
Abstract
During each step of prostate cancer metastasis, cancer displays phenotypic plasticity that is associated with the expression of both epithelial and mesenchymal properties or an epithelial to mesenchymal transition. This phenotypic transition is typically in response to microenvironment signals and is the basis for basic cancer cell survival (e.g. motility and invasion versus proliferation). In this review we discuss the loss and gain of E-cadherin expression as a marker of tumor plasticity throughout the steps of metastasis, and particularly focus on dynamic tumor–stromal interaction that induce a cancer cell-associated mesenchymal to epithelial reverting transition in the bone and liver microenvironments.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Prostate Cancer
- Prostate Cancer Cell
- Mesenchymal Phenotype
- Bone Microenvironment
- Prostate Cancer Metastasis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Epithelial to Mesenchymal Transition
Histological evidence of distinct neoplastic cell types within a tumor mass were observed as early as 1978 [1]. There are two main cell types; epithelial and mesenchymal cells, albeit most tumor cells are derived from epithelial origins, however in 1987 the term epithelial to mesenchymal transition (EMT) was utilized. This was subsequently followed by Elizabeth Hay in 1995 [2], with a cellular characterization of transitioned cells, that is still currently utilized to identify phenotypic subtypes within the tumor mass. Several hallmarks to phenotypically characterize these transitioned cells, such as cellular morphogenesis, change in shape, and tissue organization have all been associated with EMT. However, loss of cell–cell connectivity appears to an essential step feature. Normal epithelial cells comprise a sheet of cells that adhere laterally to each other by cell-to-cell junctions. In addition, epithelial cells have apical–basolateral polarization that is maintained through organization of the actin cytoskeleton, which has intimate interactions with cell membrane adhesion molecules such as cadherins, tight junctions, and certain integrins. This allows the polarized cells to maintain cell–cell junctions as a lateral belt, preventing robust cell motility, while remaining within the epithelial layer.
Mesenchymal-like cells, on the other hand, are spindle-shaped cells that exhibit end-to-end polarity, and have fibroblast morphology. Mesenchymal cells do not form an organized cell layer, nor do they have the same apical–basolateral organization, polarization of cell surface molecules, and the actin cytoskeleton as epithelial cells. Cell–cell contacts with neighboring mesenchymal cells are possible, however limited to focal adhesion only. As such this provides the freedom to migrate and interact with the surround extracellular matrix (ECM). Cell migration results from dynamic remodeling of actin into filamentous filopodia, lamellipodia, stress fibers. These cell protrusions lead to dynamic interactions with ECM substrates, which are mainly intergrins. The onset of these cell extensions are a prerequisite for maintenance of cell motility in normal and cancer cells, whether they are initiated spontaneously or induced by chemokines and growth factors. Coincidently, the migration mechanisms that occur in normal, non-neoplastic cells, such as embryonic morphogenesis, wound healing and immune-cell trafficking are identical to neoplastic cells [3, 4].
1.1 Epithelial to Mesenchymal Transition in Prostate Cancer
EMT has been shown to be a necessary step in the dissemination of cancer cell from the primary tumor mass. During this process there have been documented changes in the phenotypic expression of the cancer cells including a reduction in the cell adhesiveness. In-depth analysis showed that reduced or aberrant expression of cytokeratin levels, and cell–cell contacts related proteins are observed over multiple cancer types including breast and prostate cancer. Adhesive complexes such as ZO-1, desmoplakin, and E-cadherin are typically loss, and serve as a prerequisite for dissemination. The clinical significance of E-cadherin loss has also been well documented. Decreased expression of cell adhesion molecule E-cadherin has been largely observed to be inversely correlated clinical characteristic including grade, local invasiveness, and biochemical failure after salvage radiotherapy. Furthermore, patients with biochemical failure after prostatectomy and aberrant E-cadherin expression are likely to have subclinical disseminated disease [5]. Thus, the mechanisms responsible for such changes in adhesion complexes are of great interest.
Majority of the reports focused on of E-cadherin gene (CDH1), suggest that hypermethylation of the E-cadherin promoter [6, 7], is the main mode of downregulation, however a combination of mutations in one allele with loss or inactivation (by DNA methylation) of the remaining allele [8, 9] has been observed. However, in many types of cancer including breast and prostate cancers, E-cadherin expression is lost without mutations in the gene [10], due to transcriptional repression of E-cadherin. Concomitant with the loss of E-cadherin, N-cadherin levels increases during prostate carcinomas. This increased expression of N-cadherin has also been observed in invasive prostate cancer cell lines, and is associated with androgen deprivation [11]. The decreases in E-cadherin expression and increases in N-cadherin expression have been shown to be correlated with increased metastatic ability [12, 13]. Up-regulation of N-cadherin, and cadherin-11, and OB (osteoblasts) cadherin are typically associated with high-grade E-cadherin negative tumors. Other EMT-related changes included transition from cubodial morphology to a spindle-shaped fibroblastic morphology, and genotypic changes including loss of cytokeratin, and increased vimentin, snail, collagen I, thrombospondin-I, and other mesenchymal genes. However, the most consistent marker of EMT has been E-cadherin.
The relevance of EMT-associated markers is supported by studies describing how expression is regulated. Many transcription factors such as the family of zinc finger proteins of the Slug/Snail family, EF1/ZEB1, SIP-1, and the basic helix-loop-helix E12/E47 factor that interact with E-box sequences in the proximal E-cadherin promoter region triggering repression. Of these transcriptional repressors, forced expression of SNAIL is sufficient to induce EMT in ARCAPE and LnCaP prostate cancer cell lines [14], while Slug acts to only regulate cell proliferation [15]. However, recent reports have suggested in PC-3 cells that SNAIL inhibition alters common EMT markers, but does not affect invasiveness [16]. Other transcription factors are implicated as EMT mediators as well. TWIST, a highly conserved bHLH transcription factor, is upregulated in 90% of prostate cancer tissues. RNAi interference of TWIST expression significantly increased sensitivity to the anticancer drug taxol-induced cell death [17]. Furthermore, in addition to EMT, TWIST may also promote prostate cancer to bone metastasis by modulating prostate cancer cell-mediated bone remodeling via regulating the expression of a secretory factor, DKK-1, and enhancing osteomimicry of prostate cancer cells [18]. Thus, multiple factors contribute EMT in prostate cancer cells. Although the complex mechanisms that regulate the expression of multiple factors simultaneously in prostate cancer one common observation is that targeting individual factor is sufficient to reverse step wise events associated with EMT, thus providing targets for the development of therapeutics.
Decreased cell–cell adhesion in many cancers may not only be the result of direct transcriptional regulation. Soluble factors such as epidermal growth factor (EGF), scatter factor/hepatocyte growth factor (SF/HGF), and members of the transforming growth factor, TGFβ1, and basic fibroblast growth factor (bFGF) families have been shown to promote EMT in several model systems. Most all of these have been shown to influence the downregulate of E-cadherin expression with subsequent increased cell proliferation, dedifferentiation, and induction of cell motility [19–21]. As cancer-associated EMT is reversible, the loss of cell–cell connections creates a situation where decreased E-cadherin levels concede the tight junctions and enable apically secreted soluble growth factors to establish an autocrine loop with the basolaterally sequestered receptors. [22]. This feed-forward mechanism supports the maintenance of the mesenchymal phenotype. Although decreased levels of E-cadherin mRNA occurs at the transcriptional level, E-cadherin stability is a direct result of phosphorylated catenins. Extensive investigations have revealed that increased phosphorylation of the preferential catenins, β-catenin and p120, destabilize the cadherin complex thus inducing scattering of cancer cell lines to a more invasive phenotype [23]. We have showed that DU-145 and PC-3 cells express aberrant p120ctn and ß-catenin, and this is reversible through blockage of EGFR signaling [24]. In addition to disrupting the cell–cell junctions and enabling a more migratory phenotype, EGF upregulates secretion of matrix metalloproteinases that degrade the ECM aiding in tumor dissemination. EGF upregulates matrilysin (MMP-7) that mediates extracellular cleavage of E-cadherin, thereby further disrupting cell–cell adhesion and switching of prostate cells from a lesser to a highly invasive phenotype [25]. Thus ADAM10, ADAM9 knockdown increased E-cadherin and integrins and modulates epithelial phenotype and functional characteristics of prostate cancer cells [26], further emphasizes the vast number of pathways regulating E-cadherin expression.
Accumulating evidence suggest that growth factor-induced EMT is the result of transcriptional reprogramming and chromatin remodeling. Of the soluble growth factors mentioned, TGFß-1 is the most noted, however for the focus of this review we will focus on tyrosine kinase growth regulation of EMT. IGF-I stimulation of ARCaPE cells upregulates ZEB1 expression in prostate cancer cells exhibiting a phenotype and increased cell migration. The authors also demonstrated that this is mediated through activation of MAPK/ERK pathway [27]. Similarly EGF, which is a robust stimulator of the MAPK pathway, resulted in activation of new EMT-related marker receptor activator of NF-κB ligand (RANKL), and enhances bone resorption and bone turnover, facilitating successful bone metastasis [14]. Findings from our laboratory, support these observations in DU-145 and PC-3, both of which undergo enhanced EMT upon EGF stimulation [14, 28, 29].
It is important to note that in addition to transcriptional repression, DNA methylation of key tumor suppressor and EMT-related genes has been observed. In the case of E-cadherin, available cell culture models DU-145 and PC-3 do not exhibit methylation of E-cadherin, however this is not observed clinically, as E-cadherin is methylated in 70% of late-stage prostate [30].
More recently microRNAs (miRNAs), small non-coding RNAs regulating gene expression, control large cohort of genes post-translationally. A number of miRNAs have been identified as either oncogenes or tumor suppressor genes. The importance EMT-related miRNAs was first discovered in breast cancer model where the mir-200 family was found to indirectly regulate EMT via targeted regulation of transcription factor 8 (ZEB1, DEF1, Nil-2-A). Further evidence of miRNA involvement in E-cadherin expression is increased expression of mir-9 [31] and mir-9-1 [32], which target E-cadherin [33]. Of the miRNA 200 family return of miR-200b levels in PC3 induced to overexpress PDGF-D cells led to reversal of the EMT phenotype, which was associated with the downregulation of ZEB1, ZEB2, and Snail2 expression. Moreover, this resulted in inhibited cell migration and invasion, with concomitant repression of cell adhesion to the culture surface and cell detachment [34].
2 Tumor–Stromal Interactions Influence Tumor Plasticity
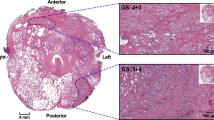
Several reports have shown that inoculation of prostate cancer cells within the bone microenvironment induces reciprocal interactions that results in seemly phenotypic and genetic changes in the cancer cells. This is evidenced by the LNCaP cells, ARCaP, and PC-3 cells after injection into the bone, yielding cell line derivatives C4-2, ARCaP M, and PC-3 M cells, which exhibit more mesenchymal phenotypes and increased growth and invasiveness [35, 36]. This general concept was described by Paget in 1889 [37] who proposed that the seeding of metastatic cancer cells is dependent upon the host organ microenvironment (the “seed and soil” concept). The realization that the host microenvironment comprises a number of stromal cell types (fibroblasts, smooth muscle cells, endothelial cells, neural endocrine cells, inflammatory cells) and a host of growth factors (VEGF, IGF, FGF, EGF, and TGFβ) and extracellular matrices (laminin, fibronectin, collagen, and proteoglycans) and the fact that these molecules support cancer growth and progression emphasizes the possible directive role of the local host tumor microenvironment at both primary and metastatic sites that could affect the overall growth and malignant potential of the transformed cancer epithelial cells. Our findings of this is indeed the case, as EMT-related cellular behavior of invasive cancer cells is subject to regulation by the tumor microenvironment [29]. To demonstrate the dynamic influence we examined paired primary and liver metastasis prostate cancer patient samples and stained for E-cadherin. Figure 7.1 shows that E-cadherin is densely expressed within epithelial compartment of primary prostate tumors and liver metastases, with only the prostate cancer cells that have invaded the local stroma exhibiting complete lack of E-cadherin expression. With the prostate cancer liver metastasis, cell appear to be less differentiated than primary tumor, and morphologically similar to the hepatocytes, through heterotypic cell–cell interactions. That prostate cancer cells would appear morphogencially similar to liver cells is rooted in the concept of prostate cancer osteomimicry within bone microenvironment [38]. This is also supported by observation that breast cancer metastases to the liver seem to recreate hepatocyte cords with carcinoma cells [39]. We further observed that DU-145 and PC-3 cells reexpress E-cadherin through heterotypic interactions and display complete reversion of the mesenchymal phenotype, with decreased vitmentin and increased cytokeratin, when cocultured with primary rat hepatocytes [29]. We further observed a lack of total and active EGFR expression in cell lines and patient tumors.

Tumor cells exhibit phenotypic plasticity within the liver microenvironment. (a) Human primary prostate cancer (left) and metastases to liver (right) show expression of E-cadherin. Formalin-fixed, paraffin-embedded tissues were obtained from two well-defined prostate adenocarcinomas with liver metastasis, and stained with E-cadherin antibody. (b) Immunofluorescence of co-cultures shows subcellular location of E-cadherin re-expression. MCF-7 RFP (red) and GFP (green) primary rat hepatocytes were stained with human-specific anti-E-cadherin for a multiday period. Top left (Day 2), top right (Day 4), bottom left (Day 8), bottom right (Day 14). Cy5 secondary antibody (blue) was used for E-cadherin primary antibody
Liver metastases express E-cadherin on tumor cells suggested that signals from the tumor microenvironment modulate E-cadherin expression. However, there was question of whether the transdifferenation was sustained during secondary tumor development. Therefore, we co-cultured E-cadherin positive RFP-MCF-7 cells with GFP primary rat hepatocytes and examined E-cadherin expression over a multiday period (Fig. 7.2). These co-cultures revealed that E-cadherin was stability expressed after 2 days, but was subsequently reverted at the leading edge after 8 days of co-culture. However, after long-term coculture (14 days) MCF-7 cells underwent three-dimensional organization. These findings are similar to our prostate cancer patient observations and provide the proof-of-principle that E-cadherin-associated EMT is the result of dynamic interactions of the tumor cell with its surrounding microenvironment.

ARCaPE cells show a growth and colony-forming capacity advantage in presence of HS-27a cells. (a) ARCaPM cells were cocultured in the presence of GFP-HS-27a cells over a 6-day period. Growth of RFP, ARCaPE or ARCaPM human prostate cancer cells was assessed by RFU (relative fluorescent units) in the presence cocultures over a 6-day period. Results are means ± SE of three independent experiments. *P, 0.05 (Student’s t test) compared to cell number at day 1 ±SEM. (b) Clongenic colony-forming capacity of ARCaPE and ARCaPM prostate cancer cell after coculture ±SEM. ARCaPM data was normalized to ARCaPM control, and ARCaPE data was normalized to ARCaPE control. (c) Clonogenic colony forming capacity of ARCaPE and ARCaPM prostate cancer cell after coculture ± SEM. ARCaPM data were normalized to ARCaPM control, and ARCaPE data were normalized to ARCaPE control (Note HS-27a induced slightly (1.35´) the growth of ARCaPM cells but markedly (8´) the growth of ARCaPE cells). (d) ARCaPE or ARCaPM cells were cocultured with HS-27a cells. Shown are phase contrast images of colonies formed in the clonogenic assay
Since we were able to observe stromal-induced reexpression of E-cadherin within the liver microenvironment, would suggest that a reepithelialization process is necessary for establishment of secondary tumors. However, given the inherent differences in the stromal parenchyma of each organ, it is likely that multiple soluble factors can achieve similar effects.
For example, exogenous BMP-7 was able to induce E-cadherin of prostate tumors within the bone microenvironment, however failed to have any effect on tumors within the lymph nodes [40]. Furthermore, differential expression of a number of angiogenesis-associated genes and their proteins between prostate cancer metastasis to bone versus liver, and lymph nodes have been observed [41]. To determine if this is case with E-cadherin-associated EMT, we cocultured the ARCaP model with bone marrow stromal cells. Cocultured ARCaPM cells displayed a reversal of E-cadherin, and the more epithelial ARCaPE cells showing increased colony-forming capacity and growth advantage in presence of bone stromal cells [42]. Clinical evidence of E-cadherin expression in bone metastasis has been observed, and interesting is associated with a reversal of E-cadherin-specific methylation pattern [43].
3 Targeting Cell Adhesion for Therapeutic Intervention
Although we are just at the beginning of understanding the directive role of the stroma, more insight into how the stroma regulates tumor cells will lead to better therapies for late-stage metastatic disease. Multiple reports have suggested the benefits of targeting E-cadherin as a therapeutic approach. For example, E-cadherin neutralizing antibody (SHEP8-7) has been shown to sensitize multi-cellular spheroids to microtubule binding therapies in the taxane family in HT29 human colorectal adenocarcinoma cells [44]. A more recent observation that survival of androgen receptor-expressing differentiated prostate cells are dependent on E-cadherin and PI3K, but not on androgen, AR or MAPK [45]. Indeed this is the case because our findings suggest the blocking E-cadherin cell–cell interaction with E-cadherin neutralizing antibody, decreased both epithelial or mesenchymal-like prostate cells from reepithelialization and colonizing the bone microenvironment. The neutralizing antibody increases their sensitivity to radiation treatment [42]. Of further clinical benefit, recently a monoconal antibody to N-cadherin has been described as an effective treatment for prostate cancer limiting local invasion and metastasis both in vitro and in vivo [12]. Thus, blocking cellular adhesions appears to be a rationale strategy limiting prostate cancer metastasis.
4 Summary
In summary, we propose that the EMT required to “escape” from the primary tumor mass is transiently “reverted” during the initial stages of metastatic seeding, enabling the alien tumor cell to incorporate into the target tissue and derive survival signals thereof. Thus, tumor–stromal-interacts induce cellular plasticity gives rise to distinct populations of cancer cells within secondary site. This plasticity gives rise to distinct population, i.e. mesenchymal phenotype and its kinetic characteristics (motility/invasive), and the epithelial characteristics necessary for secondary tumor development. Our findings that epithelial cells are more successful in establishing secondary tumor suggest that dissemination from the primary tumor mass requires the mesenchymal phenotype, however a mesenchymal to epithelial transition is associated with initial metastatic seeding and subsequent formation of a cohesive tumor mass within the bone microenvironment (Fig. 7.3). Critical to our model of phenotypic mesenchymal-to-epithelial-reverting transitions is the underling signaling mechanisms that mediate this transition. Multiple-cell signaling pathways, most likely initiated by stromal-derived soluble factors, converge on an ever-expanding set of transcriptional and post-translation factors that epigenetically regulate specific proteins that ultimately serve as markers of the epithelial vs. mesenchymal phenotype. Understanding the events may offer new opportunity to target during and the reverting transition that appear to be essential to metastatic relapse.

Diagram of mesenchymal-to-epithelial reverting transition. Epithelial carcinomas cells and carcinoma cells undergo EMT, which involves a loss of adhesion and reinforcement of autocrine signaling that drive the cancer cell to escape the tumor mass and intravasate the blood or lymphatic vessels. At the secondary site mesenchymal cells extravasate the tissue parenchyma and phenotypic reversion occurs to form heterotypic interactions within foreign microenvironment, with subsequent development of micrometastases
References
Kahn LB, Uys CJ, Dale J, Rutherfoord S (1978) Carcinoma of the breast with metaplasia to chondrosarcoma: a light and electron microscopic study. Histopathology 2:93–106
Hay ED, Zuk A (1995) Transformations between epithelium and mesenchyme: normal, pathological, and experimentally induced. Am J Kidney Dis 26:678–690
Ehrlich JS, Hansen MD, Nelson WJ (2002) Spatio-temporal regulation of Rac1 localization and lamellipodia dynamics during epithelial cell-cell adhesion. Dev Cell 3:259–270
Akhtar N, Hudson KR, Hotchin NA (2000) Co-localization of Rac1 and E-cadherin in human epidermal keratinocytes. Cell Adhes Commun 7:465–476
Ray ME, Mehra R, Sandler HM, Daignault S, Shah RB (2006) E-cadherin protein expression predicts prostate cancer salvage radiotherapy outcomes. J Urol 176:1409–1414, discussion 14
Hennig G, Behrens J, Truss M, Frisch S, Reichmann E, Birchmeier W (1995) Progression of carcinoma cells is associated with alterations in chromatin structure and factor binding at the E-cadherin promoter in vivo. Oncogene 11:475–484
Graff JR, Herman JG, Lapidus RG et al (1995) E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 55:5195–5199
Machado JC, Oliveira C, Carvalho R et al (2001) E-cadherin gene (CDH1) promoter methylation as the second hit in sporadic diffuse gastric carcinoma. Oncogene 20:1525–1528
Berx G, Becker KF, Hofler H, van Roy F (1998) Mutations of the human E-cadherin (CDH1) gene. Hum Mutat 12:226–237
Hirohashi S (1998) Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol 153:333–339
Jennbacken K, Tesan T, Wang W, Gustavsson H, Damber JE, Welen K (2010) N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr Relat Cancer 17:469–479
Tanaka H, Kono E, Tran CP et al (2010) Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med 16:1414–1420
Tran NL, Nagle RB, Cress AE, Heimark RL (1999) N-cadherin expression in human prostate carcinoma cell lines. An epithelial-mesenchymal transformation mediating adhesion with stromal cells. Am J Pathol 155:787–798
Odero-Marah VA, Wang R, Chu G et al (2008) Receptor activator of NF-kappaB ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res 18:858–870
Liu J, Uygur B, Zhang Z et al (2010) Slug inhibits proliferation of human prostate cancer cells via downregulation of cyclin D1 expression. Prostate 70:1768–1777
Emadi Baygi M, Soheili ZS, Schmitz I, Sameie S, Schulz WA (2010) Snail regulates cell survival and inhibits cellular senescence in human metastatic prostate cancer cell lines. Cell Biol Toxicol 26:553–567
Kwok WK, Ling MT, Lee TW et al (2005) Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res 65:5153–5162
Yuen HF, Chan YP, Wong ML et al (2007) Upregulation of twist in oesophageal squamous cell carcinoma is associated with neoplastic transformation and distant metastasis. J Clin Pathol 60:510–514
Downing JR, Reynolds AB (1991) PDGF, CSF-1, and EGF induce tyrosine phosphorylation of p120, a pp 60src transformation-associated substrate. Oncogene 6:607–613
Hazan RB, Norton L (1998) The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J Biol Chem 273:9078–9084
Andl CD, Mizushima T, Nakagawa H et al (2003) Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem 278:1824–1830
Kassis J, Moellinger J, Lo H, Greenberg NM, Kim HG, Wells A (1999) A role for phospholipase C-gamma-mediated signaling in tumor cell invasion. Clin Cancer Res 5:2251–2260
Nakashiro K, Okamoto M, Hayashi Y, Oyasu R (2000) Hepatocyte growth factor secreted by prostate-derived stromal cells stimulates growth of androgen-independent human prostatic carcinoma cells. Am J Pathol 157:795–803
Yates C, Wells A, Turner T (2005) Luteinising hormone-releasing hormone analogue reverses the cell adhesion profile of EGFR overexpressing DU-145 human prostate carcinoma subline. Br J Cancer 92:366–375
Davies G, Jiang WG, Mason MD (2001) HGF/SF modifies the interaction between its receptor c-Met, and the E-cadherin/catenin complex in prostate cancer cells. Int J Mol Med 7:385–388
Josson S, Anderson CS, Sung SY et al (2011) Inhibition of ADAM9 expression induces epithelial phenotypic alterations and sensitizes human prostate cancer cells to radiation and chemotherapy. Prostate 71(3):232–240
Graham TR, Zhau HE, Odero-Marah VA et al (2008) Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res 68:2479–2488
Yates C, Shepard CR, Papworth G et al (2007) Novel three-dimensional organotypic liver bioreactor to directly visualize early events in metastatic progression. Adv Cancer Res 97:225–246
Yates CC, Shepard CR, Stolz DB, Wells A (2007) Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br J Cancer 96:1246–1252
Li LC, Zhao H, Nakajima K et al (2001) Methylation of the E-cadherin gene promoter correlates with progression of prostate cancer. J Urol 166:705–709
Ma L, Teruya-Feldstein J, Weinberg RA (2007) Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 449:682–688
Lehmann U, Hasemeier B, Christgen M et al (2008) Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol 214:17–24
Dalmay T, Edwards DR (2006) MicroRNAs and the hallmarks of cancer. Oncogene 25:6170–6175
Kong D, Li Y, Wang Z et al (2009) miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells 27:1712–1721
Wu TT, Sikes RA, Cui Q et al (1998) Establishing human prostate cancer cell xenografts in bone: induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int J Cancer 77:887–894
Zhau HE, Li CL, Chung LW (2000) Establishment of human prostate carcinoma skeletal metastasis models. Cancer 88:2995–3001
Paget S (1889) The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev 8:98–101
Chung LW (2003) Prostate carcinoma bone-stroma interaction and its biologic and therapeutic implications. Cancer 97:772–778
Stessels F, Van den Eynden G, Van der Auwera I et al (2004) Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br J Cancer 90:1429–1436
Buijs JT, Rentsch CA, van der Horst G et al (2007) BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am J Pathol 171:1047–1057
Morrissey C, True LD, Roudier MP et al (2008) Differential expression of angiogenesis associated genes in prostate cancer bone, liver and lymph node metastases. Clin Exp Metastasis 25:377–388
Josson S, Sharp S, Sung SY (2010) et al. Tumor-stromal interactions influence radiation sensitivity in epithelial – versus mesenchymal-like prostate cancer cells, J Oncol
Saha B, Kaur P, Tsao-Wei D et al (2008) Unmethylated E-cadherin gene expression is significantly associated with metastatic human prostate cancer cells in bone. Prostate 68:1681–1688
Green SK, Karlsson MC, Ravetch JV, Kerbel RS (2002) Disruption of cell-cell adhesion enhances antibody-dependent cellular cytotoxicity: implications for antibody-based therapeutics of cancer. Cancer Res 62:6891–6900
Lamb LE, Knudsen BS, Miranti CK (2010) E-cadherin-mediated survival of androgen-receptor-expressing secretory prostate epithelial cells derived from a stratified in vitro differentiation model. J Cell Sci 123:266–276
Acknowledgments
I would like to acknowledge all collaborators involved in the different phases of this work. This work was funded by grant from the Department of Defense Prostate Cancer Research Program (PC073977), and NIH/RCMI G12 RR03059-21A1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Yates, C. (2011). Prostate Tumor Cell Plasticity: A Consequence of the Microenvironment. In: Rhim, J., Kremer, R. (eds) Human Cell Transformation. Advances in Experimental Medicine and Biology, vol 720. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-0254-1_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-0254-1_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-0253-4
Online ISBN: 978-1-4614-0254-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)