
Abstract
Reproductive function is tightly regulated by an intricate network of central and peripheral factors; however, the precise mechanism triggering critical reproductive events, such as puberty onset, remains largely unknown. Recently, the neuropeptides kisspeptin (encoded by Kiss1) and neurokinin B (NKB, encoded by TAC3 in humans and Tac2 in rodents) have been placed as essential gatekeepers of puberty. Studies in humans and rodents have revealed that loss-of-function mutations in the genes encoding either kisspeptin and NKB or their receptors, Kiss1r and neurokinin 3 receptor (NK3R), lead to impaired sexual maturation and infertility. Kisspeptin, NKB, and dynorphin A are co-expressed in neurons of the arcuate nucleus (ARC), so-called Kisspeptin/NKB/Dyn (KNDy) neurons. Importantly, these neurons also co-express NK3R. Compelling evidence suggests a stimulatory role of NKB (or the NK3R agonist, senktide) on LH release in a number of species. This effect is likely mediated by autosynaptic inputs of NKB on KNDy neurons to induce the secretion of gonadotropin-releasing hormone (GnRH) in a kisspeptin-dependent manner, with the coordinated actions of other neuroendocrine factors, such as dynorphin, glutamate, or GABA. Thus, we have proposed a model in which NKB feeds back to the KNDy neuron to shape the pulsatile release of kisspeptin, and hence GnRH, in a mechanism also dependent on the sex steroid level. Additionally, NKB may contribute to the regulation of the reproductive function by metabolic cues. Investigating how NKB and kisspeptin interact to regulate the gonadotropic axis will offer new insights into the control of GnRH release during puberty onset and the maintenance of the reproductive function in adulthood, offering a platform for the understanding and treatment of a number of reproductive disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction (Why Study Neurokinin B in a Kisspeptin Setting?)
Perpetuation of the species is an essential, but extremely energy costly, endeavor for most animals—especially for mammals [1, 2]. For this reason, it is not surprising that a vast array of neurotransmitters and endocrine factors are devoted to the precise control of the gonadotropic axis and, hence, translate the information of environmental and internal cues into a specific timing and pattern of gonadotropin-releasing hormone (GnRH) release. A key aspect that deserves to be emphasized is the incapability of GnRH neurons to show a direct response to some important modulators of reproductive function, e.g., the negative feedback of sex steroids [3, 4]. In recent years, Kiss1 neurons have been shown to be—directly or indirectly—receptive to numerous regulatory cues, including sex steroids and metabolic and circadian factors [5]. This has placed kisspeptin in the spotlight to play a major role as a regulator of GnRH release. Yet, it is conceivable that such a critical function for the species as reproduction cannot rely exclusively on a single molecule (kisspeptin) and, hence, a number of essential “fine-tuners” and “fail-safes” might exist to ensure reproductive success. Indeed, in 2009, the endocrine community witnessed the emergence of neurokinin B (NKB) as a critical player in the control of gonadotropin release [6]. In another example of reverse translational research—suitably called “from bedside to benchside”—human genetic studies revealed that patients bearing inactivating mutations in the gene encoding NKB (TAC3) or its receptor, neurokinin 3 receptor (NK3R, encoded by TACR3), displayed hypogonadotropic hypogonadism and closely resembled the phenotype of patients with loss-of-function mutations in the genes that encode kisspeptin (KISS1) and the kisspeptin receptor (KISS1R, also known as GPR54) [6–13]. Some of these findings have also been partially recapitulated in Tacr3 null mice [14], indicating that the NKB/NK3R system plays a role in the control of gonadotropin secretion in different species. Altogether, given the clear parallelism in the reproductive phenotype of humans (and mice) suffering from congenital inactivation of the kisspeptin/Kiss1r or the NKB/NK3R systems, it is conceivable that the actions of these two neuroendocrine systems interact to control GnRH release. As a result, significant efforts in the field have been recently devoted to puzzle out this interaction in what may constitute a nodal regulatory center in the control of reproductive function. This chapter intends to offer a concise overview of the latest achievements in the characterization of the reproductive facet of the NKB/NK3R system, with attention paid to the implications for the central mechanisms that govern GnRH release.
The NKB/NK3R System: Structure and Distribution in the Brain
Neurokinin B
NKB belongs to the tachykinin family of peptides that initially included the neuropeptides substance P (SP), neurokinin A (NKA), and NKB and more recently, endokinins and hemokinins [15]. Tachykinins are peptides comprised of 10–11 amino acid residues in length that share a common carboxy-terminal amino acid sequence (Phe-X-Gly-Leu-Met-NH2), where X corresponds to an aliphatic (NKA and NKB) or an aromatic (SP) residue [16]. The gene-encoding NKB (TAC3 in higher primates and Tac2 in rodents) is divided into seven exons, five of which encode the precursor preprotachykinin B [16–18]. Following proteolytic cleavage, this precursor leads to, first, proneurokinin B, and then NKB (initially contained in exon 5) (Fig. 15.1) [16].

Schematic representation of the TAC3 gene depicting the exon encoding NKB
The expression of NKB mRNA and protein displays a dispersed distribution in the brain of all studied species to date. In humans, prominent populations of TAC3-positive neurons have been identified in the infundibular nucleus, anterior hypothalamic area, septal region, diagonal band of Broca, bed nucleus of the stria terminalis, amygdala, and neocortex [19, 20]. In the rat, Tac2 expression has been found in the cerebral cortex, hippocampus, amygdaloid complex, bed nucleus of the stria terminalis, ventral pallidum, habenula, olfactory bulb, dorsomedial nucleus, ventromedial nucleus, lateral hypothalamic area (LHA), caudate-putamen, medial preoptic area, arcuate nucleus (ARC), lateral mammillary bodies, superior colliculus, central gray, and dorsal horn of the spinal cord (Fig. 15.2) [21–23]. Of note, mice display a roughly similar distribution of Tac2 mRNA, although, unlike rats, mice express Tac2 neither in the hippocampus nor in the nucleus of the lateral olfactory tract [24]. Immunohistochemistry studies depicting the distribution of NKB closely parallel the neuroanatomical mapping of the gene transcript, and importantly, also offer conclusive information on the localization of NKB fibers, thus pointing to potential areas of NKB action [25–31]. Focusing on the population of NKB neurons in the ARC, projections from this nucleus have been described through a combination of tract tracing and double labeling techniques with specific known co-transmitters of NKB in this neuronal site (e.g., dynorphin and kisspeptin) [25, 27, 28, 30–32]. NKB fibers have been found to form a dense network within the ARC and the median eminence. From the ARC of the rat, NKB neurons branch to innervate rostral brain areas, such as the magnocellular and parvocellular nucleus, the anteroventral periventricular nucleus (AVPV), preoptic area, septal nuclei, and the bed nucleus of the stria terminalis [30, 33]. In addition, NKB neurons have been shown to project dorsally to the dorsomedial nucleus, periventricular nucleus, ventromedial nucleus, and LHA, and also may extend caudally to the ventral premammillary nucleus [30].

Schematic representation of a coronal section of a rat brain at the arcuate level depicting Tac2 and Tacr3 expression. SON supraoptic nucleus; BLAa basolateral nucleus of the amygdala; BMAa basomedial nucleus of the amygdala; MH medial habenular nucleus; LHA lateral hypothalamus; ZI zona incerta; VMH ventromedial hypothalamic nucleus; PVH paraventricular hypothalamic nucleus; ARC arcuate nucleus; 3V third ventricle
Neurokinin 3 Receptor
Tachykinins bind a family of G-protein-coupled receptors (GPCRs)—including neurokinin receptor 1 (NK1R), NK2R, and NK3R—to mediate their biological effect. Although interactions of the three initial tachykinins with each of these receptors have been described previously, their potency of affinity varies as follows: NK1R, SP > NKA > NKB; NK2R, NKA > NKB > SP; and NK3R, NKB > NKA > SP [34, 35]. Within the context of this review, it is conceivable that this affinity of NKB for other tachykinin receptors, even though marginal, might compromise the interpretation of the studies using NKB itself to decipher its putative roles in reproductive control. Thus, in order to overcome this limitation, the synthetic peptide senktide, a highly selective and potent agonist of NK3R [16, 34], has been systematically used in the majority of the experimental studies.
All three tachykinin receptors are encoded by genes divided into five exons with identical distribution of intronic sequences. NK3R, encoded by TACR3 in humans and Tacr3 in rodents, leads to a longer amino acid sequence than NK1R or NK2R, with a slight difference in length between species (465 and 452 residues in humans and rats, respectively) [16].
NK3R is mainly located in the central nervous system (CNS) and the spinal cord [15, 16, 36], although it has been also described in uterus, mesenteric vein, gut neurons, and placenta [37, 38]. Within the CNS of the rat, Tacr3 mRNA is detected in the olfactory bulb, cortex, amygdala, hippocampus, medial habenula, zona incerta, substantia nigra, ventral tegmental area, interpeduncular nucleus, raphe nuclei, dorsal tegmental nucleus, nucleus of the solitary tract, striatum, dentate gyrus and subiculum, medial septum, diagonal band of Broca, ventral pallidum, globus pallidus, bed nucleus of the stria terminalis, ARC, paraventricular and supraoptic nuclei of the hypothalamus, dorsal and lateral regions of the posterior hypothalamus, premammillary and mammillary nuclei, midbrain central gray, cerebellum, parabrachial nuclei, nucleus of the spinal trigeminal tract, dorsal horn of the spinal cord, and the retina (Fig. 15.2) [22, 29, 39–41]. Of note, Tacr3 shows a maturational process in the distribution of the expression of the gene along postnatal development. Thus, prepubertal animals show more abundant expression in the amygdala (basolateral amygdalar nucleus and basomedial amygdalar nucleus) and LHA than peripubertal animals [40]. At the protein level, immunohistochemistry studies depicting the distribution of NK3R resemble the brain areas where the Tacr3 messenger is detected [25, 29, 42–45].
Regulation of the NKB/NK3R System by Sex Steroids
It is well established that gonadal steroids, i.e., estrogens and testosterone, feed back to the CNS to exert a key role in the central control of the reproductive axis by regulating the tonic release of GnRH (negative feedback) and the preovulatory surge-like release of GnRH in females (positive feedback) [46]. In spite of this, neurons that synthesize GnRH cannot respond directly to the homeostatic feedback of sex steroids, i.e., GnRH cells express neither estrogen receptor alpha (ERα) nor androgen receptor (AR) [47, 48] and only a subset express ERβ [49, 50]. However, the action of ERβ in GnRH neurons, while not yet well characterized, does not seem to be critical for, at least, the negative feedback control of sex steroids upon GnRH release although, of note, it may participate in the regulation of the excitability of GnRH neurons [51]. In this context, it is tenable that the actions of sex steroid hormones on the reproductive axis must target upstream modulators of GnRH neurons, such us (probably) NKB neurons in the ARC. Thus, in keeping with this proposed role, this population of NKB neurons may directly sense circulating levels of estradiol (E2) and testosterone (T) through the expression of ERα [22, 52–56] and AR [57, 58]. A number of studies in adult mammals have demonstrated that, indeed, these ARC neurons exhibit a robust inhibition of TAC3/Tac2 mRNA and NKB protein levels in the presence of E2 or T [22, 40, 52–56, 59, 60] and, consequently, the opposite (i.e., stimulation) is true in E2/T-deprived situations, such as the postmenopausal stage or post-gonadectomy [22, 28, 52, 53, 56, 59, 61–63]. Importantly, not only NKB but also its receptor, NK3R, is subjected to regulation by sex steroids. Thus, Tacr3 mRNA expression is inhibited in the presence of E2 [22, 56]. Of note, this is reminiscent of the regulation of Kiss1 mRNA expression in the ARC by sex steroids [64, 65], which indicates that the Kiss1 and NKB systems likely control GnRH release in the same direction in critical regulatory pathways, such as sex steroid negative feedback, through interactions that yet remain to be fully understood. In this sense, an elegant study published recently by Mittelman-Smith and collaborators demonstrated that ablation of Kisspeptin/NKB/Dyn (KNDy) neurons impairs the compensatory rise of LH after gonadectomy, i.e., the removal of steroid negative feedback, thus adding further support to the critical role of these ARC neurons in the control of GnRH release [66].
It is striking, nonetheless, that not all NKB neuronal populations in the hypothalamus behave similarly to circulating levels of estradiol. Thus, estrogens significantly inhibit NKB neurons in the ARC, as mentioned previously, while other populations of NKB neurons may display diametrically different regulation. In this vein, NKB neurons in the LHA exhibit remarkable stimulation of Tac2 expression in the presence of E2 [67]. Again, this fact evokes comparisons with the Kiss1 system, recalling the dual regulation by sex steroids that Kiss1 expression undergoes when comparing ARC vs. AVPV populations [64]. The LHA is known to hold neural centers that control metabolism and, therefore, indirectly, reproductive function [68, 69]; however, whether the NKB/NK3R system in the LHA exercises a role in this control remains to be assessed.
The regulation of the NKB system by circulating levels of sex steroids—at least in the ARC—is not restricted to adult individuals. Studies in mice have depicted a striking sexual dimorphism of prepubertal animals in the sensitivity of Tac2 expression (in the ARC) to E2. Thus, in the absence of E2 after gonadectomy, juvenile female mice respond with the expected compensatory rise of ARC Tac2 expression [70], which is accompanied by the rise in ARC Kiss1 mRNA and plasma LH levels, just as adult animals would respond [64]. However, their male counterparts appear indisposed to exhibit similar responses, unlike their adult male equivalents [70]. This fact not only suggests a clear sexual dimorphism in the physiology of the Kiss1/NKB neurons in the ARC prepubertally, but may also hold key aspects for the differential timing in puberty onset (later in males), which also needs to be investigated in more detail. An important phenomenon that may help to understand the contribution of the NKB system to puberty onset may rely in the apparent differential sensitivity to the negative feedback of sex steroids on the NKB system compared to the Kiss1 system. Whereas both genes (Kiss1 and Tac2) are susceptible to regulation by E2 prepubertally, Tac2 expression seems to be less sensitive to the rising levels of gonadal steroids than Kiss1. This may account for a sex steroid-independent increase in NKB levels that could, possibly, contribute to the activation of the Kiss1/GnRH axis prepubertally [71].
Finally, it is worth mentioning that the NKB system may be susceptible to the organizing effects of sex steroids during the critical period of sexual differentiation of the brain, i.e., perinatally. Under a physiologic sex steroid environment, the population of NKB neurons in the ARC is larger in females than in males [28, 55, 67, 72]; however, supraphysiological doses of E2 or T in both sexes during this critical period leads to a significant reduction in the number of NKB neurons in the ARC that persists throughout the animal’s life span [55, 67]. Given the exceedingly high hormone doses used in these studies, it remains unclear if normal physiological levels of perinatal sex steroids similarly modulate NKB neuron development.
On the whole, such a modulation of the NKB/NK3R system by sex steroids from early developmental stages to adulthood further unveils a potential role for this system in the central control of the reproductive axis.
Identification of the Hypothalamic KNDy Neuron
The emerging interest to decipher the role of NKB in the central control of reproduction arose, as mentioned above, in 2009 when human genetic studies documented hypogonadotropic hypogonadism in patients bearing loss-of-function mutations in TAC3 and TAC3R genes [6]. Admittedly, however, the potential role of NKB as modulator of GnRH release had been previously recognized in humans, primates, rodents, and sheep [25–27, 29, 32, 52, 53, 55, 57, 58, 60, 61, 63, 73–75]. Nonetheless, the concept of NKB as a regulator of reproductive function has recently advanced to now being considered part of an intricate network of central factors that govern the acquisition and maintenance of reproductive function. In this vein, despite the fact that NKB (transcript and protein) has been described in the hypothalamus for decades, the manner in which NKB interacts with other hypothalamic factors is only starting to be deciphered. Initial studies in sheep and rats documented, by immunohistochemistry, a high degree of co-localization of NKB with the endogenous opioid peptide, dynorphin A (Dyn), in the ARC [25, 27]. These findings paved the way for a seminal study by Goodman and collaborators who showed that NKB/Dyn neurons in the ARC of ewes are also kisspeptin neurons [32]. Thereafter, the co-expression of these three neuropeptides was confirmed in mice, goats, and monkeys at the levels of mRNA by in situ hybridization and protein by immunohistochemistry (Fig. 15.3) [31, 56, 76], suggesting high physiological relevance for this phenomenon, which, otherwise, would not have been maintained throughout evolution. This conserved co-expression has spurred the scientific community to rename this population of neurons as KNDy neurons [77]. In addition, of great significance for the role of NKB in reproductive biology is the fact that virtually all KNDy neurons also co-express NK3R [25, 29, 42, 43, 56]. Interestingly, studies in mice and sheep have revealed that the unambiguous co-expression of kisspeptin with NKB, dynorphin, and NK3R is exclusive to the population of Kiss1 neurons in the ARC, while the other major population of Kiss1 neurons located in the AVPV and the preoptic area (in mice and sheep, respectively) is virtually devoid of these kisspeptin co-transmitters [42, 43, 56]. Consequently, it is reasonable to infer that the role of NKB in the central control of reproductive function must be related to the role of kisspeptin in the ARC, for example, in the negative feedback of sex steroids upon the gonadotropic axis.

Representative photomicrographs of mouse ARC illustrating the co-expression of Kiss1 (labeled in red with digoxigenin coupled to vector red) and NKB (Tac2), NK3R (Tacr3), dynorphin A (Pdyn), and KOR (Oprk1) represented by silver grains. Scale bars = 50 μm
An additional aspect in the physiology of NKB neurons that merits special attention is the identification of the neuronal linage that generates the ARC population. Recent work in male mice indicates that while the vast majority of Kiss1 neurons in the ARC seem to form a homogeneous population and collectively express Tac2 [56], only approximately half of Tac2 neurons in the ARC (at least in the male mouse) appear to co-express Kiss1 [59]. This fact demonstrates a subdivision of this neuronal group with possible functional differences that yet remain to be unfolded. To note, this phenomenon is in keeping with a previous description of two populations of NKB fibers (with and without kisspeptin co-expression) in the median eminence of female rats [78].
Gonadotropin Responses to NKB
The initial characterization of the action of NKB upon gonadotropin release has been controversial. The first studies documented null, or even inhibitory, actions of NKB or the selective NK3R agonist, senktide, on LH release. A study by Sandoval-Guzmán and Rance showed that ovariectomized and estradiol-replaced rats exhibited a substantial decrease in circulating LH levels after senktide treatment [74]. To note, these animals failed to display the expected phenotype subsequent to estradiol replacement, i.e., lowering of the postcastration rise of LH, and, therefore, they should be considered as ovariectomized and sham-replaced animals. In the same vein, the latter rationale could also apply to a recent study by Kinsey-Jones et al. [79]. As a consequence, considering this limitation, the above observations would essentially be in keeping with previous reports documenting inhibitory actions of senktide (or NKB) in gonadectomized mice, rats, and goats [22, 56, 76]. These results, in fact, were clearly contradictory to the initially predicted stimulation of LH release by NKB on the basis of the human studies, in which patients suffering constitutive deficiency in the NKB system exhibited hypogonadotropic hypogonadism [6, 8–10, 13]. Interestingly, this deficiency in humans seemed to be exclusively restricted to LH release, since FSH levels appeared relatively normal [8]. This feature of NKB-null patients suggests a residual low-frequency release of GnRH (NKB-independent), which may result as a consequence of the action of high vs. low pulses of GnRH in the discrimination of LH or FSH release, respectively [80, 81]. Despite the above observations, the overall literature on this topic substantiates a remarkable discrepancy in the response to NKB or senktide in different physiological settings. However, a growing number of studies are offering irrefutable demonstrations of the robust stimulatory action of NKB upon gonadotropin release in a number of species [22, 31, 40, 43, 59, 76, 82, 83]. In this sense, while further investigation is needed to fully understand this paradox (stimulation or inhibition of LH release after senktide treatment), studies in gonadectomized and sham-replaced vs. E2-replaced animals strongly suggest the need of physiological levels of circulating sex steroids (acting on ERα, most likely, in KNDy neurons) to allow the stimulation of LH release by NKB. This is also in line with the stimulatory action of senktide on LH secretion in the follicular, but not luteal, phase in sheep [43]. Of note, continuous stimulation of NK3R with senktide leads to desensitization of the receptor [83], which might, hypothetically, imply that under the elevated levels of kisspeptin and NKB reached in gonadectomized animals, the additional stimulation of NK3R by senktide could account for the observed decrease of LH release in E2-deprived animals. Admittedly, a recent publication indicates that, in this scenario, dynorphin may be hyperstimulated by senktide and, consequently, mediate this inhibition [79]. This inhibitory action, however, remains to be carefully explored.
Notwithstanding the important role of NKB upon GnRH release, NKB’s characterization initially focused on the female. Intriguingly, subsequent studies in the male are now suggesting a more complex regulation of the NKB system than previously anticipated. In this sense, initial studies in male mice pointed to a likely sexual dimorphism in the response to senktide. Corander et al. first documented the absence of LH release after NKB administration in intact adult male mice [84]; however, shortly after, in a study performed in adult male mice, we reported a positive response to senktide administration, with a robust increase in LH release and also, to a lesser extent, FSH [59]. Although speculative, this discrepancy might be due to different efficacy in the activation of NK3R after administration of senktide [59] vs. NKB itself [84] or, purportedly, to the delivery method, i.e., limitation of NKB to dissolve into solution in saline vehicles [84].
Recently, we have expanded our knowledge of the actions of senktide on LH release in male and female rats along postnatal development. Intact female rats at every assessed developmental stage (i.e., infantile, juvenile, prepubertal, pubertal, and adult in diestrus) display conspicuous stimulation of LH secretion after senktide treatment [22, 67]. Male rats, however, progress from being responsive to senktide prepubertally to becoming irresponsive from puberty onwards, dissociating themselves from adult female rats and male mice [67]. These data unveil a striking sexual dimorphism in the rat as well as important differences in otherwise closely related species, i.e., mice and rats. The precise mechanisms underlying these differences require further investigation in order to clarify the role of NKB in the control of gonadotropin secretion.
Kiss1 Neurons as Primary Targets of NKB Action
As described previously, KNDy neurons express NK3R [25, 26], which supports the possibility of autosynaptic loops within the network of NKB/kisspeptin fibers surrounding KNDy neurons in the ARC [25, 30, 33]. The following evidence supports the contention that NKB acts upstream of, or immediately on, Kiss1 neurons in the ARC: (a) senktide and NKB strongly depolarize Kiss1 neurons in the mouse, and this effect is prevented by the NKB antagonist SB222200 [59]; (b) central administration of senktide to gonadectomized and estradiol-replaced (OVX + E2) female rats induces c-fos mRNA expression in Kiss1 neurons [22]; (c) goats treated centrally with NKB exhibit a clear increase in the frequency and amplitude of multiunit activity (MUA) volleys in the ARC, which are invariably mirrored by LH pulses [76], yet the administration of kisspeptin modifies LH pulses without modifying MUA volleys [85].
Kiss1 neurons, however, express a number of other co-transmitters (e.g., dynorphin and glutamate). For this reason, any action of NKB upon GnRH release, evoked by the activation of ARC Kiss1 neurons, could not be entirely attributed to the release of kisspeptin. For instance, these kisspeptin neurons have been also shown to innervate the tuberoinfundibular dopaminergic (TIDA) neurons in the ARC [86]. Yet, compelling evidence indicates that kisspeptin release is, indeed, necessary for the reproductive role of NKB given the following data: (a) mice bearing nonfunctional kisspeptin receptors (Kiss1r KO mice) are irresponsive to the central administration of senktide in terms of LH release [82]; (b) juvenile monkeys showing blunted LH responses due to NK3R desensitization still respond to kisspeptin; however, monkeys subjected to Kiss1r desensitization showed a significant reduction in their response to senktide treatment [31, 83], although, admittedly, they did present a marginal (perhaps kisspeptin-independent) stimulation of LH release.
On the whole, these data strongly suggest a putative action of NKB upon ARC Kiss1 neurons; however, at the same time, we cannot rule out any additional action of this peptide on other brain areas or even different neuronal populations within the ARC. Along these lines, several studies document the expression of NK3R in GnRH terminals of rats [25, 29] and Tacr3 mRNA in GnRH neurons in the mouse [87] as well as in the immortalized GT1-7 cell line—a model of differentiated GnRH neurons—[88]; however, a call of caution should be added since immortalized cell lines do not always resemble in vivo models. Despite the latter observations, recent studies document the absence of NK3R immuno-localization and Tacr3 mRNA in GnRH neurons of sheep [42] and mice [59], respectively, adding further controversy to the role of NKB in the control of GnRH secretion. Indeed, mounting evidence seems to further support the contention that GnRH neurons are devoid—or only present marginal expression—of NK3R and therefore should not be considered as the primary site of action of NKB to regulate GnRH release. For example, GnRH neurons coupled to green fluorescent protein (GnRH-GFP) and subjected to whole-cell recordings do not display signs of activation after senktide treatment in the mouse [59]. This latter study, nonetheless, shows lack of activity at the level of the GnRH cell body, but does not rule out the presence of NK3R at the level of GnRH terminals. In this sense, Corander and colleagues demonstrated that, unlike kisspeptin [89], addition of NKB to hypothalamic explants from male rats (devoid of GnRH cell bodies) did not evoke any effect on GnRH release, which, initially, would preclude any direct action at the level of these GnRH fiber terminals [84].
The Roles of NKB/Kisspeptin Interactions in the Control of Reproductive Function
Generation of GnRH Pulses
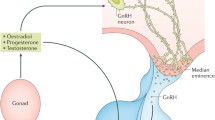
Although this book includes a specific chapter devoted to this topic, it is, however, worth highlighting the potential implications for reproductive physiology that interactions of NKB and kisspeptin (and dynorphin) may have for the normal functioning of the gonadotropic axis. There is convincing evidence to conclude that kisspeptin release is pulsatile and that those pulses correlate with GnRH/LH pulses, based on studies in monkeys and goats [76, 90, 91]. Consequently, we have proposed a model whereby NKB from KNDy neurons acts autosynaptically on the NK3R of the same neuron, through recurrent collaterals, as well as on other neighboring KNDy neurons (Fig. 15.4) [56, 76, 92, 93]. This action of NKB would evoke a synchronized release of kisspeptin within the ARC, a fact of critical importance in order to present a unique GnRH pulse after every kisspeptin pulse. This possibility is in keeping with the dense network of NKB fibers surrounding KNDy neurons in the ARC [25, 30, 33]. Next in this model, kisspeptin would reach the kisspeptin receptor at the GnRH terminals to induce GnRH release to the median eminence. Of note, there is no direct evidence of the presence of Kiss1r at the level of GnRH terminals; however, this could be inferred based on the observations that (a) kisspeptin can elicit GnRH secretion from explants of the mediobasal hypothalamus, which contains few, if any, GnRH cell bodies [89, 94–96] and (b) kisspeptin fibers form close appositions at the GnRH fiber terminals [97–99]. At the same time, dynorphin (an endogenous opioid peptide with known inhibitory action upon gonadotropin release [100, 101]), would act mainly on yet unknown intermediate neurons to eventually shut down NKB and kisspeptin release, therefore creating a kisspeptin pulse. Noteworthy, each neuropeptide released from KNDy neurons seems to target a different set of neurons. Thus, (a) kisspeptins act mainly on GnRH neurons, since KNDy neurons themselves in the ARC are devoid of Kiss1 receptor [94] and do not respond to kisspeptin [59]; (b) NKB seems to primarily activate KNDy neurons, since GnRH neurons (at least in mice and sheep) do not express detectable levels of NK3R [43, 59]; (c) GnRH neurons do not express the dynorphin receptor (κ-opioid receptor, KOR) [102, 103], and its expression in KNDy neurons seems to be marginal [56, 59]. These findings allude to intermediate neurons as the primary site of action of dynorphin in the context of kisspeptin release, which, allegedly, would play a substantial role by increasing the turnaround time of the inhibitory signal before acting back on KNDy neurons, therefore allowing for the appearance of a kisspeptin pulse. Of note, as research on the characterization of KNDy neurons advances, new co-transmitters emerge, which, in the near future, may make the descriptive term “KNDy” obsolete. In this vein, glutamate and glutamate receptors, also described in KNDy neurons [104], may constitute additional autosynaptic regulatory loops [82, 104, 105] to “fine-tune” kisspeptin release (Fig. 15.4). It has been speculated that this or a similar mechanism may be involved in the attenuation of the severe reproductive phenotype of Tacr3 null mice compared to humans described recently by Yang and colleagues [14, 106]. Moreover, recent studies also suggest an action (direct and indirect) of GnRH itself on KNDy neurons to, eventually, modulate GnRH release [107]; however, this contention requires further research. For instance, the presence of the GnRH receptor in KNDy neurons has yet to be demonstrated.

Schematic representation of a KNDy neuron depicting possible autoregulatory loops and interactions with GnRH neurons
Control of Puberty Onset
GnRH release undergoes a series of developmental changes, progressing from an activated neonatal period, followed by a dormant stage during the infantile and juvenile ages, to puberty onset, which is characterized by the appearance of GnRH pulses with increasing amplitude and frequency. A number of central and peripheral factors have been posed to mediate this awakening of the reproductive axis; however, exactly what triggers puberty onset remains elusive.
In recent years, hypothalamic kisspeptin has become a likely candidate to serve as a gatekeeper of puberty onset. Kisspeptin is the most potent secretagogue of GnRH described to date [5], and compelling evidence shows that Kiss1 expression and synaptic contacts between Kiss1 neurons in the hypothalamus increase at the time of puberty onset in rodents [92, 108, 109], suggesting that hypothalamic Kiss1 neurons play an important role during this maturational process [110]. Furthermore, chronic administration of kisspeptin to prepubertal female rats advances puberty onset [111], which is prevented in the presence of a kisspeptin antagonist [112]. In this context, if we assume that NKB has a role in the control of kisspeptin release at the onset of puberty, we could expect that impairments in the NKB/NK3R system would directly translate into disorders in the timing of puberty. Indeed, as mentioned previously, a number of studies have documented impuberism associated with hypogonadotropic hypogonadism (HH) in humans bearing inactivating mutations in the genes encoding NKB and NK3R, which has been partially recapitulated in Tacr3 knockout mice. To note, Tac2/Tacr3 expression in the hypothalamus of rats (and markedly Tacr3 in the ARC) increases prior to puberty onset [22, 40], apparently anticipating the increase of kisspeptin immunoreactivity reported in the rostral periventricular area of the hypothalamus in prepubertal female mice [113]. Importantly, despite the documented sensitivity of Tac2 expression to the negative feedback of sex steroids prepubertally in the mouse [70], the major regulatory pathway that drives the stimulation of Tac2 expression prior to puberty onset seems to be independent and, seemingly, dominates over the rising levels of gonadal steroids that occur during puberty onset [40, 71, 114].
Additionally, in line with a role for NKB in the timing of puberty, senktide induces strong secretory responses of LH in juvenile monkeys [31], prepubertal male and female rats [67] and prepubertal ewes [115], suggesting that the gonadotropic axis is responsive to NKB stimulation before puberty onset. On the whole, these observations, along with recent clinical observations suggesting that the stimulatory action NKB on the gonadotropic axis may be more prominent during early stages of sexual maturation [10], are in line with an eventual stimulatory role of NKB upon Kiss1 expression during pubertal maturation.
Moreover, studies of chronic blockade of NKB signaling using the NK3R antagonist, SB222200, to prepubertal female rats induce a slight delay in the timing of puberty onset (Fig. 15.5) [40]. The effect of this antagonist to block pubertal progression is, however, not as effective as the chronic administration of a kisspeptin antagonist, which results in a marked suppression of vaginal opening (Fig. 15.5) and gonadal weights—as indirect markers of the rise of circulating sex steroids during puberty onset [112]. In sum, based on the above observations, it is reasonable to deduce a stimulatory role of NKB in the timing of puberty, an effect which may be dependent on kisspeptin signaling.

NKB as a Signaling System of the Metabolic Status
Reproduction is an extremely energy-demanding function and, as such, subjected to regulation by metabolic cues that eventually contribute to the regulation of GnRH release [1, 116]. Mounting data during the last few years suggest that Kiss1 neurons in the ARC play a critical role conveying metabolic information onto the hypothalamic centers that control the attainment and maintenance of reproductive function. For instance, models of metabolic stress, such as acute fasting, impinge a significant restraint on the hypothalamic expression of Kiss1/kisspeptins in pubertal animals, which can be reverted by exogenous kisspeptin [117]. Given the fact that the NKB/NK3R system is also present in the metabolic conveyor that KNDy neurons in the ARC constitute, it was reasonable to assume that the action of NKB is also subjected to metabolic regulation during (and after) puberty. In this sense, a recent study documented the sensitivity of this system to metabolic cues [118]. In more detail, a later study in pubertal female rats demonstrates a significant suppression of Tacr3, and to a lesser extent Tac2, in the ARC after 48-h fasting [40], replicating the previously described expression profile of Kiss1 in both the ARC and AVPV [117]. Moreover, in this study, LH responses to senktide administration in pubertal (36-d) rats are not only preserved but even augmented in fasting conditions, suggesting a possible sensitization of its stimulatory effects under conditions of negative energy balance—again, resembling previous findings on the gonadotropin-releasing actions of kisspeptins [117, 119]. Additionally, chronic administration of senktide to prepubertal female rats subjected to caloric restriction was sufficient to partially rescue markers of puberty onset, e.g., vaginal opening and LH secretion [40], as previously reported for kisspeptin [117]. On the other hand, situations of exceedingly high caloric intake, such as rats subjected to high fat diet prepubertally, exhibited precocious puberty that correlates with the advancement in the timing of Kiss1 and Tac2 expression that, in turn, was associated with the advancement in LH pulsatility [120].
Altogether, the above observations suggest that the NKB system is subjected to modulation by metabolic cues, at least during pubertal progression, probably facilitating the transmission of the energy status of the organism on to the Kiss1 system, most likely, at the level of the ARC. However, kisspeptin-independent pathways for the stimulatory effects of NKB on the gonadotropic axis at puberty cannot be excluded—which remains to be explored. Noteworthy, recent studies associate KNDy neurons in the ARC with the regulatory (inhibition) effect that estrogens exert on body weight [66]. This finding poses the KNDy neuron as a nodal regulatory center for the integration of reproductive axis and energy balance; however, the mechanisms underlying this effect need to be deciphered.
Conclusion and Future Perspectives
Our knowledge of the neuronal interactions that potentially impinge the accurate functioning of the endocrine system is rapidly evolving. Particularly, in recent years, reproductive neuroendocrinologists are witnessing the appearance of a constellation of central factors that significantly contribute to the modulation of GnRH release. In this context, since 2003, the scientific community has enthusiastically welcomed Kiss1 neurons as key elements to answer remaining open questions in the physiology of GnRH release. More recently, the NKB system has added a new level of complexity to Kiss1 neurons. Compelling evidence suggests that NKB plays a critical role in the control of kisspeptin release, at least at the level of the ARC. Not only is NKB able to modulate gonadotropin release through, according to recent studies, its action on Kiss1 neurons but, also, serves as a conveyor of additional regulatory factors, e.g., developmental and metabolic, to Kiss1 neurons, thus contributing to the exquisite regulation of kisspeptin release.
Many aspects of the physiology of the NKB/NK3R system in the context of reproduction remain to be fully characterized. For instance, a vivid debate is currently ongoing in the field regarding the putative target/s of NKB, with evidence on both Kiss1 and GnRH neurons that may only reflect species differences or, perhaps, residual levels of redundancy that, under constitutive absence of one or another, may lead to potential compensation. Additionally, important aspects regarding the sexual differentiation of the response of gonadotropins to NKB (or senktide), as recently observed in the rat, constitute a mystery that demands to be resolved. Indeed, reconciliation of the results in male mice and rats, and a detailed comparison between both sexes, to determine which one better resembles the human phenotype would clearly help the scientific community to establish a working model with translational potential to humans. Overall, elucidating how the brain triggers the neuroendocrine events that lead to the attainment and maintenance of reproductive function will provide the intellectual platform for understanding certain disorders of reproduction, including delayed or precocious puberty—and perhaps guide us toward improved therapies for their treatment.
References
Hill JW, Elmquist JK, Elias CF (2008) Hypothalamic pathways linking energy balance and reproduction. Am J Physiol Endocrinol Metab 294:E827–E832
Ojeda SR, Advis JP, Andrews WW (1980) Neuroendocrine control of the onset of puberty in the rat. Fed Proc 39:2365–2371
Herbison A (2006) Physiology of the GnRH neuronal network. In: Neil J, Knobil E (eds) Physiology of reproduction. Academic, San Diego, CA, pp 1415–1482
Herbison AE, Robinson JE, Skinner DC (1993) Distribution of estrogen receptor-immunoreactive cells in the preoptic area of the ewe: co-localization with glutamic acid decarboxylase but not luteinizing hormone-releasing hormone. Neuroendocrinology 57:751–759
Oakley AE, Clifton DK, Steiner RA (2009) Kisspeptin signaling in the brain. Endocr Rev 30:713–743
Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Ozbek MN, Imamoglu S, Akalin NS, Yuksel B, O’Rahilly S, Semple RK (2009) TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin B in the central control of reproduction. Nat Genet 41:354–358
de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E (2003) Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A 100:10972–10976
Francou B, Bouligand J, Voican A, Amazit L, Trabado S, Fagart J, Meduri G, Brailly-Tabard S, Chanson P, Lecomte P, Guiochon-Mantel A, Young J (2011) Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS One 6:e25614
Fukami M, Maruyama T, Dateki S, Sato N, Yoshimura Y, Ogata T (2010) Hypothalamic dysfunction in a female with isolated hypogonadotropic hypogonadism and compound heterozygous TACR3 mutations and clinical manifestation in her heterozygous mother. Horm Res Paediatr 73:477–481
Gianetti E, Tusset C, Noel SD, Au MG, Dwyer AA, Hughes VA, Abreu AP, Carroll J, Trarbach E, Silveira LF, Costa EM, de Mendonca BB, de Castro M, Lofrano A, Hall JE, Bolu E, Ozata M, Quinton R, Amory JK, Stewart SE, Arlt W, Cole TR, Crowley WF, Kaiser UB, Latronico AC, Seminara SB (2010) TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab 95:2857–2867
Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O’Rahilly S, Carlton MB, Crowley WF Jr, Aparicio SA, Colledge WH (2003) The GPR54 gene as a regulator of puberty. N Engl J Med 349:1614–1627
Topaloglu AK, Tello JA, Kotan LD, Ozbek MN, Yilmaz MB, Erdogan S, Gurbuz F, Temiz F, Millar RP, Yuksel B (2012) Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N Engl J Med 366:629–635
Young J, Bouligand J, Francou B, Raffin-Sanson ML, Gaillez S, Jeanpierre M, Grynberg M, Kamenicky P, Chanson P, Brailly-Tabard S, Guiochon-Mantel A (2010) TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. J Clin Endocrinol Metab 95:2287–2295
Yang JJ, Caligioni CS, Chan YM, Seminara SB (2012) Uncovering novel reproductive defects in neurokinin B receptor null mice: closing the gap between mice and men. Endocrinology 153(3):1498–1508
Page NM (2005) New challenges in the study of the mammalian tachykinins. Peptides 26:1356–1368
Almeida TA, Rojo J, Nieto PM, Pinto FM, Hernandez M, Martin JD, Candenas ML (2004) Tachykinins and tachykinin receptors: structure and activity relationships. Curr Med Chem 11:2045–2081
Bonner TI, Affolter HU, Young AC, Young WS III (1987) A cDNA encoding the precursor of the rat neuropeptide, neurokinin B. Brain Res 388:243–249
Page NM, Morrish DW, Weston-Bell NJ (2009) Differential mRNA splicing and precursor processing of neurokinin B in neuroendocrine tissues. Peptides 30:1508–1513
Chawla MK, Gutierrez GM, Young WS III, McMullen NT, Rance NE (1997) Localization of neurons expressing substance P and neurokinin B gene transcripts in the human hypothalamus and basal forebrain. J Comp Neurol 384:429–442
Pinto FM, Almeida TA, Hernandez M, Devillier P, Advenier C, Candenas ML (2004) mRNA expression of tachykinins and tachykinin receptors in different human tissues. Eur J Pharmacol 494:233–239
Marksteiner J, Sperk G, Krause JE (1992) Distribution of neurons expressing neurokinin B in the rat brain: immunohistochemistry and in situ hybridization. J Comp Neurol 317:341–356
Navarro VM, Castellano JM, McConkey SM, Pineda R, Ruiz-Pino F, Pinilla L, Clifton DK, Tena-Sempere M, Steiner RA (2011) Interactions between kisspeptin and neurokinin B in the control of GnRH secretion in the female rat. Am J Physiol Endocrinol Metab 300:E202–E210
Warden MK, Young WS III (1988) Distribution of cells containing mRNAs encoding substance P and neurokinin B in the rat central nervous system. J Comp Neurol 272:90–113
Duarte CR, Schutz B, Zimmer A (2006) Incongruent pattern of neurokinin B expression in rat and mouse brains. Cell Tissue Res 323:43–51
Burke MC, Letts PA, Krajewski SJ, Rance NE (2006) Coexpression of dynorphin and neurokinin B immunoreactivity in the rat hypothalamus: morphologic evidence of interrelated function within the arcuate nucleus. J Comp Neurol 498:712–726
Ciofi P, Krause JE, Prins GS, Mazzuca M (1994) Presence of nuclear androgen receptor-like immunoreactivity in neurokinin B-containing neurons of the hypothalamic arcuate nucleus of the adult male rat. Neurosci Lett 182:193–196
Foradori CD, Amstalden M, Goodman RL, Lehman MN (2006) Colocalisation of dynorphin a and neurokinin B immunoreactivity in the arcuate nucleus and median eminence of the sheep. J Neuroendocrinol 18:534–541
Hrabovszky E, Molnar CS, Sipos M, Vida B, Ciofi P, Borsay BA, Sarkadi L, Herczeg L, Bloom SR, Ghatei MA, Dhillo WS, Kallo I, Liposits Z (2011) Sexual dimorphism of kisspeptin and neurokinin B immunoreactive neurons in the infundibular nucleus of aged men and women. Front Endocrinol 2:80. doi:10.3389/fendo.2011.00080
Krajewski SJ, Anderson MJ, Iles-Shih L, Chen KJ, Urbanski HF, Rance NE (2005) Morphologic evidence that neurokinin B modulates gonadotropin-releasing hormone secretion via neurokinin 3 receptors in the rat median eminence. J Comp Neurol 489:372–386
Krajewski SJ, Burke MC, Anderson MJ, McMullen NT, Rance NE (2010) Forebrain projections of arcuate neurokinin B neurons demonstrated by anterograde tract-tracing and monosodium glutamate lesions in the rat. Neuroscience 166:680–697
Ramaswamy S, Seminara SB, Ali B, Ciofi P, Amin NA, Plant TM (2010) Neurokinin B stimulates GnRH release in the male monkey (Macaca mulatta) and is colocalized with kisspeptin in the arcuate nucleus. Endocrinology 151:4494–4503
Goodman RL, Lehman MN, Smith JT, Coolen LM, de Oliveira CV, Jafarzadehshirazi MR, Pereira A, Iqbal J, Caraty A, Ciofi P, Clarke IJ (2007) Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology 148:5752–5760
Yeo SH, Herbison AE (2011) Projections of arcuate nucleus and rostral periventricular kisspeptin neurons in the adult female mouse brain. Endocrinology 152:2387–2399
Mussap CJ, Geraghty DP, Burcher E (1993) Tachykinin receptors: a radioligand binding perspective. J Neurochem 60:1987–2009
Seabrook GR, Bowery BJ, Hill RG (1995) Pharmacology of tachykinin receptors on neurones in the ventral tegmental area of rat brain slices. Eur J Pharmacol 273:113–119
Pantaleo N, Chadwick W, Park SS, Wang L, Zhou Y, Martin B, Maudsley S (2010) The mammalian tachykinin ligand-receptor system: an emerging target for central neurological disorders. CNS Neurol Disord Drug Targets 9:627–635
Valero MS, Fagundes DS, Grasa L, Arruebo MP, Plaza MA, Murillo MD (2011) Contractile effect of tachykinins on rabbit small intestine. Acta Pharmacol Sin 32:487–494
Page NM, Bell NJ (2002) The human tachykinin NK1 (short form) and tachykinin NK4 receptor: a reappraisal. Eur J Pharmacol 437:27–30
Ding YQ, Shigemoto R, Takada M, Ohishi H, Nakanishi S, Mizuno N (1996) Localization of the neuromedin K receptor (NK3) in the central nervous system of the rat. J Comp Neurol 364:290–310
Navarro VM, Ruiz-Pino F, Sanchez-Garrido MA, Garcia-Galiano D, Hobbs SJ, Manfredi-Lozano M, León S, Sangiao-Alvarellos S, Castellano JM, Clifton DK, Pinilla L, Steiner RA, Tena-Sempere M (2012) Role of neurokinin B in the control of female puberty and its modulation by metabolic status. J Neurosci 153(1):316–328
Shughrue PJ, Lane MV, Merchenthaler I (1996) In situ hybridization analysis of the distribution of neurokinin-3 mRNA in the rat central nervous system. J Comp Neurol 372:395–414
Amstalden M, Coolen LM, Hemmerle AM, Billings HJ, Connors JM, Goodman RL, Lehman MN (2009) Neurokinin 3 receptor immunoreactivity in the septal region, preoptic area and hypothalamus of the female sheep: colocalisation in neurokinin B cells of the arcuate nucleus but not in gonadotropin-releasing hormone neurones. J Neuroendocrinol 22:1–12
Billings HJ, Connors JM, Altman SN, Hileman SM, Holaskova I, Lehman MN, McManus CJ, Nestor CC, Jacobs BH, Goodman RL (2010) Neurokinin B acts via the neurokinin-3 receptor in the retrochiasmatic area to stimulate luteinizing hormone secretion in sheep. Endocrinology 151:3836–3846
Duque E, Mangas A, Salinas P, Diaz-Cabiale Z, Narvaez JA, Covenas R (2013) Mapping of alpha-neo-endorphin- and neurokinin B-immunoreactivity in the human brainstem. Brain Struct Funct 218(1):131–149
Koutcherov Y, Ashwell KW, Paxinos G (2000) The distribution of the neurokinin B receptor in the human and rat hypothalamus. Neuroreport 11:3127–3131
Fink G (2000) Neuroendocrine regulation of pituitary function: general principles. In: Conn PM, Freeman ME (eds) Neuroendocrinology in physiology and medicine. Humana Press, Totowa, pp 107–134
Herbison AE, Skinner DC, Robinson JE, King IS (1996) Androgen receptor-immunoreactive cells in ram hypothalamus: distribution and co-localization patterns with gonadotropin-releasing hormone, somatostatin and tyrosine hydroxylase. Neuroendocrinology 63:120–131
Huang X, Harlan RE (1993) Absence of androgen receptors in LHRH immunoreactive neurons. Brain Res 624:309–311
Hrabovszky E, Kallo I, Szlavik N, Keller E, Merchenthaler I, Liposits Z (2007) Gonadotropin-releasing hormone neurons express estrogen receptor-beta. J Clin Endocrinol Metab 92:2827–2830
Hrabovszky E, Shughrue PJ, Merchenthaler I, Hajszan T, Carpenter CD, Liposits Z, Petersen SL (2000) Detection of estrogen receptor-beta messenger ribonucleic acid and 125I-estrogen binding sites in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology 141:3506–3509
Chu Z, Takagi H, Moenter SM (2010) Hyperpolarization-activated currents in gonadotropin-releasing hormone (GnRH) neurons contribute to intrinsic excitability and are regulated by gonadal steroid feedback. J Neurosci 30:13373–13383
Danzer SC, Price RO, McMullen NT, Rance NE (1999) Sex steroid modulation of neurokinin B gene expression in the arcuate nucleus of adult male rats. Brain Res Mol Brain Res 66:200–204
Dellovade TL, Merchenthaler I (2004) Estrogen regulation of neurokinin B gene expression in the mouse arcuate nucleus is mediated by estrogen receptor alpha. Endocrinology 145:736–742
Eghlidi DH, Haley GE, Noriega NC, Kohama SG, Urbanski HF (2010) Influence of age and 17beta-estradiol on kisspeptin, neurokinin B, and prodynorphin gene expression in the arcuate-median eminence of female rhesus macaques. Endocrinology 151:3783–3794
Goubillon ML, Forsdike RA, Robinson JE, Ciofi P, Caraty A, Herbison AE (2000) Identification of neurokinin B-expressing neurons as an highly estrogen-receptive, sexually dimorphic cell group in the ovine arcuate nucleus. Endocrinology 141:4218–4225
Navarro VM, Gottsch ML, Chavkin C, Okamura H, Clifton DK, Steiner RA (2009) Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J Neurosci 29:11859–11866
Ciofi P, Lapirot OC, Tramu G (2007) An androgen-dependent sexual dimorphism visible at puberty in the rat hypothalamus. Neuroscience 146:630–642
Ciofi P, Leroy D, Tramu G (2006) Sexual dimorphism in the organization of the rat hypothalamic infundibular area. Neuroscience 141:1731–1745
Navarro VM, Gottsch ML, Wu M, Garcia-Galiano D, Hobbs SJ, Bosch MA, Pinilla L, Clifton DK, Dearth A, Ronnekleiv OK, Braun RE, Palmiter RD, Tena-Sempere M, Alreja M, Steiner RA (2011) Regulation of NKB pathways and their roles in the control of Kiss1 neurons in the arcuate nucleus of the male mouse. Endocrinology 152:4265–4275
Pillon D, Caraty A, Fabre-Nys C, Bruneau G (2003) Short-term effect of oestradiol on neurokinin B mRNA expression in the infundibular nucleus of ewes. J Neuroendocrinol 15:749–753
Abel TW, Voytko ML, Rance NE (1999) The effects of hormone replacement therapy on hypothalamic neuropeptide gene expression in a primate model of menopause. J Clin Endocrinol Metab 84:2111–2118
Rance NE (2009) Menopause and the human hypothalamus: evidence for the role of kisspeptin/neurokinin B neurons in the regulation of estrogen negative feedback. Peptides 30:111–122
Rance NE, Bruce TR (1994) Neurokinin B gene expression is increased in the arcuate nucleus of ovariectomized rats. Neuroendocrinology 60:337–345
Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA (2005) Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology 146:3686–3692
Smith JT, Dungan HM, Stoll EA, Gottsch ML, Braun RE, Eacker SM, Clifton DK, Steiner RA (2005) Differential regulation of KiSS-1 mRNA expression by sex steroids in the brain of the male mouse. Endocrinology 146:2976–2984
Mittelman-Smith MA, Williams H, Krajewski-Hall SJ, Lai J, Ciofi P, McMullen NT, Rance NE (2012) Arcuate kisspeptin/neurokinin B/dynorphin (KNDy) neurons mediate the estrogen suppression of gonadotropin secretion and body weight. Endocrinology 153:2800–2812
Ruiz-Pino F, Navarro VM, Bentsen A, García-Galiano D, Sánchez-Garrido MA, Clifton DK, Steiner RA, Pinilla L, Mikkelsen J, Tena-Sempere M (2012) Neurokinin B and the control of the gonadotropic axis in the rat: developmental changes, sexual dimorphism and regulation by gonadal steroids. Endocrinology 153(10):4818–4829
Nurmio M, Tena-Sempere M, Toppari J (2010) Orexins and the regulation of the hypothalamic-pituitary-testicular axis. Acta Physiol (Oxf) 198:349–354
Williamson-Hughes PS, Grove KL, Smith MS (2005) Melanin concentrating hormone (MCH): a novel neural pathway for regulation of GnRH neurons. Brain Res 1041:117–124
Kauffman AS, Navarro VM, Kim J, Clifton DK, Steiner RA (2009) Sex differences in the regulation of Kiss1/NKB neurons in juvenile mice: implications for the timing of puberty. Am J Physiol Endocrinol Metab 297:E1212–E1221
Gill JC, Navarro VM, Kwong C, Noel SD, Martin C, Xu S, Clifton DK, Carroll RS, Steiner RA, Kaiser UB (2012) Increased neurokinin B (Tac2) expression in the mouse arcuate nucleus is an early marker of pubertal onset with differential sensitivity to sex steroid negative feedback than Kiss1. Endocrinology 153(10):4883–4893
Rance NE, Krajewski SJ, Smith MA, Cholanian M, Dacks PA (2010) Neurokinin B and the hypothalamic regulation of reproduction. Brain Res 1364:116–128
Robinson JE, Birch RA, Taylor JA, Foster DL, Padmanabhan V (2002) In utero programming of sexually differentiated gonadotropin releasing hormone (GnRH) secretion. Domest Anim Endocrinol 23:43–52
Sandoval-Guzman T, Rance NE (2004) Central injection of senktide, an NK3 receptor agonist, or neuropeptide Y inhibits LH secretion and induces different patterns of Fos expression in the rat hypothalamus. Brain Res 1026:307–312
Rance NE, Young WS III (1991) Hypertrophy and increased gene expression of neurons containing neurokinin-B and substance-P messenger ribonucleic acids in the hypothalami of postmenopausal women. Endocrinology 128:2239–2247
Wakabayashi Y, Nakada T, Murata K, Ohkura S, Mogi K, Navarro VM, Clifton DK, Mori Y, Tsukamura H, Maeda K, Steiner RA, Okamura H (2010) Neurokinin B and dynorphin A in kisspeptin neurons of the arcuate nucleus participate in generation of periodic oscillation of neural activity driving pulsatile gonadotropin-releasing hormone secretion in the goat. J Neurosci 30:3124–3132
Lehman MN, Coolen LM, Goodman RL (2010) Minireview: kisspeptin/neurokinin B/dynorphin (KNDy) cells of the arcuate nucleus: a central node in the control of gonadotropin-releasing hormone secretion. Endocrinology 151:3479–3489
True C, Kirigiti M, Ciofi P, Grove KL, Smith MS (2011) Characterisation of arcuate nucleus kisspeptin/neurokinin B neuronal projections and regulation during lactation in the rat. J Neuroendocrinol 23:52–64
Kinsey-Jones JS, Grachev P, Li XF, Lin YS, Milligan SR, Lightman SL, O’Byrne KT (2012) The inhibitory effects of neurokinin B on GnRH pulse generator frequency in the female rat. Endocrinology 153(1):307–315
Ciccone NA, Xu S, Lacza CT, Carroll RS, Kaiser UB (2010) Frequency-dependent regulation of follicle-stimulating hormone beta by pulsatile gonadotropin-releasing hormone is mediated by functional antagonism of bZIP transcription factors. Mol Cell Biol 30:1028–1040
Marshall JC, Dalkin AC, Haisenleder DJ, Paul SJ, Ortolano GA, Kelch RP (1991) Gonadotropin-releasing hormone pulses: regulators of gonadotropin synthesis and ovulatory cycles. Recent Prog Horm Res 47:155–187; discussion 159–188
García-Galiano D, León-Tellez S, Manfredi-Lozano M, Romero-Ruiz A, Navarro VM, Pinilla L, Tena-Sempere M (2012) Kisspeptin signaling is indispensable for neurokinin B, but not glutamate, stimulation of gonadotropin secretion in mice. Endocrinology 153(1):316–328
Ramaswamy S, Seminara SB, Plant TM (2011) Evidence from the agonadal juvenile male rhesus monkey (Macaca mulatta) for the view that the action of neurokinin B to trigger gonadotropin-releasing hormone release is upstream from the kisspeptin receptor. Neuroendocrinology 94(3):237–245
Corander MP, Challis BG, Thompson EL, Jovanovic Z, Loraine Tung YC, Rimmington D, Huhtaniemi IT, Murphy KG, Topaloglu AK, Yeo GS, O’Rahilly S, Dhillo WS, Semple RK, Coll AP (2010) The effects of neurokinin B upon gonadotropin release in male rodents. J Neuroendocrinol 22:181–187
Ohkura S, Takase K, Matsuyama S, Mogi K, Ichimaru T, Wakabayashi Y, Uenoyama Y, Mori Y, Steiner RA, Tsukamura H, Maeda KI, Okamura H (2009) Gonadotropin-releasing hormone pulse generator activity in the hypothalamus of the goat. J Neuroendocrinol 21:813–821
Sawai N, Iijima N, Takumi K, Matsumoto K, Ozawa H (2012) Immunofluorescent histochemical and ultrastructural studies on the innervation of kisspeptin/neurokinin B neurons to tuberoinfundibular dopaminergic neurons in the arcuate nucleus of rats. Neurosci Res 74(1):10–16
Todman MG, Han SK, Herbison AE (2005) Profiling neurotransmitter receptor expression in mouse gonadotropin-releasing hormone neurons using green fluorescent protein-promoter transgenics and microarrays. Neuroscience 132:703–712
Glidewell-Kenney CA, Grove AMH, Iyer K, Mellon PL (2010) The neurokinin 3 receptor agonist, senktide, reduces gonadotropin releasing hormone synthesis and secretion in the inmortalized GT1-7 hypothalamic cell model. In: Society for Neuroscience 40th annual meeting, San Diego
Castellano JM, Navarro VM, Fernandez-Fernandez R, Castano JP, Malagon MM, Aguilar E, Dieguez C, Magni P, Pinilla L, Tena-Sempere M (2006) Ontogeny and mechanisms of action for the stimulatory effect of kisspeptin on gonadotropin-releasing hormone system of the rat. Mol Cell Endocrinol 257–258:75–83
Guerriero KA, Keen KL, Terasawa E (2012) Developmental increase in kisspeptin-54 release in vivo is independent of the pubertal increase in estradiol in female rhesus monkeys (Macaca mulatta). Endocrinology 153(4):1887–1897
Keen KL, Wegner FH, Bloom SR, Ghatei MA, Terasawa E (2008) An increase in kisspeptin-54 release occurs with the pubertal increase in luteinizing hormone-releasing hormone-1 release in the stalk-median eminence of female rhesus monkeys in vivo. Endocrinology 149:4151–4157
Navarro VM, Tena-Sempere M (2011) Neuroendocrine control by kisspeptins: role in metabolic regulation of fertility. Nat Rev Endocrinol 8(1):40–53
Navarro VM (2012) New insights into the control of pulsatile GnRH release: the role of Kiss1/neurokinin B neurons. Front Endocrinol (Lausanne) 3:48
d’Anglemont de Tassigny X, Fagg LA, Carlton MB, Colledge WH (2008) Kisspeptin can stimulate gonadotropin-releasing hormone (GnRH) release by a direct action at GnRH nerve terminals. Endocrinology 149:3926–3932
Irwig MS, Fraley GS, Smith JT, Acohido BV, Popa SM, Cunningham MJ, Gottsch ML, Clifton DK, Steiner RA (2004) Kisspeptin activation of gonadotropin releasing hormone neurons and regulation of KiSS-1 mRNA in the male rat. Neuroendocrinology 80:264–272
Thompson EL, Patterson M, Murphy KG, Smith KL, Dhillo WS, Todd JF, Ghatei MA, Bloom SR (2004) Central and peripheral administration of kisspeptin-10 stimulates the hypothalamic-pituitary-gonadal axis. J Neuroendocrinol 16:850–858
Kallo I, Vida B, Deli L, Molnar CS, Hrabovszky E, Caraty A, Ciofi P, Coen CW, Liposits Z (2012) Co-Localisation of kisspeptin with galanin or neurokinin B in afferents to mouse GnRH neurones. J Neuroendocrinol 24:464–476
Matsuyama S, Ohkura S, Mogi K, Wakabayashi Y, Mori Y, Tsukamura H, Maeda K, Ichikawa M, Okamura H (2011) Morphological evidence for direct interaction between kisspeptin and gonadotropin-releasing hormone neurons at the median eminence of the male goat: an immunoelectron microscopic study. Neuroendocrinology 94:323–332
Uenoyama Y, Inoue N, Pheng V, Homma T, Takase K, Yamada S, Ajiki K, Ichikawa M, Okamura H, Maeda KI, Tsukamura H (2011) Ultrastructural evidence of kisspeptin-gonadotropin-releasing hormone interaction in the median eminence of female rats: implication of axo-axonal regulation of GnRH release. J Neuroendocrinol 23(10):863–870
Kinoshita F, Nakai Y, Katakami H, Imura H (1982) Suppressive effect of dynorphin-(1-13) on luteinizing hormone release in conscious castrated rats. Life Sci 30:1915–1919
Schulz R, Wilhelm A, Pirke KM, Gramsch C, Herz A (1981) Beta-endorphin and dynorphin control serum luteinizing hormone level in immature female rats. Nature 294:757–759
Mitchell V, Prevot V, Jennes L, Aubert JP, Croix D, Beauvillain JC (1997) Presence of mu and kappa opioid receptor mRNAs in galanin but not in GnRH neurons in the female rat. Neuroreport 8:3167–3172
Sannella MI, Petersen SL (1997) Dual label in situ hybridization studies provide evidence that luteinizing hormone-releasing hormone neurons do not synthesize messenger ribonucleic acid for mu, kappa, or delta opiate receptors. Endocrinology 138:1667–1672
Cravo RM, Margatho LO, Osborne-Lawrence S, Donato J Jr, Atkin S, Bookout AL, Rovinsky S, Frazao R, Lee CE, Gautron L, Zigman JM, Elias CF (2011) Characterization of Kiss1 neurons using transgenic mouse models. Neuroscience 173:37–56
Gottsch ML, Popa SM, Lawhorn JK, Qiu J, Tonsfeldt KJ, Bosch MA, Kelly MJ, Ronnekleiv OK, Sanz E, McKnight GS, Clifton DK, Palmiter RD, Steiner RA (2011) Molecular properties of kiss1 neurons in the arcuate nucleus of the mouse. Endocrinology 152:4298–4309
Steiner RA, Navarro VM (2012) Tacking toward reconciliation on Tacr3/TACR3 mutations. Endocrinology 153(4):1578–1581
Moenter SM, Burger LL, Chu Z (2011) Gonadotropin-releasing hormone (GnRH) regulates the excitability of neurokinin B (NKB) neurons of the mouse arcuate nucleus. Society for Neuroscience Annual Meeting, Washington, DC
Clarkson J, Herbison AE (2006) Postnatal development of kisspeptin neurons in mouse hypothalamus; sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology 147:5817–5825
Navarro VM, Castellano JM, Fernandez-Fernandez R, Barreiro ML, Roa J, Sanchez-Criado JE, Aguilar E, Dieguez C, Pinilla L, Tena-Sempere M (2004) Developmental and hormonally regulated messenger ribonucleic acid expression of KiSS-1 and its putative receptor, GPR54, in rat hypothalamus and potent luteinizing hormone-releasing activity of KiSS-1 peptide. Endocrinology 145:4565–4574
Roa J, Navarro VM, Tena-Sempere M (2011) Kisspeptins in reproductive biology: consensus knowledge and recent developments. Biol Reprod 85(4):650–660
Navarro VM, Fernandez-Fernandez R, Castellano JM, Roa J, Mayen A, Barreiro ML, Gaytan F, Aguilar E, Pinilla L, Dieguez C, Tena-Sempere M (2004) Advanced vaginal opening and precocious activation of the reproductive axis by KiSS-1 peptide, the endogenous ligand of GPR54. J Physiol 561:379–386
Pineda R, Garcia-Galiano D, Roseweir A, Romero M, Sanchez-Garrido MA, Ruiz-Pino F, Morgan K, Pinilla L, Millar RP, Tena-Sempere M (2010) Critical roles of kisspeptins in female puberty and preovulatory gonadotropin surges as revealed by a novel antagonist. Endocrinology 151:722–730
Clarkson J, Boon WC, Simpson ER, Herbison AE (2009) Postnatal development of an estradiol-kisspeptin positive feedback mechanism implicated in puberty onset. Endocrinology 150:3214–3220
Gill JC, Wang O, Kakar S, Martinelli E, Carroll RS, Kaiser UB (2010) Reproductive hormone-dependent and -independent contributions to developmental changes in kisspeptin in GnRH-deficient hypogonadal mice. PLoS One 5:e11911
Nestor CC, Briscoe AM, Davis SM, Valent M, Goodman RL, Hileman SM (2012) Evidence of a role for kisspeptin and neurokinin B in puberty of female sheep. Endocrinology 153:2756–2765
Castellano JM, Bentsen AH, Mikkelsen JD, Tena-Sempere M (2010) Kisspeptins: bridging energy homeostasis and reproduction. Brain Res 1364:129–138
Castellano JM, Navarro VM, Fernandez-Fernandez R, Nogueiras R, Tovar S, Roa J, Vazquez MJ, Vigo E, Casanueva FF, Aguilar E, Pinilla L, Dieguez C, Tena-Sempere M (2005) Changes in hypothalamic KiSS-1 system and restoration of pubertal activation of the reproductive axis by kisspeptin in undernutrition. Endocrinology 146:3917–3925
True C, Kirigiti MA, Kievit P, Grove KL, Smith MS (2011) Leptin is not the critical signal for kisspeptin or luteinising hormone restoration during exit from negative energy balance. J Neuroendocrinol 23:1099–1112
Tovar S, Vazquez MJ, Navarro VM, Fernandez-Fernandez R, Castellano JM, Vigo E, Roa J, Casanueva FF, Aguilar E, Pinilla L, Dieguez C, Tena-Sempere M (2006) Effects of single or repeated intravenous administration of kisspeptin upon dynamic LH secretion in conscious male rats. Endocrinology 147:2696–2704
Li XF, Lin YS, Kinsey-Jones JS, O’Byrne KT (2012) High-fat diet increases LH pulse frequency and kisspeptin-neurokinin B expression in puberty-advanced female rats. Endocrinology 153(9):4422–4431
Acknowledgments
This research was supported by the Marie Curie Outgoing International Fellowship within the seventh Framework Programme of the European Union. The author is grateful to Drs. Amy Oakley and Leonor Pinilla for their constructive comments on the manuscript and Drs. Manuel Tena-Sempere, Don Clifton, and Robert Steiner for their contribution to some of the studies highlighted in this chapter.
Conflict of interest: The author reports no conflicts of interest.
Disclosure: The author has nothing to disclose.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Navarro, V.M. (2013). Interactions Between Kisspeptins and Neurokinin B. In: Kauffman, A., Smith, J. (eds) Kisspeptin Signaling in Reproductive Biology. Advances in Experimental Medicine and Biology, vol 784. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6199-9_15
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6199-9_15
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6198-2
Online ISBN: 978-1-4614-6199-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)