
Abstract
Since 1940 chemotherapy has been one of the major therapies used to kill cancer cells. However, conventional standard cytotoxic agents have a low therapeutic index and often show toxicity in healthy cells. Over the past decade, progress in molecular biology and genomics has identified signaling pathways and mutations driving different types of cancer. Genetic and epigenetic alterations that characterize tumor cells have been used in the development of targeted therapy, a very active area of cancer research. Moreover, identification of synthetic lethal interactions between two altered genes in cancer cells shows much promise to target specifically tumor cells. For a long time, apoptosis was considered the principal mechanism by which cells die from chemotherapeutic agents. Autophagy, necroptosis (a programmed cell death mechanism of necrosis), and lysosomal-mediated cell death significantly improve our understanding of how malignancy can be targeted by anticancer treatments. Autophagy is a highly regulated process by which misfolded proteins and organelles reach lysosomes for their degradation. Alterations in this cellular process have been observed in several pathological conditions, including cancer. The role of autophagy in cancer raised a paradox wherein it can act as a tumor suppressor at early stage of tumor development but can also be used by cancer cells as cytoprotection to promote survival in established tumors. It is interesting that autophagy can be targeted by anticancer agents to provoke cancer cell death. This review focuses on the role of autophagy in cancer cells and its potential to therapeutically kill cancer cells.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
8.1 Overview of the Autophagy Machinery
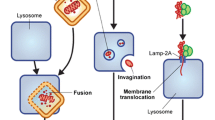
Autophagy is a self-digestive process. From the Greek auto, meaning “oneself,” and phagy, meaning “eating,” this process is highly conserved in organisms from yeast to mammals and acts to remove misfolded proteins, aggregates, lipids, and damaged organelles. To maintain cellular homeostasis, cytoplasmic cargoes are sequestered into vesicles that reach lysosomes, where the material is degraded (Yang and Klionsky 2010). There are different types of autophagy, ranging from nonselective macroautophagy to selective autophagy such as chaperone-mediated autophagy, microautophagy, and the type based on the origin of the sequestered cargo, including mitophagy for mitochondria. Chaperone-mediated autophagy targets specific proteins containing the KFERQ sequence across the lysosome membrane, whereas microautophagy involves the direct engulfment of cytoplasm at the lysosome surface by invagination of the lysosome membrane (Reggiori et al. 2012). In contrast, macroautophagy (referred to hereafter as autophagy) is mediated by the special organelle autophagosome that engulfs proteins, lipids, and damaged organelles into double-membraned vesicles. Then the autophagosome fuses with an endosome/lysosome, a single-membrane vesicle, where the cargo is degraded through lysosomal activity (Fig. 8.1) (Klionsky and Emr 2000). Autophagy is activated under physiological and pathological conditions, such as nutrient starvation, hypoxia, metabolic stress, and in response to drugs and radiation. This dynamic process generates cellular energy resources that allow a cell to adapt its metabolism to energy demand. Defects during any step of the autophagy process result in the accumulation of damaged proteins and/or genomic damage that can stimulate the development of many human diseases, including neurodegeneration, infectious disease, heart disease, and cancer (Levine and Kroemer 2008; Turcotte and Giaccia 2010).

Principal steps regulating the autophagy process. Autophagy involves the formation of double-membrane autophagosomes that fuse with lysosomes to form autolysosomes for the degradation of intracellular proteins and organelles. Under conditions of nutrient deprivation or microenvironmental stress, initiation gives rise to a phagophore, which elongates while being regulated by a series of autophagy-related genes. The phagophore closes into an autophagosome. This autophagosome then fuses with a lysosome to become an amphisome, which will mature and give rise to an autolysosome, where the encapsulated material is degraded via lysosomal activity
8.1.1 Autophagosome Formation
The unique structure of the autophagosome was first observed more than 50 years ago using electronic microscopy, and successive studies have demonstrated that autophagy is regulated through activation of autophagy-related genes (Atg) (Yang and Klionsky 2010). These genes were first identified in yeast, and many of them are found as homologs in murine and human cells (Takeshige et al. 1992). More than 15 mammalian Atg proteins have been identified and regulate the formation of autophagosomes (Table 8.1) (Mizushima et al. 2011). The initiation stage of this process engages the formation of a phagophore, followed by its elongation and closure to form an autophagosome. The origin of the phagophore is still controversial, but the endoplasmic reticulum membrane (Axe et al. 2008; Hayashi-Nishino et al. 2009; Yla-Anttila et al. 2009), mitochondrial outer membrane (Hailey et al. 2010), and plasma membrane (Ravikumar et al. 2010) have been suggested to contribute to autophagosome formation.
The activity of the autophagic machinery is regulated by different complexes: the ULK1/2 kinase complex, the vacuolar sorting protein (Vps) 34/Beclin-1 complex, the shuttling of the Atg9 protein (the only transmembrane Atg) between organelles including endosomes, and the two ubiquitin-conjugation systems, the Atg5-Atg12-Atg16 and Atg8/LC3 complexes (Fig. 8.2) (Orsi et al. 2012; Lamb et al. 2013; Rubinsztein et al. 2012). ULK1and ULK2, two Atg1 homologs, are associated with Atg13 and FIP200 in a large complex that integrates stress signals from the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) (Jung et al. 2009; Mizushima 2010). Many signals, including growth factors, amino acids, glucose, and energy status, regulate mTORC1. Upon inhibition of mTORC1 induced by starvation or chemotherapeutic agents targeting mTOR, ULK1 and ULK2 are phosphorylated and activated, initiating the autophagy cascade. Other complexes essential to autophagosome formation is Beclin-1, the Atg6 homolog, and Vps34, a class III phosphoinositide 3-kinase (PI3K), which recruit autophagy proteins such as UVRAG (ultraviolet irradiation resistance-associated gene), Ambra-1, Bif-1, and Barkor (Kroemer et al. 2010). Furthermore, Beclin-1 binds to anti-apoptotic proteins of the BCL-2 family, such as BCL-XL, through a BCL-2 homology 3 domains and inhibits autophagy (Pattingre et al. 2005; Erlich et al. 2007). In response to starvation, phosphorylation on Bcl-2 by Jun kinase 1 dissociates the binding between Bcl-2 and Beclin-1 and allow Beclin-1 to induce autophagy (Wei et al. 2008; Pattingre et al. 2009). BCL-2 homology 3 mimics can also disrupt Bcl-2 and Beclin-1 binding. Finally, there are two ubiquitin conjugation systems that have been associated with autophagosome formation: Atg12-Atg5-Atg16 and Atg8/LC3. Atg5 and Atg12 were the first Atgs identified in mammals by Mizushima et al. (1998), who reported that the Atg5-Atg12-Atg16 conjugation system was conserved. The other ubiquitin conjugation system is MAP1LC3 (also called LC3), the mammalian Atg8 homolog (Kabeya et al. 2000). In unstressed cells, LC3 is present in cytoplasm in an unprocessed form, LC3I, which is converted into a phosphatidylethanolamine-conjugated form, LC3II, associated with completed autophagosomes. LC3II remains associated with the double-membraned vesicle until fusion with lysosomes. The identification of LC3 is an important finding that is routinely used to monitor autophagy in eukaryote cells. Moreover, LC3 binds the p62/sequestome1 (SQSMT1) protein via its LC3-interactin region domain and prevents its accumulation (Pankiv et al. 2007). p62 Is an adaptor protein involved in protein trafficking to the proteasome and facilitates autophagic degradation of ubiquitinated protein aggregates. It is known to activate the nuclear factor erythroid 2-related factor 2 (NRF2) (Inami et al. 2011). This transcription factor turns on the antioxidant gene transcription that allows cells to protect themselves from oxidative stress.

Overview of the complexes involved in autophagosome formation. At least four important functional groups of autophagy-related gene proteins are required for autophagy: ULK1 protein-kinase complex and vacuolar sorting protein 34–Beclin 1 class III phosphoinositide 3-kinase (PI3K) complex regulate autophagy initiation; the Atg9-Atg2-Atg18 complex regulates the expansion of the phagophore assembly site; and the Atg5-Atg12-Atg16 and LC3 conjugation systems regulate the elongation of autophagosome membranes. Phosphatidylethanolamine (PE)-conjugated LC3 (called LC3-II) remains on the isolation membranes and autophagosome membranes, whereas the Atg12-Atg5-Atg16 complex transiently associates with the isolation membranes and dissociates from the autophagosome membranes. Pharmacological inhibitors of the autophagy process are 3-methyladenine, which inhibits PI3K, and autophagosome formation, while chloroquine (CQ) and hydroxychloroquine (HCQ) block autophagosome maturation by increasing the pH of the lysosomes
8.1.2 Maturation of the Autophagosome Through the Endocytic Pathway
Autophagosomes are subsequently transformed to an amphisome after fusion with an endosome/lysosome. During this step, endocytosis and autophagy share machinery for the maturation of the autophagosome. A functional endocytic pathway from the early endosomes to the late endosomes and including multivesicular bodies is essential to maintaining an efficient autophagic flux. Several proteins, including members of the Rab GTPase family, Vps, and endosomal sorting complexes required for transport, have been identified as regulating each step of this process and are described in recent reviews (Lamb et al. 2013). Rab7 is an important element that controls endosomal maturation and lysosome traffic, and its activity is regulated in part by its GTPase-activating proteins and by the PI3K complex formed by Rubicon-UVRAG-Rab7 (Liang et al. 2008; Sun et al. 2010). Rab7 activity is inhibited by its binding with Rubicon and UVRAG (Liang et al. 2008). However, when the level of Rab7 increases until a threshold point, binding with Rubicon is lost and UVRAG can activate the HOPS (homotypic fusion and Vps) complex, which further increases Rab7 activity, promoting fusion with lysosomes (Zlatic et al. 2011; Peralta et al. 2010).
8.1.3 The End of the Road Through the Lysosome
Lysosomes have emerged as an important platform of mTORC1 signaling and regulation. It has been shown that lysosomal genes are regulated by the transcription factor EB (TFEB), which also controls the major steps of the autophagy pathway (autophagosome formation, autophagosome fusion with lysosomes, and degradation of cargo) linking autophagy to lysosomal biogenesis (Sardiello et al. 2009; Settembre et al. 2011). Under stress or aberrant lysosomal storage conditions, TFEB translocates from the cytoplasm to the nucleus and induces lysosomal biogenesis (Settembre et al. 2012). Other groups demonstrated that the lysosomal reformation that occurs during autophagy is regulated by mTORC1 and that TFEB phosphorylation and nuclear translocation are coordinately regulated by mTORC1 (Yu et al. 2010; Pena-Llopis et al. 2011). At the peak of autophagy, lysosomes are consumed by their fusion with autophagosomes, but after a prolonged period of autophagy, mTORC1 is reactivated (inhibits autophagy) and induces lysosomal biogenesis through TFEB activation (Yu et al. 2010).
The mTORC1 pathway that regulates cell growth in response to numerous cues, including amino acids, has been found on the lysosomal surface, its site of activation (Pena-Llopis et al. 2011; Korolchuk et al. 2011). Although the mechanism that elucidates every step of this process is not completely understood, elegant studies indicate that Rag GTPases (a heterodimeric complex of RagA/B and RagC/D GTPases), also located on the lysosomes, and vacuolar-type H+-ATPase (V-ATPase) form a signaling system that is necessary for amino acid sensing by mTORC1 (Bar-Peled et al. 2012; Zoncu et al. 2011; Settembre et al. 2012; Sancak et al. 2010). Under nutrient-rich conditions, mTOR is located on peripheral lysosomes, where it becomes activated and promotes cell growth and inhibits autophagy, whereas mTOR and lysosomes are clustered in the perinuclear area during starvation, leading to induction of autophagy. This location facilitates the fusion of autophagosomes with lysosomes and autophagosome synthesis (by inhibiting mTOR activity) (Korolchuk and Rubinsztein 2011). The lysosome distribution depends, in part, on their being transported along microtubules, a process mediated by Arl8 (a small GTPase) and KIF2 (a kinesis family member) (Korolchuk et al. 2011). pHi has been shown to affect lysosome positioning, where acidification redistributes lysosomes from their predominantly perinuclear location toward the cell periphery and correlates with increased mTOR activity and inhibition of autophagy (Korolchuk et al. 2011; Heuser 1989).
8.2 Role of Autophagy in Cancer
Cells with defects in autophagy accumulate misfolded proteins, ubiquitinated aggregates, lipid droplets, and damaged organelles (mostly mitochondria, peroxisomes, and endoplasmic reticulum) that could lead to accumulation of reactive oxygen species (ROS), metabolic stress, and toxicity. Disruption of autophagy has been associated with cancer. The consequences of autophagy defects in cancer are complex, and new advances indicate that it could be linked to the tumor stages (White 2012; Mah and Ryan 2012; Janku et al. 2011). Autophagy can suppress tumors by preventing accumulation of toxic waste and tumor initiation, but it can also help cancer cells survive under metabolic stress and promote tumors once the tumor is established. Understanding the role of autophagy in cancer is critical because inhibition or activation of autophagy can be therapeutically applicable to killing cancer cells.
8.2.1 Autophagy in Tumor Suppression and Tumor Initiation
Genetic deletion of Beclin-1 is among the first evidence that autophagy can prevent tumor formation: mice with allelic loss of Beclin-1 are partially defective for autophagy and have increased spontaneous malignancies (Qu et al. 2003; Yue et al. 2003). Similarly, humans with Beclin-1 deletion have a higher frequency of leukemia, lymphomas, and tumors of the liver, lung, breast, ovarian, and prostate (Liang et al. 1999; Aita et al. 1999). Further studies of knockout mice demonstrated that basal autophagy is essential for viability because deletion of both Beclin-1 alleles induces embryonic lethality. In addition, the activation of Beclin-1 inhibits cell proliferation in vitro and tumor growth. Moreover, mice deficient in Atg4C develop fibrosarcomas (Marino et al. 2007), whereas a loss of Atg5 and Atg7 improve the risk of benign liver tumors (Takamura et al. 2011).
It has been shown that autophagy activation can prevent necrotic cell death in apoptosis-deficient cells, a process that may cause local inflammation and promote tumor growth (White et al. 2010). One explanation for the role of autophagy in tumor suppression has been linked to its ability to removed toxic waste during the initiation stage of tumorigenesis. Cells with deregulation in autophagy cause impaired mitochondria and accumulation of ROS, which promote genotoxic stress through DNA damage (Mathew et al. 2007; Degenhardt et al. 2006). This could lead to the loss of mitochondrial potential membrane, activation of phosphatase and tensin homolog–induced putative linase-1 (PINK1) and induction of PARK2, an E3 ubiquitin ligase involved in mitophagy (Arena et al. 2013). PARK2 is a tumor suppressor gene, and mutations of it have been observed in glioblastomas and colon and lung cancers (Veeriah et al. 2010; Poulogiannis et al. 2010).
Another possibility by which autophagy may prevent cancer is through p62 (Mathew et al. 2009). In unstressed cells, NRF2 activity is inhibited by its binding to kelch-like ECH-associated protein 1 (KEAP1), which inactivates the antioxidant defense genes and stimulates proteasomal degradation (Copple et al. 2010; Lau et al. 2010). In autophagy-defective cells or in the presence of oxidative stress, KEAP1 is modified and its binding with NRF2 is lost (Lau et al. 2010). Then, p62 can bind and sequester KEAP1, promoting NRF2 activation, antioxidant defense, and survival. Therefore, autophagy is necessary to prevent p62 accumulation and NRF2 activation that could promote tumorigenesis.
8.2.2 Autophagy in Tumor Progression
Autophagy is induced as an alternative source of energy and metabolites to maintain cell survival during nutrient starvation or metabolic or other stress such as hypoxia, ischemia, and proteasome inhibition. Almost all of these conditions are observed in established tumors. Under stress conditions, autophagy protects dormant cells from damage (White 2012). When the conditions are more favorable or return to normal, these cells can recover and grow. Then, autophagy can provide a survival advantage to tumor cells, allowing them to adapt to metabolic stress found in the tumor microenvironment; a variety of mechanisms have been proposed to support this. It has been shown that the Bcl-2/adenovirus E1B interacting protein (BNIP3), a downstream target of hypoxia-inducible factor (HIF)-1α, can induce autophagy by disrupting the Beclin-1–Bcl-2 complex to release Beclin-1 in response to a hypoxic microenvironment (Bellot et al. 2009). Amino acid and glucose deprivation found in the tumor microenvironment have been correlated with a higher level of autophagosomes and deletion of essential Atgs, which induces tumor cell death associated with the hypoxic regions. Recent studies indicate that human cancer tissues with a low level of Beclin-1 have been associated with worse prognosis in esophageal (Chen et al. 2009), colon (Li et al. 2009), and pancreatic cancer (Kim et al. 2011). In addition, tumors from Beclin-1-deficient mice are more aggressive under hypoxic conditions, a mechanism that could be regulated through the HIF-2α (Lee et al. 2011). Other studies reported that autophagy is triggered to protect cancer cells from nutrient deprivation by activation of AMP-activated protein kinase (AMPK), a sensor of energy status. AMPK activation limits translation initiation and protein synthesis through the inhibition of elongation factor 2 (EF2) and the inhibition of mTOR, leading to the induction of autophagy (Horbinski et al. 2010).
By studying the role of autophagy in cancer, several groups have noticed that cancer cells have a high level of basal autophagy, even in unstressed conditions. White and colleagues showed that activated cells expressing Ras are dependent on autophagy to survive starvation, and biallelic deletion of Atg5 or Atg7 decrease tumor growth of RAS-transformed epithelial cells in the kidneys of nude mice (Guo et al. 2011). This study indicated that autophagy is required to maintain functional mitochondrial and oxidative metabolism necessary to Ras-expressing tumor growth. Autophagy can also promote metastasis and cell survival in response to microenvironmental stresses (Kenific et al. 2010). High expression of LC3 and Beclin-1 are correlated with poor survival and a shorter disease-free period in pancreatic and nasopharyngeal carcinomas, respectively (Fujii et al. 2008; Wan et al. 2010). It is interesting to note that γ-aminobutyric acid type A receptor-associated protein (GABARAP), a member of the LC3 family, is a new prognostic marker for colorectal carcinoma because its overexpression is associated with reduced survival (Miao et al. 2010).
8.3 Autophagy and Cell Death as Targets for Anticancer Therapy
There are a number of molecules targeting various proteins of the apoptosis pathway. Some groups of these molecules – such as ABT-263 (www.clinicaltrials.gov identifier NCT00743028), AT-101 (NCT00275431), and GX15-070MS or Obatoclax (NCT00600964) – affect the activation or balance of the Bcl-2 protein family, tipping the scale toward apoptosis, while others block the inhibitor apoptosis proteins, including AT-406 (NCT01078649), ENZ-3042 (NCT01186328), HGS-1029 (NCT00708006), and LCL-161 (NCT01098838), thus inducing the apoptosis pathway. On the other hand, elucidation of the molecular mechanisms involved in autophagy indicates crosstalk between the apoptotic and autophagic pathways (Amelio et al. 2011; Ouyang et al. 2012). For example, inhibition of apoptosis can induce autophagy, whereas inhibition of autophagy can stimulate apoptosis (Maiuri et al. 2007). In addition, both pathways can be activated through similar proteins, among them, the complex formed by Beclin-1 and Bcl-2 (Kang et al. 2011). Depending on the anticancer agents and the cell type, drugs can have a lethal effect in response to autophagy induction through the influence of the anti-apoptotic effect of Bcl-2 or the phosphorylation of Jun kinase (Wei et al. 2008). Among other proteins that could be involved in the crosstalk between apoptosis and autophagy are the activation of p53, which transcriptionally increases the signaling of AMPK; death-associated protein kinase (DAPK1); tuberous sclerosis protein 2 (TSC2); and ULK1/2 (Feng 2010). Autophagy may also protect against tumorigenesis by limiting necrosis and chronic inflammation in response to metabolic stress, which is associated with the release of the proinflammatory HMGB1 (Degenhardt et al. 2006). A hypoxic tumor microenvironment, nutrient or amino acid levels, as well as the signaling pathway can influence the final outcome between cell death and survival when autophagy is induced. Whether cells can die from autophagy (autophagic cell death) or as a consequence of autophagy induction needs to be addressed.
8.3.1 Autophagy to Induce Cell Death
Various chemotherapeutic agents have been shown to induce autophagy and participate in the induction of cell death. Therefore, inhibition of autophagy using small interfering RNA targeting Atg5, Atg7, or Beclin-1 reduces death, suggesting that autophagy can eliminate tumor cells (Amaravadi et al. 2011; Janku et al. 2011). Table 8.2 summarize agents that have been reported to have anticancer effects as monotherapies. One of the most targeted approaches to killing cancer cells in response to autophagy is through the mTOR pathway. This process regulates cell proliferation and protein translation, and its inhibition induces autophagy as well as cell cycle arrest and apoptosis. The strong induction of autophagy in vivo in response to everolimus, a chemotherapeutic agent targeting mTOR, reduces the growth of advanced pancreatic tumors (Yao et al. 2010) and leukemia (Crazzolara et al. 2009). Furthermore, temsirolimus and everolimus have been approved for the treatment of renal cell carcinoma (RCC). In addition, radiation as well as many chemotherapeutic agents inducing DNA damage and p53 activation have demonstrated a synergic effect in combination with everolimus to kill cancer cells (O’Reilly et al. 2011). Other drugs inhibiting Bcl-2 and activating Beclin-1 in apoptosis-defective cells show a potential effect on cell killing by the formation of autophagosomes. Obatoclax is a Bcl-2 inhibitor that induces cell death. However, when apoptosis is functional, Obatoclax could promote both autophagy and apoptosis to kill acute lymphoblastic leukemia and non-small-lung cancer (Heidari et al. 2010; McCoy et al. 2010).
8.3.2 Inhibition of Autophagy to Improve Anticancer Treatments
As an alternative, autophagy could be associated with chemoresistance by protecting the survival of cancer cells. Thus, inhibition of the autophagic flux synergized the killing effect of chemotherapeutic agents in many tumor types. The mechanism by which autophagy inhibition increases cell death could be associated with a switch toward other types of cell death, such as apoptosis, necrosis, or necroptosis. Chloroquine (CQ) and its analog hydroxychloroquine (HCQ) are antimalarial agents that increase the pH of the lysosome and then inhibit the fusion between autophagosome and lysosome (Amaravadi et al. 2011) (Table 8.3). For example, administration of the Akt inhibitor MK2206 in combination with HCQ is in clinical trials of pancreatic, kidney, and many advanced tumors. HCQ with everolimus is in a phase I clinical trial of RCC. The combination of HCQ, radiation, and temozolomide are in clinical trials of patients with glioblastomas (www.ClinicalTrials.gov identifier NCT00486603). In chronic myelogenous leukemia, cell death is observed by the combined treatment with CQ and the histone deacetylase inhibitor suberoylanilide hydroxamic acid (Carew et al. 2007). Finally, HCQ has been shown to potentiate the anticancer effect of 5-fluorouracil in colon cancer (Sasaki et al. 2010). Two other autophagy inhibitors have recently been identified in preclinical trials. The first inhibitor is lucanthone, or Myricil D, an existing drug that is used for the treatment of schistosomal parasites (Clarkson and Erasmus 1984). While earlier investigations have shown that lucanthone inhibits topoisomerase 2 activity, a more recent study defined a novel mechanism of action for lucanthone that includes the disruption of lysosomal function, inhibition of autophagy, and induction of apoptosis (Carew et al. 2011). In breast carcinoma cell lines, lucanthone is tenfold more potent that CQ and shows a better safety profile than CQ or HCQ. The second autophagy inhibitor is Lys05. This new drug accumulates more easily within the lysosome, increasing pH more effectively compared to HCQ (McAfee et al. 2012). Similar to lucanthone, Lys05 displayed significantly higher anticancer activity than CQ or HCQ in preclinical models, without inducing significant observable toxicity. These two new autophagy inhibitors need to be further investigated as potential therapeutic anticancer agents.
8.4 Synthetic Lethality and Autophagy in Anticancer Drug Discovery
8.4.1 Synthetic Lethality in the Context of Cancer
Advances in cell and molecular biology have improved our knowledge of the mechanism by which cells escape death to become cancerous. The expansion of “omics” technology, from genomic through metabolomic, have identified specific mutations of genes or altered RNA and protein signaling that are responsible for different types of cancer. As discussed earlier, targeted therapy is an active area of research that has expanded the type and modality of treatments (alone or in combination). It is unfortunate that few of them show clinical efficacy, but the ones that received approval from the US Food and Drug Administration have improved survival of inflexible cancers, including RCC (Motzer et al. 2006, 2007, 2008; Gu et al. 2005; Hudes et al. 2007; Escudier et al. 2007a, b), pancreatic cancers (Moore et al. 2007), and non-small-cell lung cancers (Ansari et al. 2009; Shepherd et al. 2005). One promising approach to develop targeted therapy against tumor cells and spare normal tissue is based on synthetic lethality, which targets specific mutations in cancer genes that are not altered in normal cells (Chan and Giaccia 2011). Synthetic lethality is the genetic interaction of two genes, both of which are involved in essential processes (Hartman et al. 2001). When either gene is mutated alone, the cell remains viable. However, the combination of these two mutations induces cell death (Hartman et al. 2001; Kaelin 2005; Hartwell et al. 1997). Chemical or RNA interference screens have made it possible to search for synthetic lethal interactions in mammalian cells (Farmer et al. 2005; Jiang et al. 2009). Thus, deregulation of an oncogene or inactivation of a tumor suppressor gene can be specifically targeted through synthetic lethality to kill tumor cells. This approach could be advantageous and facilitate the development of treatment with a single agent because only cancer cells with the specific mutation will die. The normal cells will not be affected by the therapy, and side effects from chemotherapy will be reduced. Synthetic lethality could also be used in combination with drugs and/or radiation or in patients with relapsed cancer, providing the opportunity to use lower doses of cytotoxic drugs, improve the therapeutic index of cytotoxic drugs, and reduce off-target effects. Driving mutation in cancer cells can change at different stages of tumor development – from the primary tumor to metastases – and therefore synthetic lethality could be useful to target the epithelial-to-mesenchymal transition as well as metastatic disease for which there are few options of effective treatment.
The first example of synthetic lethal interaction in cancer cells came from the mutation affecting the gene BRCA1/2 and the enzyme poly (ADP ribose) polymerase (PARP). The tumor suppressor protein BRCA is an important player in the reparation of double-strand DNA breaks, and mutations affecting these genes have been reported in breast and ovarian cancers (Hall et al. 1992; Casey et al. 1993; Parikh and Advani 1996). In addition, PARP is an important protein that repairs single-stand DNA breaks (Petermann et al. 2005). By using pharmacological inhibitors or small interfering/small hairpin RNA targeting PARP in BRCA-mutated cells, studies indicate that these cells were not able to repair double-strand DNA breaks and recombination lesions and that they die by apoptosis (Bryant et al. 2005; Farmer et al. 2005). The identification of the lethal interaction between BRCA mutations and PARP inhibitors has been investigated in cancer cells, and several PARP inhibitors are currently in clinical trials (phase I/II/III) for the treatment of breast and ovarian cancer with the inactivated BRCA1/2 gene (Tutt et al. 2010; Fong et al. 2009; Hutchinson 2010). These studies demonstrated proof of the concept that synthetic lethality can be useful in (and are possible for) targeting cancer cells. Some researchers and pharmaceutical companies are working to develop this killing approach in association with other oncogenes that are frequently disrupted in cancer, such as the oncogenes Ras and Myc (Chan and Giaccia 2011). New drugs (triphenyltetrazolium and a sulphinylcytidine derivative) (Torrance et al. 2001), the inhibitor apoptosis protein survivin (Sarthy et al. 2007), and cyclin-dependent kinase 4 (Puyol et al. 2010) have been identified by independent screening and demonstrate some potential as KRAS inhibitors. Otherwise, other large screens performed in Ras-mutated cells and pathways governing the mitotic machinery or the proteasomes showed synthetic lethal interaction with Ras (Scholl et al. 2009; Luo et al. 2009). Among other examples of synthetic lethal interaction, inhibition of aurora kinase B or death receptor 5 agonists induced killing in cells overexpressing Myc (Wang et al. 2004; Yang et al. 2010).
8.4.2 Synthetic Lethality and Autophagy in RCC
RCC, the most common form of kidney cancer, is particularly challenging because it is resistant to standard cytotoxic therapies. The overall 5-year survival rate ranges from 85 % in patients with local tumors treated by partial or total nephrectomy to 10 % in patients with advanced or metastatic RCC (Motzer et al. 1996). There is no curative treatment for RCC, and these patients are diagnosed at an advanced stage because no symptoms are associated with kidney tumors until they are quite large. Current targeted therapies used to treat RCC (e.g., bevacizumab, sunitinib) have focused on anti-angiogenic agents targeting vascular endothelial growth factor and its receptor and agents that inhibit mTOR (e.g., temsirolimus, everolimus). Although these agents demonstrate efficiency in RCC, the clinical response to these therapies is generally short-lived, suggesting that tumor growth might be supported by alternative sources of nutrients, such as autophagy (Patel et al. 2006).
Biallelic inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene arises in up to 85 % of RCC cases. Mutation and/or hypermethylation, which inactivate the VHL gene, are also responsible for the hereditary VHL cancer syndrome that affects 1 in 36,000 individuals (Maher 2004). These patients inherit a faulty allele of VHL and are predisposed to the development of renal cysts, RCC, retinal and central nervous system hemangioblastomas, and pheochromocytomas (Maher 2004; Kaelin 2008). Tumor development is caused by somatic inactivation of the remaining wild-type allele (Young et al. 2009; Nickerson et al. 2008; Patard et al. 2009). Because VHL is a common and early event in the development of RCC, targeting its inactivation represents a promising target for the development of new therapies. High-throughput screening using a small interfering RNA library or small molecules have been performed in VHL-deficient RCC in two independent studies. The first approach used a library of small hairpin RNA against about 100 different kinases and distinguished CDK6, hepatocyte growth factor receptor (also known as MET), and mitogen-activated protein kinase 1 (MAP2K1), which have the ability to reduce growth of VHL-inactivated cells (Bommi-Reddy et al. 2008). A recent study reported that microRNA-1826 reduced the expression of β-catenin and MAP2K1 in RCC and inhibits the proliferation of VHL-deficient cells by inducing G1 arrest and apoptosis (Hirata et al. 2012).
The second approach used a library of 64,000 small molecules to find drugs that specifically kill RCC lacking VHL without affecting the viability of the cells with the functional VHL gene (Turcotte et al. 2008). This study identified two classes of compounds: ST-31 inhibited the survival of VHL-deficient cells through GLUT1 and HIF-1α (Chan et al. 2011), whereas STF-62247 killed VHL-mutated cells by inducing autophagy (Turcotte et al. 2008). Moreover, they showed that reducing levels of Atg5, Atg7, and Atg9 rescued the survival of VHL-deficient cells in response to STF-62247, indicating that autophagy induction is required for cell death. Turcotte et al. recently investigated the autophagy machinery and found that the in vitro and in vivo sensitivity of VHL-deficient RCC in response to STF-62247 is associated with a default in the autophagic process involving lysosomal degradation, which ultimately leads to cell death. In accordance with this, cells lacking VHL expression accumulate autophagic vacuoles that are not degraded by lysosomes, thus interfering with the clearance of damaged organelles and misfolded or aggregated proteins in response to STF-62247. Furthermore, lysosomes in these cells undergo labialization or lysosome permeabilization, which also contributes to cell death. Production of ROS that are not detoxified by the cells, lysosomotropic agents, microtubule-stabilizing agents, protein kinase C, phospholipase A2, and lipids are the mechanisms speculated to induce lysosome permeabilization (Kreuzaler and Watson 2012).
8.5 Conclusion and Future Directions
The field of cancer research has made significant progress in recent years. New techniques have identified genetic alterations associated with different types of cancer. In parallel, advances in drug screening using small interfering RNA libraries and/or small molecules have expanded drug design and the development of targeted therapies. Using these approaches, new anticancer agents or novel uses of existing drugs are in clinical trials or have been approved for treatment. Exciting drugs exploiting synthetic lethality have gained attention as a new type of anticancer therapy. Searching for synthetic lethal interaction between two genes or drug-gene interactions represent a promising approach to kill tumor cells and leave normal cells healthy. Cancer cells evade programmed cell death to initiate tumor formation, and research has identified nonapoptotic mechanisms for how cells survive or die in response to a drug. The role of autophagy in cancer is complex: it can help prevent tumor initiation, overcome resistance to anticancer therapy, promote cytoprotection in established tumors, and may help to eradicate malignant cells. Inhibitors of the autophagic flux, including CQ and HCQ, used alone or in combination with chemotherapeutic agents and/or radiation, are currently in clinical trials of several types of cancer. In addition, drugs that induce autophagy and provoke cell death show encouraging results. Overall, other screens using synthetic lethality and our knowledge of cell death mechanisms could open a new field of oncology, helping to design monotherapy agents or a combination of cytotoxic chemotherapy and radiation.
References
Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B (1999) Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 59(1):59–65. doi:10.1006/geno.1999.5851
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E (2011) Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 17(4):654–666. doi:10.1158/1078-0432.CCR-10-2634
Amelio I, Melino G, Knight RA (2011) Cell death pathology: cross-talk with autophagy and its clinical implications. Biochem Biophys Res Commun 414(2):277–281. doi:10.1016/j.bbrc.2011.09.080
Ansari J, Palmer DH, Rea DW, Hussain SA (2009) Role of tyrosine kinase inhibitors in lung cancer. Anticancer Agents Med Chem 9(5):569–575
Arena G, Gelmetti V, Torosantucci L, Vignone D, Lamorte G, De Rosa P, Cilia E, Jonas EA, Valente EM (2013) PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. doi:10.1038/cdd.2013.19
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182(4):685–701. doi:10.1083/jcb.200803137
Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM (2012) Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150(6):1196–1208. doi:10.1016/j.cell.2012.07.032
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29(10):2570–2581. doi:10.1128/MCB.00166-09
Bommi-Reddy A, Almeciga I, Sawyer J, Geisen C, Li W, Harlow E, Kaelin WG Jr, Grueneberg DA (2008) Kinase requirements in human cells: III. Altered kinase requirements in VHL−/− cancer cells detected in a pilot synthetic lethal screen. Proc Natl Acad Sci U S A 105(43):16484–16489. doi:10.1073/pnas.0806574105
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434(7035):913–917. doi:10.1038/nature03443
Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P, Giles FJ, Cleveland JL (2007) Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 110(1):313–322. doi:10.1182/blood-2006-10-050260
Carew JS, Espitia CM, Esquivel JA 2nd, Mahalingam D, Kelly KR, Reddy G, Giles FJ, Nawrocki ST (2011) Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J Biol Chem 286(8):6602–6613. doi:10.1074/jbc.M110.151324
Casey G, Plummer S, Hoeltge G, Scanlon D, Fasching C, Stanbridge EJ (1993) Functional evidence for a breast cancer growth suppressor gene on chromosome 17. Hum Mol Genet 2(11):1921–1927
Chan DA, Giaccia AJ (2011) Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov 10(5):351–364. doi:10.1038/nrd3374
Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, SolowCordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP, Giaccia AJ (2011) Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med 3(94):94ra70. doi:10.1126/scitranslmed.3002394
Chen Y, Lu Y, Lu C, Zhang L (2009) Beclin-1 expression is a predictor of clinical outcome in patients with esophageal squamous cell carcinoma and correlated to hypoxia-inducible factor (HIF)-1alpha expression. Pathol Oncol Res 15(3):487–493. doi:10.1007/s12253-008-9143-8
Clarkson J, Erasmus DA (1984) Schistosoma mansoni: an in vivo study of drug-induced autophagy in the gastrodermis. J Helminthol 58(1):59–68
Copple IM, Lister A, Obeng AD, Kitteringham NR, Jenkins RE, Layfield R, Foster BJ, Goldring CE, Park BK (2010) Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem 285(22):16782–16788. doi:10.1074/jbc.M109.096545
Crazzolara R, Bradstock KF, Bendall LJ (2009) RAD001 (Everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy 5(5):727–728
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E (2006) Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10(1):51–64. doi:10.1016/j.ccr.2006.06.001
Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R, Pinkas-Kramarski R (2007) Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 3(6):561–568
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM (2007a) Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 356(2):125–134. doi:10.1056/NEJMoa060655
Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, Chevreau C, Filipek M, Melichar B, Bajetta E, Gorbunova V, Bay JO, Bodrogi I, Jagiello-Gruszfeld A, Moore N (2007b) Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet 370(9605):2103–2111. doi:10.1016/S0140-6736(07)61904-7
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434(7035):917–921. doi:10.1038/nature03445
Feng Z (2010) p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb Perspect Biol 2(2):a001057. doi:10.1101/cshperspect.a001057
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361(2):123–134. doi:10.1056/NEJMoa0900212
Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M, Kinoshita T, Ueno T, Esumi H, Ochiai A (2008) Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci 99(9):1813–1819. doi:10.1111/j.1349-7006.2008.00893.x
Gu J, Ruppen ME, Cai P (2005) lipase-catalyzed regioselective esterification of rapamycin: synthesis of temsirolimus (CCI-779). Org Lett 7(18):3945–3948. doi:10.1021/ol0514395
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25(5):460–470. doi:10.1101/gad.2016311
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141(4):656–667. doi:10.1016/j.cell.2010.04.009
Hall JM, Friedman L, Guenther C, Lee MK, Weber JL, Black DM, King MC (1992) Closing in on a breast cancer gene on chromosome 17q. Am J Hum Genet 50(6):1235–1242
Hartman JL, Garvik B, Hartwell L (2001) Principles for the buffering of genetic variation. Science 291(5506):1001–1004
Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH (1997) Integrating genetic approaches into the discovery of anticancer drugs. Science 278(5340):1064–1068
Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A (2009) A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11(12):1433–1437. doi:10.1038/ncb1991
Heidari N, Hicks MA, Harada H (2010) GX15-070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death Dis 1:e76. doi:10.1038/cddis.2010.53
Heuser J (1989) Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol 108(3):855–864
Hirata H, Hinoda Y, Ueno K, Nakajima K, Ishii N, Dahiya R (2012) MicroRNA-1826 directly targets beta-catenin (CTNNB1) and MEK1 (MAP2K1) in VHL-inactivated renal cancer. Carcinogenesis 33(3):501–508. doi:10.1093/carcin/bgr302
Horbinski C, Mojesky C, Kyprianou N (2010) Live free or die: tales of homeless (cells) in cancer. Am J Pathol 177(3):1044–1052. doi:10.2353/ajpath.2010.091270
Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O’Toole T, Lustgarten S, Moore L, Motzer RJ (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356(22):2271–2281. doi:10.1056/NEJMoa066838
Hutchinson L (2010) Targeted therapies: PARP inhibitor olaparib is safe and effective in patients with BRCA1 and BRCA2 mutations. Nat Rev Clin Oncol 7(10):549. doi:10.1038/nrclinonc.2010.143
Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee MS, Tanaka K, Komatsu M (2011) Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol 193(2):275–284. doi:10.1083/jcb.201102031
Janku F, McConkey DJ, Hong DS, Kurzrock R (2011) Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 8(9):528–539. doi:10.1038/nrclinonc.2011.71
Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT (2009) The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev 23(16):1895–1909. doi:10.1101/gad.1815309
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20(7):1992–2003. doi:10.1091/mbc.E08-12-1249
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19(21):5720–5728. doi:10.1093/emboj/19.21.5720
Kaelin WG Jr (2005) The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5(9):689–698
Kaelin WG Jr (2008) The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer 8(11):865–873
Kang R, Zeh HJ, Lotze MT, Tang D (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18(4):571–580. doi:10.1038/cdd.2010.191
Kenific CM, Thorburn A, Debnath J (2010) Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol 22(2):241–245. doi:10.1016/j.ceb.2009.10.008
Kim HS, Lee SH, Do SI, Lim SJ, Park YK, Kim YW (2011) Clinicopathologic correlation of beclin-1 expression in pancreatic ductal adenocarcinoma. Pathol Res Pract 207(4):247–252. doi:10.1016/j.prp.2011.02.007
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290(5497):1717–1721
Korolchuk VI, Rubinsztein DC (2011) Regulation of autophagy by lysosomal positioning. Autophagy 7(8):927–928
Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, O’Kane CJ, Deretic V, Rubinsztein DC (2011) Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 13(4):453–460. doi:10.1038/ncb2204
Kreuzaler P, Watson CJ (2012) Killing a cancer: what are the alternatives? Nat Rev Cancer 12(6):411–424. doi:10.1038/nrc3264
Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40(2):280–293. doi:10.1016/j.molcel.2010.09.023
Lamb CA, Dooley HC, Tooze SA (2013) Endocytosis and autophagy: shared machinery for degradation. Bioessays 35(1):34–45. doi:10.1002/bies.201200130
Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD (2010) A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 30(13):3275–3285. doi:10.1128/MCB.00248-10
Lee SJ, Kim HP, Jin Y, Choi AM, Ryter SW (2011) Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 7(8):829–839
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132(1):27–42
Li BX, Li CY, Peng RQ, Wu XJ, Wang HY, Wan DS, Zhu XF, Zhang XS (2009) The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy 5(3):303–306
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402(6762):672–676. doi:10.1038/45257
Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU (2008) Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10(7):776–787. doi:10.1038/ncb1740
Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ (2009) A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137(5):835–848. doi:10.1016/j.cell.2009.05.006
Mah LY, Ryan KM (2012) Autophagy and cancer. Cold Spring Harb Perspect Biol 4(1):a008821. doi:10.1101/cshperspect.a008821
Maher ER (2004) Von Hippel-Lindau disease. Curr Mol Med 4(8):833–842
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8(9):741–752. doi:10.1038/nrm2239
Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C (2007) Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem 282(25):18573–18583. doi:10.1074/jbc.M701194200
Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E (2007) Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 21(11):1367–1381. doi:10.1101/gad.1545107
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137(6):1062–1075. doi:10.1016/j.cell.2009.03.048
McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, Winkler JD, Amaravadi RK (2012) Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A 109(21):8253–8258. doi:10.1073/pnas.1118193109
McCoy F, Hurwitz J, McTavish N, Paul I, Barnes C, O’Hagan B, Odrzywol K, Murray J, Longley D, McKerr G, Fennell DA (2010) Obatoclax induces Atg7-dependent autophagy independent of beclin-1 and BAX/BAK. Cell Death Dis 1:e108. doi:10.1038/cddis.2010.86
Miao Y, Zhang Y, Chen Y, Chen L, Wang F (2010) GABARAP is overexpressed in colorectal carcinoma and correlates with shortened patient survival. Hepatogastroenterology 57(98):257–261
Mizushima N (2010) The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22(2):132–139. doi:10.1016/j.ceb.2009.12.004
Mizushima N, Sugita H, Yoshimori T, Ohsumi Y (1998) A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 273(51):33889–33892
Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132. doi:10.1146/annurev-cellbio-092910-154005
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada clinical trials group. J Clin Oncol 25(15):1960–1966. doi:10.1200/JCO.2006.07.9525
Motzer RJ, Bander NH, Nanus DM (1996) Renal-cell carcinoma. N Engl J Med 335(12):865–875. doi:10.1056/NEJM199609193351207
Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI (2006) Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol 24(1):16–24. doi:10.1200/JCO.2005.02.2574
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA (2007) Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 356(2):115–124. doi:10.1056/NEJMoa065044
Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A (2008) Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372(9637):449–456. doi:10.1016/S0140-6736(08)61039-9
Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, Navratilova M, Szeszenia-Dabrowska N, Mates D, Mukeria A, Holcatova I, Schmidt LS, Toro JR, Karami S, Hung R, Gerard GF, Linehan WM, Merino M, Zbar B, Boffetta P, Brennan P, Rothman N, Chow WH, Waldman FM, Moore LE (2008) Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res 14(15):4726–4734
O’Reilly T, McSheehy PM, Wartmann M, Lassota P, Brandt R, Lane HA (2011) Evaluation of the mTOR inhibitor, everolimus, in combination with cytotoxic antitumor agents using human tumor models in vitro and in vivo. Anticancer Drugs 22(1):58–78. doi:10.1097/CAD.0b013e3283400a20
Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA (2012) Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 23(10):1860–1873. doi:10.1091/mbc.E11-09-0746
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK (2012) Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 45(6):487–498. doi:10.1111/j.1365-2184.2012.00845.x
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282(33):24131–24145. doi:10.1074/jbc.M702824200
Parikh B, Advani S (1996) Pattern of second primary neoplasms following breast cancer. J Surg Oncol 63(3):179–182. doi:10.1002/(SICI)1096-9098(199611)63:3<179::AID-JSO8>3.0.CO;2-A
Patard JJ, Rioux-Leclercq N, Masson D, Zerrouki S, Jouan F, Collet N, Dubourg C, Lobel B, Denis M, Fergelot P (2009) Absence of VHL gene alteration and high VEGF expression are associated with tumour aggressiveness and poor survival of renal-cell carcinoma. Br J Cancer 101(8):1417–1424
Patel PH, Chadalavada RS, Chaganti RS, Motzer RJ (2006) Targeting von Hippel-Lindau pathway in renal cell carcinoma. Clin Cancer Res 12(24):7215–7220
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122(6):927–939. doi:10.1016/j.cell.2005.07.002
Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P (2009) Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem 284(5):2719–2728. doi:10.1074/jbc.M805920200
Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, Brugarolas J (2011) Regulation of TFEB and V-ATPases by mTORC1. EMBO J 30(16):3242–3258. doi:10.1038/emboj.2011.257
Peralta ER, Martin BC, Edinger AL (2010) Differential effects of TBC1D15 and mammalian Vps39 on Rab7 activation state, lysosomal morphology, and growth factor dependence. J Biol Chem 285(22):16814–16821. doi:10.1074/jbc.M110.111633
Petermann E, Keil C, Oei SL (2005) Importance of poly(ADP-ribose) polymerases in the regulation of DNA-dependent processes. Cell Mol Life Sci 62(7–8):731–738. doi:10.1007/s00018-004-4504-2
Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, Arends MJ (2010) PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci U S A 107(34):15145–15150. doi:10.1073/pnas.1009941107
Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, Guerra C, Santamaria D, Barbacid M (2010) A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 18(1):63–73. doi:10.1016/j.ccr.2010.05.025
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112(12):1809–1820. doi:10.1172/JCI20039
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC (2010) Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 12(8):747–757. doi:10.1038/ncb2078
Reggiori F, Komatsu M, Finley K, Simonsen A (2012) Selective types of autophagy. Int J Cell Biol 2012:156272. doi:10.1155/2012/156272
Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11(9):709–730. doi:10.1038/nrd3802
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141(2):290–303. doi:10.1016/j.cell.2010.02.024
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325(5939):473–477. doi:10.1126/science.1174447
Sarthy AV, Morgan-Lappe SE, Zakula D, Vernetti L, Schurdak M, Packer JC, Anderson MG, Shirasawa S, Sasazuki T, Fesik SW (2007) Survivin depletion preferentially reduces the survival of activated K-Ras-transformed cells. Mol Cancer Ther 6(1):269–276. doi:10.1158/1535-7163.MCT-06-0560
Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, Kaneko M, Kitayama J, Takahashi K, Nagawa H (2010) Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 10:370. doi:10.1186/1471-2407-10-370
Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, Dohner K, Bullinger L, Sandy P, Boehm JS, Root DE, Jacks T, Hahn WC, Gilliland DG (2009) Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 137(5):821–834. doi:10.1016/j.cell.2009.03.017
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332(6036):1429–1433. doi:10.1126/science.1204592
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31(5):1095–1108. doi:10.1038/emboj.2012.32
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabarbara P, Seymour L (2005) Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 353(2):123–132. doi:10.1056/NEJMoa050753
Sun Q, Westphal W, Wong KN, Tan I, Zhong Q (2010) Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A 107(45):19338–19343. doi:10.1073/pnas.1010554107
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25(8):795–800. doi:10.1101/gad.2016211
Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 119(2):301–311
Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW (2001) Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol 19(10):940–945. doi:10.1038/nbt1001-940
Turcotte S, Giaccia AJ (2010) Targeting cancer cells through autophagy for anticancer therapy. Curr Opin Cell Biol 22(2):246–251. doi:10.1016/j.ceb.2009.12.007
Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ (2008) A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell 14(1):90–102. doi:10.1016/j.ccr.2008.06.004
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376(9737):235–244. doi:10.1016/S0140-6736(10)60892-6
Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB, Chan TA (2010) Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 42(1):77–82. doi:10.1038/ng.491
Wan XB, Fan XJ, Chen MY, Xiang J, Huang PY, Guo L, Wu XY, Xu J, Long ZJ, Zhao Y, Zhou WH, Mai HQ, Liu Q, Hong MH (2010) Elevated Beclin 1 expression is correlated with HIF-1alpha in predicting poor prognosis of nasopharyngeal carcinoma. Autophagy 6(3):395–404
Wang Y, Engels IH, Knee DA, Nasoff M, Deveraux QL, Quon KC (2004) Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell 5(5):501–512
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30(6):678–688. doi:10.1016/j.molcel.2008.06.001
White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12(6):401–410. doi:10.1038/nrc3262
White E, Karp C, Strohecker AM, Guo Y, Mathew R (2010) Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol 22(2):212–217. doi:10.1016/j.ceb.2009.12.008
Yang Z, Klionsky DJ (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12(9):814–822. doi:10.1038/ncb0910-814
Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM (2010) Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci U S A 107(31):13836–13841. doi:10.1073/pnas.1008366107
Yao JC, Lombard-Bohas C, Baudin E, Kvols LK, Rougier P, Ruszniewski P, Hoosen S, St Peter J, Haas T, Lebwohl D, Van Cutsem E, Kulke MH, Hobday TJ, O’Dorisio TM, Shah MH, Cadiot G, Luppi G, Posey JA, Wiedenmann B (2010) Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol 28(1):69–76. doi:10.1200/JCO.2009.24.2669
Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL (2009) 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 5(8):1180–1185
Young AC, Craven RA, Cohen D, Taylor C, Booth C, Harnden P, Cairns DA, Astuti D, Gregory W, Maher ER, Knowles MA, Joyce A, Selby PJ, Banks RE (2009) Analysis of VHL gene alterations and their relationship to clinical parameters in sporadic conventional renal cell carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res 15(24):7582–7592. doi:10.1158/1078-0432.CCR-09-2131
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465(7300):942–946. doi:10.1038/nature09076
Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 100(25):15077–15082. doi:10.1073/pnas.2436255100
Zlatic SA, Tornieri K, L’Hernault SW, Faundez V (2011) Metazoan cell biology of the HOPS tethering complex. Cell Logist 1(3):111–117. doi:10.4161/cl.1.3.17279
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334(6056):678–683. doi:10.1126/science.1207056
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this paper
Cite this paper
Reyjal, J., Cormier, K., Turcotte, S. (2014). Autophagy and Cell Death to Target Cancer Cells: Exploiting Synthetic Lethality as Cancer Therapies. In: Koumenis, C., Hammond, E., Giaccia, A. (eds) Tumor Microenvironment and Cellular Stress. Advances in Experimental Medicine and Biology, vol 772. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5915-6_8
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5915-6_8
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5914-9
Online ISBN: 978-1-4614-5915-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)