
Abstract
Neurodegenerative disorders are devastating human diseases that include Parkinson’s, Huntington’s and Alzheimer’s disease, amyotrophic lateral sclerosis, and the frontal temporal dementias. Although the clinical manifestations of these disorders have been known for some time, our understanding of the molecular underpinnings is only starting to emerge. Protein misfolding and aggregation is a common hallmark among these diseases, and these events are likely to produce a number of cellular and functional alterations. In Parkinson’s disease (PD), alpha-synuclein misfolds and forms intracellular inclusions known as Lewy bodies (LBs). Accumulating evidence suggests that these events lead to the disruption of intracellular trafficking which will, undoubtedly, lead to defects in synaptic transmission. Dysfunction in the basal ganglia circuitry is strongly associated with PD, with loss of dopaminergic neurons in the substantia nigra. Here, we present an overview of how misfolding and aggregation of alpha-synuclein might be related to synaptic dysfunction in PD. A deeper understanding of the molecular basis of PD will enable us to devise novel strategies for therapeutic intervention which may be applied to several neurodegenerative disorders that seem to have similar roots.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Amyotrophic Lateral Sclerosis
- Multiple System Atrophy
- Dementia With Lewy Body
- Synaptic Dysfunction
- Neurodegeneration With Brain Iron Accumulation
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Protein Misfolding Diseases
Neurodegenerative disorders are devastating human diseases that include Parkinson’s, Huntington’s and Alzheimer’s disease, amyotrophic lateral sclerosis, and the frontal temporal dementias (Forman et al. 2004). Invariably, these disorders are correlated with aging, which is the major risk factor known to date. While very different in their pathophysiology, they are collectively referred to as protein-misfolding disorders because a common hallmark is the presence of misfolded and aggregated forms of various proteins in the brains of affected individuals (Caughey and Lansbury 2003; Dobson 2001; Thomas et al. 1995).
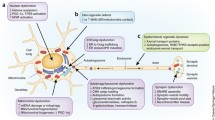
Despite the well-known connection between protein misfolding, aggregation, and disease, the manner by which misfolding results in disease is not clearly understood. In some cases, it seems that the deposition of protein aggregates may physically disrupt the functioning of specific cell groups and the respective tissues and organs where those cells are located. In other cases, it seems that the lack of functional protein, due to its recruitment into the aggregates, results in the failure of crucial cellular processes (Muchowski 2002; Soto et al. 2006; Thomas et al. 1995). However, for neurodegenerative diseases, such as Alzheimer’s, Parkinson’s or the Prion diseases, it appears that the symptoms arise from the destruction of cells by a “gain of toxic function” that results from the aggregation process (via oligomers, protofibrils, amyloid fibrils or other intermediates) or by a combination of both this gain of toxic function and a loss of normal function of the protein (Fig. 13.1) (Caughey and Lansbury 2003; Ross and Poirier 2004).

Schematic representation of the folding/unfolding/misfolding reactions. Once proteins are synthesized, they face a wide variety of challenges in the crowded environment of the cell. Throughout their lives, proteins need to change their conformations, allowing unwanted species to accumulate, which may lead to cytotoxicity (gain of toxic function). U unfolded, I intermediate, N native
The “amyloid hypothesis” (developed originally for Alzheimer’s disease) posits that the aggregation of proteins into an ordered fibrillar structure is causally related to aberrant protein interactions that culminate in neuronal dysfunction and ultimately neurodegeneration (Chiti and Dobson 2006; Hardy and Selkoe 2002). Proteins, as the main effectors in the cell, play underpinning roles in all biological processes. Thus, it is not surprising that the list of these diseases is continuously expanding, as new proteins are identified and their functions understood. Although different proteins are associated with different disorders, their ability to misfold and form amyloid fibrils seems to be a shared property. Nevertheless, this property is not sufficient to explain the differences in pathology observed in the disorders (Dobson 2003).
2 Parkinson’s Disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, exceeded only by Alzheimer’s disease (Dawson and Dawson 2003; Forman et al. 2005; Forno 1996; Gasser 2001; Lynch et al. 1997; Moore et al. 2005; Nussbaum and Polymeropoulos 1997; Vila and Przedborski 2004). PD affects about 2% of people over 65 years old and 4–5% of people over 85. Clinical manifestations of PD include severe motor defects characterized by resting muscle tremor (“pill-rolling”), muscle rigidity, bradykinesia, and postural instability (Goedert 2001).
LBs, the pathological hallmark of PD, are concentric hyaline cytoplasmic inclusions that can be visualized by histological analysis. LBs contain the protein alpha-synuclein (aSyn), as well as proteasomal subunits, lysosomal enzymes and molecular chaperones (Irizarry et al. 1998; Spillantini et al. 1997). It is still unclear whether the formation of LBs represents a mechanism developed by neurons to preclude the accumulation of the pathogenic intermediates, which have been proposed to be aSyn oligomers (Ding et al. 2002).
The majority of PD cases arise sporadically and only approximately 10% are familial. These have been genetically linked to the genes encoding aSyn, parkin, UCH-L1, DJ-1, PINK1, LRRK2, and ATP13A2 (Gasser 2007). While these cases are rare, it is hoped that understanding the molecular mechanisms underpinning the genetic forms of the disease will provide insight into the pathogenesis of the sporadic forms as well. Therefore, intense research is focused on investigating the genes and proteins involved in PD, both in terms of their normal physiological role as well as their contribution to disease.
3 The Basal Ganglia Circuitry
The basal ganglia are a group of related subcortical nuclei and include the neostriatum (caudate nucleus and caudate putamen), the external and internal pallidal segments (GPe, GPi), the subthalamic nucleus (STN), and the substantia nigra with its pars reticulate (SNr) and pars compacta (SNc) (Alexander et al. 1986). They form a complex network of parallel loops that involve specific thalamic and cortical areas. These parallel circuits are divided into “motor,” “associative” and limbic loops depending on the function of the cortical area involved (for a review see (Galvan and Wichmann 2008)).
Both the striatum and STN receive glutamatergic afferents from specific areas of the cerebral cortex or thalamus, and transfer the information to the basal ganglia output nuclei, GPi and SNr. The projections between the striatum and GPi/SNr are divided into two separate pathways, a “direct” (monosynaptic) connection and an “indirect” projection, via the intercalated GPe and STN (Fig. 13.2, left). Neurons in the “direct” pathway express dopamine D1 receptors and coexpress the peptides substance P and dynorphin, whereas those in the “indirect” pathway contain dopamine D2 receptors and the peptide enkephalin (Gerfen et al., 1990). Movement-related output is directed from GPi and SNr to the ventral nucleus of the thalamus which in turn projects back to the primary motor cortex. GPi and SNr also project to noncholinergic neurons in the pedunculolantine nucleus (PPN) in the brainstem and to CM/Pf (Galvan and Wichmann 2008).

Simplified diagram of the motor “loop” in the basal ganglia in normal (left panel) and in Parkinson’s condition (right panel). Black arrows indicate inhibitory connections and gray arrows indicate excitatory connections. The thickness of the arrows is an indication of their rate of activity. Dopamine is thought to inhibit neuronal activity in the “indirect” pathway and to excite neurons in the “direct” pathway. In the parkinsonian state, dopamine depletion leads to disinhibition of dopamine D2 receptor striatal neurons in the “indirect” pathway leading ultimately to over-inhibition of the thalamo-cortical and brainstem motor centers. The nigrostriatal neurons affected are highlighted in the top right magnification. Abbreviations: CM centromedian nucleus of thalamus, CMA cingulated motor area, GPe external segment of the globus pallidus, GPi internal segment of the globus pallidus, PPN pedunculopontine nucleus, SNc substantia nigra pars compacta, SNr substantia nigra pars reticulate, STN subthalamic nucleus, VA ventral anterior nucleus of thalamus, VL ventrolateral nucleus of thalalamus. Adapted from (Bender et al. 2006)
Activation of the direct pathway neurons in the striatum will inhibit basal ganglia output neurons, which, in turn, will disinhibit the connected thalamocortical neurons and facilitate movement. In contrast, the activation of indirect pathway neurons in the striatum will lead to increased basal ganglia output and, presumably, to suppression of movement. Because most GPi neurons increase their firing rate with movement, the main role of the basal ganglia motor circuit is to inhibit and to stabilize the activity of the thalamocortical neurons. Thus, the direct and indirect pathways have opposing effects on the output function of the basal ganglia (Alexander et al. 1986; DeLong 1990). Dopamine modulates these glutamatergic effects on corticostriatal inputs by exerting a dual effect on striatal neurons: excitatory action by activating D1 receptors in the “direct” pathway and inhibitory action through D2 receptors in the “indirect” pathway (Cepeda et al. 1993). Thus, dopamine’s net action may be to reduce GPi/SNr activity, thereby increasing the activity in thalamocortical projection neurons and, through activation of the cerebral cortex, facilitating movement (DeLong and Wichmann 2007) (Fig. 13.2, left).
4 Functional Alterations in PD
The prominent motor abnormalities in PD arise in large part from degeneration of neurons in the SNc with the resulting loss of dopamine in the basal ganglia. Abnormal activity in the “motor” loop of the basal ganglia is strongly implicated in the development of parkinsonism (Albin et al. 1989; DeLong 1990; Obeso et al. 2000). Recordings of firing rates in the basal ganglia in animal models of parkinsonism show that spontaneous neuronal discharge in STN, GPi and SNr is increased as compared with normal controls, neuronal discharge in GPe is decreased (Fig. 13.2, right panel). Striatal dopamine loss results in reduced activity in the direct pathway, resulting in disinhibition of the basal ganglia output nuclei. This dopamine loss also leads to greater inhibition of GPe, and consequently, in disinhibition of STN and GPi. The net effect of these changes is an increase in basal ganglia output to brainstem and thalamus which has been postulated to result in over-inhibition of thalamocortical neurons and reduced responsiveness of brainstem and cortical mechanisms involved in motor control (Galvan and Wichmann 2008).
Imaging studies have also suggested that the striatal dopamine concentrations steeply decline preceding the onset of clinical parkinsonism, which is only seen when more than 70% of striatal dopamine is lost. In early phases of PD, dopamine loss affects primarily the posterior putamen (the striatal motor area), but later spreads to involve other nigrostriatal regions. In later stages, more widespread dopamine loss and neuronal degeneration in non-dopaminergic systems, such as the locus coeruleus and the raphe nuclei, may account for some of the non-motor aspects of PD (Galvan and Wichmann 2008).
5 aSyn and Parkinson’s Disease
Accumulating evidence points to a causative role for the presynaptic protein aSyn in the pathogenesis of PD. aSyn, is a member of the synuclein family of proteins, which includes β- and γ-synuclein. It was initially identified in the electric lobe of Torpedo californica for reacting with an antiserum raised against purified cholinergic vesicles (Maroteaux et al. 1988), but its function remains unclear. aSyn is a 14.5 kDa protein, abundant in the brain, which has been involved in a variety of cellular processes (Fig. 13.3) (Cookson 2006; Lucking and Brice 2000).

Putative functions and interacting partners of aSyn. The binding partners of aSyn are indicated by arrows. “–” and “+” indicate enzyme inhibition or activation by aSyn, respectively. Boxes describe potential functions of aSyn interacting with the respective partner. SIRT2 sirtuin 2, PLD2 phospholipase D2, PKC protein kinase C, PKA protein kinase A, ERK extracellular-regulated kinase. Adapted from (Lucking and Brice 2000)
After an initial report of a non-Aβ component of AD amyloid (NAC), consisting of a 35 amino acid polypeptide generated by cleavage of aSyn, the protein has been associated to several neurodegenerative disorders, as described below.
The PARK1 locus, which encodes for aSyn, became associated with PD, when a point mutation was found in an Italian kindred afflicted by autosomal dominant PD. The mutation causes a threonine for alanine substitution at position 53 (A53T) (Polymeropoulos et al. 1997). This discovery was then followed by a report identifying aSyn in LBs (Irizarry et al. 1998; Spillantini et al. 1997). Afterwards, another familial form of PD was linked to a mutation in aSyn causing a proline for alanine substitution at position 30 (A30P) (Kruger et al. 1998). More recently, a third mutation consisting of a lysine for glutamate substitution at position 46 (E46K) was discovered to be associated with familial PD (Zarranz et al. 2004). Additionally, duplications and triplications of the PARK1 locus have also been linked to familial PD (Singleton et al. 2003). The dominant nature of the inherited mutants is thought to reflect a gain rather than a loss of function in the aSyn proteins.
After the initial discovery of aSyn in LBs in PD, the protein was detected in cellular inclusions in several other neurodegenerative diseases, including dementia with Lewy bodies (DLB), multiple system atrophy (MSA), and Hallervorden–Spatz syndrome, now called neurodegeneration with brain iron accumulation type 1 (NBIA). The neurodegenerative diseases that share aSyn pathology as a primary feature are collectively known as synucleinopathies (Spillantini and Goedert 2000). It is intriguing that the same protein is associated in different disorders, but the exact molecular mechanisms involved are still unknown.
5.1 aSyn: Gain or Loss of Function?
Several genetically tractable model organisms have been employed to study the role of aSyn in PD (Feany and Bender 2000; Masliah et al. 2000; Outeiro and Lindquist 2003). Over-expression studies of aSyn, or its PD-linked mutants, in mouse, rat, fly, nematode, and yeast have all converged on the conclusion that increased levels of aSyn leads to neurotoxicity, possibly due to a gain of toxic function associated with protein aggregation (Auluck et al. 2002; Feany and Bender 2000; Lakso et al. 2003; Lo Bianco et al. 2002; Masliah et al. 2000; Outeiro and Lindquist 2003). However, the presence of LBs in different disorders raises the possibility that sequestration of aSyn into Lewy bodies may decrease effective aSyn levels in nerve terminals and may contribute to neurodegeneration, also due to a loss of normal function (Chandra et al. 2005; Cookson and van der Brug 2007). Nevertheless, despite intense study of aSyn, frustratingly little is known about its normal cellular function and how it contributes to the various diseases (Fig. 13.3) (Gitler and Shorter 2007; Lucking and Brice 2000).
5.2 aSyn, Intracellular Trafficking and Synaptic Dysfunction
Through the use of a variety of cellular and animal models, we are gaining insight into the normal and abnormal roles of aSyn in neurons. Although the function of aSyn is still poorly understood, it has been extensively implicated in the regulation of presynaptic secretion. Indeed, aSyn is present in the cytosol of the presynaptic terminal in close proximity to synaptic vesicles (Iwai et al. 1995). Studies focusing specifically on the dopamine system showed that mice lacking aSyn exhibit an increased release of dopamine from nigrostriatal terminals that is mimicked by elevated Ca2+ and the reduction of striatal dopamine (Abeliovich et al. 2000). In these mice, however, there are no deficits or abnormalities in neuronal number, morphology or dopaminergic inervation pattern (Abeliovich et al. 2000). This is probably due to the fact that aSyn decreases the dopamine vesicles refilling rate, maintaining a stable dopamine pool (Yavich et al. 2004). Corroborating this, mice with aSyn null mutation or over-expressing A30P aSyn show decreased dopamine release in the striatum in response to prolonged stimulation, a situation in which the releasable pool of dopamine is more rapidly depleted (Yavich et al. 2004, 2005). More recently, it was also shown that A30P mutants display reduced activity of the vesicular monoamine transporter 2 (VMAT2) (Yavich et al. 2005) which is known to induce progressive nigrostriatal neurodegeneration (Goetz et al. 2005).
Studies using the budding yeast, Saccharomyces cerevisiae, as a model organism have shown that an increase in the levels of accumulation of aSyn, similar to that observed in patients with multiplications of the PARK1 gene, leads to intracellular trafficking defects, especially between the ER and Golgi (Cooper et al. 2006; Gitler et al. 2008; Outeiro and Lindquist 2003). Subsequent studies in Caenorhabditis elegans models of PD where aSyn was expressed pan-neuronally, confirmed the deleterious effects of aSyn on trafficking and endocytosis (Cooper et al. 2006; Lakso et al. 2003). In addition, a genetic study using mice demonstrated that aSyn ameliorates the inhibition of SNARE complex assembly, the latter being important in exocytosis of neurotransmitter (Chandra et al. 2005; Chua and Tang 2006).
6 Concluding Remarks
Endocytosis, an essential process for the recycling of cell surface molecules, has been implicated in the etiopathogenesis not only of PD, but also of AD. On the other hand, tight control of exocytosis is crucial in neurotransmitter release at the synapse. These cellular trafficking pathways are closely linked and under similar regulatory mechanisms. Therefore, aSyn-mediated disruption of essential trafficking events will, undoubtedly, have important consequences on synaptic dysfunction.
One outstanding issue is whether the increased vulnerability of dopaminergic neurons in the substantia nigra is due to the direct impact of aSyn mainly on monoamine pathways, either in vesicle recycling or exocytosis or, alternatively, in presynaptic secretory function in general, irrespective of the neurotransmitter involved. In the latter case, aSyn accumulation in dopaminergic neurons might be secondary and subsequent to neuronal degeneration.
In conclusion, the understanding of the role played by aSyn in synaptic dysfunction promises to open novel avenues for therapeutic intervention in PD and other synucleinopathies.
References
Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W.H., Castillo, P.E., Shinsky, N., Verdugo, J.M., Armanini, M., Ryan, A., Hynes, M., Phillips, H., Sulzer, D. and Rosenthal, A. (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system, Neuron, 25, 239–252.
Albin, R.L., Aldridge, J.W., Young, A.B. and Gilman, S. (1989) Feline subthalamic nucleus neurons contain glutamate-like but not GABA-like or glycine-like immunoreactivity, Brain Res, 491, 185–188.
Alexander, G.E., DeLong, M.R. and Strick, P.L. (1986) Parallel organization of functionally segregated circuits linking basal ganglia and cortex, Annu Rev Neurosci, 9, 357–381.
Auluck, P.K., Chan, H.Y., Trojanowski, J.Q., Lee, V.M. and Bonini, N.M. (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease, Science, 295, 865–868.
Bender, A., Koch, W., Elstner, M., Schombacher, Y., Bender, J., Moeschl, M., Gekeler, F., Muller-Myhsok, B., Gasser, T., Tatsch, K. and Klopstock, T. (2006) Creatine supplementation in Parkinson disease: a placebo-controlled randomized pilot trial, Neurology, 67, 1262–1264.
Caughey, B. and Lansbury, P.T. (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders, Annu Rev Neurosci, 26, 267–298.
Cepeda, C., Buchwald, N.A. and Levine, M.S. (1993) Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated, Proc Natl Acad Sci U S A, 90, 9576–9580.
Chandra, S., Gallardo, G., Fernandez-Chacon, R., Schluter, O.M. and Sudhof, T.C. (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration, Cell, 123, 383–396.
Chiti, F. and Dobson, C.M. (2006) Protein misfolding, functional amyloid, and human disease, Annu Rev Biochem, 75, 333–366.
Chua, C.E. and Tang, B.L. (2006) alpha-synuclein and Parkinson’s disease: the first roadblock, J Cell Mol Med, 10, 837–846.
Cookson, M.R. (2006) Hero versus antihero: the multiple roles of alpha-synuclein in neurodegeneration, Exp Neurol, 199, 238–242.
Cookson, M.R. and van der Brug, M. (2007) Cell systems and the toxic mechanism(s) of alpha-synuclein, Exp Neurol, 209, 5–11.
Cooper, A.A., Gitler, A.D., Cashikar, A., Haynes, C.M., Hill, K.J., Bhullar, B., Liu, K., Xu, K., Strathearn, K.E., Liu, F., Cao, S., Caldwell, K.A., Caldwell, G.A., Marsischky, G., Kolodner, R.D., Labaer, J., Rochet, J.C., Bonini, N.M. and Lindquist, S. (2006) {alpha}-Synuclein blocks ER-golgi traffic and Rab1 rescues neuron loss in Parkinson’s models, Science, 313, 3234–328.
Dawson, T.M. and Dawson, V.L. (2003) Molecular pathways of neurodegeneration in Parkinson’s disease, Science, 302, 819–822.
DeLong, M.R. (1990) Primate models of movement disorders of basal ganglia origin, Trends Neurosci, 13, 281–285.
DeLong, M.R. and Wichmann, T. (2007) Circuits and circuit disorders of the basal ganglia, Arch Neurol, 64, 20–24.
Ding, T.T., Lee, S.J., Rochet, J.C. and Lansbury, P.T., Jr. (2002) Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes, Biochemistry, 41, 10209–10217.
Dobson, C.M. (2001) The structural basis of protein folding and its links with human disease, Philos Trans R Soc Lond B Biol Sci, 356, 133–145.
Dobson, C.M. (2003) Protein folding and misfolding, Nature, 426, 884–890.
Feany, M.B. and Bender, W.W. (2000) A Drosophila model of Parkinson’s disease, Nature, 404, 394–398.
Forman, M.S., Lee, V.M. and Trojanowski, J.Q. (2005) Nosology of Parkinson’s disease: looking for the way out of a quagmire, Neuron, 47, 479–482.
Forman, M.S., Trojanowski, J.Q. and Lee, V.M. (2004) Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs, Nat Med, 10, 1055–1063.
Forno, L.S. (1996) Neuropathology of Parkinson’s disease, J Neuropathol Exp Neurol, 55, 259–272.
Galvan, A. and Wichmann, T. (2008) Pathophysiology of parkinsonism, Clin Neurophysiol, 119, 1459–1474.
Gasser, T. (2001) Genetics of Parkinson’s disease, J Neurol, 248, 833–840.
Gasser, T. (2007) Update on the genetics of Parkinson’s disease, Mov Disord, 22 Suppl 17, S343–S350.
Gerfen, C.R., Engber, T.M., Mahan, L.C., Susel, Z., Chase, T.N., Monsma, F.J., Jr. and Sibley, D.R. (1990) D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons, Science, 250, 1429–1432.
Gitler, A.D., Bevis, B.J., Shorter, J., Strathearn, K.E., Hamamichi, S., Su, L.J., Caldwell, K.A., Caldwell, G.A., Rochet, J.C., McCaffery, J.M., Barlowe, C. and Lindquist, S. (2008) The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis, Proc Natl Acad Sci U S A, 105, 145–150.
Gitler, A.D. and Shorter, J. (2007) Prime time for alpha-synuclein, J Neurosci, 27, 2433–2434.
Goedert, M. (2001) Alpha-synuclein and neurodegenerative diseases, Nat Rev Neurosci, 2, 492–501.
Goetz, C.G., Poewe, W., Rascol, O. and Sampaio, C. (2005) Evidence-based medical review update: pharmacological and surgical treatments of Parkinson’s disease: 2001 to 2004, Mov Disord, 20, 523–539.
Hardy, J. and Selkoe, D.J. (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics, Science, 297, 353–356.
Irizarry, M.C., Growdon, W., Gomez-Isla, T., Newell, K., George, J.M., Clayton, D.F. and Hyman, B.T. (1998) Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity, J Neuropathol Exp Neurol, 57, 334–337.
Iwai, A., Masliah, E., Yoshimoto, M., Ge, N., Flanagan, L., de Silva, H.A., Kittel, A. and Saitoh, T. (1995) The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system, Neuron, 14, 467–475.
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., Przuntek, H., Epplen, J.T., Schols, L. and Riess, O. (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease, Nat Genet, 18, 106–108.
Lakso, M., Vartiainen, S., Moilanen, A.M., Sirvio, J., Thomas, J.H., Nass, R., Blakely, R.D. and Wong, G. (2003) Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein, J Neurochem, 86, 165–172.
Lo Bianco, C., Ridet, J.L., Schneider, B.L., Deglon, N. and Aebischer, P. (2002) alpha -Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease, Proc Natl Acad Sci U S A, 99, 10813–10818.
Lucking, C.B. and Brice, A. (2000) Alpha-synuclein and Parkinson’s disease, Cell Mol Life Sci, 57, 1894–1908.
Lynch, T., Farrer, M., Hutton, M. and Hardy, J. (1997) Genetics of Parkinson’s disease, Science, 278, 1212–1213.
Maroteaux, L., Campanelli, J.T. and Scheller, R.H. (1988) Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal, J Neurosci, 8, 2804–2815.
Masliah, E., Rockenstein, E., Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., Sagara, Y., Sisk, A. and Mucke, L. (2000) Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders, Science, 287, 1265–1269.
Moore, D.J., West, A.B., Dawson, V.L. and Dawson, T.M. (2005) Molecular pathophysiology of Parkinson’s disease, Annu Rev Neurosci, 28, 57–87.
Muchowski, P.J. (2002) Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron, 35, 9–12.
Nussbaum, R.L. and Polymeropoulos, M.H. (1997) Genetics of Parkinson’s disease, Hum Mol Genet, 6, 1687–1691.
Obeso, J.A., Rodriguez-Oroz, M.C., Rodriguez, M., Lanciego, J.L., Artieda, J., Gonzalo, N. and Olanow, C.W. (2000) Pathophysiology of the basal ganglia in Parkinson’s disease, Trends Neurosci, 23, S8–19.
Outeiro, T.F. and Lindquist, S. (2003) Yeast cells provide insight into alpha-synuclein biology and pathobiology, Science, 302, 1772–1775.
Polymeropoulos, M.H., Lavedan, C., Leroy, E., Ide, S.E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenroos, E.S., Chandrasekharappa, S., Athanassiadou, A., Papapetropoulos, T., Johnson, W.G., Lazzarini, A.M., Duvoisin, R.C., Di Iorio, G., Golbe, L.I. and Nussbaum, R.L. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease, Science, 276, 2045–2047.
Ross, C.A. and Poirier, M.A. (2004) Protein aggregation and neurodegenerative disease, Nat Med, 10 Suppl, S10–S17.
Singleton, A.B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R., Lincoln, S., Crawley, A., Hanson, M., Maraganore, D., Adler, C., Cookson, M.R., Muenter, M., Baptista, M., Miller, D., Blancato, J., Hardy, J. and Gwinn-Hardy, K. (2003) alpha-Synuclein locus triplication causes Parkinson’s disease, Science, 302, 841.
Soto, C., Estrada, L. and Castilla, J. (2006) Amyloids, prions and the inherent infectious nature of misfolded protein aggregates, Trends Biochem Sci, 31, 150–155.
Spillantini, M.G. and Goedert, M. (2000) The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy, Ann N Y Acad Sci, 920, 16–27.
Spillantini, M.G., Schmidt, M.L., Lee, V.M., Trojanowski, J.Q., Jakes, R. and Goedert, M. (1997) Alpha-synuclein in Lewy bodies, Nature, 388, 839–840.
Thomas, P.J., Qu, B.H. and Pedersen, P.L. (1995) Defective protein folding as a basis of human disease, Trends Biochem Sci, 20, 456–459.
Vila, M. and Przedborski, S. (2004) Genetic clues to the pathogenesis of Parkinson’s disease, Nat Med, 10 Suppl, S58–S62.
Yavich, L., Oksman, M., Tanila, H., Kerokoski, P., Hiltunen, M., van Groen, T., Puolivali, J., Mannisto, P.T., Garcia-Horsman, A., MacDonald, E., Beyreuther, K., Hartmann, T. and Jakala, P. (2005) Locomotor activity and evoked dopamine release are reduced in mice overexpressing A30P-mutated human alpha-synuclein, Neurobiol Dis, 20, 303–313.
Yavich, L., Tanila, H., Vepsalainen, S. and Jakala, P. (2004) Role of alpha-synuclein in presynaptic dopamine recruitment, J Neurosci, 24, 11165–11170.
Zarranz, J.J., Alegre, J., Gomez-Esteban, J.C., Lezcano, E., Ros, R., Ampuero, I., Vidal, L., Hoenicka, J., Rodriguez, O., Atares, B., Llorens, V., Tortosa, E.G., Del Ser, T., Munoz, D.G. and De Yebenes, J.G. (2004) The new mutation, E46K, of alpha-synuclein causes parkinson and Lewy body dementia, Ann Neurol, 55, 164–173.
Acknowledgements
TFO is supported by the Michael J. Fox Foundation, Calouste Gulbenkian Foundation, Fundação para a Ciência e Tecnologia and by a Marie Curie International Reintegration Grant from the European Commission. LVL is supported by Fundação para a Ciência e Tecnologia.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Outeiro, T.F., Lopes, L.V. (2011). Synaptic Dysfunction in Parkinson’s Disease: From Protein Misfolding to Functional Alterations. In: Wyttenbach, A., O'Connor, V. (eds) Folding for the Synapse. Springer, Boston, MA. https://doi.org/10.1007/978-1-4419-7061-9_13
Download citation
DOI: https://doi.org/10.1007/978-1-4419-7061-9_13
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4419-7060-2
Online ISBN: 978-1-4419-7061-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)