
Abstract
Tropomyosin (Tm) is an essential component in the regulation of striated muscle contraction. Questions about Tm functional role have been difficult to study because sarcomere Tm content is not as easily manipulated as Troponin (Tn). Here we describe the method we recently developed to replace Tm-Tn of skeletal and cardiac myofibrils from animals and humans to generate an experimental model of homogeneous Tm composition and giving the possibility to measure a wide range of mechanical parameters of contraction (e.g. maximal force and kinetics of force generation). The success of the exchange was determined by SDS–PAGE and by mechanical measurements of calcium dependent force activation on the reconstituted myofibrils. In skeletal and cardiac myofibrils, the percentage of Tm replacement was higher than 90%. Maximal isometric tension was 30–35% lower in the reconstituted myofibrils than in control myofibrils but the rate of force activation (kACT) and that of force redevelopment (kTR) were not significantly changed. Preliminary results show the effectiveness of Tm replacement in human cardiac myofibrils. This approach can be used to test the functional impact of Tm mutations responsible for human cardiomyopathies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Contraction and relaxation of striated muscles are regulated by the thin filament proteins tropomyosin (Tm) and troponin (Tn). Tm is a dimer of two α-helical chains (each 284 residues long and about 33 kDa molecular mass) that wrap around one-another to form a continuous unbroken coiled-coil cable that winds around the actin helix. Tn is an etherotrimeric complex made of the calcium-binding subunit Troponin C (TnC), the inhibitory subunit Troponin I (TnI), and the TnC-TnI-Tm binding subunit Troponin T (TnT). Tn binds to Tm 1:1. The Tm filaments which are formed by head to tail assembly with a short overlap play a pivotal role in regulating the actin-myosin interaction in striated muscle and stabilizing actin structure (Gordon et al. 2000). In the last years, several methods proved to efficiently replace whole Tn complex or its subunits in both skeletal and cardiac muscle preparations (Brandt et al. 1984; Moss et al. 1986; Brenner et al. 1999; Piroddi et al. 2003; Kruger et al. 2005; Narolska et al. 2006; Yang et al. 2009). A significant advancement in understanding the thin filament regulatory system should be achieved then by extending to native Tm a replacement protocol for Tm isoforms and mutants, associated with myopathies (or engineered forms). As Tm is a filamentous protein strongly attached to actin, extracting and replacing with exogenous proteins is a challenge and the few attempts made met with little success (Chang et al. 2005). Recently, a successful method for the Tm-Tn replacement has been developed for skeletal myofibrils that preserve the viability of the preparation for mechanical studies (Siththanandan et al. 2009).
Replacement of regulatory proteins in single myofibrils offers a number of advantages, as compared to more conventional muscle preparations (She et al. 2000). The smaller diffusion distances allow a more complete and homogeneous exchange of proteins in a much shorter time (Piroddi et al. 2003). Furthermore, fast solution switching methods in single myofibrils (Tesi et al. 1999) can be used (1) to abruptly change the concentration of Ca2+ and investigate activation and relaxation kinetics (2) to abruptly change the concentrations of substrate and products of the acto-myosin ATPase and dissect some specific steps of the cross-bridge (CB) mechanical cycle (Tesi et al. 1999, 2000, 2002a, b).
A major goal of our recent work has been to learn how to completely replace myofibril endogenous Tm with isoforms and mutants (associated with myopathies or engineered) to get mechanistic insight on Tm’s role in regulation. To this aim, we developed an effective Tm-Tn extraction-reconstitution technique for both skeletal and cardiac myofibrils, producing sarcomeres homogeneous for Tm composition; these allow measurement of a wide range of mechanical parameters of contraction (maximal force, kinetics of force generation and relaxation at maximal and sub-maximal Ca2+-activation, myofilament Ca2+-sensitivity and cooperativity). This will improve the resolution of previous studies performed in vitro (Clemmens et al. 2005) or in skinned fibers from transgenic mouse models (Pieples et al. 2002; Jagatheesan et al. 2009) and gelsolin treated/reconstituted systems (Fujita et al. 2002).
With this experimental approach we will measure the functional effects of: (1) selected Tm isoforms in the form of homogeneous dimers (alpha-alpha or beta-beta), (2) cardiac and skeletal myopathy-linked Tm mutants, (3) engineered Tm mutants with modified structural properties.
2 Methods and Results
Single myofibrils are the smallest units of the contractile apparatus of striated muscle that retain the organized myofilament lattice and ensemble of associated proteins. Single myofibrils or bundles of two to three myofibrils were prepared from fast skeletal or cardiac muscle by homogenization in rigor solution of glycerinated (rabbit psoas) or triton treated (mouse left ventricle) fiber bundles or tissue strips (Tesi et al. 1999; Piroddi et al. 2006). Recently, we also used myofibrils isolated from human atrial and ventricular biopsies prepared as previously described (Piroddi et al. 2007). All solutions to which the samples and myofibrils were exposed contained a cocktail of protease inhibitors including leupeptin (10 μM), pepstatin (5 μM), phenylmethylsulphonylfluoride (200 μM), E64 (10 μM), NaN3 (500 μM), and dithiothreitol (500 µm).
Myofibril suspensions were centrifuged (1,600 × g, 8 min, 4°C) and resuspended in a low ionic strength solution (2 mM Tris–HCl, pH 8) to remove native Tm and Tn. To increase the amount of extraction, the cycle was repeated five to seven times (or more in cardiac samples which were found more resistant to Tm removal). Extracted myofibrils were then washed in a 200 mM ionic strength rigor solution (100 mM KCl, 2 mM MgCl2, 1 mM EGTA, 50 mM Tris–HCl, pH 7) and reconstituted with exogenous Tm and Tn following a 2-steps protocol.
First, the extracted myofibrils were collected and incubated in rigor solution containing 5 μM Tm (0°C, 1 h at least). Tm was either rabbit skeletal (sTm), bovine cardiac (cTm) and human cardiac (hcTm), depending on the experimental model used.
Secondly, Tm-reintroduced myofibrils were incubated in rigor solution containing 2 µM Tn (0°C, 1 h at least). Tn was either rabbit skeletal (sTn), bovine cardiac (cTn) or human cardiac (hcTn), depending on the experimental model used. Skeletal and cardiac Tn and Tm, both extracted and purified from rabbit fast skeletal and bovine cardiac muscles were kindly provided by Dr. E. Homsher (UCLA University, Los Angeles, USA). Human cardiac Tm was purified from human samples; recombinant human cardiac Tn was kindly provided by Dr. F. Matsumoto (Quantum Beam Science Directorate, Ibaraki, Japan). Finally, reconstituted myofibrils were washed and stored in 200 mM ionic strength rigor solution at 4°C, and used within 4 days. At each stage of the protocol, samples were retained from both supernatant and pellet fractions and then used to determine the extent of the Tm-Tn extraction and replacement procedure by 12% SDS–PAGE analysis (de Tombe et al. 2007).
Measurements of myofibrillar maximal isometric force and kinetics of force development (kACT) and redevelopment (kTR) were also used to evaluate the mechanical viability of myofibrils following the reconstitution, as well as to establish the effectiveness of the technique.
All activating (pCa 4.5) and relaxing solutions (pCa 9.0), which were calculated as described previously (Tesi et al. 2000), were at pH 7.0. The solutions contained: 10 mM total EGTA (CaEGTA/EGTA ratio set to obtain the different values of pCa), 5 mM MgATP, 1 mM free Mg2+, 10 mM MOPS, propionate and sulphate to adjust the final solution to an ionic strength of 200 mM and a monovalent cation concentration of 155 mM. Although continuous solution flow minimizes alterations in the concentration of MgATP and its hydrolysis products in the myofibrillar space, the measurements were made in the presence of creatine phosphate (10 mM) and creatine kinase (200 units ml−1) to prevent any ADP gradients. Contaminant [Pi] (around 170 mM in standard solutions) was reduced in some experiments to less than 5 mM (Pi-free solutions) by a Pi-scavenging enzyme system (purine-nucleoside-phosphorylase with substrate 7-methyl-guanosine; Tesi et al. 2000).
After the Tm-Tn depletion step, both skeletal and cardiac myofibrils remained stable in rigor solution but underwent an irreversible Ca2+-independent contraction when perfused with Mg-ATP containing solutions (“relaxing solution”, pCa 9.0). This behavior is expected from a successful depletion of Tn. After the two steps protocol for Tm-Tn replacement, skeletal and cardiac myofibrils were stable in relaxing solution and could be activated only by the concomitant presence of micromolar Ca2+ (“activating solution”, pCa 4.5). These observations are confirmed by the SDS–PAGE analysis.
Figure 1 shows representative gels of skeletal (a) and cardiac (b) myofibril samples retained at each step of the extraction-reconstitution protocol, run on a 12% Tris–HCl SDS–PAGE gel, after Comassie blue staining. For each lane, the amount of protein loaded (4–10 µg) is different due to protein loss during the preparation. The degree of Tm replacement in each lane was assessed from the intensity profile of scanned gels using UN-SCAN-IT gel 6.0 software (Silk Scientific, Inc., UT, USA) and determining the ratio of α-Tm to actin band intensities. As in Siththanandan et al. (2009), only α-Tm is used in these measurements because β-Tm (which is significantly present in skeletal myofibrils) migrates more slowly, within the TnT band (see lane 1 and 4; Cummins and Perry 1973; Salviati et al. 1982). For extracted rabbit skeletal myofibrils (Fig. 1a), the α-Tm/actin ratio (lane 2, Ext) is 0.04 compared to 0.35 in the controls (lane 1, Ctrl) and 0.33 in the reconstituted myofibrils (lane 3, Rec), representing about 89% removal and replacement. For mouse extracted cardiac myofibrils (Fig. 1b), the α-Tm/actin ratio is 0.03 (lane 2, Ext) compared to 0.21 in the controls (lane 1, Ctrl) and 0.19 in the reconstituted myofibrils (lane 3, Rec), representing about 86% removal and replacement.

12 percent Tris–HCl SDS–PAGE gel analysis of Tm-Tn extraction and reconstitution in rabbit skeletal and mouse left ventricle myofibrils. (a) Comassie-stained SDS–PAGE analysis of myofilament proteins from rabbit psoas myofibrils before (lane 1; Ctrl) and after (lane 2; Ext) removal of endogenous sTm-sTn complex and following reconstitution (lane 3; Rec) with exogenous sTm-sTn. sTm (lane 4); sTn complex (lane 5). (b) Comassie-stained SDS–PAGE analysis of myofilament proteins from mouse ventricle myofibrils before (lane 1; Ctrl) and after (lane 2; Ext) removal of endogenous cTm-cTn complex and following reconstitution (lane 3; Rec) with exogenous cTm-cTn. cTm (lane 4); cTn complex (lane 5). cTnC and LC2 bands are here fused. Only parts of gels containing Tm and Tn subunits are shown
As shown in Fig. 2, Tm (and Tn) replacement works equally well in skeletal (a) and cardiac (b) myofibrils. Mean values of Tm extraction-replacement were: sTm extraction 90% ± 2 and sTm reintroduction 96% ± 3 for rabbit skeletal myofibrils (mean ± SEM; n = 5); cTm extraction 90% ± 5 and cTm reintroduction 91% ± 5 in mouse cardiac myofibrils (mean ± SEM; n = 5). In both cases, Tm was always expressed relative to actin.

Replacement of Tm in rabbit skeletal and mouse cardiac myofibrils. Mean values of Tm extraction and reconstitution in rabbit skeletal (a) and mouse ventricle (b) myofibrils from the evaluation of the ratio of α-Tm to actin band intensities of correspondent gel lanes (Ctrl control; Ext extracted; Rec reconstituted). Values are given as mean ± SEM of five gels. Average levels of sTm extraction and replacement in rabbit psoas samples were 90% ± 2 and 96% ± 3, respectively. Average levels of cTm extraction and replacement in mouse ventricle samples were 90% ± 5 and 91% ± 5, respectively
The critical test to assess the success of the Tm and Tn removal and reconstitution protocol using exogenous proteins is the preservation of the mechanical properties of myofibril contraction and its overall Ca2+ switching mechanism. To this aim, small bundles of rabbit skeletal and mouse cardiac myofibrils were mounted in a force recording apparatus in an experimental bath filled with relaxing solution of pCa 9.0. The system used to record force from single myofibrils and for rapid solution changes has been described earlier (Colomo et al. 1997; Tesi et al. 1999). Briefly, myofibrils selected for use were mounted horizontally between two glass micro-tools. One tool is a calibrated cantilevered force probe while the other is connected to a length control motor. The myofibrils strongly adhered to the glass tools, which were positioned using micromanipulators to maximize the attachment area. The initial length of reconstituted myofibrils between attachments averaged 63.8 ± 2.7 μm, at a sarcomere length of 2.38 ± 0.03 µm (mean ± SEM, n = 24) for rabbit psoas myofibrils and 65.1 ± 5.3 µm at a sarcomere length of 2.17 ± 0.02 µm (mean ± SEM, n = 18) for mouse ventricle myofibrils. Isometric force was measured from the deflection of the force probe shadow projected on a split photodiode. Myofibrils were activated and relaxed by rapid translation between two continuous streams of relaxing (pCa 9.0) and activating (pCa 4.5) solutions flowing by gravity from a double-barreled glass pipette placed at a right angle to, and within 1 mm of, the preparation. Solution changes after the start of the paired-pipette movement (driven by a stepper-motor controlled system) occurred with a time constant of 2–4 ms and were complete within 10 ms (Tesi et al. 1999, 2000). Experiments were performed at 15°C. Release-restretch protocols were used to measure kTR (Brenner 1988). Values of the rate of rise of tension following a step decrease in pCa by fast solution switching (kACT) were estimated from the time required to reach 50% of the maximal isometric force.
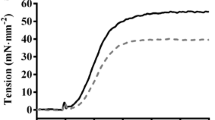
Typical force records of Fig. 3 show that both rabbit skeletal (a, b) and mouse cardiac (c, d) Tm-Tn replaced myofibrils preserve the basic property of being completely relaxed at high pCa, as expected from the proper reintroduction of the Tn calcium switch in the sarcomere suggested by SDS–PAGE analysis.

Maximal calcium activated tension in rabbit skeletal and mouse cardiac myofibrils before and after Tm-Tn replacement at 15°C. Representative force records from myofibrils maximally Ca2+ activated by rapid solution switching (pCa changes marked by lines over traces). The rate of tension generation (kACT) was measured form the kinetics of force development following fast Ca2+ activation. Fast length changes (bottom traces, release-restretch protocol) are applied to myofibrils under conditions of steady tension generation in order to measure the rate constant of tension redevelopment (kTR). Upper panels: Representative force responses from rabbit psoas myofibrils. (a) Control myofibril (sl 2.5 μm, P0 420 mN mm−2, kACT 7.2 s−1, kTR 7.3 s−1). (b) sTm/sTn-reconstituted myofibril (sl 2.4 μm, P0 406 mN mm−2, kACT 7.7 s−1, kTR 7.9 s−1). Lower panels: Representative force responses from mouse ventricle myofibrils. (c) Control myofibril (sl 2.1 μm, P0122 mN mm−2, kACT 5.6 s−1, kTR 4.4 s−1).; (d) cTm/cTn-reconstituted myofibril (sl 2.1 μm, P0 79 mN mm−2, kACT 5.2 s−1, kTR 4.1 s−1). Time scale, force and length calibrations as indicated by bars
Mean data of force development in control and Tm-Tn reconstituted rabbit skeletal and mouse cardiac myofibrils are reported in Table 1. Following calcium activation, maximal isometric tension was significantly lower (about 30–35%; p < 0.05 – Student t test) in both the Tm-Tn reconstituted skeletal and cardiac myofibrils than in correspondent controls. In both experimental models, following Tm-Tn reconstitution, no significant difference was found for the rate of force activation (kACT) and that of force redevelopment (kTR), showing that skeletal and cardiac myofibril remains mechanically viable and capable of producing substantial forces with normal kinetics. Sarcomere length, maximal calcium activated force, kACT and kTR of reconstituted myofibrils were in the range of control values previously observed in skeletal (Piroddi et al. 2003) and cardiac (Piroddi et al. 2006) myofibrils.
Recently, the method worked also in human cardiac myofibrils from left ventricular and atrial biopsies. Protein exchange assessment in human cardiac myofibrils, treated as described above and replaced with hcTm-hcTn, is shown by SDS–PAGE gel electrophoresis of Fig. 4a. Preliminary results indicate that the level of hcTm extraction ranged 65% and that of hcTm reconstitution 96%. Preliminary data indicate that after reconstitution, human cardiac myofibrils (both atrial and ventricular) can develop substantial amount of calcium activated tension, as shown in Fig 4b and c with kinetics unchanged by the Tm-Tn extraction and reconstitution procedure. These results in human cardiac myofibrils are promising for the full development of a method for the study of functional impact of Tm mutations associated with cardiomyopathies in humans.

Tm-Tn extraction and reconstitution in human ventricle myofibrils. (a) Comassie-stained 12% Tris–HCl SDS–PAGE gel of human ventricle myofilament proteins before (lane 3; Ctrl) and after (lane 2; Ext) removal of endogenous hcTm-hcTn complex and following reconstitution (lane 1; Rec) with exogenous hcTm-hcTn. Right panels: Representative recordings of tension development following maximal Ca2+-activation by rapid solution switching (upper traces; pCa changes marked by lines over traces) from a control myofibril (b) and from a myofibril whose endogenous Tm and Tn had been extracted and replaced with exogenous hcTm and recombinant hcTn (c). Bottom traces: length changes. Control myofibril: sl 2.2 µm, P0 104 mN mm−2, kACT 0.98 s−1, kTR 0.70 s−1. hcTm/hcTn-reconstituted myofibril: sl 2.1 µm, P0 83 mN mm−2, kACT 0.64 s−1, kTR 0.50 s−1. Time scale, force and length calibrations as indicated by bars
3 Discussion
The present work demonstrates that we can substantially replace Tm-Tn in skeletal and cardiac myofibrils, from both animal and human source, with low impact on myofibril mechanics. This is important as little is known about the functional role of Tm in the cooperative mechanisms of force activation and relaxation in cardiac and skeletal muscle. It is also important because several mutations in the protein have been associated with cardiomyopathies and skeletal muscle disease (Gunning et al. 2008) such as nemaline myopathy or distal arthrogriposis (Ochala et al. 2008). Production of an experimental model with homogeneous Tm composition gives us a system in which to compare Tm forms over a wide range of mechanical parameters of contraction (maximal force, kinetics of force generation and relaxation at maximal and sub-maximal Ca2+-activation, myofilament Ca2+-sensitivity and cooperativity) in both skeletal and cardiac muscle.
This will improve the resolution of previous studies performed in vitro (Clemmens et al. 2005) or in skinned fibers from transgenic models (Pieples et al. 2002) and gelsolin treated/reconstituted systems (Fujita et al. 2002). Until recently, replacement of Tm in a skinned cardiac muscle preparation was only possible using a technique that selectively removed the entire thin filaments by gelsolin (Fujita et al. 1996, 2002) followed by sequential reconstitution of the actin filaments with exogenous G actin and then with Tm and Tn. Though the technique is powerful, control of the length of the reconstituted thin filaments is quite empirical and the final structure of the sarcomere may be rather different than the original. Genetic techniques, making use of transgenic animals for Tm isoforms (Wolska et al. 1999) or mutants (Muthuchamy et al. 1999; see also Gunning et al. 2008) also show important limitations regarding the control of the expression level of exogenous transgenes that can dramatically influence the morphological and/or physiological phenotype of the tissue.
Several decades ago, a method to deplete native Tm from myofibrils was introduced by Perry and Corsi (1958) using low ionic strength alkaline solutions. In an attempt to extract all proteins from myofibrils, Perry and Corsi (1958) found that the total amount of protein extracted increased with the alkalinity of the buffers used but not for actin. Kominz and Yoshioka (1969) later used sodium carbonate/bicarbonate buffers to remove Tm from actomyosin preparations.
In the last years, the successful development in our lab of a method for substantial and non-invasive Tn replacement in myofibrils, suggested an approach to the challenging aim of replacing the whole Tm-Tn complex in skeletal and cardiac myofibrils. We based our initial attempts on a protocol published in 2005 (Chang et al. 2005) that claimed to selectively remove and replace all thin filament regulatory proteins in porcine cardiac myofibrils. Though myofibril suspensions treated by this Tm-Tn extraction and reconstitution method exhibited normal biochemical activity (Chang et al. 2005), in our hands they were no longer able to develop substantial levels of Ca2+-activated tension (unpublished observations). Only very recently, starting from Chang et al. (2005) approach but modifying the pH buffer (Tris instead of bicarbonate) and reducing alkalinity (pH 8 instead of 9 for the Tm removal step), Homsher and collaborators (Siththanandan et al. 2009) succeeded in designing a Tm-Tn depletion/reconstitution protocol producing skeletal rabbit psoas myofibrils viable for mechanical studies. Here we report improvements of this method for the replacement of native Tm and Tn with exogenous regulatory proteins not only in skeletal but also in mouse and human cardiac myofibrils with a significant recovery of the tension generation capability.
This Tm-Tn protocol has a certain degree of variability with an overall reduction of maximal force to about 65–70%. This could be a consequence of the reconstitution process itself and of a possible loss in actomyosin during the extraction stage (Siththanandan et al. 2009). When only Tn (or its subunits) are replaced in myofibrils, low or no decrease in maximal force relative to controls is observed (de Tombe et al. 2007; Kreutziger et al. 2008), especially when a “replacement by exchange” approach instead of an “extraction-reconstitution” protocol is used. In the first case, instead of extracting Tn, wash myofibril suspension, and then replace/reconstitute the thin filament with an exogenous protein, native Tn is replaced by mass action by incubating myofibrils in a rigor solution containing 3 mM calcium in presence of an excess (0.5 mg/ml) of the exogenous Tn isoform (pH 6.9, 4°C, overnight; de Tombe et al. 2007). This treatment is much milder and reduces myofibril damage because at no time are the myofibrils without their complement of all the Tn subunits. We are now working on a further development of the Tm-Tn protocol towards finding conditions of pH, pH buffering and ionic strength promoting a whole Tm-Tn complex replacement by exchange. A milder treatment to replace endogenous Tm and Tn, besides better preserving myofibrils and their force generating capacity, could be particularly relevant in the case of human cardiac myofibrils because of the limited availability of suitable control tissue samples.
In conclusion, the development of this Tm-Tn replacement protocol in striated sarcomere with exogenous Tm or Tn of different structure and properties in association with the advantages offered by the use of single myofibrils for mechanical studies will offer unique opportunities (1) to better understand the structural mechanism through which the regulation signal of Ca2+ binding passes between Tn subunits to the Tm-actin filament, also in its cooperative aspects (2) to investigate whether and how regulatory proteins modulate CB function.
References
Brandt PW, Diamond MS, Schachat FH (1984) The thin filament of vertebrate skeletal muscle co-operatively activates as a unit. J Mol Biol 180:379–84
Brenner B (1988) Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci U S A 85:3265–3269
Brenner B, Kraft T, Yu L, Chalovich JM (1999) Thin filament activation probed by fluorescence of N-((2-(Iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1, 3-diazole-labeled troponin I incorporated into skinned fibers of rabbit psoas muscle. Biophys J 77:2677–2691
Chang AN, Harada K, Ackerman MJ, Potter JD (2005) Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in alpha-tropomyosin. J Biol Chem 280:34343–3439
Clemmens EW, Entezari M, Martyn DA, Regnier M (2005) Different effects of cardiac versus skeletal muscle regulatory proteins on in vitro measures of actin filament speed and force. J Physiol 566:737–46
Colomo F, Piroddi N, Poggesi C, te Kronnie G, Tesi C (1997) Active and passive forces of isolated myofibrils from cardiac and fast skeletal muscle of the frog. J Physiol 500:535–548
Cummins P, Perry SV (1973) The subunits and biological activity of polymorphic forms of tropomyosin. Biochem J 133:765–777
de Tombe PP, Belus A, Piroddi N, Scellini B, Walker JS, Martin AF, Tesi C, Poggesi C. (2007) Myofilament calcium sensitivity does not affect cross-bridge activation-relaxation kinetics. Am J Physiol Regul Integr Comp Physiol 292:R1129–1136
Fujita H, Yasuda K, Niitsu S, Funatsu T, Ishiwata S (1996) Structural and functional reconstitution of thin filaments in the contractile apparatus of cardiac muscle. Biophys J 71:2307–2318
Fujita H, Sasaki D, Ishiwata S, Kawai M (2002) Elementary steps of the cross-bridge cycle in bovine myocardium with and without regulatory proteins. Biophys J 82:915–28
Gordon AM, Homsher E, Regnier M (2000) Regulation of contraction in striated muscle. Physiol Rev 80:853–924
Gunning P, O’Neill G, Hardeman E (2008) Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol Rev 88: 1–835
Jagatheesan G, Rajan S, Schulz EM, Ahmed RP, Petrashevskaya N, Schwartz A, Boivin GP, Arteaga GM, Wang T, Wang YG, Ashraf M, Liggett SB, Lorenz J, Solaro RJ, Wieczorek DF (2009) An internal domain of {beta}-tropomyosin increases myofilament Ca2+ sensitivity. Am J Physiol Heart Circ Physiol 297:H181–190
Kominz DR, Yoshioka K (1969) The influence of native tropomyosin on the ATP threshold for turbidity development of actomyosin and myofibril suspensions. Arch Biochem Biophys 129:609–614
Kreutziger KL, Piroddi N, Scellini B, Tesi C, Poggesi C, Regnier M (2008) Thin filament Ca2+ binding properties and regulatory unit interactions alter kinetics of tension development and relaxation in rabbit skeletal muscle. J Physiol 586:3683–700
Kruger M, Zittrich S, Redwood C, Blaudeck N, James J, Robbins J, Pfitzer G, Stehle R (2005) Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in alpha-tropomyosin. J Physiol 564:347–357
Moss RL, Allen JD, Greaser ML (1986) Effects of partial extraction of troponin complex upon the tension-pCa relation in rabbit skeletal muscle. Further evidence that tension development involves cooperative effects within the thin filament. J Gen Physiol 87:761–74
Muthuchamy M, Pieples K, Rethinasamy P, Hoit B, Grupp IL, Boivin GP, Wolska B, Evans C, Solaro RJ, Wieczorek DF (1999) Mouse model of a familial hypertrophic cardiomyopathy mutation in alpha-tropomyosin manifests cardiac dysfunction. Circ Res 85:47–56
Narolska NA, Piroddi N, Belus A, Boontje NM, Scellini B, Deppermann S, Zaremba R, Musters RJ, dos Remedios C, Jaquet K, Foster DB, Murphy AM, van Eyk JE, Tesi C, Poggesi C, van der Velden J, Stienen GJ (2006) Effects of the mutation R145G in human cardiac troponin I on the kinetics of the contraction-relaxation cycle in isolated cardiac myofibrils. Circ Res 99:1012–1020
Ochala J, Li M, Ohlsson M, Oldfors A, Larsson L (2008) Defective regulation of contractile function in muscle fibres carrying an E41K beta-tropomyosin mutation. J Physiol 586:2993–3004
Perry SV, Corsi A (1958) Extraction of proteins other than myosin from the isolated rabbit myofibril. Biochem J. 68:5–12
Pieples K, Arteaga G, Solaro RJ, Grupp I, Lorenz JN, Boivin GP, Jagatheesan G, Labitzke E, DeTombe PP, Konhilas JP, Irving TC, Wieczorek DF (2002) Tropomyosin 3 expression leads to hypercontractility and attenuates myofilament length-dependent Ca(2+) activation. Am J Physiol Heart Circ Physiol 283:H1344–1353
Piroddi N, Tesi C, Pellegrino MA, Tobacman LS, Homsher E, Poggesi C (2003) Contractile effects of the exchange of cardiac troponin for fast skeletal troponin in rabbit psoas single myofibrils. J Physiol 552:917–931
Piroddi N, Belus A, Eiras S, Tesi C, van der Velden J, Poggesi C, Stienen GJ (2006) No direct effect of creatine phosphate on the cross-bridge cycle in cardiac myofibrils. Pflugers Arch 452:3–6
Piroddi N, Belus A, Scellini B, Tesi C, Giunti G, Cerbai E, Mugelli A, Poggesi C (2007) Tension generation and relaxation in single myofibrils from human atrial and ventricular myocardium. Pflügers Arch 454:63–73
Salviati G, Betto R, Danieli Betto D (1982) Polymorphism of myofibrillar proteins of rabbit skeletal-muscle fibres. An electrophoretic study of single fibres. Biochem J. 207:261–272
She M, Trimble D, Yu L, Chalovich JM (2000) Factors contributing to troponin exchange in myofibrils and in solution. J Mus Res Cell Mot 21:737–745
Siththanandan VB, Tobacman LS, Van Gorder N, Homsher E (2009) Mechanical and kinetic effects of shortened tropomyosin reconstituted into myofibrils. Pflugers Arch 458:761–776
Tesi C, Colomo F, Nencini S, Piroddi N, Poggesi C (1999) Modulation by substrate concentration of maximal shortening velocity and isometric force in single myofibrils from frog and rabbit fast skeletal muscle. J Physiol 516:847–853
Tesi C, Colomo F, Nencini S, Piroddi N, Poggesi C (2000) The effect of inorganic phosphate on force generation in single myofibrils from rabbit skeletal muscle. Biophys J 78:3081–3092
Tesi C, Colomo F, Piroddi N, Poggesi C (2002a) Characterization of the cross-bridge force-generating step using inorganic phosphate and BDM in myofibrils from rabbit skeletal muscles. J Physiol. 541:187–99
Tesi C, Piroddi N, Colomo F, Poggesi C (2002b) Relaxation kinetics following sudden Ca(2+) reduction in single myofibrils from skeletal muscle. Biophys J 83:2142–2151
Wolska BM, Keller RS, Evans CC, Palmiter KA, Phillips RM, Muthuchamy M, Oehlenschlager J, Wieczorek DF, de Tombe PP, Solaro RJ (1999) Correlation between myofilament response to Ca2+ and altered dynamics of contraction and relaxation in transgenic cardiac cells that express beta-tropomyosin. Circ Res 1999 84:745–51
Yang Z, Yamazaki M, Shen QW, Swartz DR (2009) Differences between cardiac and skeletal troponin interaction with the thin filament probed by troponin exchange in skeletal myofibrils. Biophys J 97:183–94
Acknowledgement
This work was supported by 7th Framework Programs of the European Union (STREP Project “BIG-HEART”, grant agreement 241577) and by Telethon-Italy (GGP07133). Authors gratefully acknowledge Dr. Earl Homsher for helpful discussion and technical advice.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Scellini, B., Piroddi, N., Poggesi, C., Tesi, C. (2010). Extraction and Replacement of the Tropomyosin–Troponin Complex in Isolated Myofibrils. In: Rassier, D. (eds) Muscle Biophysics. Advances in Experimental Medicine and Biology, vol 682. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-6366-6_9
Download citation
DOI: https://doi.org/10.1007/978-1-4419-6366-6_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-6365-9
Online ISBN: 978-1-4419-6366-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)