
Abstract
The TNF family members RANKL and its receptor RANK have initially been described as factors expressed on T cells and dendritic cells (DCs), respectively, and have been shown to augment the ability of DCs to stimulate naive T cell proliferation and enhance DC survival. Since another, yet soluble receptor for RANKL, namely OPG, was initially characterized as a factor inhibiting osteoclast development and bone resorption, it was somewhat enigmatic at first why one and the same genes would be essential both for the immune system and bone development – two processes that on first sight do not have much in common. However, in a series of experiments it was conclusively shown that RANKL-expressing T cells can also activate RANK-expressing osteoclasts, and thereby in principal mimicking RANKL-expressing osteoblasts. These findings lead to a paradigm shift and helped to coin the term osteoimmunology in order to account for the crosstalk of immune cells and bone. More importantly was that these findings also provided a rationale for the bone loss observed in patients with a chronically activated immune system such as in rheumatoid arthritis, leukemias, or the like, arguing that T cells, which were activated during the course of the disease to fight it off, also express RANKL, which induces osteoclastogenesis and thereby shifts the intricate balance of bone deposition and resorption in favor of the latter. Through knockout mice it became also clear that the RANKL-RANK-OPG system is involved in other processes such as in controlling autoimmunity or immune responses in the skin. We will briefly summarize the role of RANK(L) signaling in the immune system before we discuss some of the recent data we and others have obtained on the role of RANK(L) in controlling autoimmunity and immune responses in the skin.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- RANK
- RANKL
- OPG
- Osteoimmunology
- Osteoclast
- Osteoclastogenesis
- Immune system
- Rheumatoid arthritis
- Osteoporosis
- Autoimmunity
1 RANK(L) Signaling in the Immune System
1.1 Lymph Node Development
Surprisingly, the detailed analysis of both Rankl–/– and Rank–/– revealed an entirely unexpected phenotype – the complete absence of all lymph nodes [1–3]. Other TNF family molecules have also been implicated in the development and organization of secondary lymphoid tissues, such as lymphotoxin-α (LTα) [4], LTβ [5, 6], TNFR1 [7], or LTβ receptor (LTβR) [8]. It is important to note that LTα knockout mice lack all lymph nodes (LNs), Peyer’s patches (PPs), and follicular dendritic cells; they also show altered splenic architecture [6]. Since LTα not only forms (soluble) homotrimers but also heterotrimers with LTβ, the disruption of LTα alone and the effect on peripheral lymphoid organs cannot rule out a critical involvement of LTβ. Thus, LTβ knockout mice have been generated, which show similar defects in the organogenesis of the lymphoid system as LTα: they lack PPs, peripheral lymph nodes, splenic germinal centers, and follicular dendritic cells, but most LTβ–/– mice still retain mesenteric and cervical lymph nodes, suggesting an LTβ-independent function of LTα in the development of these LNs [5, 6].
However, LTβR knockout mice lack PPs, colon-associated lymphoid tissues, and importantly all lymph nodes [8]. The lack of lymph nodes in LTα mutant mice is best explained by defective LTβR signaling that is further strengthened by the observation that LTβR–/– mice also lack all lymph nodes. The observation of some lymph nodes in LTβ-deficient mice would, however, argue that another cytokine co-operates with LTα in the development of mesenteric and cervical lymph nodes. Since a common denominator of mice with disrupted LT signaling is the lack of PPs, it was initially assumed that PP and LN development are alwaysgenetically linked. However, the analysis of Rankl- and Rank-deficient mice showed that these mutant mice display intact splenic architecture and lack all lymph nodes but develop PPs normally, providingevidence, for the first time, that development of LNs and PPs can be genetically uncoupled [1–3]. In patients with an osteoclast-poor form of autosomal recessive osteopetrosis (ARO), various mutations in RANKL have been identified as the cause of the disease [9]. These patients presented with no palpable lymph nodes, suggesting that RANKL-RANK signaling also controls lymph node formation in humans [9].
Several distinct cell lineages such as fibroblasts, macrophages, reticular cells, and endothelial cells are required for primordial lymph node development [10]. These primordial lymph nodes are subsequently seeded by T and B cells and CD4+CD3–LTβ,+ cells that differentiate into NK cells, antigen presenting cells, and follicular cells to form mature compact nodes [11]. Of noted importance is that RANKL- and RANK-expressing cells are present in the “mature” lymph nodes, located mainly in the cortical areas adjacent to subcapsular sinuses [12]. Since RANK and RANKL are also expressed in the spleen and PPs, restricted RANKL-RANK expression cannot account for the selective lack of lymph nodes. Moreover, transfer experiments of Rankl–/– fetal liver cells into Rag1–/– mice indicated that the lack of lymph nodes in Rankl–/– is not due to a cellular homing defect of Rankl–/– lymphocytes; transfer of normal bone marrow cells into newborn Rankl knockout mice did also not rescue lymph node formation [1]. Another study could show that defective LN development in Rankl–/– mice correlates with a significant reduction of α4β7 +CD45+CD4+CD3– cells in developing LN anlagen and their failure to cluster.
In LTα–/– mice, these hematolymphoid precursor cells also showed impaired colonization and cluster formation in the LN anlage. Transgenic overexpression of RANKL could not restore LN development in LTα–/– mice, showing that LTα1β2 expression is required on α4β7 +CD45+CD4+CD3– cells [13]. Thus, these results indicate that both RANKL and lymphotoxin ligands regulate the LN genesis by controlling the colonization and cluster formation of α4β7 +CD45+CD4+CD3– cells during LN development. Since the defects in the RANKL/RANK or LT/LTβR system did not affect the initiation of colonization by α4β7 +CD45+CD4+CD3– cells, the results would also suggest that neither RANKL nor LT are intrinsically required for the initiation of the formation of LN anlage, but rather they are required for further development of the LN anlage [13]. Although some light has been shed on LN development over the last years, the exact cellular and molecular mechanism of RANKL-RANK-regulated LN organogenesis as well as the link between LN and Peyer’s patch formation still await resolution.
1.2 Dendritic Cells
Dendritic cells (DCs) are cells specialized to capture and process antigens. In most tissues, DCs are present in an immature state unable to stimulate T cells. Contact with antigen leads to their maturation in response to inflammatory stimuli. Mature DCs that have captured antigens then migrate to T cell zones of secondary lymphoid organs by afferent lymphatics and present antigen to antigen-specific T cells. The T cell areas of secondary lymphoid organs represent the microenvironment to allow interactions between DCs, T cells, and B cells to initiate adaptive immune responses [14]. Antigen-bearing DCs are in direct contact with naive antigen-specific T cells within the T cell areas of lymph nodes and after interaction with T cells these DCs are rapidly eliminated [15]. For DC elimination to occur, activated T cells induce apoptosis of DCs by producing the TNF family molecules TRAIL, FasL, and TNF-α. The relevance of DC death after antigen presentation could be shown in patients with autoimmune lymphoproliferative syndrome (ALPS), an inherited disease of lymphocyte homeostasis and defective apoptosis. ALPS patients manifest lymphocytosis and autoimmune disorders. In the type II form of ALPS, DCs accumulation and prolonged DC survival were shown to be due to a caspase 10 mutation that rendered DCs resistant to TRAIL-induced cell death [16]. These results indicated that mature DCs presenting antigens to T cells have to be effectively eliminated in order to avoid excessive immune responses. The life span of DCs might thus be an important checkpoint to control for the induction of tolerance, priming, and chronic inflammation.
RANKL is not expressed on resting CD4+ or CD8+ T cells, but 4 h after anti-CD3/CD28 stimulation, surface RANKL is detected on CD4+ T cells with a peak at 48 h and sustained high levels until at least 96 h. RANKL expression on CD8+ T cells follows similar kinetics upon stimulation but generally with lower levels of surface RANKL than CD4+ T cells [17]. RANK surface expression can be detected on DCs from various sources – on mature bone marrow-derived DCs, freshly isolated lymph node DCs, or freshly isolated splenic Dcs, which can even be increased by overnight culture in vitro, but RANK cannot be detected on freshly isolated LN B cells, LN-derived T cells, thymocytes, or peritoneal macrophages [18]. In fact, interactions between RANKL on activated T cells and RANK on DCs have been shown to mediate DC survival via Bcl-xL induction and upregulation of the costimulatory molecule CD40 on DCs [18–20]. Since RANKL activates the anti-apoptotic Ser/Thr kinase Akt/PKB, NF-κB and ERK through a signaling complex involving TRAF6 and c-Src on mature DCs and osteoclasts [21], activation of anti-apoptotic molecules seems to be at least partially responsible for RANKL-mediated DC survival [22].
Moreover, it has been shown that treatment of antigen-pulsed mature DCs with soluble RANKL in vitro before immunization enhances the number and persistence of antigen-presenting DCs in the draining LNs in vivo, and RANKL treatment also increased antigen-specific primary T cell responses. Interestingly, significant memory responses were observed only in mice injected with RANKL-treated DCs [20]. Since RANKL can induce multiple cytokines in DCs like IL-1, IL-6, IL-12 and IL-15 [17, 23], it could well be that these increased primary and memory T cells responses following vaccination with RANKL-treated DCs are due to enhanced/altered cytokine production. Alternatively, the enhanced T cell responses by RANKL might also be due to increased numbers of antigen-pulsed DCs found in draining LNs [20]. CD40L, a TNF family member closely related to RANKL, shows functional similarity to RANKL, in that both CD40L and RANKL are expressed on activated T cells and enhance the activation and survival of DCs [18, 19]. However, in contrast to the CD40L-CD40 system, RANKL-RANK signaling from T cells to DCs does not alter the expression of cell surface molecules such as MHC class II, CD80, CD86, and CD54. Whereas CD40L is primarily expressed on activated CD4+ T cells, RANKL is expressed on both activated CD4+ and CD8+ T cells [17, 24]. However, the kinetics of RANKL and CD40L expression are somewhat different: maximal RANKL levels following initial T cell activation peaked around 48 h and remained high at least until 96 h, while CD40L reaches maximal levels between 6 and 8 h and is downregulated to resting levels between 24 and 48 h [17, 25]. Thus, CD40L-CD40 interactions might primarily control the initial priming stage whereas RANKL-RANK might act at later time points than CD40L during the immune response.
Although CD40L-CD40 interactions are crucial for the generation of antigen-specific T cell responses in vivo [26], the finding that CD40- and CD40L-deficient mice can still mount protective CD4+ T cell responses upon viral challenge in vivo [27] would suggest that some pathogens can activate CD4+ T cells independent of CD40L signaling. The factors that might mediate the CD40(L)-independent CD4+ T cell priming could be RANKL and RANK: inhibition of RANKL in vivo by soluble RANK-Fc did not block the priming of lymphocytic choriomeningitis virus-specific T cells but impaired proliferation of CD4+ T cells to the viral antigen at later time points after infection and was especially apparent in the absence of CD40-expression [23]. Thus, at later stages of the immune response, RANKL can regulate CD40L-independent activation of CD4+ T helper cells. These observations suggested that although CD40L and RANKL have functional similarity and might cooperate, RANKL and CD40L might also have fundamentally different functions in the control of immune responses: CD40L might regulate T and B cell responses while RANKL appears to have a role in memory T cell responses.
The decoy receptor for RANKL, OPG, can also be found on the cell surface of DCs and was furthermore shown to bind to the TNF-family molecule TRAIL, which is produced by activated T cells to induce apoptosis of DCs [28]. Thus, it seems as if the balance between RANKL and TRAIL – both produced from activated T cells – could influence DC survival and OPG might modulate that regulatory loop. However, since the binding affinity of OPG to TRAIL is rather low (∼10,000 times less binding to TRAIL than to RANKL) [12] , it is still unclear whether OPG-TRAIL interactions have any functional relevance in vivo. Based on these studies, various groups are currently trying to control the DC fate via RANKL-RANK and OPG to modulate in vivo DC survival and to enhance the efficacy of DC-based vaccinations for anti-tumor therapy or the treatment of autoimmune diseases. However, in the final analyses of all the published genetic and functional studies on RANKL, RANK, and OPG, it appears that although these molecules can influence some aspects of lymphocyte and DC functions, none of these molecules plays an essential role in T, B, or dendritic cells that cannot be compensated for by other molecules such as CD40L/CD40. Since we and others have shown that expression of RANKL and OPG molecules can be controlled by sex hormones [29], RANKL-RANK might control gender specific differences in immunity and could be involved in the higher incidence of autoimmune diseases like arthritis in women.
1.3 RANK(L) Signaling in the Thymus
In our initial RANKL knock-out paper, we had actually observed altered T cell development and expression of RANKL on primarily CD4–8– thymocyte progenitors. However, since RANK mutant mice exhibited apparently normal thymocyte development it was unclear whether this effect was secondary to other in vivo phenotypes in our mice and whether RANKL-RANK might indeed have some primary functions in the thymus. Interestingly, a recent study provided evidence for such a primary function of RANKL-RANK in the thymus; surprisingly, RANKL-RANK controls the development of AIRE+ thymic epithelial cells.
Cortical thymic epithelial cells (cTECs) positively select thymocytes, which subsequently migrate to the medulla where thymocytes interact with medullary thymic epithelial cells (mTECs), expressing costimulatory molecules and self-tissue-restricted antigens (TRAs) [30]. TRA expression has been shown to be regulated in part by the transcription factor AIRE (autoimmune regulator). AIRE mutations lead to multiorgan autoimmune disease in humans and autoimmunity in AIRE gene targeted mice. This phenotype could be mapped to a subtype of thymic medullary epithelial cells, thereby demonstrating the importance of Aire+ mTECs in maintaining self-tolerance [31]. Despite the identification of a common progenitor of cTECs and mTECs [32], the developmental pathway leading to Aire+ mTECs was not entirely clear.
However, it has recently been demonstrated that an intrathymic CD4+3– lymphoid tissue inducer (LTi) cell population expresses RANKL and that RANKL signals from thymic LTi cells to CD80-Aire- mTECs are required for their development into CD80+Aire+ mTECs [33]. In line with these data, Aire expression is absent in thymi from Rank–/– mice and transplantation of Rank–/– thymic tissue under the kidney capsule of nude mice resulted in inflammatory infiltrates in liver and autoantibodies to several tissues, which parallels the phenotype observed after transplantation of Aire–/– thymic stroma [31, 33]. These findings suggest a key role of RANK signals in the regulation of central tolerance for which LTα-LTβR signals have also been shown to be important [34, 35]. However, Aire+ mTECs can develop in the absence of LTβR and LTα [33, 35], suggesting that some aspects of mTEC development and organization may involve both RANK and LTβR signals. Moreover, in the absence of Traf6, the key downstream signaling adaptor of RANK, Aire+ mTECs also do not develop leading to the onset of organ-specific autoimmunity [36] . Thus, RANKL expressed by inducer cells, and possibly other thymic cell types, can activate RANK on thymic epithelial progenitor cells to develop into AIRE+ thymic medullary epithelial cells.
In three recent studies, further evidence for a crucial role of RANKL-RANK interactions in mediating mTEC development was presented. In one study, the authors could demonstrate that mTEC development required both RANK and CD40 signaling [37]. Interestingly, RANK signaling was essential only for mTEC development during embryogenesis, since mTECs failed to develop properly in embryonic thymus while mTECs – albeit at reduced numbers – were detected in postnatal mice even in the complete absence of RANKL. The signal capable of taking over for RANKL in postnatal thymus was shown to be CD40L, since development of mTECs was almost completely abolished in CD40-RANKL double mutant mice. Moreover, CD40L- or RANKL-mediated mTEC development was also shown to be dependent on Traf6, NIK, and IKKβ, respectively. These results show that developmental-stage-dependent co-operation between RANK and CD40 promotes mTEC development, thereby establishing self-tolerance [37].
In the second study, the authors could show that positively selected thymocytes express RANKL and expand mTEC cellularity to form the mature thymic medulla [38]. This effect, importantly, effect depended on interaction with RANK and OPG expressed by mTECs, since neutralization of RANKL by RANK-Fc expression perturbed mTEC cellularity and RANKL expression in mice deficient for positive selection restored thymic medulla. These results suggest that RANKL produced by positively selected thymocytes can also foster thymic medulla formation, thereby establishing central tolerance [38].
In order to address the role of single-positive CD4+ and CD8+ thymocytes in the process of postnatal mTEC development, a recent third study went on to show that although either CD4+ or CD8+ thymocytes were sufficient to sustain formation of a well-defined medulla expansion of the mature mTEC population required autoantigen-specific interactions between positively selected CD4+ thymocytes bearing autoreactive T cell receptor (TCR) and mTECs displaying cognate self-peptide-MHC class II complexes. These interactions were shown to involve the engagement of CD40 on mTECs by CD40L induced on the positively selected CD4+ thymocytes. This antigen-specific TCR-MHC class II-mediated crosstalk between CD4+ thymocytes and mTECs thus defines a unique checkpoint in thymic stromal development that is pivotal for generating a mature mTEC population competent for ensuring central T cell tolerance [39].
1.4 Extramedullary Haematopoiesis and B Cell Phenotypes
In addition to T cells, Rankl–/– and Rank–/– mice have reduced numbers of mature B220+IgD+ and B220+IgM+ B cells in the spleen and lymph nodes and slightly disorganized B cell areas in primary splenic follicles [1, 2, 46]. Since Rankl–/– and Rank–/– mice have no bone marrow cavities, the reduced cellularity of B cells could be due to an altered microenvironment or due to changes in the composition of stromal cells outside the bone marrow cavity that affect B cell differentiation. For example, Rankl–/– mice form an ectopically organized extramedullary haematopoietic tissue localized at the outer surfaces of vertebral bodies [1]. This tissue exhibits morphological and phenotypic features characteristic of haematopoiesis and proliferating precursor cells. Whether these haematopoietic islands in Rankl–/– mice represent a defect in the homing of precursors during the switch from hepatic to bone marrow haematopoiesis or an event secondary to osteopetrosis, which interferes with the seeding of bone marrow cavities, remains to be determined. In fetal liver cell chimeras, RANKL was found to regulate early B cell differentiation from the B220+CD43+25– pro-B cell to the B220+CD43-25+ pre-B cell stage of development, indicating suggesting the TNF-family cytokine RANKL is indeed a regulator of early B lymphocyte development [1]. Evidence in an Opg mutant mouse strain confirmed the notion that the interplay RANKL-RANK and the molecular decoy receptor OPG may regulate the development and possibly the function of B lymphocytes [47]. Ex vivo, Opg–/– pro-B cells have enhanced proliferation to IL-7 and type 1 transitional B cells accumulate in the spleens of Opg–/– mice. Thus, loss of OPG may control B cell maturation. Moreover, it should be noted that OPG is a CD40-regulated gene in B cells and dendritic cells and that prostaglandin E2 treatment can increase the amount of RANKL messenger RNA in B220+ B cells in an estrogen-dependent manner [47, 48].
2 RANKL-RANK Signaling Can Mediate UV-Induced Immunosuppression
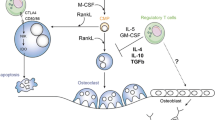
Recently, together with the group of Stefan Beissert, we could show that RANKL expression in the skin can control the number of regulatory T cells (Tregs). Tregs, in particular CD4+25+ Tregs expressing the transcription factor Foxp3, are a functionally distinct T cell subpopulation. Tregs maintain immunological self-tolerance and suppress excessive immune responses to self-antigens such as those in autoimmune diseases or allergies [40]. Despite the importance of DCs in inducing immunity to infections, it has been shown that DCs can also induce expansion of CD4+25+ Tregs and thereby induce T cell tolerance [41]. Given that activation of epidermal Langerhans cells (LCs; dendritic cells of the skin) by CD40L, a TNF family member closely related to RANKL, can induce severe systemic autoimmunity [42] and the importance of RANKL-RANK signals in T cell-DC interactions, we speculated that RANK signaling might also be important for immune homeostasis in the skin. Moreover, skin is the main site for sun light-induced Vitamin D3 production; Vitamin D3 is one of the key triggers of RANKL expression during osteoclastogenesis.
RANKL expression was indeed evident in keratinocytes of the skin and strongly upregulated following UV irradiation. Importantly, in stark contrast to transgenic overexpression of CD40L, RANKL overexpression in keratinocytes abrogated cutaneous contact hypersensitivity responses and concomitantly resulted in a marked increase of Tregs [43]. The receptor for RANKL, RANK, is expressed on LCs and enhanced signaling between RANKL-overexpressing keratinocytes and RANK-expressing LCs increased their survival and rendered LCs more effective in enhancing Treg proliferation [43]. Moreover, RANKL overexpression in keratinocytes could rescue the autoimmunity phenotype caused by CD40L overexpression in K14-RANKL/CD40L double transgenic mice [43]. Taken together, these findings provide a rationale for the long known immunosuppressive effect of ultraviolet exposure: UV irradiation is thought to upregulate RANKL in keratinocytes which in turn activates RANK-expressing LCs through RANKL-RANK interactions. RANKL-activated LCs preferentially trigger expansion of Tregs and thereby suppress immune reactions in the skin and other tissues. Importantly, UV-mediated immunosuppression, as determined by a DTH reaction in the ear, is impaired in mice transplanted with RANKL knock-out skin. Thus, RANKL-RANK might be the missing links to solve a long known conundrum – how sun exposure (sun burns) can be immunosuppressive.
These findings have several important clinical implications: for instance, local induction of the RANKL-RANK system in the skin could be used as a new approach for the treatment of allergies or systemic autoimmunity through increasing Treg numbers while avoiding systemic side effects [43]. The importance of Tregs and the influence of RANKL-RANK signals on their number and regulatory capacity, respectively, has also been previously suggested in an inflammation-induced model of type 1 diabetes. In this mouse model, islet-specific expression of TNF-α can be switched off upon doxycycline administration (Tet-TNF-α); in Tet-TNF-α/CD80 double transgenic mice, which constitutively co-express TNF-α and the costimulatory molecule CD80 on β cells in the islets of Langerhans, progression to diabetes depends on the duration of TNF-α expression [44, 45]. In this model, inflammation activates self-reactive CD8+ T cells to induce autoimmunity and diabetes, respectively, but CD4+25+ Tregs can successfully prevent β cell destruction. These Tregs have been shown to accumulate preferentially in the pancreatic lymph nodes (PLN) and islets and their capacity to prevent diabetes development appears to depend on RANKL signals [45]. Blockade of RANKL-RANK signaling by application of a RANK-Fc resulted in a decreased frequency of CD4+25+ Tregs in the PLN, consequently resulting in intra-islet differentiation of CD8+ T cells into cytotoxic T cells and rapid progression to diabetes [45]. In summary, inflammation may result in the RANKL-RANK-dependent generation and activation of CD4+25+ Treg cells, which then localize to the inflamed tissue and draining lymph nodes for the prevention of tissue destruction and autoimmunity by autoaggressive T cells.
3 T Cells and Bone Loss
Bone remodeling and bone loss are controlled by the RANKL-RANK-OPG axis. Moreover, RANKL is also induced in T cells following antigen receptor engagement. While piecing these findings together, it was intriguing to ask if T cell-derived RANKL could also regulate the development and activation of osteoclasts (i.e., would activated T cells modulate bone turnover via RANKL?). In an in vitro cell culture system of haematopoietic bone marrow precursors, we were indeed able to show that activated CD4+ T cells can induce osteoclastogenesis. Conversely, osteoclastogenesis could be blocked by addition of the physiological decoy receptor of RANKL, and OPG and was not dependent on T-cell-derived cytokines such as IL-1 or TNF-α, which could also upregulate RANKL expression in stromal cells [24]. Activated T cells also affect bone physiology in vivo, as judged by the severe osteoporotic phenotype of Ctla4 knockout mice in which T cells are spontaneously activated. Likewise, transferred Ctla4–/– T cells led to a decrease in bone mineral density in lymphocyte-deficient Rag1–/– mice and continued OPG administration to Ctla4–/– mice diminished their osteoporotic phenotype [24, 49]. These results unequivocally established the pivotal role of systemically activated T cells in resorbing bone through upregulation of RANKL, thereby stressing the importance of T cells as crucial mediators of bone loss in vivo. The results provided a novel paradigm for immune cells as regulators of bone physiology and gave birth to the field of osteoimmunology to account for the interplay between the adaptive immune system and bone metabolism. It also gave a new perspective to certain inflammatory or autoimmune diseases such as rheumatoid arthritis.
4 RANKL-RANK as Key Triggers of Bone Loss in Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a common human autoimmune disease and affects around 1% of people. RA is characterized by chronic inflammation of synovial joints, progressive destruction of cartilage and bone, severe joint pain, and ultimately life-long crippling [50]. Since osteoclasts are found at areas of bone erosion in RA patients [51], it was tempting to speculate that RANKL might be a key mediator of bone erosion in RA patients. Moreover, in an adjuvant-induced arthritis model (AdA), activated, RANKL-expressing T cells specific for the eliciting antigen can transfer the disease [52]. Consequently, we initially analyzed the contribution of RANKL to RA in an AdA model in Lewis rats. The AdA condition in rats mimics many of the clinical and pathological features of human RA (i.e., severe inflammation in bone marrow and soft tissues surrounding joints, accompanied by extensive local bone and cartilage destruction, loss of bone mineral density and crippling) [53]. In addition, T cells present in the inflamed joints and draining lymph nodes produce many pro-inflammatory cytokines [54]. Although inhibition of RANKL through OPG did not influence the severity of inflammation, OPG treatment nonetheless abolished the loss of mineral bone in inflamed joints of arthritic rats in a dose-dependent manner. Bone destruction in untreated arthritic animals correlated with a dramatic increase in osteoclast numbers, which was not observed in OPG-treated rats [24]. As a consequence, OPG-treated arthritic rats exhibited minimal loss of cortical and trabecular bone, whereas untreated AdA animals developed severe bone lesions characterized by partial to complete destruction of cortical and trabecular bone and positively affected erosion of the articular cartilages. In further pre-clinical studies in AdA rats, a single OPG injection was in fact shown to inhibit joint erosions for several days and produced sustained antierosive activity after a short course, but is most effective when initiated early in the disease [55]. These results demonstrated the importance of RANKL in mediating joint destruction and bone loss in AdA arthritis.
An important step in the etiology of arthritis is the alteration of cartilage structures leading to cartilage collapse in the joints. It is not quite clear whether cartilage destruction occurs independently of bone loss or whether damage to the subchondral bone indirectly causes cartilage deterioration [56]. In AdA rats, partial or complete erosion of cartilage in the central and peripheral regions of joint surfaces is observed, which can be preserved by OPG administration. Neither cartilage erosion nor matrix degeneration in the centers of joint surfaces occurred in OPG-treated AdA rats [24]. OPG could protect the cartilage by maintaining the underlying subchondral bone and insulating the overlying cartilage from the inflammatory cell infiltrates in the bone marrow. Since both RANKL and RANK are expressed on chondrocytes [12, 46], and Rankl–/– and Rank–/– mice exhibit significant changes in the columnar alignment of chondrocytes at the growth plate [1, 3], it is possible that the RANKL-RANK pathway plays a direct role in cartilage growth and cartilage homeostasis. These data provided the first evidence that inhibition of RANKL activity by OPG can also prevent cartilage destruction, a critical, irreversible step in the pathogenesis of arthritis. It should be noted that arthritis can also develop in the absence of activated T cells, as shown in the K/B×N serum transfer model of spontaneous autoimmunity [57]. Although RANKL-deficient mice still develop inflammation in the K/B×N serum transfer arthritic model, Rankl–/– mice showed a dramatic reduction in bone erosion – in line with the absolute requirement of RANKL for osteoclastogenesis [58]. However, cartilage damage was still observed in both arthritic Rankl–/– and arthritic control mice, but a trend toward milder cartilage damage in the Rankl–/– mice was noted. Thus, it appears that RANKL is not essential for cartilage destruction but clearly plays an as yet unidentified modulatory role [58].
In every rodent model of arthritis examined such as in TNF-α- or collagen-induced arthritis [59], inhibition of RANKL prevented bone erosion [60]. Of importance is that RANKL expression could be detected in inflammatory cells isolated from the synovial fluid of patients with adult or juvenile RA and patients with osteoarthritis while OPG was not detectable [24]. The correlation between RANKL expression in inflamed joints and arthritis appears to be absolute. In order to precisely define the cells producing RANKL, inflammatory synovial fluids were separated into T and non-T cell populations. Consistent with results obtained in rats, both synovial T and non-T cell populations from RA patients expressed RANKL but not OPG, and the capacity of human T cells expressing RANKL to directly induce osteoclastogenesis from human monocytes has been confirmed [61]. Moreover, RANKL expression is also upregulated in rheumatoid synovial fibroblasts, which in turn can efficiently induce osteoclastogenesis in vitro [62]. These data confirm the findings in rodent adjuvant arthritis, and suggest that RANKL signals from T cells and synoviocytes are the principal mediators of bone destruction in human arthritis. Taken together, RANKL is the trigger of bone loss and crippling in all animal models of arthritis studied so far, making RANKL a prime drugf candidate for therapeutic intervention in different forms of arthritis. Recent phase II clinical trials in humans suggest that inhibition of RANKL in human RA patients have no apparent effects on the inflammation but prevent bone loss at the site of inflammation [63].
These findings also provided a molecular explanation for the observed bone loss in many other humans diseases with chronic activation of the immune system such as adult and childhood leukemia [64], chronic infections such as hepatitis C or HIV [65], autoimmune disorders such as diabetes mellitus and lupus erythematosus [66], allergic diseases such as asthma [67], or lytic bone metastases in multiple cancers such as breast cancer [68]. These bone disorders can all cause irreversible crippling and thereby pose a tremendous burden on the quality of life of a huge number of patients. For example, many patients with lupus require hip replacement surgery and essentially all children that survive leukemia experience severe bone loss and growth retardation. In addition, T cell-derived RANKL also contributes to alveolar bone resorption and tooth loss in an animal model that mimics periodontal disease in humans. This was shown by transplanting human peripheral blood lymphocytes from periodontitis patients into immune-compromised NOD/SCID mice and by challenging these mice with a bacterial strain (Actinobacillus actinomycetemcomitans) that can cause periodontitis in humans. In response to stimulation by that microorganism, CD4+ T cells upregulated RANKL and induced osteoclastogenesis and bone destruction, respectively. Most importantly, inhibition of RANKL significantly reduced alveolar bone resorption around the teeth [69]. Further experiments showed that blocking RANKL might also help to prevent periodontitis in diabetic patients which are at high risk of developing periodontitis Nonobese diabetic (NOD) mice – the analog of human type 1 diabetes – were orally infected with
A. actinomycetemcomitans and it turned out that diabetic NOD mice manifested significantly higher alveolar bone loss than non-diabetic control mice. The observed bone loss was correlated with pathogen-specific proliferation and RANKL expression in local CD4+ T cells and could be reduced to baseline levels by RANKL inhibition [70]. Taken together, these findings suggest that specific interference with RANKL signaling pathways might be of great therapeutic value for treating inflammatory bone disorders such as human periodontitis or even bone loss in diabetic patients at high risk.
Since disease pathogenesis correlates with the activation of T cells in many osteopenic disorders; the obvious question then arises of why T cells in our body – of which a certain proportion is activated at any time due to fighting off the universe of foreign antigens to which we are permanently exposed – do not cause extensive bone loss? Likewise, in some chronic T cell and TNF-α-mediated diseases such as ankylosis spondylitis [71], T cell activation does not result in bone loss. One mechanism that counteracts RANKL-mediated bone resorption of activated T cells is the upregulation of interferon-γ (IFN-γ) in certain T cell subsets. IFN-γ blocks RANKL-induced osteoclastogenesis in vitro and IFN-γR–/– mice are more prone to osteoclast formation in a model of endotoxin-induced bone resorption than their wildtype littermates [72]. In line with this study, IFN-γ receptor knockout mice also exhibited enhanced severity in the collagen-induced model of T cell-mediated autoimmune arthritis [73]. Mechanistically, IFN-γ activates the ubiquitin-proteasome pathway in osteoclasts, resulting in TRAF6 degradation and therefore blocks RANK signaling. Thus, it appears that IFN-γ can prevent uncontrolled bone loss during inflammatory T-cell responses. Moreover, T cell-derived IL12 alone and IL12 in synergy with IL18 inhibits osteoclast formation in vitro [74], and IL-4 can abrogate osteoclastogenesis through STAT6-dependent inhibition of NF-κB signaling [75]. Thus, multiple T cell-derived cytokines might be able to interfere with RANK(L) signaling and therefore block osteoclastogenesis and osteoclast functions.
A recent report showed that a certain subset of CD4+ T helper cells, namely Th17, function as osteoclastogenic helper T cells [76]. Derived from naive T cells by a distinct mechanism than Th1 or Th2 cells [77], Th17 cells produce IL-17 and are thus responsible for a variety of autoimmune inflammatory effects [78]. Since IL-17 is also a potent inducer of RANKL expression and can found in the synovial fluid from RA patients [79], Th17 cells seem to be the prime candidate for the osteoclastogenic Th cell subset. Indeed, Th17 cells, but not Th1, Th2, or Treg cells, can stimulate osteoclastogenesis in vitro [76]. This study indicates that Th17 cells act as key mediators of bone destruction in RA patients by different means such as stimulation of local inflammation through IL-17, expression of RANKL on themselves, and induction of RANKL on osteoblasts or synovial fibroblasts, thereby contributing to accelerated bone erosin. The positive effect of Th17 cells on osteoclastogenesis is believed to be balanced by Th1 and Th2 cells mainly through their production of the cytokines IFN-γ and IL-4, respectively [76]. Thus, targeting Th17 might also be a powerful approach to prevent bone destruction associated with T cell activation in RA and other inflammatory bone diseases. Further studies will have to clarify the precise relationship and regulatory crosstalk of Th1, Th2, and Th17 subsets.
5 RANKL Inhibition as a New Therapy to Control Bone Loss in Human Patients
Several years ago, a fully human monoclonal IgG2 antibody to human RANKL, Denosumab, wasdeveloped and is currently in late-stage clinical trials for post-menopausal osteoporosis, cancer-metastases-induced bone loss, and RA [63, 80, 81]. Significantly, the binding of denosumab to RANKL is selective, and Denosumab does not show any signs of cross-reactivity to TNF-α, TNF-β, CD40L, or TRAIL [82]. Subcutaneous application of Denosumab at 3- or 6-month intervals over a period of 12 months to 412 postmenopausal women with low bone mineral density (BMD) in a randomized, placebo-controlled, dose-ranging phase 2 study resulted in a sustained decrease in bone turnover and a rapid increase in BMD [83]. In another 2-year randomized, double-blind, placebo-controlled study with 332 osteoporotic, postmenopausal women, twice-yearly subcutaneous application of Denosumab significantly increased BMD and decreased bone turnover markers in early and later postmenopausal women [80]. In a similar study in patients with breast cancer (n = 29) and multiple myeloma (n = 25) with radiologically confirmed bone lesions, a single dose of Denosumab resulted in the rapid and sustained decrease of bone turnover [84]. Lastly, in a multicenter, randomized, double-blind, placebo-controlled, phase II study with 218 patients with RA receiving methotrexate treatment, RANKL inhibition by Denosumab also increased BMD and protected from bone loss at the site of the inflamed joint without affecting inflammation per se [63]. In all cases, Denosumab administration was well tolerated and at least as good or superior to current standard medication. However, considering the various in vivo functions of RANKL-RANK, further clinical trials will be required to substantiate the benefits of RANKL inhibition on suppressing bone destruction.
6 Conclusions
The identification of RANKL, its receptor RANK, and the decoy receptor OPG as the key regulators for osteoclast development and the activation of mature osteoclasts has provided the key molecular framework to understand bone physiology and has opened the doors for the development of highly effective and rational drugs to treat bone loss in millions of patients. The finding that RANKL is produced by activated T cells; that activated T cells, in turn, can directly induce osteoclastogenesis also provided a novel molecular paradigm for bone loss associated with diseases having immune system involvement such as T cell leukemias, autoimmunity, various viral infections, RA, or periodontitis. In addition, RANKL-RANK control development of mammary glands in pregnancy and the formation of lymph nodes and AIRE+ thymic medullary epithelial cells. Moreover, RANKL might be the missing link between sun exposure and Treg mediated immunosuppression. Based on all available data, an important notation is that the inhibition of RANKL function might be the most rational therapy to ameliorate many osteopenic conditions and prevent bone destruction and cartilage damage (e.g., in osteoporosis and arthritis, thereby dramatically enhancing the lives of millions of patients).
Abbreviations
- TNF:
-
Tumor Necrosis Factor
- RANKL:
-
Receptor Activator of Nuclear Factor-κB (NF-κB) Ligand
- RANK:
-
Receptor Activator of Nuclear Factor-κB (NF-κB)
- OPG:
-
Osteoprotegerin
- DCs:
-
Dendritic Cells
- LT:
-
Lymphotoxin
- PPs:
-
Peyer’s Patches
- LNs:
-
Lymph Nodes
- ALPS:
-
Autoimmune Lymphoproliferative Syndrome
- cTEC:
-
Cortical Thymic Epithelial Cell
- mTEC:
-
Medullary Thymic Epithelial Cell
- TRA:
-
Tissue Restricted Antigen
- AIRE:
-
Autoimmune Regulator
- LTi:
-
Lymphoid Tissue Inducer
- Tregs:
-
eRgulatory T Cells
- LCs:
-
Langerhans Cells
- RA:
-
Rheumatoid Arthritis
- BMD:
-
Bone Mineral Density
References
Abu-Amer, Y. (2001). IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kappaB. J Clin Invest, 107: 1375–1385.
Akiyama, T., Shimo, Y., Yanai, H. et al. (2008). The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity, 29: 423–437.
Alimzhanov, M.B., Kuprash, D.V., Kosco-Vilbois, M.H. et al. (1997). Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc Natl Acad Sci U S A, 94: 9302–9307.
Anderson, G., Lane, P.J., & Jenkinson, E.J. (2007). Generating intrathymic microenvironments to establish T-cell tolerance. Nat Rev Immunol, 7: 954–963.
Anderson, D.M., Maraskovsky, E., Billingsley, W.L. et al. (1997). A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature, 390: 175–179.
Anderson, M.S., Venanzi, E.S., Klein, L. et al. (2002). Projection of an immunological self shadow within the thymus by the aire protein. Science, 298: 1395–1401.
Bachmann, M.F., Wong, B.R., Josien, R. et al. (1999). TRANCE, a tumor necrosis factor family member critical for CD40 ligand-independent T helper cell activation. J Exp Med, 189: 1025–1031.
Banchereau, J., & Steinman, R.M. (1998). Dendritic cells and the control of immunity. Nature, 392: 245–252.
Bendele, A., McComb, J., Gould, T. et al. (1999). Animal models of arthritis: relevance to human disease. Toxicol Pathol, 27: 134–142.
Bendixen, A.C., Shevde, N.K., Dienger, K.M. et al. (2001). IL-4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator-activated receptor gamma 1. Proc Natl Acad Sci USA, 98: 2443–2448; Akiyama, T., Maeda, S., Yamane, S. et al. (2005). Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science, 308: 248–251.
Boehm, T., Scheu, S., Pfeffer, K. et al. (2003). Thymic medullary epithelial cell differentiation, thymocyte emigration, and the control of autoimmunity require lympho-epithelial cross talk via LTbetaR. J Exp Med, 198: 757–769.
Body, J.J., Facon, T., Coleman, R.E. et al. (2006). A study of the biological receptor activator of nuclear factor-kappaB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res, 12: 1221–1228.
Bone, H.G., Bolognese, M.A., Yuen, C.K. et al. (2008). Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab, 93: 2149–2157.
Brandt, J., Haibel, H., Cornely, D. et al. (2000). Successful treatment of active ankylosing spondylitis with the anti-tumor necrosis factor alpha monoclonal antibody infliximab. Arthritis Rheum, 43: 1346–1352.
Campagnuolo, G., Bolon, B., & Feige, U. (2002). Kinetics of bone protection by recombinant osteoprotegerin therapy in Lewis rats with adjuvant arthritis. Arthritis Rheum, 46: 1926–1936; Bolon, B., Campagnuolo, G., & Feige, U. (2002). Duration of bone protection by a single osteoprotegerin injection in rats with adjuvant-induced arthritis. Cell Mol Life Sci, 59: 1569–1576.
Chin, R.K., Lo, J.C., Kim, O. et al. (2003). Lymphotoxin pathway directs thymic Aire expression. Nat Immunol, 4: 1121–1127.
Cohen, S.B., Dore, R.K., Lane, N.E. et al. (2008). Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum, 58: 1299–1309.
De Togni, P., Goellner, J., Ruddle, N.H. et al. (1994). Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science, 264: 703–707.
Dong, C. (2006). Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol, 6: 329–333.
Dougall, W.C., & Chaisson, M. (2006). The RANK/RANKL/OPG triad in cancer-induced bone diseases. Cancer Metastasis Rev, 25: 541–549.
Dougall, W.C., Glaccum, M., Charrier, K. et al. (1999). RANK is essential for osteoclast and lymph node development. Genes Dev, 13: 2412–2424.
Ebeling, P.R., Erbas, B., Hopper, J.L. et al. (1998). Bone mineral density and bone turnover in asthmatics treated with long-term inhaled or oral glucocorticoids. J Bone Miner Res, 13: 1283–1289.
Emery, J.G., McDonnell, P., Burke, M.B. et al. (1998). Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem, 273: 14363–14367.
Fata, J.E., Kong, Y.Y., Li, J. et al. (2000). The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell, 103: 41–50.
Feldmann, M., Brennan, F.M., & Maini, R.N. (1996). Rheumatoid arthritis. Cell, 85: 307–310.
Feldmann, M., Brennan, F.M., & Maini, R.N. (1996). Role of cytokines in rheumatoid arthritis. Annu Rev Immunol, 14: 397–440.
Futterer, A., Mink, K., Luz, A. et al. (1998). The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity, 9: 59–70.
Fu, Y.X., Chaplin, D.D. (1999). Development and maturation of secondary lymphoid tissues. Annu Rev Immunol, 17: 399–433.
Green, E.A., Choi, Y., & Flavell, R.A. (2002). Pancreatic lymph node-derived CD4(+)CD25(+) Treg cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity, 16: 183–191.
Green, E.A., & Flavell, R.A. (2000). The temporal importance of TNFalpha expression in the development of diabetes. Immunity, 12: 459–469.
Harrington, L.E., Hatton, R.D., Mangan, P.R. et al. (2005). Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol, 6: 1123–1132.
Hikosaka, Y., Nitta, T., Ohigashi, I. et al. (2008). The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity, 29: 438–450.
Hofbauer, L.C., Khosla, S., Dunstan, C.R. et al. (1999). Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology, 140: 4367–4370.
Horwood, N.J., Elliott, J., Martin, T.J. et al. (2001). IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J Immunol, 166: 4915–4921.
Hsu, H., Lacey, D.L., Dunstan, C.R. et al. (1999). Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A, 96: 3540–3545.
Ingulli, E., Mondino, A., Khoruts, A. et al. (1997). In vivo detection of dendritic cell antigen presentation to CD4(+) T cells. J Exp Med, 185: 2133–2141
Irla, M., Hugues, S., Gill, J. et al. (2008). Autoantigen-specific interactions with CD4+ thymocytes control mature medullary thymic epithelial cell cellularity. Immunity, 29: 451–463.
Josien, R., Li, H.L., Ingulli, E. et al. (2000). TRANCE, a tumor necrosis factor family member, enhances the longevity and adjuvant properties of dendritic cells in vivo. J Exp Med, 191: 495–502.
Josien, R., Wong, B.R., Li, H.L. et al. (1999). TRANCE, a TNF family member, is differentially expressed on T cell subsets and induces cytokine production in dendritic cells. J Immunol, 162: 2562–2568.
Kanematsu, M., Sato, T., Takai, H., et al. (2000). Prostaglandin E2 induces expression of receptor activator of nuclear factor-kappa B ligand/osteoprotegrin ligand on pre-B cells: implications for accelerated osteoclastogenesis in estrogen deficiency. J Bone Miner Res, 15: 1321–1329.
Keffer, J., Probert, L., Cazlaris, H. et al (1991). Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. Embo J, 10: 4025–4031,
Kim, D., Mebius, R.E., MacMicking, J.D. et al. (2000). Regulation of peripheral lymph node genesis by the tumor necrosis factor family member TRANCE. J Exp Med, 192: 1467–1478.
Kong, Y.Y., Feige, U., Sarosi, I. et al. (1999). Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature, 42: 304–309.
Kong, Y.Y., Yoshida, H., Sarosi, I. et al. (1999). OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature, 397: 315–323.
Koni, P.A., Sacca, R., Lawton, P. et al. (1997). Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity, 6: 491–500.
Korganow, A.S., Ji, H., Mangialaio, S. et al. (1999). From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity, 10: 451–461.
Kotake, S., Udagawa, N., Hakoda, M. et al. (2001). Activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum, 44: 1003–1012.
Kotake, S., Udagawa, N., Takahashi, N. et al. (1999). IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest, 103: 1345–1352.
Kouskoff, V., Korganow, A.S., Duchatelle, V., &et al. (1996). Organ-specific disease provoked by systemic autoimmunity. Cell, 87: 811–822.
Kyewski, B., & Klein, L. (2006). A central role for central tolerance. Annu Rev Immunol, 24: 571–606.
Lacey, D.L., Timms, E., Tan, H.L. et al. (1998). Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell, 93: 165–176.
Li, J., Sarosi, I., Yan, X.Q. et al. (2000). RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A, 97: 1566–1571.
Lipton, A. (2006). Future treatment of bone metastases. Clin Cancer Res, 12: 6305s–6308s.
Lipton, A., Steger, G.G., Figueroa, J. et al. (2007). Randomized active-controlled phase II study of denosumab efficacy and safety in patients with breast cancer-related bone metastases. J Clin Oncol, 25: 4431–4437.
Loser, K., Mehling, A., Loeser, S. et al. (2006). Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat Med, 12: 1372–1379.
Mahamed, D.A., Marleau, A., Alnaeeli, M. et al. (2005). G(-) anaerobes-reactive CD4+ T-cells trigger RANKL-mediated enhanced alveolar bone loss in diabetic NOD mice. Diabetes, 54: 1477–1486.
Manoury-Schwartz, B., Chiocchia, G., Bessis, N. et al. (1997). High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J Immunol, 158: 5501–5506.
Matsumoto, M., Mariathasan, S., Nahm, M.H. et al. (1996). Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science, 271: 1289–1291.
Mebius, R.E. (2003). Organogenesis of lymphoid tissues. Nat Rev Immunol, 3: 292–303.
Mebius, R.E., Rennert, P., & Weissman, I.L. (1997). Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity, 7: 493–504.
Mehling, A., Loser, K., Varga, G. et al. (2001). Overexpression of CD40 ligand in murine epidermis results in chronic skin inflammation and systemic autoimmunity. J Exp Med, 194: 615–628.
McClung, M.R., Lewiecki, E.M., Cohen, S.B. et al. (2006). Denosumab in postmenopausal women with low bone mineral density. N Engl J Med, 354: 821–831.
Mori, H., Kitazawa, R., Mizuki, S. et al. (2002). RANK ligand, RANK, and OPG expression in type II collagen-induced arthritis mouse. Histochem Cell Biol, 117: 283–292.
Muller-Ladner, U., Gay, R.E., & Gay, S. (1998). Molecular biology of cartilage and bone destruction. Curr Opin Rheumatol, 10: 212–219.
Nakashima, T., Wada, T., & Penninger, J.M. (2003). RANKL and RANK as novel therapeutic targets for arthritis. Curr Opin Rheumatol, 15: 280–287.
Oliveri, M.B., Mautalen, C.A., Rodriguez Fuchs, C.A. et al. (1991). Vertebral compression fractures at the onset of acute lymphoblastic leukemia in a child. Henry Ford Hosp Med J, 39: 45–48.
Oxenius, A., Campbell, K.A., Maliszewski, C.R. et al. (1996). CD40-CD40 ligand interactions are critical in T-B cooperation but not for other anti-viral CD4+ T cell functions. J Exp Med, 183: 2209–2218.
Oliveri, MB., Mautalen, C.A., Rodriguez Fuchs, C.A. et al. (1991). Vertebral compression fractures at the onset of acute lymphoblastic leukemia in a child. Henry Ford Hosp Med J, 39: 45–48.
Oxenius, A., Campbell, K.A., Maliszewski, C.R. et al. (1996). CD40-CD40 ligand interactions are critical in T-B cooperation but not for other anti-viral CD4+ T cell functions. J Exp Med, 183: 2209–2218.
Panayi, G.S., Lanchbury, J.S., & Kingsley, G.H. (1992). The importance of the T cell in initiating and maintaining the chronic synovitis of rheumatoid arthritis. Arthritis Rheum, 35: 729–735.
Park, H., Li, Z., Yang, X.O. et al. (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol, 6: 1133–1141.
Pettit, A.R., Ji, H., von Stechow, D. et al. (2001). TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol, 159: 1689–1699.
Piepkorn, B., Kann, P., Forst, T. et al. (1997). Bone mineral density and bone metabolism in diabetes mellitus. Horm Metab Res, 29: 584–591.
Quezada, S.A., Jarvinen, L.Z., Lind, E.F. et al. (2004). CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol, 22: 307–328.
Redlich, K., Hayer, S., Maier, A. et al. (2002). Tumor necrosis factor alpha-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum, 46: 785–792.
Rennert, P.D., Browning, J.L., & Hochman, P.S. (1997). Selective disruption of lymphotoxin ligands reveals a novel set of mucosal lymph nodes and unique effects on lymph node cellular organization. Int Immunol, 9: 1627–1639.
Rennert, P.D., James, D., Mackay, F. et al. (1998). Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity, 9: 71–79.
Romas, E., Sims, N.A., Hards, D.K. et al. (2002). Osteoprotegerin reduces osteoclast numbers and prevents bone erosion in collagen-induced arthritis. Am J Pathol, 161: 1419–1427.
Rossi, S.W., Jenkinson, W.E., Anderson, G. et al. (2006). Clonal analysis reveals a common progenitor for thymic cortical and medullary epithelium. Nature, 441: 988–991.
Rossi, S.W., Kim, M.Y., Leibbrandt, A. et al. (2007). RANK signals from CD4(+)3(-) inducer cells regulate development of Aire-expressing epithelial cells in the thymic medulla. J Exp Med, 204: 1267–1272.
Roy, M., Waldschmidt, T., Aruffo, A. et al. (1993). The regulation of the expression of gp39, the CD40 ligand, on normal and cloned CD4+ T cells. J Immunol, 151: 2497–2510.
Sakaguchi, S. (2005). Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol, 6: 345–352.
Sato, K., Suematsu, A., Okamoto, K. et al. (2006). Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med, 203: 2673–2682.
Seitz, M., & Hunstein, W. (1985). Enhanced prostanoid release from monocytes of patients with rheumatoid arthritis and active systemic lupus erythematosus. Ann Rheum Dis, 44: 438–445.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Science+Business Media, LLC
About this paper
Cite this paper
Leibbrandt, A., Penninger, J.M. (2009). Novel Functions of RANK(L) Signaling in the Immune System. In: Choi, Y. (eds) Osteoimmunology. Advances in Experimental Medicine and Biology, vol 658. Springer, Boston, MA. https://doi.org/10.1007/978-1-4419-1050-9_9
Download citation
DOI: https://doi.org/10.1007/978-1-4419-1050-9_9
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4419-1049-3
Online ISBN: 978-1-4419-1050-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)