulation, perhaps for the reasons discussed in relation to Fig. 15.1. From birth our microbiota are constituted by colonization with organisms from our mothers, from other social contacts [101, 102], and from the environment, and then further modified by factors such as diet and antibiotics [103–106]. Thus lifestyle has major effects on an individual’s microbiota and on its diversity. The gut microbiota of children from traditional villages in Burkina Faso is totally different from that of Europeans, and shows greater diversity [104]. There is abundant evidence that diversity of gut microbiota is associated with wellbeing. Mice exhibit at least two enterotypes (bacterial ecosystems in the gut microbiota), one of which has low biodiversity, and correlates with biomarkers of inflammation [107]. In humans reduced biodiversity of the gut microbiota has been associated with a range of inflammatory states, including allergic disorders [108], inflammatory bowel diseases [109–111] and obesity [112]. Loss of diversity in later life is associated with increases in circulating CRP and IL-6 levels [113]. This implies that diversity is associated with effective immunoregulation.
In agreement with this, allergies are less common in children exposed to sources of microbial biodiversity such as farms [40], dogs [114], or the natural environment [115, 116]. Similarly, allergies are reduced where there is evidence of social interactions that promote exchange of microbiota. Indicators of such exchange include infection with orofecally transmitted organisms such as enteroviruses [117], H. pylori, T. gondii, and hepatitis A virus [118]. There is some evidence that these orofecally transmitted pathogens are themselves immunoregulatory. However it might be much more important that these organisms are markers of transfer of microbiota between individuals. Interestingly mothers who clean their baby’s dummy/pacifier by sucking it rather than by sterilizing it have children with less allergic problems [119].
In sharp contrast, lifestyle events that are likely to restrict the diversity of gut microbiota are associated with increased risk of chronic inflammatory disorders. Birth by caesarian section may be a risk factor for allergic disorders [120, 121]. Similarly, excessive antibiotic use during pregnancy [122] or in early childhood is a risk factor for allergic disorders [123, 124] and IBD [125, 126]. And as discussed above, living in a high-income rather than in a low-income country is a risk factor for all of these disorders.
Organisms from the Natural Environment and Microbiota
Clearly microbiota from other people (and animals) can colonize our guts. But do organisms from the natural environment also colonize, or are these organisms “pseudocommensals” that impinge on the skin [116], airways and gut, and have independent immunoregulatory properties? Both mechanisms probably occur, though there are rather limited data on these issues. An interesting animal experiment compared piglets that were housed in a natural outdoor environment, with genetically similar piglets that had been reared in a very clean indoor facility. Firmicutes, in particular Lactobacillus strains were dominant in the gut microbiotas of the outdoor piglets, whereas the hygienic indoor piglets had reduced Lactobacillus and more potentially pathogenic phylotypes [127]. The indoor piglets also had less diverse gut microbiota, and a more inflammatory pattern of gene expression in ileal biopsies [127]. For example they had increased Type 1 interferon activity, increased MHC Class 1, and upregulation of many chemokines [127], again confirming the correlation between reduced gut microbial biodiversity and poor control of inflammation discussed above.
Were these effects due to direct colonization by immunoregulation-inducing organisms from the outdoor environment [pathway (A) in Fig. 15.3], or did these organisms fail to colonize, but exert indirect effects on the immune system? The answer is unclear, but indirect effects certainly can occur in several ways. Some organisms compete with, or antagonize established organisms [pathway (B)] and so alter the microbiota [128]. Others alter the immune system directly [pathway (D)], or modulate the immune system in ways that lead secondarily to a change in the host-microbiota relationship, which in turn leads to changes in the microbiota [pathway (C) in Fig. 15.3].

Environmental organisms and immunoregulation. Microbial biodiversity from the environment can modulate immunoregulation by (D) directly interacting with the immune system, or (A, B, C) by leading secondarily to altered microbiota. The environmental organism may cause secondary changes to the microbiota by (A) colonizing, or (B) antagonizing or competing with established microbiota or (C) modulating the host immune system-microbiota relationship
The last mechanism is well established in experimental models. Genetic manipulations of the innate immune system that have profound effects on immune function (such as gene knockout) often operate indirectly by altering the gut microbiota. The phenotypic effects can then be transferred to wild-type mice that have not been genetically modified, by transferring the altered microbiota [129, 130]. It is the altered microbiota that is the proximate cause of the altered immunoregulation [129–133].
Is the Gut Still Involved in Immunoregulation by Organisms That Do Not Enter the Gut?
Does this mean that all immunoregulation by “Old Friends” operates indirectly by modulating the immune system, and so secondarily causing changes in the gut microbiota? It is likely that such indirect effects occur, but there could be direct effects too. For example the skin microbiota has at least some immunoregulatory role independent of the gut [134]. Indeed components of the skin microbiota extend into the subepidermal compartments, suggesting subtle and unexplored mechanisms [135]. Moreover psychological stressors cause increased bacterial translocation to lymphoid tissue from both the gut and the skin so the nature of the organisms present on skin is likely to be directly relevant to subsequent effects on immune function [136]. Interestingly, in the British field mouse (Apodemus), the burden of the louse Polyplax serrata correlated with the state of activation of the innate immune system in the spleen, implying that ectoparasites (fleas, lice, mites, ticks) might also have immunoregulatory roles [137, 138].
The blood nematodes are also of interest because these do not enter the gut at any phase of their life cycles, but they are powerfully immunoregulatory [9]. They cause impaired induction of T-bet and GATA-3 mRNA, Th1/Th2 deficiency and increased Foxp3, TGF-β, CTLA-4, PD-1, ICOS and IDO. Some blood nematodes secrete identifiable immunoregulatory molecules [139, 140].
Nevertheless, in view of the experiments listed in the previous section, it is still possible that in addition to direct effects on the immune system [pathway (D) in Fig. 15.3] these effects also operate indirectly via secondary modification of the gut microbiota [pathway (C) in Fig. 15.3].
Genetics and “Inflammatory Overshoot” in High-Income Countries
In parts of the world where there was a heavy load of organisms that drive potent immunoregulation (such as helminths) there has been selection for single nucleotide polymorphisms (SNP) or other variants to partially compensate for excessive immunoregulation, or to combat new infections such as malaria that spread from gorilla to man about 10,000 years ago [141, 142]. Such proinflammatory SNPs are seen for several proinflammatory cytokines [143], IgE [144] and STAT6, a transcription factor involved in Th2 responses [145]. There is also an increased frequency of the short allele of the serotonin transporter promoter that also has a marked proinflammatory effect [146]. However this results in a dangerous situation. As soon as the immunoregulation-inducing organisms are withdrawn by the modern lifestyle, or after immigration to a high-income country, these genetic variants lead to inflammatory overshoot. The proinflammatory variants become risk factors for chronic inflammatory disorders [143–146].
This is important because work that identifies proximate “causes” for diseases that were rare or nonexistent before the late nineteenth or early twentieth centuries, and that remain rare in low-income countries, may merely be unraveling a gene-environment interaction that would be irrelevant if the microbial status could be returned to that seen in the paleolithic age. For instance, the recent claim to have discovered that the “cause” of Crohn’s disease is a genetically determined defect in the homing of neutrophils [147] is difficult to reconcile with the fact that 100 years ago the disease barely existed. But recent environmental changes could conceivably have caused this phenotype to become a risk factor.
How Does Stress Cause Inflammation?
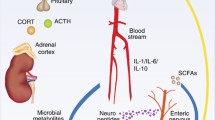
Two major issues were avoided in the discussion of the role of immunoregulation in determining stress resilience (Fig. 15.2). First we did not discuss why stress causes release of inflammatory mediators, and secondly we did not discuss why such mediators trigger depression. Stress leads to activation of the hypothalamic-pituitary adrenal axis (HPA) and the sympathetic nervous system (SNS), and to changes in the microbiota and gut permeability. How do these mechanisms combine to result in raised proinflammatory cytokine levels?
GC Resistance
Depression is commonly associated with hypercortisolaemia and glucocorticoid resistance (GCR) [148]. A recent analysis has revealed that persistently raised levels of inflammatory cytokines cause GCR by impairing the function of glucocorticoid receptors [148], leading to further loss of control of inflammation. This was the suggested mechanism in individuals with recent exposure to severe psychosocial stressors who developed GCR and subsequently released more proinflammatory cytokines in response to an inflammatory stimulus (virus challenge to airways) [149].
Sympathetic Nervous System (SNS)
There is an increase in plasma concentrations of norepinephrine following exposure to a standardized laboratory stressor, the Trier Social Stress Test. This is accompanied by activation of the master regulator of inflammation, nuclear factor-kappa beta (NF-κB), in peripheral blood monocyte cells (PBMCs) [150]. Blocking the effects of activation of the SNS with the β-adrenergic receptor antagonist propranolol blocked the stressor-induced increases in proinflammatory cytokines, and reduced the development of GCR [151].
Corticotropin-Releasing Hormone (CRH)
Increased expression of corticotropin-releasing hormone (CRH) is found in CSF and in the limbic brain regions in depression [152, 153], but CRH is also involved in the control of gut permeability [154–156]. For example, chronic administration of CRH via minipumps caused colonic barrier dysfunction in rats [155]. Moreover when released in the periphery by T cells, CRH is not only a regulator of intestinal permeability [154, 155], but also a potent pro-inflammatory cytokine [157]. In many cell types CRH activates NF-κB, and stimulates expression of IL-1β, IL-6, and TNF mRNAs [158], so some of the effects of CRH on permeability are secondary to the release of proinflammatory cytokines. Paracellular permeability is controlled by tight junctions, intermediate junctions and desmosomes, which constitute a size- and cation-selective filter for small molecules. However, TNF, IL-17, IFN-γ and nitric oxide (NO) increase permeability. IFN-γ disrupts the tight junctions, and can modify para-epithelial traffic of inflammatory cells. By contrast, the immunoregulatory cytokine TGF-β decreases permeability [154, 155].
The composition of the microbiota, particularly Lactobacillus strains and helminth “Old Friends” also modulate permeability. When idiopathic chronic diarrhea in rhesus monkeys was treated with the whipworm Trichuris trichiura, clinical improvement was accompanied by striking changes in the microbiota attached to the mucosa [159]. Similarly in a mouse model of IBD, infection with H. polygyrus caused an increase in lactobacilli.
Indirect Effects of Psychosocial Stress via the Microbiota
Stress induces changes in the composition of the microbiota of rodents [72], and induces bacterial translocation from gut and skin [136]. The same is true in humans. When sampled within hours of admission to the emergency room fecal bacterial counts were decreased 1,000-fold compared to control subjects, and obligate anaerobes and Lactobacillus species were significantly decreased [160]. Similarly, a large change in the microbiota after allogeneic bone marrow transfer was identified as a risk factor for subsequent inflammation and graft-versus host disease [161], implying that stress alters immunoregulation at least partly by altering the microbiota.
The gut microbiota is involved in the activation of the HPA axis by stress, and in the systemic release of cytokines. A social disruption stressor caused increases in circulating IL-6 and MCP-1 that correlated with changes in the composition of the microbiota. But this response was greatly attenuated by pre-treatment with an antibiotic cocktail to deplete the microbiota [72]. Therefore much of the systemic cytokine response to stress might be secondary to uptake of LPS and other proinflammatory microbial components. Uptake of LPS was measured in another study. Animals subjected to restraint stress show increased portal blood LPS, together with HPA axis activation manifested as increased plasma ACTH and corticosterone, increased hypothalamic CRF and increased IL-1β, IL-6 and TNF. All of these manifestations were blocked by treatment with Lactobacillus farciminis, which blocks leakiness due to HPA axis activation [162].
Stress Resilience and the Microbiota
The microbiota might also be involved in the observation that poor stress resilience and an exaggerated cytokine response to environmental [31] or laboratory [22] stressors is characteristic of people who have suffered increased early life stress. We know that early life stress can modulate the microbiota [30, 163]. This interpretation is in agreement with the observation that in a low-income country population the adults who had a high level of microbial exposure in infancy were resistant to the long-term proinflammatory effects of a very severe childhood stressor [32]. In addition to the role in immunoregulation, the microbiota in the first weeks of life also modulates the development of the HPA axis and stress response [164], and the development of the brain [165].
How Does Inflammation Alter Behavior and Cognition?
The mechanisms that cause inflammatory responses to alter behavior and cognition have been extensively reviewed recently [166] and are summarized briefly here. From an evolutionary point of view this link between immunity and psychiatry is explained by the fact that during an acute infection, withdrawal (to conserve resources, fight infection and heal wounds) and hypervigilance (to detect danger) are adaptive responses [167]. Withdrawal and hypervigilance probably result from inflammation-mediated signals to distinct parts of the brain. Positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) have been used to identify brain regions affected by inflammatory cytokines, and the list includes the basal ganglia, the dorsal anterior cingulate cortex (dACC), amygdala, hippocampus, insula, dorsolateral prefrontal cortex, and subgenual ACC [reviewed in 166]. It is suggested that involvement of the basal ganglia is important for withdrawal, while the effects on the dACC are important for hypervigilance. However these states are not sustainable and if prolonged, withdrawal becomes depression and hypervigilance becomes anxiety. This relationship between prolonged inflammatory stimuli and behavioral changes that resemble depression and anxiety was postulated long ago following observations of “sickness behavior” in mice [168].
Inflammation-associated depression and anxiety are more likely in situations where there is poor control of inflammation [169]. This is seen in high-income countries where persistently high CRP is common [170], as discussed earlier in the context of the Old Friends mechanism. Similarly, depression and anxiety often accompany the chronic inflammatory disorders that are increasing in high-income countries. They are also seen in obesity where fat tissue contains cytokine-secreting activated macrophages, and in people who underwent traumatic childhoods, perhaps because of developmental changes in the HPA axis, brain, and microbiota [31, discussed in 169]. Genetic factors also play a role. For example individuals who have the short allele of the serotonin transporter-linked polymorphic region were more likely to get depression after IFN-α treatment [171].
Cytokines and Cellular Infiltration
Recent studies of patients receiving IFN-α have confirmed in humans many of the mechanisms previously reported in animal studies. For example, recipients of IFN-α develop raised CSF levels of IL-6 and MCP-1, proving that the inflammatory signal is transmitted to the brain [172]. Signals from the inflamed periphery enter the brain via several pathways. First, cytokines can enter the brain in areas such as the circumventricular organs where there is no blood-brain barrier. Perhaps these areas should be regarded as sensory organs of which one role is the detection of inflammation. Signals also pass via activation of endothelial cells within the cerebral vasculature, leading secondarily to release of prostaglandins and NO in the CNS. Furthermore brain endothelium expresses specific cytokine transporters. Cytokines in the periphery can also signal via afferent fibres within the vagus and other sensory nerves. This has been called the “facsimile” mechanism, because the cytokine stimulating the peripheral nerve terminals may subsequently be synthesized de novo and released within the brain.
These various inflammatory signals may secondarily activate local CNS cell populations. The microglia are derived from a subset of CD45+ monocytic cells and enter the CNS during embryogenesis in utero and during early post-natal life. These stable long-lived cells form a network with surveillance functions within the brain parenchyma, and under normal conditions are in a non-activated, non-terminally differentiated state, with low expression of major histocompatibility complex class II [173]. However inflammation propagated to the brain by the pathways listed above can cause these cells to express inflammatory cytokines and to release reactive oxygen and nitrogen species [166]. Activated microglia may also be a source of the MCP-1 mentioned earlier, which recruits monocytes into the brain [174].
Recruited leukocytes can enter the CNS by several routes. In healthy individuals there is background traffic via the choroid plexus or through postcapillary venules located in the subarachnoid space [175, 176]. However in inflammatory states cells can cross the endothelium of parenchymal post-capillary venules, and so enter the perivascular space. The relevant adhesion molecules are not expressed on these endothelial cells under resting conditions but they are induced by peripheral inflammatory signals such as LPS or TNF, facilitating the MCP-1-driven recruitment [176].
Indoleamine-2,3-Dioxygenase (IDO)
Inflammatory cytokines also activate the enzyme, indoleamine-2,3-dioxygenase (IDO), converting tryptophan to kynurenine. It has been suggested that this can deplete tryptophan sufficiently to cause depression. Recent mouse work showed that inflammation induced by peripheral administration of lipopolysaccharide (LPS) activated IDO and caused mice to display a depression-like behavioral syndrome that could be inhibited by blocking IDO [reviewed in 177]. However, the syndrome could be reproduced by administering kynurenine, which is taken up into the brain by the large amino acid transporter, where it can be further metabolized in microglia, astrocytes and macrophages to quinolinic acid, kynurenic acid and other metabolites [178]. This suggested that depletion of tryptophan was not the primary mechanism of the depression-like syndrome [179] (though this cannot be ruled out as an additional factor because there is evidence that kynurenine can compete with tryptophan for transport into the brain [180]). These mouse findings have been confirmed in patients with hepatitis C who were being treated with IFN-α, in whom there were depressive symptoms, but no reduction in CSF concentrations of tryptophan [181]. The treatment did however cause increased CSF levels of kynurenine, quinolinic acid and kynurenic acid, and also of IFN-α, soluble tumor necrosis factor-α receptor 2 and MCP-1 [181]. This suggested that as in the mouse, the depressogenic effect might involve transport of kynurenine into the brain followed by local generation of further active metabolites, rather than depletion of tryptophan [181]. There is increasing evidence that these neuro-active tryptophan metabolites are important in depression and in schizophrenia, and this topic has been reviewed in detail recently [177].
Transporters and Reuptake
Inflammatory cytokines also increase the expression of the transporters responsible for reuptake of dopamine, norepinephrine and serotonin. For example, in mice, IL-1, TNF and LPS all cause increased expression of the serotonin transporter paralleled by depression-like behavior [182]. In humans administration of IFN-α increases the reuptake and decreases the release of radiolabeled L-DOPA, the precursor of dopamine [183].
Tetrahydrobiopterin (BH4)
Inflammation also disturbs the availability of tetrahydrobiopterin (BH4), which is an essential cofactor for tryptophan hydroxylase and tyrosine hydroxylase. These are the rate-limiting enzymes for the synthesis of serotonin, dopamine and norepinephrine [166]. It is possible that the BH4 gets used up when cytokines drive NO synthase to generate NO, but this pathway is less active in man than in mouse. However BH4 can also be degraded by oxygen radicals and nitrogen radicals. Evidence for depletion of BH4 in the human brain in the presence of inflammatory mediators has recently been obtained in patients receiving IFN-α [184]. In the CSF of treated patients levels of the inactive oxidized form BH2 were increased, while levels of BH4 were inversely correlated with increased IL-6 [184].
Anti-Inflammatory Neurotransmitter Pathways
Inflammation in the CNS may also interfere with neurotransmitter pathways that have anti-inflammatory roles, and so weaken negative feedback on the inflammatory response. For example neural signals transmitted to the periphery via the vagus nerve inhibit cytokine release through a mechanism that requires the α7-subunit-containing nicotinic acetylcholine receptor [185]. This cholinergic anti-inflammatory pathway can be inhibited centrally by mediators such as IL-1 that increase neuronal acetylcholinesterase activity [185]. Similarly IL-1β might reduce signaling by γ-aminobutyric acid (GABA), and so enhance local inflammation [186]. This is due to the fact that GABA-ergic tone is anti-inflammatory because it inhibits the NF-κB and p38 MAPK pathways, and so reduces the response of microglia to LPS or IFN-γ [186].
Abnormal Brain Development
In addition to the postnatal mechanisms described above, inflammation can also act in utero to cause developmental defects in the foetal CNS that lead later to psychiatric problems. The likelihood of this phenomenon occurring will be influenced by the efficiency of immunoregulation in mother and child during pregnancy (Fig. 15.4).

Reduced efficiency of immunoregulation during pregnancy could predispose to inflammatory episodes in utero that lead to neurodevelopmental abnormalities. Such abnormalities are seen in ASD and schizophrenia, and both disorders are accompanied by evidence of failing immunoregulation that is most striking in ASD and in family members. The immunological points listed in the boxes at lower left and right are taken from, and fully explained within, the references in the main text
The Old Friends mechanism will be one factor that determines this immunoregulation, though as discussed below, there are also genetic factors. Interestingly autism spectrum disorders (ASD) are increased in towns [39] and in second generation immigrants [52]. These findings parallel the simultaneous increases in chronic inflammatory disorders, and depression in which the Old Friends mechanism likely plays a role [169].
Maternal Infection, Immunoregulation, Fetal Inflammation and ASD
There has been a significant and genuine increase in the prevalence of autism spectrum disorders (ASD) that cannot be explained only by increased awareness [187]. There is debate about the relative contribution of genetics and environment. A very recent study suggested that “Susceptibility to ASD has moderate genetic heritability and a substantial shared twin environmental component” [188]. Rather than worrying about the relative importance of genes and environment it is important to note that known autism susceptibility genes include a neuronal module and a module enriched for immune genes and glial markers [189]. Another study of interactome networks associated with highly expressed ASD-candidate genes found that immune signaling through NF-κB, TNF, and Jnk were strongly represented and that these interactomes involved glia in addition to neurons [190]. Thus the genetics point to inflammation and the immune system, which could modulate susceptibility to the types of environmental influence discussed in this chapter. A recent study suggests that there might be an underlying immune phenotype (whether genetic or environmental): the immune systems of autistic children and their healthy siblings were found to have similar immune dysregulation, when compared to the immune systems of matched healthy children [191].
An obvious link between immunoregulation and ASD is provided by evidence that maternal infection during pregnancy increases the risk of ASD in the infant. In one study 13 % of infants developed ASD following exposure to congenital rubella [192]. However it seems that any infection increases the risk, particularly if severe enough to require hospitalization during pregnancy [193]. A study of 1.2 million births in Finland showed that raised maternal CRP early in gestation was associated with increased risk, whatever the cause [194]. Animal work proves that maternal inflammation during pregnancy is transmitted to the foetus. TNF-α and IL-1β expression was upregulated in a dose dependent manner in the fetuses of pregnant rats exposed to LPS [discussed in 195]. Similarly I125-labelled IL-6 administered i.v. to pregnant rats was found in the fetal compartment [196].
Experimental animals exposed to maternal immune activation in utero also display developmental changes measured by MRI, and behavioral changes suggestive of ASD [197]. In pregnant mouse models these effects are dependent upon IL-6, and can be mimicked by administering IL-6 itself rather than an indirect inflammatory stimulus [25]. Interestingly these abnormalities are accompanied by a systemic deficit in CD4+ TCRβ+ Foxp3+ CD25+ T regulatory cells, and by increased IL-6 and IL-17 production by CD4+ T cells [198]. The behavioural abnormalities can be attenuated by transplanting normal bone-marrow, implicating the immune system both in the development of the syndrome, and in its subsequent maintenance [198].
Inflammation and Faulty Immunoregulation in ASD
The role of inflammation in utero in the development of the CNS abnormalities that accompany at least some cases of human ASD is not in doubt. But there is increasing evidence for an ongoing immunoregulatory deficit in human ASD [199, 200], as in the mouse model mentioned above [198]. ASD patients have increased circulating levels of proinflammatory cytokines, and reduced levels of TGF-β [200], and there is an increased prevalence of asthma and autoimmunity in family members, reinforcing the view that there is an hereditary, or at least familial, immunoregulatory deficit [191, 199, 200]. This tendency to autoimmunity is manifested as brain autoantibodies in plasma from children with ASD, and from their mothers [200].
Studies of autistic brains reveal activation of microglia and astroglia and increased expression of a range of proinflammatory mediators such as TNF, IFN-γ, IL-8 and IL-6 [201–203]. IL-6 is normally expressed at very low levels in the brain, but it is able to cross the placenta [196] and induce an ASD-like state in the offspring when administered to pregnant mice [25]. Overexpression of IL-6 in transgenic mice causes neuroanatomical and neurophysiological alterations associated with neurological disease [204]. Immunohistochemistry studies confirmed that IL-6 is raised in the cerebellum of autistic brain [205].
Autoantibodies to Brain in ASD
There is an alternative way of interpreting some of the data. There is no doubt that autoantibodies present during fetal life [206], or during inflammatory episodes when blood-brain barrier function is compromised [176], can alter CNS function [207]. Such antibodies have been shown in autism [206] and, as suggested by a murine model, might explain the high frequency of learning disorders in the offspring of mothers suffering from systemic lupus erythematosus (SLE) [208]. The association between autoimmune disease and psychiatric disease has been confirmed in a massive recent study [209]. Patients with MDD have not only a higher frequency of brain-reactive antibodies, but also an increased circulating Th17/Treg ratio [21]. In short, the relationship between immunodysregulation and psychiatric disease might involve additional autoantibody-mediated damage when autoimmunity is one of the forms of chronic inflammatory disorder present in a given individual, even if subclinical. The evidence for this in some cases of ASD is strong [206].
GI Symptoms and Immunoregulation in ASD
Gastrointestinal symptoms are common in ASD. These include diarrhea, constipation, vomiting/reflux, abdominal pain/discomfort, gaseousness, and unusually foul-smelling stools [210] and are similar to the symptoms of irritable bowel syndrome (IBS), which affects 10–20 % of the US population [discussed in 211]. While symptoms are not in doubt, there is controversy about whether these correlate with detectable inflammation in the gut mucosa [discussed in 211]. The observation that low-grade endotoxemia occurs in patients with severe autism tends to suggest that some gut inflammation might be present [212]. Meanwhile the use of modern culture-independent methods has revealed that the gut microbiota of autistics is abnormal [213–215]. Some authors argue that this abnormality might lead to excessive production and absorption of short chain fatty acids (SCFA; for example propionic acid) that in experimental animals induce autism-like states [216].
Brain Development and Schizophrenia
The etiology of schizophrenia is not known but the predominant view is that, like at least some cases of ASD, it can involve abnormalities in brain development occurring during fetal/neonatal life (Fig. 15.4) long before manifestation of the illness in adolescence or early adulthood [173, 217]. There is correlational evidence for this. Raised maternal levels of IL-8 and TNF (but not of IL-6 and IL-1β) in mid gestation were associated with psychosis in the children [173, 218, 219]. As in ASD there is evidence for immunoregulatory problems in adults with schizophrenia, though these are less extreme than in ASD [199]. Interestingly a relationship between ASD and schizophrenia has been postulated that attributes the differing disease manifestations to relative expression of maternal or paternal copies of imprinted genes [220]. It might be possible to reconcile this hypothesis with the differing degrees of immunodysregulation characteristic of the two conditions [199]. It is of particular interest that some genes that predispose to ASD do not need to be expressed in the fetus, involve the immune system and probably act during pregnancy [221].
Conclusions
The study of the gut microbiota by modern molecular methods, and the modulation of the microbiota by modern lifestyle and dietary habits, have led to a vast expansion of our knowledge, and to a tendency for the study of the microbiota to be seen as an independent medical discipline. Meanwhile, the original hygiene hypothesis was seen as a narrow concept dealing mostly with factors affecting allergic disorders in childhood, while an offshoot of the hygiene hypothesis, sometimes known as the “helminth hypothesis” has been applied mostly to the increases in IBD and MS. In this chapter we suggest that these concepts need to be studied together, or even unified under one heading such as the “Old Friends”, or “biodiversity” mechanism. They all deal with the issue of the education and regulation of the immune system by microbial contact. The gut microbiota is certainly a major component of this system, but it acts in concert with other environmental inputs that regulate the immune system, including organisms that never enter the gut. By seeing the whole picture we may be able to determine whether immunodysregulation due to divergence of our microbial exposure from that with which we evolved is able to explain the worrying and parallel increases in chronic inflammatory disorders and inflammation-linked psychiatric disorders in high-income countries.
Abbreviations
- ASD:
-
Autism spectrum disorders
- BH4:
-
Tetrahydrobiopterin
- CD:
-
Crohn’s disease
- CNS:
-
Central nervous system
- CRH:
-
Corticotropin-releasing hormone
- CRP:
-
C-reactive protein
- dACC:
-
Dorsal anterior cingulate cortex
- DC:
-
Dendritic cells
- DCreg:
-
Regulatory dendritic cells
- fMRI:
-
Functional magnetic resonance imaging
- GABA:
-
g-Aminobutyric acid
- GCR:
-
Glucocorticoid resistance
- HPA:
-
Hypothalamic-pituitary adrenal axis
- IBD:
-
Inflammatory bowel disease
- IBS:
-
Irritable bowel syndrome
- IDO:
-
Indoleamine-2,3-dioxygenase
- IFN-α:
-
Interferon-alpha
- IL:
-
Interleukin
- LPS:
-
Lipopolysaccharide
- MCP-1:
-
Monocyte chemoattractant protein-1
- MS:
-
Multiple sclerosis
- NO:
-
Nitric oxide
- Nod1:
-
Nucleotide-binding oligomerization domain-containing protein-1
- PBMCs:
-
Peripheral blood monocyte cells
- PET:
-
Positron emission tomography
- SCFA:
-
Short chain fatty acids
- SLE:
-
Systemic lupus erythematosus
- SNP:
-
Single nucleotide polymorphisms
- SNS:
-
Sympathetic nervous system
- SSRI:
-
Selective serotonin reuptake inhibitors
- T1D:
-
Type 1 diabetes
- TNF:
-
Tumor necrosis factor
- Treg:
-
Regulatory T cells
- UC:
-
Ulcerative colitis
- XLAAD:
-
X-linked autoimmunity-allergic dysregulation syndrome
References
Blackley CH (1873) Experimental researches on the causes and nature of catarrhus aestivus (Hay-fever and Hay-asthma). Baillière Tindall and Cox, London
Radon K, Windstetter D, Poluda AL, Mueller B, von Mutius E, Koletzko S (2007) Contact with farm animals in early life and juvenile inflammatory bowel disease: a case-control study. Pediatrics 120(2):354–361
Schaub B, Liu J, Hoppler S, Schleich I, Huehn J, Olek S et al (2009) Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J Allergy Clin Immunol 123(4):774–782.e5
Ege MJ, Mayer M, Normand AC, Genuneit J, Cookson WO, Braun-Fahrlander C et al (2011) Exposure to environmental microorganisms and childhood asthma. N Engl J Med 364(8):701–709
Stene LC, Nafstad P (2001) Relation between occurrence of type 1 diabetes and asthma. Lancet 357:607
Weinstock JV, Elliott DE (2009) Helminths and the IBD hygiene hypothesis. Inflamm Bowel Dis 15(1):128–133
Rook GAW (2010) 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: Darwinian medicine and the ‘hygiene’ or ‘old friends’ hypothesis. Clin Exp Immunol 160(1):70–79
von Hertzen L, Hanski I, Haahtela T (2011) Natural immunity. Biodiversity loss and inflammatory diseases are two global megatrends that might be related. EMBO Rep 12(11):1089–1093
Babu S, Blauvelt CP, Kumaraswami V, Nutman TB (2006) Regulatory networks induced by live parasites impair both Th1 and Th2 pathways in patent lymphatic filariasis: implications for parasite persistence. J Immunol 176(5):3248–3256
Wildin RS, Smyk-Pearson S, Filipovich AH (2002) Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet 39(8):537–545
Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM (2013) Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity 38(2):373–383
Gurven M, Kaplan H, Winking J, Finch C, Crimmins EM (2008) Aging and inflammation in two epidemiological worlds. J Gerontol A Biol Sci Med Sci 63(2):196–199
McDade TW, Tallman PS, Madimenos FC, Liebert MA, Cepon TJ, Sugiyama LS et al (2012) Analysis of variability of high sensitivity C-reactive protein in lowland Ecuador reveals no evidence of chronic low-grade inflammation. Am J Hum Biol 24:675–681
Rook G, Raison CL, Lowry CA (2013) Childhood microbial experience, immunoregulation, inflammation and adult susceptibility to psychosocial stressors and depression in rich and poor countries. Evol Med Public Health 2013:14–17
McDade TW, Rutherford J, Adair L, Kuzawa CW (2010) Early origins of inflammation: microbial exposures in infancy predict lower levels of C-reactive protein in adulthood. Proc Biol Sci 277(1684):1129–1137
Gimeno D, Kivimaki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A et al (2009) Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol Med 39(3):413–423
Rook GAW, Lowry CA (2008) The hygiene hypothesis and psychiatric disorders. Trends Immunol 29:150–158
Raison CL, Lowry CA, Rook GAW (2010) Inflammation, sanitation and consternation: loss of contact with co-evolved, tolerogenic micro-organisms and the pathophysiology and treatment of major depression. Arch Gen Psychiatry 67(12):1211–1224
Maes M, Scharpe S, Van Grootel L, Uyttenbroeck W, Cooreman W, Cosyns P et al (1992) Higher alpha 1-antitrypsin, haptoglobin, ceruloplasmin and lower retinol binding protein plasma levels during depression: further evidence for the existence of an inflammatory response during that illness. J Affect Disord 24(3):183–192
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65(9):732–741
Chen Y, Jiang T, Chen P, Ouyang J, Xu G, Zeng Z et al (2011) Emerging tendency towards autoimmune process in major depressive patients: a novel insight from Th17 cells. Psychiatry Res 188(2):224–230
Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH et al (2006) Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163(9):1630–1633
Musselman DL, Lawson DH, Gumnick JF, Manatunga AK, Penna S, Goodkin RS et al (2001) Paroxetine for the prevention of depression induced by high-dose interferon alfa. N Engl J Med 344(13):961–966
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF et al (2013) A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70:31–41
Smith SE, Li J, Garbett K, Mirnics K, Patterson PH (2007) Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 27(40):10695–10702
Marsland AL, Gianaros PJ, Abramowitch SM, Manuck SB, Hariri AR (2008) Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol Psychiatry 64(6):484–490
Braida D, Sacerdote P, Panerai AE, Bianchi M, Aloisi AM, Iosue S et al (2004) Cognitive function in young and adult IL (interleukin)-6 deficient mice. Behav Brain Res 153(2):423–429
Sparkman NL, Buchanan JB, Heyen JR, Chen J, Beverly JL, Johnson RW (2006) Interleukin-6 facilitates lipopolysaccharide-induced disruption in working memory and expression of other proinflammatory cytokines in hippocampal neuronal cell layers. J Neurosci 26(42):10709–10716
McEwen BS, Gianaros PJ (2010) Central role of the brain in stress and adaptation: links to socioeconomic status, health, and disease. Ann N Y Acad Sci 1186:190–222
O’Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho AM, Quigley EM et al (2009) Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry 65(3):263–267
Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A (2008) Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry 65(4):409–415
McDade TW, Hoke M, Borja JB, Adair LS, Kuzawa CW (2013) Do environments in infancy moderate the association between stress and inflammation in adulthood? Preliminary evidence from a birth cohort in the Philippines. Brain Behav Immun 31:23–30
Compton WM, Conway KP, Stinson FS, Grant BF (2006) Changes in the prevalence of major depression and comorbid substance use disorders in the United States between 1991–1992 and 2001–2002. Am J Psychiatry 163(12):2141–2147
Ustun TB, Ayuso-Mateos JL, Chatterji S, Mathers C, Murray CJ (2004) Global burden of depressive disorders in the year 2000. Br J Psychiatry 184:386–392
McDade TW, Borja JB, Adair L, Kuzawa CW (2013) Depressive symptoms are not associated with inflammation in younger and older adults in the Philippines. Evol Med Public Health 2013:18–23
Peen J, Schoevers RA, Beekman AT, Dekker J (2010) The current status of urban-rural differences in psychiatric disorders. Acta Psychiatr Scand 121(2):84–93
Kovess-Masfety V, Lecoutour X, Delavelle S (2005) Mood disorders and urban/rural settings: comparisons between two French regions. Soc Psychiatry Psychiatr Epidemiol 40(8):613–618
McGrath J, Saha S, Welham J, El Saadi O, MacCauley C, Chant D (2004) A systematic review of the incidence of schizophrenia: the distribution of rates and the influence of sex, urbanicity, migrant status and methodology. BMC Med 2:13
Lauritsen MB, Pedersen CB, Mortensen PB (2005) Effects of familial risk factors and place of birth on the risk of autism: a nationwide register-based study. J Child Psychol Psychiatry 46(9):963–971
Riedler J, Braun-Fahrlander C, Eder W, Schreuer M, Waser M, Maisch S et al (2001) Exposure to farming in early life and development of asthma and allergy: a cross-sectional survey. Lancet 358(9288):1129–1133
Ege MJ, Herzum I, Buchele G, Krauss-Etschmann S, Lauener RP, Roponen M et al (2008) Prenatal exposure to a farm environment modifies atopic sensitization at birth. J Allergy Clin Immunol 122(2):407–412, 12 e1–e4
Nicolaou N, Siddique N, Custovic A (2005) Allergic disease in urban and rural populations: increasing prevalence with increasing urbanization. Allergy 60(11):1357–1360
Hou JK, El-Serag H, Thirumurthi S (2009) Distribution and manifestations of inflammatory bowel disease in Asians, Hispanics, and African Americans: a systematic review. Am J Gastroenterol 104(8):2100–2109
Beebe GW, Kurtzke JF, Kurland LT, Auth TL, Nagler B (1967) Studies on the natural history of multiple sclerosis. 3. Epidemiologic analysis of the army experience in World War II. Neurology 17(1):1–17
Antonovsky A, Leibowitz U, Smith HA, Medalie JM, Balogh M, Kats R et al (1965) Epidemiologic Study of Multiple Sclerosis in Israel. I. An overall review of methods and findings. Arch Neurol 13:183–193
Lowis GW (1990) The social epidemiology of multiple sclerosis. Sci Total Environ 90:163–190
Rottem M, Szyper-Kravitz M, Shoenfeld Y (2005) Atopy and asthma in migrants. Int Arch Allergy Immunol 136(2):198–204
Soderstrom U, Aman J, Hjern A (2012) Being born in Sweden increases the risk for type 1 diabetes – a study of migration of children to Sweden as a natural experiment. Acta Paediatr 101(1):73–77
Ahlgren C, Oden A, Lycke J (2012) A nationwide survey of the prevalence of multiple sclerosis in immigrant populations of Sweden. Mult Scler 18:1099–1107
Breslau J, Borges G, Tancredi D, Saito N, Kravitz R, Hinton L et al (2011) Migration from Mexico to the United States and subsequent risk for depressive and anxiety disorders: a cross-national study. Arch Gen Psychiatry 68(4):428–433
Dealberto MJ (2010) Ethnic origin and increased risk for schizophrenia in immigrants to countries of recent and longstanding immigration. Acta Psychiatr Scand 121(5):325–339
Keen DV, Reid FD, Arnone D (2010) Autism, ethnicity and maternal immigration. Br J Psychiatry 196(4):274–281
Breslau J, Borges G, Hagar Y, Tancredi D, Gilman S (2009) Immigration to the USA and risk for mood and anxiety disorders: variation by origin and age at immigration. Psychol Med 39(7):1117–1127
Vega WA, Sribney WM, Aguilar-Gaxiola S, Kolody B (2004) 12-month prevalence of DSM-III-R psychiatric disorders among Mexican Americans: nativity, social assimilation, and age determinants. J Nerv Ment Dis 192(8):532–541
Coid JW, Kirkbride JB, Barker D, Cowden F, Stamps R, Yang M et al (2008) Raised incidence rates of all psychoses among migrant groups: findings from the East London first episode psychosis study. Arch Gen Psychiatry 65(11):1250–1258
Veling W, Hoek HW, Selten JP, Susser E (2011) Age at migration and future risk of psychotic disorders among immigrants in the Netherlands: a 7-year incidence study. Am J Psychiatry 168(12):1278–1285
Cantor-Graae E, Selten JP (2005) Schizophrenia and migration: a meta-analysis and review. Am J Psychiatry 162(1):12–24
Milo R, Kahana E (2010) Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev 9(5):A387–A394
Gale CR, Martyn CN (1995) Migrant studies in multiple sclerosis. Prog Neurobiol 47(4–5):425–448
Cabre P (2009) Environmental changes and epidemiology of multiple sclerosis in the French West Indies. J Neurol Sci 286(1–2):58–61
Dean G (1967) Annual incidence, prevalence, and mortality of multiple sclerosis in white South-African-born and in white immigrants to South Africa. Br Med J 2(5554):724–730
Hjern A, Rasmussen F, Hedlin G (1999) Age at adoption, ethnicity and atopic disorder: a study of internationally adopted young men in Sweden. Pediatr Allergy Immunol 10(2):101–106
Eldeirawi K, McConnell R, Furner S, Freels S, Stayner L, Hernandez E et al (2009) Associations of doctor-diagnosed asthma with immigration status, age at immigration, and length of residence in the United States in a sample of Mexican American School Children in Chicago. J Asthma 46(8):796–802
Pereg D, Tirosh A, Lishner M, Goldberg A, Shochat T, Confino-Cohen R (2008) Prevalence of asthma in a large group of Israeli adolescents: influence of country of birth and age at migration. Allergy 63(8):1040–1045
Li X, Sundquist J, Hemminki K, Sundquist K (2011) Risk of inflammatory bowel disease in first- and second-generation immigrants in Sweden: a nationwide follow-up study. Inflamm Bowel Dis 17(8):1784–1791
Carr I, Mayberry JF (1999) The effects of migration on ulcerative colitis: a three-year prospective study among Europeans and first- and second-generation South Asians in Leicester (1991–1994). Am J Gastroenterol 94(10):2918–2922
Rook GAW (2009) The broader implications of the hygiene hypothesis. Immunology 126:3–11
Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9(5):313–323
Osada Y, Kanazawa T (2010) Parasitic helminths: new weapons against immunological disorders. J Biomed Biotechnol 2010:743–758
Karimi K, Inman MD, Bienenstock J, Forsythe P (2009) Lactobacillus reuteri-induced regulatory T cells protect against an allergic airway response in mice. Am J Respir Crit Care Med 179(3):186–193
Allen RG, Lafuse WP, Galley JD, Ali MM, Ahmer BM, Bailey MT (2012) The intestinal microbiota are necessary for stressor-induced enhancement of splenic macrophage microbicidal activity. Brain Behav Immun 26(3):371–382
Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M (2011) Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun 25(3):397–407
Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN (2010) Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16(2):228–231
Kim OY, Monsel A, Bertrand M, Cavaillon JM, Coriat P, Adib-Conquy M (2009) Translocation of bacterial NOD2 agonist and its link with inflammation. Crit Care 13(4):R124
Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8(12):958–969
Loke P, MacDonald AS, Robb A, Maizels RM, Allen JE (2000) Alternatively activated macrophages induced by nematode infection inhibit proliferation via cell to cell contact. Eur J Immunol 30:2669–2678
Elliott DE, Weinstock JV (2012) Helminth-host immunological interactions: prevention and control of immune-mediated diseases. Ann N Y Acad Sci 1247:83–96
Schnoeller C, Rausch S, Pillai S, Avagyan A, Wittig BM, Loddenkemper C et al (2008) A helminth immunomodulator reduces allergic and inflammatory responses by induction of IL-10-producing macrophages. J Immunol 180(6):4265–4272
Smith P, Mangan NE, Walsh CM, Fallon RE, McKenzie ANJ, van Rooijen N et al (2007) Infection with a helminth parasite prevents experimental colitis via a macrophage-mediated mechanism. J Immunol 178:4557–4566
Mangan NE, Fallon RE, Smith P, van Rooijen N, McKenzie AN, Fallon PG (2004) Helminth infection protects mice from anaphylaxis via IL-10-producing B cells. J Immunol 173(10):6346–6356
Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF (2008) Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest 118(10):3420–3430
Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF (2008) A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 28(5):639–650
Amu S, Saunders SP, Kronenberg M, Mangan NE, Atzberger A, Fallon PG (2010) Regulatory B cells prevent and reverse allergic airway inflammation via FoxP3-positive T regulatory cells in a murine model. J Allergy Clin Immunol 125(5):1114–1124 e8
Yanaba K, Bouaziz JD, Matsushita T, Magro CM, St Clair EW, Tedder TF (2008) B-lymphocyte contributions to human autoimmune disease. Immunol Rev 223:284–299
Correale J, Farez M, Razzitte G (2008) Helminth infections associated with multiple sclerosis induce regulatory B cells. Ann Neurol 64(2):187–199
Di Giacinto C, Marinaro M, Sanchez M, Strober W, Boirivant M (2005) Probiotics ameliorate recurrent Th1-mediated murine colitis by inducing IL-10 and IL-10-dependent TGF-beta-bearing regulatory cells. J Immunol 174(6):3237–3246
Hart AL, Lammers K, Brigidi P, Vitali B, Rizzello F, Gionchetti P et al (2004) Modulation of human dendritic cell phenotype and function by probiotic bacteria. Gut 53(11):1602–1609
Braat H, van den Brande J, van Tol E, Hommes D, Peppelenbosch M, van Deventer S (2004) Lactobacillus rhamnosus induces peripheral hyporesponsiveness in stimulated CD4+ T cells via modulation of dendritic cell function. Am J Clin Nutr 80(6):1618–1625
Smits HH, Engering A, van der Kleij D, de Jong EC, Schipper K, van Capel TM et al (2005) Selective probiotic bacteria induce IL-10-producing regulatory T cells in vitro by modulating dendritic cell function through dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin. J Allergy Clin Immunol 115(6):1260–1267
Le Bert N, Chain BM, Rook G, Noursadeghi M (2011) DC priming by M. vaccae inhibits Th2 responses in contrast to specific TLR2 priming and is associated with selective activation of the CREB pathway. PLoS One 6(4):e18346
Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL et al (2008) Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med 205(9):2139–2149
Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR et al (2007) Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204(8):1775–1785
Mann ER, Landy JD, Bernardo D, Peake ST, Hart AL, Al-Hassi HO et al (2013) Intestinal dendritic cells: their role in intestinal inflammation, manipulation by the gut microbiota and differences between mice and men. Immunol Lett 150(1–2):30–40
Smelt MJ, de Haan BJ, Bron PA, van Swam I, Meijerink M, Wells JM et al (2012) L. plantarum, L. salivarius, and L. lactis attenuate Th2 responses and increase Treg frequencies in healthy mice in a strain dependent manner. PLoS One 7(10):e47244
Correale J, Farez M (2007) Association between parasite infection and immune responses in multiple sclerosis. Ann Neurol 61(2):97–108
Correale J, Farez MF (2011) The impact of parasite infections on the course of multiple sclerosis. J Neuroimmunol 233:6–11
Fleming J, Isaak A, Lee J, Luzzio C, Carrithers M, Cook T et al (2011) Probiotic helminth administration in relapsing-remitting multiple sclerosis: a phase 1 study. Mult Scler 17(6):743–754
Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA et al (2011) The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332(6032):974–977
Grainger JR, Smith KA, Hewitson JP, McSorley HJ, Harcus Y, Filbey KJ et al (2010) Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J Exp Med 207(11):2331–2341
Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y et al (2011) Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331:337–341
Meadow JF, Bateman AC, Herkert KM, O’Connor TK, Green JL (2013) Significant changes in the skin microbiome mediated by the sport of roller derby. Peer J. Doi:10.7717/peerj.53.
Song SJ, Lauber C, Costello EK, Lozupone CA, Humphrey G, Berg-Lyons D et al (2013) Cohabiting family members share microbiota with one another and with their dogs. Elife 2:e00458
Maslowski KM, Mackay CR (2011) Diet, gut microbiota and immune responses. Nat Immunol 12(1):5–9
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S et al (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 107(33):14691–14696
Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6(11):e280
Cani PD, Delzenne NM (2011) The gut microbiome as therapeutic target. Pharmacol Ther 130(2):202–212
Hildebrand F, Nguyen TL, Brinkman B, Yunta RG, Cauwe B, Vandenabeele P et al (2013) Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol 14(1):R4
Abrahamsson TR, Jakobsson HE, Andersson AF, Bjorksten B, Engstrand L, Jenmalm MC (2012) Low diversity of the gut microbiota in infants with atopic eczema. J Allergy Clin Immunol 129(2):434–440, 40 e1–e2
Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L et al (2006) Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55(2):205–211
Nemoto H, Kataoka K, Ishikawa H, Ikata K, Arimochi H, Iwasaki T et al (2012) Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig Dis Sci 57(11):2955–2964
Rehman A, Lepage P, Nolte A, Hellmig S, Schreiber S, Ott SJ (2010) Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J Med Microbiol 59(Pt 9):1114–1122
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al (2009) A core gut microbiome in obese and lean twins. Nature 457(7228):480–484
Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, Cusack S et al (2012) Gut microbiota composition correlates with diet and health in the elderly. Nature 488(7410):178–184
Aichbhaumik N, Zoratti EM, Strickler R, Wegienka G, Ownby DR, Havstad S et al (2008) Prenatal exposure to household pets influences fetal immunoglobulin E production. Clin Exp Allergy 38(11):1787–1794
Ege MJ, Mayer M, Schwaiger K, Mattes J, Pershagen G, van Hage M et al (2012) Environmental bacteria and childhood asthma. Allergy 67:1565–1571
Hanski I, von Hertzen L, Fyhrquist N, Koskinen K, Torppa K, Laatikainen T et al (2012) Environmental biodiversity, human microbiota, and allergy are interrelated. Proc Natl Acad Sci U S A 109(21):8334–8339
Korhonen L, Kondrashova A, Tauriainen S, Haapala AM, Huhtala H, Ilonen J et al (2013) Enterovirus infections in early childhood and the risk of atopic disease – a nested case-control study. Clin Exp Allergy 43(6):625–632
Matricardi PM, Rosmini F, Riondino S, Fortini M, Ferrigno L, Rapicetta M et al (2000) Exposure to foodborne and orofecal microbes versus airborne viruses in relation to atopy and allergic asthma; epidemiological study. BMJ 320:412–417
Hesselmar B, Sjoberg F, Saalman R, Aberg N, Adlerberth I, Wold AE (2013) Pacifier cleaning practices and risk of allergy development. Pediatrics 131:e1829–e1837
Magnus MC, Haberg SE, Stigum H, Nafstad P, London SJ, Vangen S et al (2011) Delivery by Cesarean section and early childhood respiratory symptoms and disorders: the Norwegian mother and child cohort study. Am J Epidemiol 174(11):1275–1285
Guibas GV et al (2013) Conception via in vitro fertilization and delivery by cesarean section are associated with pediatric asthma incidence. Clin Exp Allergy 43:1058–1066
Stensballe LG, Simonsen J, Jensen SM, Bonnelykke K, Bisgaard H (2013) Use of antibiotics during pregnancy increases the risk of asthma in early childhood. J Pediatr 162(4):832–838 e3
Russell G, Helms PJ (1997) Trend in occurrence of asthma among children and young adults. Reporting of common respiratory and atopic symptoms has increased. BMJ 315(7114):1014–1015
Metsala J, Lundqvist A, Virta LJ, Kaila M, Gissler M, Virtanen SM (2013) Mother’s and offspring’s use of antibiotics and infant allergy to cow’s milk. Epidemiology 24(2):303–309
Hviid A, Svanstrom H, Frisch M (2011) Antibiotic use and inflammatory bowel diseases in childhood. Gut 60(1):49–54
Shaw SY, Blanchard JF, Bernstein CN (2010) Association between the use of antibiotics in the first year of life and pediatric inflammatory bowel disease. Am J Gastroenterol 105(12):2687–2692
Mulder IE, Schmidt B, Stokes CR, Lewis M, Bailey M, Aminov RI et al (2009) Environmentally-acquired bacteria influence microbial diversity and natural innate immune responses at gut surfaces. BMC Biol 7:79
Hemarajata P, Versalovic J (2013) Effects of probiotics on gut microbiota: mechanisms of intestinal immunomodulation and neuromodulation. Therap Adv Gastroenterol 6(1):39–51
Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC et al (2008) Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455(7216):1109–1113
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ et al (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145(5):745–757
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S et al (2010) Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328(5975):228–231
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T et al (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482(7384):179–185
Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S et al (2007) Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131(1):33–45
Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W et al (2012) Compartmentalized control of skin immunity by resident commensals. Science 337(6098):1115–1119
Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL (2013) The microbiome extends to subepidermal compartments of normal skin. Nat Commun 4:1431
Bailey MT, Engler H, Sheridan JF (2006) Stress induces the translocation of cutaneous and gastrointestinal microflora to secondary lymphoid organs of C57BL/6 mice. J Neuroimmunol 171(1–2):29–37
Friberg IM, Bradley JE, Jackson JA (2010) Macroparasites, innate immunity and immunoregulation: developing natural models. Trends Parasitol 26:540–549
Jackson JA, Friberg IM, Bolch L, Lowe A, Ralli C, Harris PD et al (2009) Immunomodulatory parasites and toll-like receptor-mediated tumour necrosis factor alpha responsiveness in wild mammals. BMC Biol 7:16
Harnett MM, Melendez AJ, Harnett W (2010) The therapeutic potential of the filarial nematode-derived immunodulator, ES-62 in inflammatory disease. Clin Exp Immunol 159(3):256–267
Kron MA, Metwali A, Vodanovic-Jankovic S, Elliott D (2013) Nematode AsnRS resolves intestinal inflammation in murine T-cell transfer colitis. Clin Vaccine Immunol 20:276–281
Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF et al (2010) Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature 467(7314):420–425
Sotgiu S, Angius A, Embry A, Rosati G, Musumeci S (2008) Hygiene hypothesis: innate immunity, malaria and multiple sclerosis. Med Hypotheses 70(4):819–825
Fumagalli M, Pozzoli U, Cagliani R, Comi GP, Riva S, Clerici M et al (2009) Parasites represent a major selective force for interleukin genes and shape the genetic predisposition to autoimmune conditions. J Exp Med 206(6):1395–1408
Barnes KC, Grant AV, Gao P (2005) A review of the genetic epidemiology of resistance to parasitic disease and atopic asthma: common variants for common phenotypes? Curr Opin Allergy Clin Immunol 5(5):379–385
Moller M, Gravenor MB, Roberts SE, Sun D, Gao P, Hopkin JM (2007) Genetic haplotypes of Th-2 immune signalling link allergy to enhanced protection to parasitic worms. Hum Mol Genet 16(15):1828–1836
Fredericks CA, Drabant EM, Edge MD, Tillie JM, Hallmayer J, Ramel W et al (2010) Healthy young women with serotonin transporter SS polymorphism show a pro-inflammatory bias under resting and stress conditions. Brain Behav Immun 24:350–357
Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW et al (2009) Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med 206(9):1883–1897
Pace TW, Hu F, Miller AH (2007) Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun 21(1):9–19
Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS et al (2012) Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci U S A 109:5995–5999
Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D et al (2003) A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A 100(4):1920–1925
Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF (2012) Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun 26(7):1150–1159
Merali Z, Du L, Hrdina P, Palkovits M, Faludi G, Poulter MO et al (2004) Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J Neurosci 24(6):1478–1485
Lee R, Geracioti TD Jr, Kasckow JW, Coccaro EF (2005) Childhood trauma and personality disorder: positive correlation with adult CSF corticotropin-releasing factor concentrations. Am J Psychiatry 162(5):995–997
Gareau MG, Silva MA, Perdue MH (2008) Pathophysiological mechanisms of stress-induced intestinal damage. Curr Mol Med 8(4):274–281
Teitelbaum AA, Gareau MG, Jury J, Yang PC, Perdue MH (2008) Chronic peripheral administration of corticotropin-releasing factor causes colonic barrier dysfunction similar to psychological stress. Am J Physiol 295(3):G452–G459
Wallon C, Yang PC, Keita AV, Ericson AC, McKay DM, Sherman PM et al (2008) Corticotropin-releasing hormone (CRH) regulates macromolecular permeability via mast cells in normal human colonic biopsies in vitro. Gut 57(1):50–58
Calcagni E, Elenkov I (2006) Stress system activity, innate and T helper cytokines, and susceptibility to immune-related diseases. Ann N Y Acad Sci 1069:62–76
Zbytek B, Slominski AT (2007) CRH mediates inflammation induced by lipopolysaccharide in human adult epidermal keratinocytes. J Invest Dermatol 127(3):730–732
Broadhurst MJ, Ardeshir A, Kanwar B, Mirpuri J, Gundra UM, Leung JM et al (2012) Therapeutic helminth infection of macaques with idiopathic chronic diarrhea alters the inflammatory signature and mucosal microbiota of the colon. PLoS Pathog 8(11):e1003000
Hayakawa M, Asahara T, Henzan N, Murakami H, Yamamoto H, Mukai N et al (2011) Dramatic changes of the gut flora immediately after severe and sudden insults. Dig Dis Sci 56(8):2361–2365
Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA et al (2012) Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med 209:903–911
Ait-Belgnaoui A, Durand H, Cartier C, Chaumaz G, Eutamene H, Ferrier L et al (2012) Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology 37(11):1885–1895
Bailey MT, Lubach GR, Coe CL (2004) Prenatal stress alters bacterial colonization of the gut in infant monkeys. J Pediatr Gastroenterol Nutr 38(4):414–421
Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN et al (2004) Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 558(Pt 1):263–275
Heijtz RD, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A et al (2011) Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A 108(7):3047–3052
Miller AH, Haroon E, Raison CL, Felger JC (2013) Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety 30(4):297–306
Raison CL, Miller AH (2013) The evolutionary significance of depression in Pathogen Host Defense (PATHOS-D). Mol Psychiatry 18:15–37
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9(1):46–56
Rook GAW, Lowry CA, Raison CL (2013) Microbial old friends, immunoregulation and stress resilience. Evol Med Pubic Health 2013(1):46–64. Doi: 10.1093/emph/eot004
McDade TW (2012) Early environments and the ecology of inflammation. Proc Natl Acad Sci U S A 109(Suppl 2):17281–17288
Lotrich FE, Ferrell RE, Rabinovitz M, Pollock BG (2009) Risk for depression during interferon-alpha treatment is affected by the serotonin transporter polymorphism. Biol Psychiatry 65(4):344–348
Raison CL, Borisov AS, Majer M, Drake DF, Pagnoni G, Woolwine BJ et al (2009) Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry 65(4):296–303
Hagberg H, Gressens P, Mallard C (2012) Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol 71(4):444–457
D’Mello C, Le T, Swain MG (2009) Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factor alpha signaling during peripheral organ inflammation. J Neurosci 29(7):2089–2102
Kivisakk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, Ransohoff RM et al (2009) Localizing central nervous system immune surveillance: meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann Neurol 65(4):457–469
Engelhardt B, Sorokin L (2009) The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol 31(4):497–511
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13(7):465–477
Schwarcz R, Pellicciari R (2002) Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther 303(1):1–10
O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N et al (2009) Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry 14(5):511–522
Kita T, Morrison PF, Heyes MP, Markey SP (2002) Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the L-kynurenine and quinolinic acid pools in brain. J Neurochem 82(2):258–268
Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G et al (2010) CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry 15(4):393–403
Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA (2010) Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 35(13):2510–2520
Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ et al (2012) Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry 69(10):1044–1053
Felger JC, Li L, Marvar PJ, Woolwine BJ, Harrison DG, Raison CL et al (2013) Tyrosine metabolism during interferon-alpha administration: association with fatigue and CSF dopamine concentrations. Brain Behav Immun 31:153–160
Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K et al (2009) Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun 23(1):41–45
Lee M, Schwab C, McGeer PL (2011) Astrocytes are GABAergic cells that modulate microglial activity. Glia 59(1):152–165
Hertz-Picciotto I, Delwiche L (2009) The rise in autism and the role of age at diagnosis. Epidemiology 20(1):84–90
Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T et al (2011) Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 68(11):1095–1102
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474(7351):380–384
Ziats MN, Rennert OM (2011) Expression profiling of autism candidate genes during human brain development implicates central immune signaling pathways. PLoS One 6(9):e24691
Saresella M, Marventano I, Guerini FR, Mancuso R, Ceresa L, Zanzottera M et al (2009) An autistic endophenotype results in complex immune dysfunction in healthy siblings of autistic children. Biol Psychiatry 66(10):978–984
Desmond MM, Montgomery JR, Melnick JL, Cochran GG, Verniaud W (1969) Congenital rubella encephalitis. Effects on growth and early development. Am J Dis Child 118(1):30–31
Atladottir HO, Thorsen P, Schendel DE, Ostergaard L, Lemcke S, Parner ET (2010) Association of hospitalization for infection in childhood with diagnosis of autism spectrum disorders: a Danish cohort study. Arch Pediatr Adolesc Med 164(5):470–477
Brown AS, Sourander A, Hinkka-Yli-Salomaki S, McKeague IW, Sundvall J, Surcel HM (2014) Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry 19:259–264
Howerton CL, Bale TL (2012) Prenatal programing: at the intersection of maternal stress and immune activation. Horm Behav 62:237–242
Dahlgren J, Samuelsson AM, Jansson T, Holmang A (2006) Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res 60(2):147–151
Willette AA, Lubach GR, Knickmeyer RC, Short SJ, Styner M, Gilmore JH et al (2011) Brain enlargement and increased behavioral and cytokine reactivity in infant monkeys following acute prenatal endotoxemia. Behav Brain Res 219(1):108–115
Hsiao EY, McBride SW, Chow J, Mazmanian SK, Patterson PH (2012) Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc Natl Acad Sci U S A 109(31):12776–12781
Meyer U, Feldon J, Dammann O (2011) Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res 69(5 Pt 2):26R–33R
Onore C, Careaga M, Ashwood P (2012) The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun 26:383–392
Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA (2005) Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 57(1):67–81
Pardo CA, Vargas DL, Zimmerman AW (2005) Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry 17(6):485–495
Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM et al (2009) Elevated immune response in the brain of autistic patients. J Neuroimmunol 207(1–2):111–116
Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB et al (1993) Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci U S A 90(21):10061–10065
Wei H, Zou H, Sheikh AM, Malik M, Dobkin C, Brown WT et al (2011) IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation 8:52
Nordahl CW, Braunschweig D, Iosif AM, Lee A, Rogers S, Ashwood P et al (2013) Maternal autoantibodies are associated with abnormal brain enlargement in a subgroup of children with autism spectrum disorder. Brain Behav Immun 30:61–65
Margutti P, Delunardo F, Ortona E (2006) Autoantibodies associated with psychiatric disorders. Curr Neurovasc Res 3(2):149–157
Lee JY, Huerta PT, Zhang J, Kowal C, Bertini E, Volpe BT et al (2009) Neurotoxic autoantibodies mediate congenital cortical impairment of offspring in maternal lupus. Nat Med 15(1):91–96
Benros ME, Waltoft BL, Nordentoft M, Ostergaard SD, Eaton WW, Krogh J et al (2013) Autoimmune diseases and severe infections as risk factors for mood disorders: a nationwide study. JAMA Psychiatry 12:1–9
Nikolov RN, Bearss KE, Lettinga J, Erickson C, Rodowski M, Aman MG et al (2009) Gastrointestinal symptoms in a sample of children with pervasive developmental disorders. J Autism Dev Disord 39(3):405–413
Critchfield JW, van Hemert S, Ash M, Mulder L, Ashwood P (2011) The potential role of probiotics in the management of childhood autism spectrum disorders. Gastroenterol Res Pract 2011:161358
Emanuele E, Orsi P, Boso M, Broglia D, Brondino N, Barale F et al (2010) Low-grade endotoxemia in patients with severe autism. Neurosci Lett 471(3):162–165
Finegold SM, Dowd SE, Gontcharova V, Liu C, Henley KE, Wolcott RD et al (2010) Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 16(4):444–453
Adams JB, Johansen LJ, Powell LD, Quig D, Rubin RA (2011) Gastrointestinal flora and gastrointestinal status in children with autism–comparisons to typical children and correlation with autism severity. BMC Gastroenterol 11:22
Williams BL, Hornig M, Parekh T, Lipkin WI (2012) Application of novel PCR-based methods for detection, quantitation, and phylogenetic characterization of Sutterella species in intestinal biopsy samples from children with autism and gastrointestinal disturbances. MBio 3(1)
Thomas RH, Meeking MM, Mepham JR, Tichenoff L, Possmayer F, Liu S et al (2012) The enteric bacterial metabolite propionic acid alters brain and plasma phospholipid molecular species: further development of a rodent model of autism spectrum disorders. J Neuroinflammation 9:153
Fatemi SH, Folsom TD (2009) The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull 35(3):528–548
Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Wagner RL, Yolken RH (2001) Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav Immun 15(4):411–420
Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V et al (2004) Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry 161(5):889–895
Crespi B, Badcock C (2008) Psychosis and autism as diametrical disorders of the social brain. Behav Brain Sci 31(3):241–261, discussion 61–320
Johnson WG, Buyske S, Mars AE, Sreenath M, Stenroos ES, Williams TA et al (2009) HLA-DR4 as a risk allele for autism acting in mothers of probands possibly during pregnancy. Arch Pediatr Adolesc Med 163(6):542–546
Acknowledgements
GAWR is supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. CLR receives grant support from the National Center for Complementary and Alternative Medicine (R01AT004698, R01AT004698-01A1S1), the Depressive and Bipolar Disorder Alternative Treatment Foundation and The Brain and Behavior Research Foundation. He reports the following activities for the previous 2 years: advisory board participation and related travel funds for Pamlab, Lilly and North American Center for Continuing Education; development and presentation of disease state slides for Pamlab, Pfizer and Johnson & Johnson, as well as related travel funds for these activities; development of continuing medical education material for North American Center for Continuing Education and for CME Incite. CAL receives grant support from the National Institute of Mental Health (R01MH065702, R01MH086539, R01DA019921, R01MH075968), the National Science Foundation (NSF- IOS 0921969), the Depressive and Bipolar Disorder Alternative Treatment Foundation, and is the recipient of an NSF CAREER Award (NSF-IOS 0845550) and a NARSAD, Brain & Behavior Research Foundation 2010 Young Investigator Award. He reports the following activities for the previous 2 years: consultant for Enlight Biosciences.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer New York
About this chapter
Cite this chapter
Rook, G.A.W., Raison, C.L., Lowry, C.A. (2014). Microbiota, Immunoregulatory Old Friends and Psychiatric Disorders. In: Lyte, M., Cryan, J. (eds) Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease. Advances in Experimental Medicine and Biology(), vol 817. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0897-4_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0897-4_15
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0896-7
Online ISBN: 978-1-4939-0897-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)