
Abstract
Platelet activation during thrombotic events is closely associated with complement and contact system activation, which in turn leads to inflammation . Here we review the interactions between activated platelets and the complement and contact activation systems in clotting blood. Chondroitin sulfate A (CS-A), released from alpha granules during platelet activation, is a potent mediator of crosstalk between platelets and the complement system. CS-A activates complement in the fluid phase, generating anaphylatoxins that mediate leukocyte activation. No complement activation seems to occur on the activated platelet surface, but C3 in the form of C3(H2O) is bound to the surfaces of activated platelets . This finding is consistent with the strong expression of membrane-bound complement regulators present at the platelet surface. CS-A exposed on the activated platelets is to a certain amount responsible for recruiting soluble regulators to the surface. Platelet-bound C3(H2O) acts as a ligand for leukocyte CR1 (CD35), potentially enabling platelet–leukocyte interactions. In addition, platelet activation leads to the activation of contact system enzymes, which are specifically inhibited by antithrombin, rather than by C1INH, as is the case when contact activation is induced by material surfaces. Thus, in addition to their traditional role as initiators of secondary hemostasis, platelets also act as mediators and regulators of inflammation in thrombotic events.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Background
1.1 The Complement System
The complement system is part of the innate immune system and one of the main effector mechanisms of antibody-mediated immunity. As many as 30 soluble and membrane-bound glycoproteins are involved in the complement system (Liszewski et al. 1996; Sunyer and Lambris 2001; Zipfel and Skerka 2009), whose central protein is complement component 3 (C3).
Complement activation is a target surface-oriented process that is centered around the activation of C3. C3 can be activated by three activation pathways: the classical pathway (CP), which in general responds to immune complexes (ICs); alternative pathway (AP), which is triggered by interaction with bacteria, viruses; and the lectin pathway (LP), which responds to the presence of certain carbohydrates on microbial surfaces. Once activated, the complement system mediates at least three traditionally described major functions: opsonization of pathogens, and thus enhancement of phagocytosis; attraction and activation of phagocytes to the site of inflammation ; and lysis of foreign pathogens by damaging their cell membranes. Other more recently described functions of the complement system are elimination of apoptotic cell debris, enhancement of humoral immunity (Molina et al. 1996), modification of T-cell responses (Köhl 2006), and regulation of tolerance to self-antigens (Carroll 2000).
The complement system is strictly regulated in solution and on cell surfaces, such as endothelial cells and platelets , by both soluble and membrane-bound regulator proteins. Deficiencies in complement regulation can cause tissue damage as a result of uncontrolled inflammation and can contribute to the pathology of many diseases. Soluble complement regulators include C1 inhibitor (C1INH), factor H, C4 binding protein (C4BP), and factor I, as well as vitronectin and clusterin. The membrane-bound complement regulators include complement receptor 1 (CR1), CD59, membrane cofactor protein (MCP), decay accelerating factor (DAF), and CRIg (Liszewski et al. 1996; Meri and Jarva 1998; Helmy et al. 2006; He et al. 2008; Zipfel and Skerka 2009).
1.2 The Contact System
In the 1950s, it was observed that blood added to glass tubes clotted and the concept of contact activation was thus born (Margolis 1958; Waaler 1959). The mechanism of contact activation was initially studied in plasma or using purified proteins, with different negatively charged substances such as glass, kaolin, dextran sulfate, ellagic acid, endotoxins, and glycosaminoglycans (GAGs) to initiate the activation. More recently, studies of the interaction and activation of the contact components on endothelial cells have indicated alternative mechanisms of activation for these components.
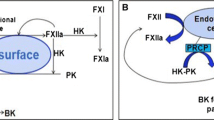
In the presence of a negatively charged surface, factor (F) XII binds to the surface and becomes autoactivated as a result of conformational changes that uncover its catalytic serine protease region (Miller et al. 1980). This activated surface-bound form of FXII, α-FXIIa, cleaves and activates plasma prekallikrein and FXI, generating active plasma kallikrein (KK) and FXIa (Revak et al. 1978). FXIa then activates FIX, initiating the process of thrombin generation. Plasma KK enhances contact activation by cleaving both FXII, converting it to FXIIa, and surface-bound α-FXIIa, liberating soluble β-FXIIa, which produces more KK. Thus, contact activation is an autocatalytic process. KK also cleaves high molecular weight kininogen (HK), liberating the vasoactive non-apeptide bradykinin (BK) (Mori and Nagasawa 1981). HK is a required cofactor in the contact activation process, since both prekallikrein and FXI bind to negatively charged surfaces in conjunction with HK.
Multiple protease inhibitors, such as C1INH, antithrombin (AT), α1-antitrypsin, α2-antiplasmin, protein C inhibitor, and α2-macroglobulin, are able to inhibit the enzymes of the contact system (Forbes et al. 1970; Heck and Kaplan 1974; Saito et al. 1979; van der Graaf et al. 1983; de Agostini et al. 1984; Pixley et al. 1985b; Meijers et al. 1988; Wuillemin et al. 1995). Most of these proteins belong to the superfamily of serine protease inhibitors (serpins). Several studies have concluded that C1INH is the predominant inhibitor of all enzymes of the contact system, followed by α-2-macroglobulin, α-2-antiplasmin, and AT (Harpel et al. 1985; Pixley et al. 1985b; Wuillemin et al. 1995, 1996). GAGs such as heparin and heparan sulfate enhance the inhibitory effect of AT on all contact enzymes (Holmer et al. 1981; Pixley et al. 1985a; Olson et al. 1993; Gozzo et al. 2003) and have also been reported to enhance, to a lesser degree, the inhibitory effect of C1INH on KK and FXIa but not FXIIa (Wuillemin et al. 1996; Gozzo et al. 2006).
In addition to its ability to initiate coagulation, the proteins of the contact system also participate in the initiation of the inflammatory response via kinin formation and possibly also via complement activation. Contact system factors have also been reported to influence fibrinolysis.
1.3 Platelets
Apart from the traditional view of platelets as mediators of hemostasis, evidence is emerging that platelets and platelet-derived microparticles (PMPs) bring together complement and contact system activation at the site of thrombotic reactions. For instance, activated complement components have been demonstrated in many types of atherosclerotic and thrombotic vascular lesions, and platelets are involved in many of the inflammatory diseases that are mediated by complement dysregulation (Torzewski et al. 1997).
Platelets become activated when they contact any thrombogenic surface, such as injured endothelium or subendothelium, or an artificial surface such as a stent, vascular graft, or cardiopulmonary or hemodialysis equipment (Gorbet and Sefton 2004). Platelets also respond to stimulation by physiological agonists, including thrombin, ADP, collagen, platelet activating factor (PAF), and thromboxane A2 (TxA2). In these situations, platelet activation is initiated by the interaction of an extracellular stimulus with receptors at the platelet surface (Blockmans et al. 1995).
Under both physiological and pathological conditions, platelet activation in the human body is triggered by contact with a disrupted vascular wall and subendothelium, which exposes collagen, von Willebrand factor (vWF), and/or fibrin. Platelets adhere to the subendothelium through the binding of vWF to the glycoprotein receptor (GP) Ib/IX/V complex present on the platelet surface (Savage et al. 1996). Binding of vWF is principally mediated through the receptor GPIb (Ruggeri et al. 1983). The binding of collagen and vWF to the receptors activates an intracellular signaling pathway, which leads to contraction and release of storage granules (Holmsen 1989; Colman et al. 1994). The key feature in the extension of platelet aggregates is the presence of receptors on the platelet surface that can respond directly to some of the released agents, such as thrombin, ADP, and TxA2 (Brass 2003). Aggregation is mediated by fibrinogen, which binds to the integrin GPIIb/IIIa on the platelet surface. When fibrinogen binds to platelets , fibrinogen forms a bridge between adjacent platelets . The affinity of GPIIb/IIIa for fibrinogen increases as a result of stimulation by ADP and TxA2 which induce as conformational change in the fibrinogen receptor (Shattil and Newman 2004).
When platelets are stimulated by agonists such as collagen that induce secretion of granule contents, a trans-bilayer flipping of the membrane phospholipids occurs that brings procoagulant phospholipids to the platelet surface. The exposed phospholipids greatly accelerate the tenase (FIXa/FVIIIa) and prothrombinase (FXa/FVa) reactions of the coagulation pathway, resulting in the generation of thrombin, the most potent platelet agonist (Zwaal et al. 1998; Rand et al. 2003). Thrombin induces further platelet stimulation, aggregation, and secretion. Thrombin also converts fibrinogen to fibrin, which is deposited around the mass of aggregated platelets and confers stability on the formed hemostatic plug.
Human platelets express two protease-activated receptors (PAR), PAR1 and PAR4, that are proteolytically cleaved and activated by thrombin. PAR1 responds to thrombin levels of approximately 1 nmol/L, while PAR4 requires a 10-fold higher thrombin concentration (Brass 2003). Thrombin-mediated activation involves binding to the ectodomain of the PAR molecule and proteolytical cleavage between Arg41 and Ser42 (Scarborough et al. 1992). This reaction exposes a new amino-terminus, which acts as a “tethered ligand” to activate the receptor. Synthetic thrombin receptor-activating peptides (TRAP) such as SFLLRN, derived from thesequence of the new amino-terminus of the cleaved PAR,can mimic thrombin receptor activation and act as full agonists for plateletactivation (Shankar et al. 1994). TRAP acts by binding to PAR1, mimicking the N-terminal ectodomain of the receptor, and thereby activating it without the proteolytic action of thrombin.
ICs are potent activators of the complement system. Therefore, they contribute to the development of acute and chronic inflammation , which can produce the tissue damage that is associated with autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus (SLE). ICs are also strong platelet agonists that bind to Fc receptors FcγRII (CD32) and FcγRIII (CD64), which are expressed on the platelet surface (Schattner et al. 1993; McKenzie 2002; Reilly 2003). The FcRγ-chain of CD32 is a common signal transducer with GPVI and CD36, which are receptors involved in collagen-induced platelet activation. The platelet activation that occurs in response to the IC–FcγRII interaction is therefore expected to be the same as that seen in collagen-mediated activation.
1.4 Proteoglycans
In nature, all GAGs except hyaluronic acid are covalently linked to a core protein to form a proteoglycan (PG). The linkage of GAGs to the protein core involves a specific trisaccharide composed of two galactose residues and a xylose residue. The saccharide residues are coupled to the protein core through an O-glycosidic bond to a serine residue (Gandhi and Mancera 2008). The substituted serine residues in the core protein are adjacent to glycine, and the Ser-Gly dipeptide seems to be a basic requirement for recognition by xylosyl transferase enzymes (Kolset and Gallagher 1990). The number of GAG chain substituents on a protein core may vary from one to over 100, producing wide variation in the type and function of PGs (Kjellén and Lindahl 1991).
Serglycin is the PG, most commonly found in hematopoietic cells (Kolset et al. 2004). In the various types of blood cells, chondroitin sulfate (CS) is the major GAG, with CS-A as the dominant form (Kolset and Gallagher 1990; Kolset et al. 2004). Serglycin is stored in the granules of hematopoietic cells, where it is believed to be involved in the generation of storage granules (Abrink et al. 2004; Grujic et al. 2005; Niemann et al. 2007). In a recent study, Woulfe et al. (2008) have demonstrated defects in platelet function and aggregation in serglycin-knockout mice(Woulfe et al. 2008).
1.5 Chondroitin Sulfate (CS)
CS consists of the repeating disaccharide units N-acetyl-d-galactosamine (GalNAc) and d-glucuronic acid (GlcA). Dermatan sulfate (DS) differs from CS in its frequent epimerization of glucuronic acid to iduronic acid (Kolset and Gallagher 1990). Both CS and DS may be sulfated at carbon 2 of the uronic acid and at carbon 4 and/or 6 of the amino sugar (Kjellén and Lindahl 1991). The pattern of sulfation is usually used to name the CS GAGs: CS monosulfated at carbon 4 is known as CS-4 or CS-A; CS sulfated in carbon 6 as CS-6 or CS-C; and CS disulfated at carbons 4 and 6 as CS-4,6 or CS-E. DS, formerly known as CS-B, is sulfated at carbon 4 of the galactosamine and 2 of the uronic acid (Kolset and Gallagher 1990).
CS is the most abundant GAG in human plasma (≈70–80%), with CS-A being the major component (Lamari and Karamanos 2006). It has also been well established that CS-A is the predominant GAG in platelets (Okayama et al. 1986), where it is stored in the α-granules of platelets and released during the activation and degranulation process (Barber et al. 1972; Hagen 1972). It is also rapidly expressed on the surface of activated platelets (Ward and Packham 1979). The release of CS-A in response to different agonists, including ADP, collagen, and thrombin, is very rapid and raises the concentration of plasma CS to 2–4 µg/mL, with considerably higher concentrations in the immediate vicinity of the platelets (Donato et al. 1994). Unlike the CS in blood plasma, the CS in platelets is fully sulfated, and its molecular mass has been estimated to be ~28 kDa (Okayama et al. 1986). This high degree of sulfation is thought to be linked to immune-related processes and inflammatory conditions (Uhlin-Hansen et al. 1989).
CS has been shown to bind and interact with C1q (Silvestri et al. 1981; Kirschfink et al. 1997) and has been implicated as a C1q inhibitor in several studies (Silvestri et al. 1981; Ghebrehiwet and Hamburger 1982; Galanakis and Ghebrehiwet 1994). Its binding is thought to be mediated through the globular heads of C1q and to involve ionic interactions (Ghebrehiwet and Galanakis 1993).
1.6 Platelet–Leukocyte Interactions
When platelets are activated in whole blood by various agonists, they form complexes with leukocytes, both granulocytes and monocytes (Jurk and Kehrel 2005). Such complexes are often detected in patients with stroke, unstable angina pectoris, or myocardial infections, and may thus be of pathological significance (Marquardt et al. 2009; Zhang et al. 2007). Platelets and leukocytes interact via direct cell–cell contact or indirectly via soluble mediators, following the activation of either platelets or leukocytes (Ruef et al. 2008).
Complex formation between activated platelets and leukocytes has been reported to be mediated by P-selectin, which is expressed on the surface of activated platelets , and its ligand, P-selectin glycoprotein ligand-1 (PSGL-1), on leukocytes. The interaction of P-selectin with PSGL-1 mediates the initial tethering of leukocytes to activated platelets through a mechanism resembling the interaction of leukocytes with endothelial cells (Zarbock et al. 2009). This P-selectin-mediated interaction induces the activation and up-regulation of β2-integrin αMβ2 (CD11b/CD18) on leukocytes (da Costa Martins et al. 2006). Firm attachment is subsequently mediated by the interaction of CD11b/CD18 with various ligands and adhesion molecules on platelets (Kuijper et al. 1998). GPIIb/IIIa expressed on platelets is often considered to be essential for platelet–leukocyte interactions (Zhao et al. 2003). GPIIb/IIIa has been proposed to interact with leukocytes either by direct binding to CD11b/CD18 or through fibrinogen, which binds to GPIIb/IIIa on platelets and CD11b/CD18 on leukocytes (Lishko et al. 2004). Other proteins that are exposed on platelets and reported to take part in platelet–leukocyte complex formation are CD40L, which interacts with CD40 on leukocytes (Zhao et al. 2003), and GPIbα, which binds to CD11b/CD18 (Ehlers et al. 2003). However, Konstantopoulos et al. (1998) and Ruef et al. (2008) have shown that platelet–leukocyte attachment occurs independent of GPIIb/IIIa. Thus, the ligand(s) for leukocyte–platelet inteaction via CD11b/CD18 are still unclear.
Upon activation, platelets shed microparticles (PMPs) from the plasma membrane. PMPs have high procoagulant activity because of the negatively charged phospholipids exposed on their surface. Various receptors that are present on the surface of activated platelets are also found on the surface of PMPs, enabling them to interact with cells (Bode and Hickerson 2000; Forlow et al. 2000). It has been well established that platelets and PMPs contain tissue factor (TF), which is the most important initiator of intravascular coagulation (Scholz et al. 2002; Siddiqui et al. 2002; Camera et al. 2003).
2 Platelet–Complement Interactions
2.1 Activation of Platelets by Complement Activation Products
Complement and platelets appear to activate each other in a reciprocal manner that may have physiological and pathological implications. Complement -induced platelet activation has been studied since the 1980s, when Sims et al. demonstrated an increased procoagulatory activity of platelets exposed to sC5b-9 complexes (Sims and Wiedmer 1991). These observations suggested that the sC5b-9 complex mimics platelet stimulation by thrombin and other agonists. However, it has been reported that platelet activation by sC5b-9 is under the control of the complement regulatory protein CD59, which is present on the surface of platelets (Sims et al. 1989). Blocking CD59 with an antibody augments the sC5b-9-mediated procoagulatory response of platelets .
2.2 Binding of Complement Components to Activated Platelets
Platelets are thought to play a significant role in the activation and regulation of complement. In agreement with this concept, several studies have shown that complement components bind to platelets (Polley and Nachman 1978; Endresen and Mellbye 1984; Sandvik et al. 1984). Platelets have also been found to store, secrete, and express complement proteins and regulators upon activation. It was recently reported that C3 is present in platelet lysates (Del Conde et al. 2005). Already in 1981, Kenney et al. had shown that platelet homogenates can inhibit the formation and accelerate the decay of the C3bBb convertase, as a result of the presence of factor H (Kenney and Davis 1981). Factor H was found to bind to washed human platelets via thrombospondin-1 (TSP-1) (Vaziri-Sani et al. 2005) or GPIIb/IIIa (Mnjoyan et al. 2008).
Complement activation has previously been reported to occur on the surface of activated platelets (Del Conde et al. 2005; Peerschke et al. 2006). Del Conde et al. (2005) reported an activation of complement and deposition of C3b on activated platelets that was dependent on P-selectin expression and mediated by the AP. Using an assay with platelets fixed to microtiter plates, Peerschke et al. (2006) demonstrated a complement activation involving CP components that was due in part to the expression of the receptor for the globular heads of C1q (gC1qR) on the surface of activated platelets . However, there is somelack of congruity between these observations and the previously demonstrated abundant expression of the membrane-bound complement regulators DAF (Nicholson-Weller et al. 1985), MCP (Yu et al. 1986), and CD59 (Morgan 1992) and the interactions of soluble regulators as C1INH (Schmaier et al. 1993), clusterin (Tschopp et al. 1993), and factor H with the activated platelet surface. One would have expected to see a well-controlled complement equilibrium on the surface of platelets under physiological conditions, as supported by the following examples that illustrate the critical importance of physiological complement regulation:
-
1.
A potential role for the complement system in the thrombotic episodes associated with paroxysmal nocturnal hemoglobinuria (PNH) has been postulated on the basis of the increased sensitivity of platelets to activation by sC5b-9 complexes as a result of a diminished surface expression of CD59 on their surfaces (Wiedmer et al. 1993). Similarly, a correlation between complement activation and thrombotic events has been demonstrated in various autoimmune conditions, including SLE, and antiphospholipid syndrome (Peerschke et al. 2010).
-
2.
Platelet activation and a lowering of platelet counts have also been noted in atypical hemolytic uremic syndrome (aHUS). aHUS is associated with mutations in the C-terminus of factor H (Zipfel et al. 2001); this relationship would suggest that dysregulation of the complement system plays a crucial part in the pathogenesis of aHUS. Stahl et al. (2008) have shown that aHUS patients with a mutated factor H have higher levels of deposition of C3 and C9 on platelets than do healthy controls. Combining aHUS patient sera containing mutated factor H with normal platelets results in complement activation and the activation and aggregation of platelets . These reactions can be abrogated by preincubation of the platelets with normal factor H or when normal serum is used (Stahl et al. 2008). Karpman et al. (2006) have suggested that the binding of factor H to human platelets can protect them from complement activation.
The choice of anticoagulant used in studies of the interaction between platelets and complement is of utmost importance. Most of the previous studies have been performed in citrated blood, where complement function is impaired to a certain extent. Similarly, heparin even at low doses interferes with complement. In order to avoid these side effects, we have developed a model system using blood containing the specific thrombin inhibitor lepirudin in which the plateles are activated byTRAP (Mollnes et al. 2002). By doing so, we can stimulate the platelets in whole blood via PAR1 without inducing clot formation.
In a recent study, using this system designed to elucidate how complement components bind to platelets , we used flow cytometry to monitor the binding of complement proteins to activated platelets in platelet-rich plasma (PRP) and in whole blood (Hamad et al. 2010b). C1q, C4, C3, and C9 were found to bind to the activated platelets in both PRP and whole blood. However, binding of complement components does not necessarily mean that these components are bound as a result of complement activation: In fact, blocking complement activation completely by using EDTA or at the level of C1q with an inhibitory antiglobular head C1q-85 monoclonal antibody (mAb) or C3 with Compstatin did not affect the binding of C3 or C9. Western blot analysis of platelet surface-associated C3 and flow cytometry using mAbs specific for different conformational forms of C3 showed that the bound C3 consisted mainly of C3(H2O), with no C3b present. This result indicated that the bound C3 was non-proteolytically activated and therefore that complement activation does not take place on the surface of activated platelets under physiological conditions. The transition from C3(H2O) to iC3(H2O) confirmed that regulation of C3 function by factor I is fully active on the platelet surface. This form of C3 was able to bind soluble CR1 (CD35), indicating that it may act as a receptor ligand.
2.3 Complement Activation Triggered by CS-A Released from Activated Platelets
It is well known that complement is activated in clotting blood, and significantly higher levels of complement activation products, for example, C3a and sC5b-9, are found in human serum than in EDTA-, heparin-, or lepirudin-anticoagulated blood. So how does platelet activation lead to complement activation? We addressed this issue in a recent study, which showed that clotting, initiated by either the TF pathway or the contact activation system, could trigger fluid-phase complement activation, as identified by the generation of C3a and sC5b-9 (Hamad et al. 2008). The level of complement activation was highly correlated with the generation of thrombin, that is, the formation of thrombin–AT (TAT) complexes.
Our hypothesis was that CS-A released from activated platelets was acting as a trigger of complement activation. To test this hypothesis, we incubated platelet-poor plasma (PPP) with exogenously added CS-A, either commercially available or in the form of platelet supernatants. Generation of C3a and sC5b-9 increased in a dose-dependent fashion, and digestion of either CS-A or the platelet supernatants with chondroitinase ABC totally abrogated the reaction.
To further investigate the role of CS-A in complement activation, we immobilized CS-A on microtiter plates; after adding plasma or serum to the wells, we were able to detect the binding of C1q. As a functional verification of this binding reaction, we quantified the complement activation that occurred in C1q depleted-serum, with or without the addition of purified C1q, by measuring the binding of C3 fragments. No binding was seen in the C1q-depleted serum, but activation was restored by the addition of purified C1q. These results suggest that complement is activated via the CP and that C1q serves as the recognition molecule for CS-A. CS-A has previously been suggested to act as a specific C1q inhibitor in plasma (Silvestri et al. 1981; Ghebrehiwet and Hamburger 1982). These results were obtained by measuring the ability of CS to inhibit C1q-specific hemolytic activity. We demonstrated that this inhibition is accomplished by CP activation, depleting the plasma of intact complement components and thereby inhibiting the hemolytic activity of C1q (Hamad et al. 2008).
In addition, CS-A-triggered complement activation induced the activation of monocytes and granulocytes, as indicated by the expression of CD11b and the formation of complexes between platelets and monocytes/granulocytes. Both CD11b expression and platelet–leukocyte conjugate formation were significantly inhibited by the addition of the complement inhibitors Compstatin or C5aR antagonist.
2.4 Interaction Between C1q, Complement Regulators, and CS-A
Subsequently, we further characterized the binding of C1q and the complement regulators C1INH, C4BP, and factor H to CS-A exposed on the surface of activated platelets or immobilized on various matrices (Hamad et al. 2010a). C1q was shown to be the main protein that specifically bound to CS-A. After depletion of C1q from the serum, binding of C4BP and factor H to immobilized CS-A as well as to activated platelets was increased, while C1INH binding was absent.
This binding was inhibited by soluble CS-A or by a CS-A-specific mAb, thereby linking the binding of C1q, C4BP, and factor H to exposure of CS-A on activated platelets . The need for large amounts of soluble CS-A or anti-CS-A mAb to inhibit the binding of C1q to activated platelets indicated that the binding of C1q to CS-A is multivalent. C1q contains six globular heads, each of which contains a single binding site for CS-A. The affinity between a monomeric globular head and CS-A is not known, but assuming that it is similar to that of IgG (≈103 M) (Kishore et al. 1998), the avidity (i.e., the combined affinities of all of the globular heads binding to CS-A) would be strong enough to explain the extremely high binding of C1q to CS-A.
In addition, CS-A-bound C1q was also shown to amplify the binding of model IC to both microtiter plate-bound CS-A and to activated platelets , suggesting a role for activated platelets in IC diseases.
2.5 Model for the Interaction Between Platelets and Complement
Physiological consequences of the platelet-mediated complement activation described in Sects. 2.2–2.4 above were an increased expression of CD11b on leukocytes and an increased generation of platelet–leukocyte complexes (Hamad et al. 2008). Platelets apparently participate in this mechanism through at least two different mechanisms: the generation of C5a and subsequent activation of leukocytes, and the binding of ligands C3(H2O)/iC3(H2O) for the CD11b/CD18 integrin on their surface.
The steps involved in the progression from platelet activation to complement activation and inflammation , as deduced from our model, can be summarized as follows: During the activation phase (1) platelets are activated and release CS-A in the form of serglycin and PMPs; (2) CS-A binds C1q and activates complement in the fluid phase, generating C3a and C5a; (3) complement components, including C3(H2O), are deposited on the surface of the activated platelets . During the effector phase (1) leukocytes become activated and up-regulate surface CD11b/CD18, which may enable (2) subsequent binding of platelets and PMPs via C3(H2O) or other ligands.
3 Interactions of Cells and Cellular Components with the Contact Activation System
3.1 Cell-Induced Contact Activation
The idea that contact activation might occur in the vicinity of platelets was raised in the 1960s, when data indicating the presence of activated FXII and FXI on the platelet surface were published (Iatridis and Ferguson 1965; Iatridis et al. 1964). In the 1980s, it was reported that isolated platelets in buffer systems containing purified proteins could promote the activation of FXII (Walsh and Griffin 1981a,b). Washed platelets have been shown to bind HK and FXI in a zinc-dependent manner (Greengard and Griffin 1984; Greengard et al. 1986; Bradford et al. 1997). Apart from the observation that FXIIa binds to washed platelets via GPIb, which competes with HK (Bradford et al. 1997, 2000; Joseph et al. 1999), little has been published regarding the direct binding of FXII to platelets and platelet-triggered contact activation . Recent work using blood plasma has only demonstrated that platelets are able to amplify the contact activation induced by a negatively charged substance or material, such as high molecular weight dextran sulfate (Johne et al. 2006).
The interaction between cells and the proteins of the contact system has thus far focused on endothelial cells. Both HK and FXII bind to human umbilical vein endothelial cells (HUVECs) in a zinc-dependent manner (Reddigari et al. 1993; Schousboe 2001). The urokinase plasminogen activator receptor (u-PAR), gC1qR, and cytokeratin 1 have all been reported to be important for the binding of both HK and FXII (Herwald et al. 1996; Joseph et al. 1996, 2001; Mahdi et al. 2002). FXII is slowly auto-activated when bound to endothelial cells (Shibayama et al. 1998). Prekallikrein can also be activated, initiating bradykinin generation independent of FXII on endothelial cells (Rojkjaer et al. 1998; Rojkjaer and Schmaier 1999). The assembly of contact proteins on the cell receptors on endothelial cell membranes has been thought to provide a physiological negatively charged surface for activation, and it has been suggested that this FXII-independent activation does not initiate coagulation but rather functions as a mediator of vascular biology, above all via the generation of BK (Schmaier 1997, 1998, 2008).
In in vitro systems, several negatively charged surfaces have been shown to bind and activate FXII, and many biological substances also promote FXII auto-activation, but it has been difficult to identify such activating compounds in the intravascular compartment in non-disease states. Negatively charged phospholipids and sulfatides expressed in platelets may be expected to activate FXII, but this phenomenon has never been convincingly shown in vivo (Schousboe 1988).
Extracellular nucleic acids, particularly RNA, enhance coagulation and have been found to be associated with fibrin-rich thrombi, thereby contributing to arterial thrombus formation in mice (Kannemeier et al. 2007). FXII binds to and is auto-activated on extracellular RNA, and therefore contact activation has been suggested as a mechanism by which RNAs can exhibit their procoagulant properties (Kannemeier et al. 2007). Thus, extracellular RNA derived from damaged or necrotic cells, particularly under pathological conditions or in severe tissue damage, may represent an in vivo inducer of FXII-mediated contact activation .
Another recently proposed mechanism for the in vivo activation of FXII is the release of inorganic polyphosphate from dense granules during platelet activation (Ruiz et al. 2004). Inorganic polyphosphate has been shown to accelerate blood clotting by triggering FXII activation and promoting FV activation in in vitro models (Smith et al. 2006).
FXII is also activated by mis-folded protein aggregates, and increased levels of FXIIa and KK have been found in the blood of patients with amyloidosis (Maas et al. 2008). In addition, heparin derived from human mast cells activates FXII, inducing contact activation , and is thought to be responsible for the generation of kinins in allergic reactions (Noga et al. 1999).
3.2 Platelet-Induced Contact Activation
Studies in the 1970s and 1980s have shown that isolated platelets in buffer systems containing purified proteins can promote the activation of FXII (Walsh and Griffin 1981b) and that platelets are able to amplify the contact activation induced by a negatively charged substance or material (Johne et al. 2006). Nevertheless, the contact activation system was considered to have no significance for normal hemostasis. This view has been challenged by the results of our analysis of the interaction between platelets and contact proteins (using the TRAP-mediated activation of platelets in lepirudin-treated PRP and whole blood in the system described above) and the influence of this interaction on platelet aggregation and clot formation (Bäck et al. 2010).
In our study, TRAP-mediated activation of platelets in lepirudin-treated PRP led to the formation of FXIIa–C1INH, FXIIa–AT, FXIa–C1INH, and FXIa–AT complexes in plasma in the presence of a TF-inhibitory mAb, demonstrating that activation of these proteins had occurred as a consequence of specific platelet activation. The presence of FXII, FXI, prekallikrein, and HMWK on activated platelets was then demonstrated by flow cytometry. Furthermore, using chromogenic substrates, we showed that the bound FXIIa, FXIa, and KK possessed enzymatic activity.
We also found that clotting triggered either by the TF pathway or in a shear force model (Nilsson et al. 1998) generates substantial amounts of FXIIa–AT and FXIa–AT, demonstrating that contact activation occurs in clotting blood. The amount of FXIIa–AT was found to correlate with both thrombin activity and TAT complexes, indicating that it contributes to thrombin formation. Neither FXIIa–C1INH nor FXIa–C1INH complexes were detected, suggesting that C1INH did not primarily regulate these enzymes in clotting blood. In PRP, TRAP-activated platelets exhibited shorter clotting times than did non-activated controls. Addition of the specific FXIIa inhibitor corn trypsin inhibitor (CTI) prolonged the clotting time, whereas a TF-blocking mAb had no effect.
In summary, our study demonstrated that FXII-mediated contact activation occurs on the surface, or in the vicinity, of activated platelets and contributes to platelet aggregation and clot formation. The low levels of contact system proteins on the platelet surface, combined with the substantial formation of FXIIa–AT and FXIa–AT in plasma, suggest that FXII is activated on the surface or in the vicinity of the platelets and then released into the fluid phase. Activated platelets may thus constitute a nidus for contact activation inside the blood vessels and can recruit proteins of the contact system into the clot formation process.
In a related study, we demonstrated that the regulation of FXII-mediated contact activation differs between material-induced activation and physiological activation by activated platelets (Bäck et al. 2009). When platelets were activated in lepirudin-treated whole blood and PRP by TRAP, ADP, collagen, or shear force, we detected FXIIa–AT, FXIa–AT, and low amounts of KK–AT, but no C1INH-complexes, in the resulting plasma. Thus, the platelet-induced activation of FXII resulted in activation of both FXI and prekallikrein. In contrast, contact activation triggered by kaolin, glass, or inorganic polyphosphate in PPP, PRP, or whole blood generated high amounts of FXIIa–C1INH. Regardless of which activating compound was used, low or only negligible amounts of FXIIa–AT were generated.
The effects of polyphosphate were also analyzed using the shear force model with non-anticoagulated blood. As was seen for lepirudin-treated blood, polyphosphate produced a strong activation of FXII, as detected by high levels of FXIIa–C1INH, but no further activation of FXI or prekallikrein occurred. A remarkable difference between platelet-induced and material-induced FXII activation was that CTI totally abolished the generation of FXIIa–C1INH after activation with glass, kaolin, or polyphosphate, but the generation of FXIIa–AT after platelet activation was only decreased to some extent.
Finally, we measured contact activation enzyme–serpin complexes in samples from patients treated for severe trauma (e.g., after traffic accidents). Blood was drawn at admission to the hospital and at repeated time points for up to 10 days. At the time points closest to the trauma, the highest amounts of FXIIa–AT and FXI–AT were detected, and the levels subsequently decreased. The levels of these complexes were correlated with the amounts of thrombospondin-1 that were released by activated platelets . In contrast, FXIIa–C1INH complexes were found days after admission, when FXIIa–AT levels had disappeared. No association between FXIIa–C1INH and TSP-1 was found. FXIIa–C1INH appeared later in the treatment and may be generated on artificial surfaces such as extracorporeal circuits and after treatments involving biomaterials, for example, plasma expanders (dextran) or intravenous lipids.
In conclusion, in this study (Bäck et al. 2009) we demonstrated that contact activation triggered by activated platelets is regulated by AT and by C1INH on artificial material surfaces. Also, the inhibition of FXIIa by CTI seems to differ according to the specific activation site. CTI had minor effects on platelet-mediated contact activation , and the activation of the contact enzymes is being studied in such models, AT complexes should be assessed, rather than C1INH complexes. Since enzyme–AT complexes, which are correlated with TSP-1, are generated during platelet-mediated contact activation , FXIIa–AT should be considered an interesting new biomarker candidate for platelet activation.
4 Conclusions
Recent studies demonstrating that activated platelets trigger both complement and contact system activation have raised questions about the extent to which these cascade systems are involved in thrombotic diseases. The focus of this review article is the crosstalk between platelets and the cascade systems in clotting blood. CS-A released by activated platelets is a major mediator of complement activation, ultimately resulting in inflammation and leukocyte activation. C3 binds to the surface of activated platelets independent of complement activation and with the thiolester disrupted, enabling interaction with leukocyte CR1. In addition, activated platelets also trigger the contact system, thereby accelerating the clotting process. This process leads to the generation of enzyme–AT complexes, but no enzyme–C1INH complexes. We propose that these complexes, in particular FXIIa–AT, could be useful as markers for platelet activation. In conclusion, in addition to their traditional role in hemostasis, platelets have been shown to have many other functions, including involvement in the regulation of the complement and contact systems.
References
Abrink M, Grujic M, Pejler G (2004) Serglycin is essential for maturation of mast cell secretory granule. J Biol Chem 279,40897-40905
Barber AJ, Kaser-Glanzmann R, Jakabova M, Luscher EF (1972) Characterization of a chondroitin 4 -sulfate proteoglycan carrier for heparin neutralizing activity (platelet factor 4 ) released from human blood platelets. Biochim Biophys Acta 286,312-329
Blockmans D, Deckmyn H, Vermylen J (1995) Platelet activation. Blood Rev 9,143-156
Bode AP, Hickerson DH (2000) Characterization and quantitation by flow cytometry of membranous microparticles formed during activation of platelet suspensions with ionophore or thrombin. Platelets 11,259-271
Bradford HN, Dela Cadena RA, Kunapuli SP, Dong JF, Lopez JA, Colman RW (1997) Human kininogens regulate thrombin binding to platelets through the glycoprotein Ib-IX-V complex. Blood 90,1508-1515
Bradford HN, Pixley RA, Colman RW (2000) Human factor XII binding to the glycoprotein Ib-IX-V complex inhibits thrombin-induced platelet aggregation. J Biol Chem 275,22756-22763
Brass LF (2003) Thrombin and platelet activation. Chest 124,18S-25S
Bäck J, Huber-Lang M, Elgue G, Kalbitz M, Sanchez J, Ekdahl KN, Nilsson B (2009) Distinctive regulation of contact activation by antithrombin and C1-inhibitor on activated platelets and material surfaces. Biomaterials 30,6573-6580
Bäck J, Sanchez J, Elgue G, Ekdahl KN, Nilsson B (2010) Activated human platelets induce factor XIIa-mediated contact activation. Biochem Biophys Res Commun 391,11-17
Camera M, Frigerio M, Toschi V, Brambilla M, Rossi F, Cottell DC, Maderna P, Parolari A, Bonzi R, De Vincenti O, Tremoli E (2003) Platelet activation induces cell-surface immunoreactive tissue factor expression, which is modulated differently by antiplatelet drugs. Arterioscler Thromb Vasc Biol 23,1690-1696
Carroll MC (2000) The role of complement in B cell activation and tolerance. Adv Immunol 74,61-88
Colman R, Hirsh, J., Marder, V., Salzman, E. (1994) Overview of Hemostasis. In, Haemostasis and Thrombosis Basic Principles and Clinical Practice (Colman, R., Hirsh, J., Marder, V., Salzman, E., ed), pp 3-18 Philadelphia, Lippincott Williams & Walkins
da Costa Martins PA, van Gils JM, Mol A, Hordijk PL, Zwaginga JJ (2006) Platelet binding to monocytes increases the adhesive properties of monocytes by up-regulating the expression and functionality of beta1 and beta2 integrins. J Leukoc Biol 79,499-507
de Agostini A, Lijnen HR, Pixley RA, Colman RW, Schapira M (1984) Inactivation of factor XII active fragment in normal plasma. Predominant role of C-1-inhibitor. J Clin Invest 73,1542-1549
Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V (2005) Platelet activation leads to activation and propagation of the complement system. J Exp Med 201,871-879
Donato JL, Nogueira MD, Marcondes S, Antunes E, Nader HB, Dietrich CP, de Nucci G (1994) The kinetics of chondroitin 4-sulfate release from stimulated platelets and its relation to thromboxane A2 formation and granule secretion. Braz J Med Biol Res 27,2163-2167
Ehlers R, Ustinov V, Chen Z, Zhang X, Rao R, Luscinskas FW, Lopez J, Plow E, Simon DI (2003) Targeting platelet-leukocyte interactions, identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibalpha. J Exp Med 198,1077-1088
Endresen GK, Mellbye OJ (1984) Studies on the binding of complement factor C3 to the surface of human blood platelets. Haemostasis 14,269-280
Forbes CD, Pensky J, Ratnoff OD (1970) Inhibition of activated Hageman factor and activated plasma thromboplastin antecedent by purified serum C1 inactivator. J Lab Clin Med 76,809-815
Forlow SB, McEver RP, Nollert MU (2000) Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood 95,1317-1323
Galanakis DK, Ghebrehiwet B (1994) A unique property of a plasma proteoglycan, the C1q inhibitor. An anticoagulant state resulting from its binding to fibrinogen. J Clin Invest 93,303-310
Gandhi NS, Mancera RL (2008) The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des 72,455-482
Ghebrehiwet B, Galanakis DK (1993) C1q inhibitor (chondroitin-4-sulfate proteoglycan), structure and function. Behring Inst Mitt 214-223
Ghebrehiwet B, Hamburger M (1982) Purification and partial characterization of a C1q inhibitor from the membranes of human peripheral blood lymphocytes. J Immunol 129,157-162
Gorbet MB, Sefton MV (2004) Biomaterial-associated thrombosis, roles of coagulation factors, complement, platelets and leukocytes. Biomaterials 25,5681-5703
Gozzo AJ, Nunes VA, Cruz-Silva I, Carmona AK, Nader HB, Faljoni-Alario A, Sampaio MU, Araujo MS (2006) Heparin modulation of human plasma kallikrein on different substrates and inhibitors. Biol Chem 387,1129-1138
Gozzo AJ, Nunes VA, Nader HB, Dietrich CP, Carmona AK, Sampaio MU, Sampaio CA, Araujo MS (2003) Glycosaminoglycans affect the interaction of human plasma kallikrein with plasminogen, factor XII and inhibitors. Braz J Med Biol Res 36,1055-1059
Greengard JS, Griffin JH (Receptors for high molecular weight kininogen on stimulated washed human platelets. Biochemistry 23,6863-6869.1984).
Greengard JS, Heeb MJ, Ersdal E, Walsh PN, Griffin JH (1986) Binding of coagulation factor XI to washed human platelets. Biochemistry 25,3884-3890
Grujic M, Braga T, Lukinius A, Eloranta ML, Knight SD, Pejler G, Abrink M (2005) Serglycin-deficient cytotoxic T lymphocytes display defective secretory granule maturation and granzyme B storage. J Biol Chem 280,33411-33418
Hagen I (1972) The release of glycosaminoglycans during exposure of human platelets to thrombin and polystyrene latex particles. Biochim Biophys Acta 273,141-148
Hamad OA, Ekdahl KN, Nilsson PH, Andersson J, Magotti P, Lambris JD, Nilsson B (2008) Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J Thromb Haemost 6,1413-1421
Hamad OA, Nilsson PH, Lasaosa M, Ricklin D, Lambris JD, Nilsson B, Ekdahl KN (2010a) Contribution of chondroitin sulfate A to the binding of complement proteins to activated platelets. PLoS One 5,e12889
Hamad OA, Nilsson PH, Wouters D, Lambris JD, Ekdahl KN, Nilsson B (2010b) Complement component C3 binds to activated normal platelets without preceding proteolytic activation and promotes binding to complement receptor 1. J Immunol 185,2686-2692
Harpel PC, Lewin MF, Kaplan AP (1985) Distribution of plasma kallikrein between C-1 inactivator and alpha 2-macroglobulin in plasma utilizing a new assay for alpha 2-macroglobulin-kallikrein complexes. J Biol Chem 260,4257-4263
He JQ, Wiesmann C, van Lookeren Campagne M (2008) A role of macrophage complement receptor CRIg in immune clearance and inflammation. Mol Immunol 45,4041-4047
Heck LW, Kaplan AP (1974) Substrates of Hageman factor. I. Isolation and characterization of human factor XI (PTA) and inhibition of the activated enzyme by alpha 1-antitrypsin. J Exp Med 140,1615-1630
Helmy KY, Katschke KJ, Jr., Gorgani NN, Kljavin NM, Elliott JM, Diehl L, Scales SJ, Ghilardi N, van Lookeren Campagne M (2006) CRIg, a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 124,915-927
Herwald H, Dedio J, Kellner R, Loos M, Muller-Esterl W (1996) Isolation and characterization of the kininogen-binding protein p33 from endothelial cells. Identity with the gC1q receptor. J Biol Chem 271,13040-13047
Holmer E, Kurachi K, Soderstrom G (1981) The molecular-weight dependence of the rate-enhancing effect of heparin on the inhibition of thrombin, factor Xa, factor IXa, factor XIa, factor XIIa and kallikrein by antithrombin. Biochem J 193,395-400
Holmsen H (1989) Physiological functions of platelets. Ann Med 21,23-30
Iatridis PG, Ferguson JH (1965) The Plasmatic Atmosphere of Blood Platelets. Evidence That Only Fibrinogen, Acg, and Activated Hageman Factor Are Present on the Surface of Platelets. Thromb Diath Haemorrh 13,114-125
Iatridis PG, Ferguson JH, Iatridis SG (1964) Surface Factor Mechanisms in Relation to Blood Platelets, Evidence That Activated Hageman Factor Is Present on the Surface of Platelets. Thromb Diath Haemorrh 11,355-371
Johne J, Blume C, Benz PM, Pozgajova M, Ullrich M, Schuh K, Nieswandt B, Walter U, Renne T (2006) Platelets promote coagulation factor XII-mediated proteolytic cascade systems in plasma. Biol Chem 387,173-178
Joseph K, Ghebrehiwet B, Peerschke EI, Reid KB, Kaplan AP (1996) Identification of the zinc-dependent endothelial cell binding protein for high molecular weight kininogen and factor XII, identity with the receptor that binds to the globular “heads” of C1q (gC1q-R). Proc Natl Acad Sci U S A 93,8552-8557
Joseph K, Nakazawa Y, Bahou WF, Ghebrehiwet B, Kaplan AP (1999) Platelet glycoprotein Ib, a zinc-dependent binding protein for the heavy chain of high-molecular-weight kininogen. Mol Med 5,555-563
Joseph K, Shibayama Y, Ghebrehiwet B, Kaplan AP (2001) Factor XII-dependent contact activation on endothelial cells and binding proteins gC1qR and cytokeratin 1. Thromb Haemost 85,119-124
Jurk K, Kehrel BE (2005) Platelets, physiology and biochemistry. In, Semin Thromb Hemost, vol. 31, pp 381-392.
Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, von Bruehl ML, Sedding D, Massberg S, Gunther A, Engelmann B, Preissner KT (2007) Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A 104,6388-6393
Karpman D, Manea M, Vaziri-Sani F, Stahl AL, Kristoffersson AC (2006) Platelet activation in hemolytic uremic syndrome. Semin Thromb Hemost 32,128-145
Kenney DM, Davis AE, 3rd (1981) Association of alternative complement pathway components with human blood platelets, secretion and localization of factor D and beta 1H Globulin. Clin Immunol Immunopathol 21,351-363
Kirschfink M, Blase L, Engelmann S, Schwartz-Albiez R (1997) Secreted chondroitin sulfate proteoglycan of human B cell lines binds to the complement protein C1q and inhibits complex formation of C1. J Immunol 158,1324-1331
Kishore U, Leigh LE, Eggleton P, Strong P, Perdikoulis MV, Willis AC, Reid KB (1998) Functional characterization of a recombinant form of the C-terminal, globular head region of the B-chain of human serum complement protein, C1q. Biochem J 333 ( Pt 1),27-32
Kjellén L, Lindahl U (1991) Proteoglycans, structures and interactions. Annu Rev Biochem 60,443-475
Köhl J (2006) The role of complement in danger sensing and transmission. Immunol Res 34,157-176
Kolset SO, Gallagher JT (1990) Proteoglycans in haemopoietic cells. Biochim Biophys Acta 1032,191-211
Kolset SO, Prydz K, Pejler G (2004) Intracellular proteoglycans. Biochem J 379,217-227
Konstantopoulos K, Neelamegham S, Burns AR, Hentzen E, Kansas GS, Snapp KR, Berg EL, Hellums JD, Smith CW, McIntire LV, Simon SI (1998) Venous levels of shear support neutrophil-platelet adhesion and neutrophil aggregation in blood via P-selectin and beta2-integrin. Circulation 98,873-882
Kuijper PH, Gallardo Torres HI, Houben LA, Lammers JW, Zwaginga JJ, Koenderman L (1998) P-selectin and MAC-1 mediate monocyte rolling and adhesion to ECM-bound platelets under flow conditions. In, J Leukoc Biol, vol. 64, pp 467-473.
Lamari FN, Karamanos NK (2006) Structure of chondroitin sulfate. Adv Pharmacol 53,33-48
Lishko VK, Podolnikova NP, Yakubenko VP, Yakovlev S, Medved L, Yadav SP, Ugarova TP (2004) Multiple binding sites in `fibrinogen for integrin alphaMbeta2 (Mac-1). J Biol Chem 279,44897-44906
Liszewski MK, Farries TC, Lublin DM, Rooney IA, Atkinson JP (1996) Control of the complement system. Adv Immunol 61,201-283
Maas C, Govers-Riemslag JW, Bouma B, Schiks B, Hazenberg BP, Lokhorst HM, Hammarstrom P, ten Cate H, de Groot PG, Bouma BN, Gebbink MF (2008) Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest 118,3208-3218
Mahdi F, Madar ZS, Figueroa CD, Schmaier AH (2002) Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood 99,3585-3596
Margolis J (1958) Activation of plasma by contact with glass, evidence for a common reaction which releases plasma kinin and initiates coagulation. J Physiol 144,1-22
Marquardt L, Anders C, Buggle F, Palm F, Hellstern P, Grau AJ (2009) Leukocyte-platelet aggregates in acute and subacute ischemic stroke. Cerebrovasc Dis 28,276-282
McKenzie SE (2002) Humanized mouse models of FcR clearance in immune platelet disorders. Blood Rev 16,3-5
Meijers JC, Kanters DH, Vlooswijk RA, van Erp HE, Hessing M, Bouma BN (1988) Inactivation of human plasma kallikrein and factor XIa by protein C inhibitor. Biochemistry 27,4231-4237
Meri S, Jarva H (1998) Complement regulation. Vox Sang 74 Suppl 2,291-302
Miller G, Silverberg M, Kaplan AP (1980) Autoactivatability of human hageman factor (factor XII). Biochem Biophys Res Commun 92,803-810
Mnjoyan Z, Li J, Afshar-Kharghan V (2008) Factor H binds to platelet integrin alphaIIbbeta3. Platelets 19,512-519.
Molina H, Holers VM, Li B, Fung Y, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, Chaplin DD (1996) Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc Natl Acad Sci U S A 93,3357-3361
Morgan BP (1992) Isolation and characterization of the complement-inhibiting protein CD59 antigen from platelet membranes. Biochem J 282 ( Pt 2),409-413
Mollnes TE, Brekke OL, Fung M, Fure H, Christiansen D, Bergseth G, Videm V, Lappegard KT, Köhl J, Lambris JD (2002) Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by anovel lepirudin-based human whole blood model of inflammation. Blood 100,1869-1877
Mori K, Nagasawa S (1981) Studies on human high molecular weight (HMW) kininogen. II. Structural change of HMW kininogen by the action of human plasma kallikrein. J Biochem 89,1465-1473
Nicholson-Weller A, March JP, Rosen CE, Spicer DB, Austen KF (1985) Surface membrane expression by human blood leukocytes and platelets of decay-accelerating factor, a regulatory protein of the complement system. Blood 65,1237-1244
Niemann CU, Abrink M, Pejler G, Fischer RL, Christensen EI, Knight SD, Borregaard N (2007) Neutrophil elastase depends on serglycin proteoglycan for localization in granules. Blood 109,4478-4486
Nilsson B, Hong J, Larsson R, Elgue G, Ekdahl KN, Sahu A, Lambris, J D (1998) Compstatin inhibits complement and cellular activation in whole blood in models for extracorpereal circulation. Blood 92,1661-1667
Noga O, Brunnee T, Schaper C, Kunkel G (1999) Heparin, derived from the mast cells of human lungs is responsible for the generation of kinins in allergic reactions due to the activation of the contact system. Int Arch Allergy Immunol 120,310-316
Okayama M, Oguri K, Fujiwara Y, Nakanishi H, Yonekura H, Kondo T, Ui N (1986) Purification and characterization of human platelet proteoglycan. Biochem J 233,73-81
Olson ST, Sheffer R, Francis AM (1993) High molecular weight kininogen potentiates the heparin-accelerated inhibition of plasma kallikrein by antithrombin, role for antithrombin in the regulation of kallikrein. Biochemistry 32,12136-12147
Peerschke EI, Yin W, Ghebrehiwet B (2010) Complement activation on platelets, Implication for vascular infalmmation and thrombosis. Mol Immunol 47,2170-2175
Peerschke EI, Yin W, Grigg SE, Ghebrehiwet B (2006) Blood platelets activate the classical pathway of human complement. J Thromb Haemost 4,2035-2042
Pixley RA, Schapira M, Colman RW (1985a) Effect of heparin on the inactivation rate of human activated factor XII by antithrombin III. Blood 66,198-203
Pixley RA, Schapira M, Colman RW (1985b) The regulation of human factor XIIa by plasma proteinase inhibitors. J Biol Chem 260,1723-1729
Polley MJ, Nachman R (1978) The human complement system in thrombin-mediated platelet function. J Exp Med 147,1713-1726
Rand ML, Leung R, Packham MA (2003) Platelet function assays. Transfus Apheresis Sci 28,307-317
Reddigari SR, Kuna P, Miragliotta G, Shibayama Y, Nishikawa K, Kaplan AP (1993) Human high molecular weight kininogen binds to human umbilical vein endothelial cells via its heavy and light chains. Blood 81,1306-1311
Reilly RF (2003) The pathophysiology of immune-mediated heparin-induced thrombocytopenia. Semin Dial 16,54-60
Revak SD, Cochrane CG, Bouma BN, Griffin JH (1978) Surface and fluid phase activities of two forms of activated Hageman factor produced during contact activation of plasma. J Exp Med 147,719-729
Rojkjaer R, Hasan AA, Motta G, Schousboe I, Schmaier AH (1998) Factor XII does not initiate prekallikrein activation on endothelial cells. Thromb Haemost 80,74-81
Rojkjaer R, Schmaier AH (1999) Activation of the plasma kallikrein/kinin system on endothelial cells. Proc Assoc Am Physicians 111,220-227
Ruef J, Kuehnl P, Meinertz T, Merten M (2008) The complement factor properdin induces formation of platelet-leukocyte aggregates via leukocyte activation. Platelets 19,359-364
Ruggeri ZM, De Marco L, Gatti L, Bader R, Montgomery RR (1983) Platelets have more than one binding site for von Willebrand factor. J Clin Invest 72,1-12
Ruiz FA, Lea CR, Oldfield E, Docampo R (2004) Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem 279,44250-44257
Saito H, Goldsmith GH, Moroi M, Aoki N (1979) Inhibitory spectrum of alpha 2-plasmin inhibitor. Proc Natl Acad Sci U S A 76,2013-2017
Sandvik T, Endresen GK, Forre O (1984) Studies on the binding of complement factor C4 in human platelets. Complement activation by means of cold agglutinins. Int Arch Allergy Appl Immunol 74,152-157
Savage B, Saldivar E, Ruggeri ZM (1996) Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 84,289-297
Scarborough RM, Naughton MA, Teng W, Hung DT, Rose J, Vu TK, Wheaton VI, Turck CW, Coughlin SR (1992) Tethered ligand agonist peptides. Structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. J Biol Chem 267,13146-13149
Schattner M, Lazzari M, Trevani AS, Malchiodi E, Kempfer AC, Isturiz MA, Geffner JR (1993) Activation of human platelets by immune complexes prepared with cationized human IgG. Blood 82,3045-3051
Schmaier AH (1997) Contact activation, a revision. Thromb Haemost 78,101-107
Schmaier AH (1998) Plasma contact activation, a revised hypothesis. Biol Res 31,251-262
Schmaier AH (2008) Assembly, activation, and physiologic influence of the plasma kallikrein/kinin system. Int Immunopharmacol 8,161-165
Schmaier AH, Amenta S, Xiong T, Heda GD, Gewirtz AM (1993) Expression of platelet C1 inhibitor. Blood 82,465-474
Scholz T, Temmler U, Krause S, Heptinstall S, Losche W (2002) Transfer of tissue factor from platelets to monocytes, role of platelet-derived microvesicles and CD62P. Thromb Haemost 88,1033-1038
Schousboe I (1988) In vitro activation of the contact activation system (Hageman factor system) in plasma by acidic phospholipids and the inhibitory effect of beta 2-glycoprotein I on this activation. Int J Biochem 20,309-315
Schousboe I (2001) Rapid and cooperative binding of factor XII to human umbilical vein endothelial cells. Eur J Biochem 268,3958-3963
Shankar R, de la Motte CA, Poptic EJ, DiCorleto PE (1994) Thrombin receptor-activating peptides differentially stimulate platelet-derived growth factor production, monocytic cell adhesion, and E-selectin expression in human umbilical vein endothelial cells. J Biol Chem 269,13936-13941
Shattil SJ, Newman PJ (2004) Integrins, dynamic scaffolds for adhesion and signaling in platelets. Blood 104,1606-1615
Shibayama Y, Reddigari SR, Teruya M, Nakamura K, Fukunaga Y, Ienaga K, Nishikawa K, Suehiro S, Kaplan AP (1998) Effect of neurotropin on the binding of high molecular weight kininogen and Hageman factor to human umbilical vein endothelial cells and the autoactivation of bound Hageman factor. Biochem Pharmacol 55,1175-1180
Siddiqui FA, Desai H, Amirkhosravi A, Amaya M, Francis JL (2002) The presence and release of tissue factor from human platelets. Platelets 13,247-253
Silvestri L, Baker JR, Roden L, Stroud RM (1981) The C1q inhibitor in serum is a chondroitin 4-sulfate proteoglycan. J Biol Chem 256,7383-7387
Sims PJ, Rollins SA, Wiedmer T (1989) Regulatory control of complement on blood platelets. Modulation of platelet procoagulant responses by a membrane inhibitor of the C5b-9 complex. J Biol Chem 264,19228-19235
Sims PJ, Wiedmer T (1991) The response of human platelets to activated components of the complement system. Immunol Today 12,338-342
Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH (2006) Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci U S A 103,903-908
Stahl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, Gutierrez A, Beringer O, Zipfel PF, Karpman D (2008) Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 111,5307-5315
Sunyer J, Lambris JD (2001) Complement. Encyclopeida of Life Science 1-9
Torzewski J, Bowyer DE, Waltenberger J, Fitzsimmons C (1997) Processes in atherogenesis, complement activation. Atherosclerosis 132,131-138
Tschopp J, Jenne DE, Hertig S, Preissner KT, Morgenstern H, Sapino AP, French L (1993) Human megakaryocytes express clusterin and package it without apolipoprotein A-1 into alpha-granules. Blood 82,118-125
Uhlin-Hansen L, Eskeland T, Kolset SO (1989) Modulation of the expression of chondroitin sulfate proteoglycan in stimulated human monocytes. J Biol Chem 264,14916-14922
Van Der Graaf F, Koedam JA, Bouma BN (1983) Inactivation of kallikrein in human plasma. J Clin Invest 71,149-158
Vaziri-Sani F, Hellwage J, Zipfel PF, Sjoholm AG, Iancu R, Karpman D (2005) Factor H binds to washed human platelets. J Thromb Haemost 3,154-162
Waaler BA (1959) Contact activation in the intrinsic blood clotting system; studies on a plasma product formed on contact with glass and similar surfaces. Scand J Clin Lab Invest 11,1-133
Walsh PN, Griffin JH (1981a) Contributions of human platelets to the proteolytic activation of blood coagulation factors XII and XI. Blood 57,106-118
Walsh PN, Griffin JH (1981b) Platelet-coagulant protein interactions in contact activation. Ann N Y Acad Sci 370,241-252
Ward JV, Packham MA (1979) Characterization of the sulfated glycosaminoglycan on the surface and in the storage granules of rabbit platelets. Biochim Biophys Acta 583,196-207
Wiedmer T, Hall SE, Ortel TL, Kane WH, Rosse WF, Sims PJ (1993) Complement-induced vesiculation and exposure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglobinuria. Blood 82,1192-1196
Woulfe DS, Lilliendahl JK, August S, Rauova L, Kowalska MA, Abrink M, Pejler G, White JG, Schick BP (2008) Serglycin proteoglycan deletion induces defects in platelet aggregation and thrombus formation in mice. Blood 111,3458-3467
Wuillemin WA, Eldering E, Citarella F, de Ruig CP, ten Cate H, Hack CE (1996) Modulation of contact system proteases by glycosaminoglycans. Selective enhancement of the inhibition of factor XIa. J Biol Chem 271,12913-12918
Wuillemin WA, Minnema M, Meijers JC, Roem D, Eerenberg AJ, Nuijens JH, ten Cate H, Hack CE (1995) Inactivation of factor XIa in human plasma assessed by measuring factor XIa-protease inhibitor complexes, major role for C1-inhibitor. Blood 85,1517-1526
Yu GH, Holers VM, Seya T, Ballard L, Atkinson JP (1986) Identification of a third component of complement-binding glycoprotein of human platelets. J Clin Invest 78,494-501
Zarbock A, Muller H, Kuwano Y, Ley K (2009) PSGL-1-dependent myeloid leukocyte activation. J Leukoc Biol 86,1119-1124
Zhang SZ, Jin YP, Qin GM, Wang JH (2007) Association of platelet-monocyte aggregates with platelet activation, systemic inflammation, and myocardial injury in patients with non-st elevation acute coronary syndromes. Clin Cardiol 30,26-31
Zhao L, Bath PM, May J, Losche W, Heptinstall S (2003) P-selectin, tissue factor and CD40 ligand expression on platelet-leucocyte conjugates in the presence of a GPIIb/IIIa antagonist. Platelets 14,473-480
Zipfel PF, Skerka C (2009) Complement regulators and inhibitory proteins. Nat Rev Immunol 9,729-740
Zipfel PF, Skerka C, Caprioli J, Manuelian T, Neumann HH, Noris M, Remuzzi G (2001) Complement factor H and hemolytic uremic syndrome. Int Immunopharmacol 1,461-468
Zwaal RF, Comfurius P, Bevers EM (1998) Lipid-protein interactions in blood coagulation. Biochim Biophys Acta 1376,433-453
Acknowledgments
This work was supported by grants from the Swedish Research Council (VR) 2009-4675, 2009-4462, from the Swedish Research Council, and Swedish Research Council/SSF/Vinnova contract grant number 60761701, and by faculty grants from the Linnæus University. We thank Dr. Deborah McClellan for excellent editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Hamad, O.A., Bäck, J., Nilsson, P.H., Nilsson, B., Ekdahl, K.N. (2012). Platelets, Complement, and Contact Activation: Partners in Inflammation and Thrombosis. In: Lambris, J., Hajishengallis, G. (eds) Current Topics in Innate Immunity II. Advances in Experimental Medicine and Biology, vol 946. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-0106-3_11
Download citation
DOI: https://doi.org/10.1007/978-1-4614-0106-3_11
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-0105-6
Online ISBN: 978-1-4614-0106-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)