
Abstract
Wernicke’s encephalopathy (WE) is a potentially reversible metabolic brain dysfunction resulting from thiamine deficiency. It is generally characterized by ataxia, ophthalmoplegia and global confusion. Described in Berlin in 1881 by Carl Wernicke, it was initially known as polioencephalitis hemorrhagica superioris and considered a fatal syndrome. The first reported cases were three patients, two with alcoholism and one with persistent vomiting after the ingestion of sulfuric acid in a suicide attempt. The common feature shared by these cases upon post-mortem exam consisted of punctate hemorrhages in the grey matter of the walls of the third and fourth ventricles and mammillary bodies (Cirignotta et al., 2000; Truswell, 2000). In 1935, Strauss discovered that the cause of Wernicke’s findings was vitamin B1 (thiamine) deficiency (Chiossi et al., 2006). Bonhoeffer posited that Wernicke’s encephalopathy and the psychosis described by Korsakoff actually represented two phases of the same pathological process (Cirignotta et al., 2000). The observation that Wernicke’s encephalopathy and Korsakoff’s psychosis have identical neuropathology supported this belief (Charness, 1999). Thus Wernicke’s encephalopathy (WE) and Korsakoff’s psychosis (KP) are often used interchangeably as the Wernicke-Korsakoff Syndrome (WKS).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Vasogenic Edema
- Apparent Diffusion Coefficient Mapping
- Thiamine Deficiency
- Alcoholic Patient
- Hyperemesis Gravidarum
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Wernicke’s encephalopathy (WE) is a potentially reversible metabolic brain dysfunction resulting from thiamine deficiency. It is generally characterized by ataxia, ophthalmoplegia and global confusion. Described in Berlin in 1881 by Carl Wernicke, it was initially known as polioencephalitis hemorrhagica superioris and considered a fatal syndrome. The first reported cases were three patients, two with alcoholism and one with persistent vomiting after the ingestion of sulfuric acid in a suicide attempt. The common feature shared by these cases upon post-mortem exam consisted of punctate hemorrhages in the grey matter of the walls of the third and fourth ventricles and mammillary bodies (Cirignotta et al., 2000; Truswell, 2000). In 1935, Strauss discovered that the cause of Wernicke’s findings was vitamin B1 (thiamine) deficiency (Chiossi et al., 2006). Bonhoeffer posited that Wernicke’s encephalopathy and the psychosis described by Korsakoff actually represented two phases of the same pathological process (Cirignotta et al., 2000). The observation that Wernicke’s encephalopathy and Korsakoff’s psychosis have identical neuropathology supported this belief (Charness, 1999). Thus Wernicke’s encephalopathy (WE) and Korsakoff’s psychosis (KP) are often used interchangeably as the Wernicke-Korsakoff Syndrome (WKS).
Korsakoff psychosis is a chronic amnestic state, characterized by retrograde amnesia (loss of memory), anterograde amnesia (defective learning) and confabulation. Loss of short-term memory is a predominant feature, while immediate and long-term memories are usually intact. (Harrison et al., 2006; Chiossi et al., 2005; Sivolap, 2005). Patients with KP have difficulty remembering events and facts that occur after the onset of disease (Hochhalter and Joseph, 2001). It occurs most commonly in patients with WE, usually secondary to alcoholism, but may be seen in patients without a previous diagnosis of WE (Truswell, 2000). It has been suggested that some of these patients may have had subacute cases of WE, where signs and symptoms were either mild or absent. Nevertheless, on autopsy, patients with KP have the same brain pathology as patients with WE, as mentioned earlier (Charness, 2006).
Pathologically, WE may be classified into acute (17%), subacute (17%) and chronic (66%) states. It has been postulated that WE may be a progressive disorder, where multiple acute and/or subclinical episodes of thiamine deficiency cause cumulative damage. These subclinical events may be devoid of the traditional symptoms associated with WE (Gui et al., 2006; Harper, 1983). This may explain the findings of brain lesions at postmortem without antecedent symptoms in some patients. Analogously, Korsakoff’s psychosis may occur in patients who have experienced multiple acute and/or subclinical events of WE. This may explain the discrepancy between the relatively high numbers of alcoholic patients who develop Korsakoff’s psychosis as compared to nonalcoholic patients. There are at least two hypotheses to explain this observation. The first hypothesis presumes that the latter group’s exposure to a thiamine-deficient state was a single event, while the former group is likely to have had long-standing thiamine deficiency and/or multiple events of WE, increasing their chances of developing KP (Homewood and Bond, 1999). The second hypothesis is that perhaps ethanol neurotoxicity and thiamine deficiency work in concert in the development of KP (Charness, 2006).
Victor et al. (1989), in classic studies, followed 186 alcoholic patients with Wernicke’s encephalopathy for up to ten years and documented that 84% developed Korsakoff’s syndrome. A subsequent study of 32 alcoholic patients followed for a period of 33 months showed the rate of progression to KP to be 56% (Wood et al., 1984). Full recovery from Korsakoff’s psychosis occured in only 20%; the majority of KP patients required some level of supervision and social support (Reuler et al., 1985; Charness, 2006).
Wernicke’s encephalopathy is associated with a mortality rate of 10–20%, predominantly as a result of sepsis, respiratory infection and decompensated liver disease; Korsakoff’s psychosis is associated with a mortality rate of approximately 17% (Harrison et al., 2006; Ogershok et al., 2002; Merkin-Zaborsky et al., 2001).
Prevalence
The prevalence rates of 0.8% –2.8% for WE come mainly from four autopsy-based studies: Norway (0.8%), New York (1.7%), Cleveland (2.2%) and Australia (2.8%) (Harper, 1983; Ogershok et al., 2002). Australia’s higher prevalence of WE was puzzling, as the country is not ranked high on the world league table of alcohol consumption. After conducting interviews with patients in alcohol rehabilitation units, Price concluded that Australian alcoholics were more likely to lack female social support, which may otherwise provide them with food containing thiamine (Truswell, 2000). Seventy-five percent of patients are male and the peak age incidence is in the sixth decade (41%) (Harper, 1983). Wernicke’s encephalopathy remains a profoundly under-diagnosed disease, which if left untreated can progress to Korsakoff syndrome or death, as a result of irreversible cytotoxic effects (Loh et al., 2004; Weidauer et al., 2004). Harper found that only 20% of cases reviewed in a necropsy study had been diagnosed with WE or Wernicke-Korsakoff syndrome prior to death (Harper, 1983). Thus a high index of suspicion is the key to diagnosis.
Clinical Features
The classic triad of symptoms in WE includes ophthalmoplegia, ataxia and global confusion (see Table 14.1). However, the clinical presentation is often incomplete (Foster et al., 2005). In a retrospective study, it was found that only 16.5% of patients exhibited all three signs and 19% exhibited none of these (Harper et al., 1986). One study found nystagmus to be present in 85%, bilateral paralysis of the lateral rectus muscles in 54% and conjugate gaze palsies in 45% of cases (Ogershok et al., 2002)). Other reported symptoms include apathy, lightheadedness, disorientation, poor memory, diplopia, inability to stand, nausea, vomiting and coma (Liu et al., 2006; Giglioli et al., 2004; Morcos et al., 2004; Harper et al., 1986; Ogershok et al., 2002). In addition, WE can affect the sympathetic system, resulting in postural hypotension and syncope, and the temperature-regulating center, resulting in mild hypothermia (Worden, 1984; Reuler et al., 1985). The lag time from onset of thiamine-deficiency to the start of symptoms is approximately 4–6 weeks (Harrison et al., 2006). Symptoms may range from a period of 2 days to 2 weeks before presentation for evaluation (Giglioli et al., 2004; Lacasse and Lum, 2004).
Wernicke’s encephalopathy is most commonly associated with alcoholism. It has been suggested that there may be a synergistic effect of alcoholism and thiamine deficiency, where a brain affected by alcoholism may be more susceptible to injury caused by thiamine deficiency (Homewood and Bond, 1999). However, WE may be found in any clinical state associated with malnutrition or thiamine deficiency (see Table 14. 2), including hyperemesis gravidarum (Chiossi et al., 2006), anorexia nervosa (Morcos et al., 2004), prolonged parenteral feeding without micronutrient supplementation (Attard et al., 2006), renal disease with hemodialysis or peritoneal dialysis and gastric or bariatric surgery (Attard et al., 2006; Worden and Allen, 2006; Loh et al., 2004; Cirignotta et al., 2000; Ogershok et al., 2002). Although studies have found that 23–50% of cases are actually not associated with alcohol abuse, the index of suspicion for thiamine deficiency is still low in non-alcoholic patients (Ogershok et al., 2002).
The incidence of thiamine deficiency in alcoholics is 30–80% (Homewood and Bond, 1999). Factors that promote thiamine deficiency in alcoholics include poor thiamine intake, decreased activation of thiamine to thiamine pyrophosphate(TPP), decreased hepatic storage, decreased intestinal thiamine transport and impairment of thiamine absorption (see Table 14.3) (Breen et al, 1985; Hoyumpa, 1980). Although thiamine is stored in various sites, including skeletal muscles, heart, kidneys and brain, the liver remains the main storage site. Due to the reasons cited above, hepatic thiamine content may be reduced by 73% in patients with severe, chronic alcoholic liver disease. In addition, ethanol has been shown to promote thiamine release from the liver (Hoyumpa, 1980).
Chiossi et al. (2006) reviewed 49 case reports of WE due to hyperemesis gravidarum. The duration of vomiting and/or poor intake was 7.7 +/ − 2.8 weeks. The mean gestational age was 14.3 +/ − 3.4 weeks and the amount of weight loss ranged from 6–25 kg. Thirty-two percent of these patients were primigravida (Chiossi et al., 2006). In laboratory rats, thiamine deficiency is a known cause of intrauterine growth retardation. In a German study, lower erythrocyte thiamine concentrations were found in patients whose pregnancies were complicated by intrauterine growth retardation than in patients with normal pregnancies (Heinze and Weber, 1990), although a causal relationship is uncertain.
Chronic renal failure patients on hemodialysis and peritoneal dialysis are at risk for thiamine deficiency due to inadequate nutrition in part and possible thiamine loss during the dialysis process. Renal failure patients are often on a diet restricted in protein and potassium, which increases the risk of thiamine deficiency (Masud, 2002; Piccoli et al., 2006). Studies with detailed dietary surveys have shown poor oral intake of thiamine in chronic renal failure patients (Hung et al., 2001). There is no convincing evidence that thiamine levels are significantly altered by either hemodialysis or peritoneal dialysis (Reuler et al., 1985). DeBari et al. (1984) measured thiamine levels of granulocytes, erythrocytes and plasma. They found no significant differences in thiamine levels in dialysis patients compared to controls. Further research in this area would benefit chronic renal failure patients and help determine possible need for supplementation of water-soluble vitamins.
Due to the obesity epidemic in the United States, bariatric surgery is becoming increasingly more common. As a result, nutrient malabsorption is becoming more common as well. A common procedure, Roux-en-y gastric bypass (RYGBP), causes both food restriction and malabsorption. Malabsorption is caused by the bypass of the distal stomach, duodenum and the first part of the jejunum. Vomiting is a common side effect of RYGBP and vitamin B12 and iron deficiency are frequently seen. Thiamine deficiency may occur in this setting as a result of bypass of the duodenum, as this is where thiamine is predominantly absorbed (Worden and Allen, 2006). Some authors routinely advocate starting parenteral thiamine administration six weeks postoperatively in malnourished patients (Loh et al., 2004). Prolonged parenteral feeding without thiamine supplementation is a well-documented cause of Wernicke’s encephalopathy.
Administration of intravenous glucose activates glycolysis, a process which utilizes thiamine and may enhance thiamine deficiency (Koguchi et al., 2004). Thus common emergency room practice includes the administration of intravenous thiamine before intravenous glucose in order to prevent the precipitation of WE. Whether or not this practice is warranted is somewhat controversial. In a review of 49 published cases of hyperemesis gravidarum as the leading cause of WE, Chiossi et al. felt that approximately 30% of the cases were provoked by intravenous glucose administration without thiamine (Chiossi et al., 2006). Many other case reports have also noted such a phenomenon but good evidence-based studies are lacking. Harrison notes this to be a “theoretical concern. ” (Harrison et al., 2006). Pazirandeh et al. (2006) point out that cellular thiamine uptake is actually slower than glucose uptake. As such, cellular thiamine repletion may not occur prior to cellular glucose exposure.
Wernicke’s encephalopathy may appear inconspicuously in psychiatric patients, as it may be obscured by mental illness. Patients with schizophrenia, for example, may be particularly at risk due to poor dietary intake, high rates of homelessness and high prevalence of alcoholism (Harrison et al., 2006).
Infantile WE may be found in developing countries, primarily among breast-fed infants, usually in the second to fifth months of development. Wernicke’s encephalopathy is very rare in developed nations. However, in 2003, Israel was faced with an epidemic of WE due to a batch of defective soy-based vegetarian infant formula. WE was documented in 20 out of an estimated 3500 infants who were fed the formula, later found to be deficient in thiamine (Kesler et al., 2005).
Diagnosis
Although the diagnosis of WE is generally considered to be a clinical one, supporting laboratory tests and neuroimaging data may be important. Generally, routine laboratory tests, such as liver profile and renal function, urinalysis, chest x-rays, electrocardiograms and echocardiograms are normal, as are cerebrospinal fluid tests. Serum lactic acid, however, has been shown to be elevated in the setting of thiamine deficiency, particularly in children (Liu et al., 2006; Attard et al., 2006; Weidauer et al., 2004). In one case study, an electromyelogram showed diffuse sensorimotor neuropathy (Cirignotta et al., 2000); in another case, an electroencephalography revealed diffuse slow activity or dysrhythmia (Chiossi et al., 2006).
Serum thiamine levels may be misleading and thus should not be employed for the diagnosis of Wernicke’s encephalopathy. There are two laboratory tests which are used as surrogates for body thiamine stores. The erythrocyte transketolase test (ETKA) is a reflection of thiamine reserves at a cellular level. The thiamine pyrophosphate effect (TPPE) test, expressed as a coefficient, is a measure of transketolase activity before and after the addition of thiamine. Values before added thiamine reflect the amount of coenzyme present in the cell; the values measured after thiamine addition is a reflection of the amount of apoenzyme present that lacks a coenzyme. Diagnosis is made by either a low ETKA and/or a high TPPE. Normal TPPE values range from 0% –14%. TPPE values between 15% and 24% signify marginal thiamine deficiency and values greater than 25% signify severe deficiency (Kesler et al., 2005; Chiossi et al., 2006). Unfortunately, ETKA and TPPE are not readily commercially available in the United States. Thus patients should be treated once suspected clinically of WE. Clinical response to treatment is the ultimate persuasive diagnostic test.
By performing thorough clinical histories, neurological and psychological exams, as well as pathological evaluations, Caine et al. (1997) developed a set of diagnostic criteria for WE in alcoholic patients. The diagnostic criteria, published in 1997, require two of the following four signs for the diagnosis of WE: oculomotor abnormalities, malnutrition, cerebellar dysfunction and either mild memory impairment or altered mental status (see Table 14.4). Validity testing of this approach demonstrated improved diagnostic sensitivity from 31% (using the classic triad) to about 100%. They report that sensitivity decreased to 50% in patients with concurrent hepatic encephalopathy. This is not unexpected, as two of the elements in the above criteria, malnutrition and altered mental status, are commonly seen in patients with hepatic encephalopathy. Proper management of these conditions (i.e. serum ammonia levels and response to appropriate therapy) should elucidate the proper diagnosis. The Caine criteria were developed based on studies of patients with alcoholism and should not be applied, at present, to a general population. Future studies should assess these criteria in a nonalcoholic population.
Neuroimaging
Computed tomography (CT) and magnetic resonance imaging (MRI), and single-photon emission computed tomography (SPECT) have been studied with regard to their evaluation of WE. CT appears to be helpful only in cases with hemorrhagic lesions, which include only approximately 5% of cases (Chiossi et al., 2006; Mascalchi et al., 1999). Computed tomography was reported by Antunez et al. (1998) to have quite a low sensitivity (13%) in the diagnosis of WE.
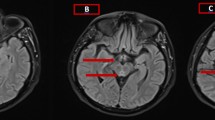
MRI sequences typically include, pre- and post-contrast (gadolinium) T1-weighted images with gadolinium, T2-weighted images, fluid-attenuated inversion recovery (FLAIR), diffusion-weighted imaging (DWI), and apparent diffusion coefficient (ADC) mapping. MRI has a sensitivity and specificity of 53% and 93%, respectively, for WE (Ogershok et al., 2002). The sensitivity may actually be higher, but MRI is often performed after the patient has been suspected of WE and empirically treated, resulting in a non-diagnostic study (Celik and Kaya, 2004). Good correlation has been found between contrast mediated magnetic resonance imaging (MRI) and neuropathological findings (Liu et al., 2006). Classically, T2-weighted and FLAIR MRI images reveal symmetrical increased signal intensity of areas including the third paraventricular regions of the thalamus and hypothalamus, periaqueductal regions of the midbrain and mammillary bodies. These lesions may sometimes be enhanced with gadolinium on T1-weighted images during an acute event and may dissipate with treatment (Chiossi et al, 2006; Mascalchi et al., 1999; Doherty et al., 2002; Halavaara et al., 2003). In one case, mammillary body enhancement was the only sign of acute WE (Shogry and Curnes, 1994). FLAIR sequences are reviewed to ensure that the cerebrospinal fluid has not masked high signal lesions on T2-weighted imaging (Chung et al., 2003). The chronic stage of WE may be depicted by brain atrophy and diffuse signal-intensity changes in the cerebral white matter (White et al., 2005).
Some of the lesion identified on MRI may be seen in other conditions, such as inferolateral and anterolateral thalamic infarcts, multiple sclerosis, Cytomegalovirus encephalitis, Behcet’s disease, primary cerebral lymphoma, central pontine myelinosis, Lyme disease, Leigh’s disease and variant Creutzfeldt-Jokob disease. These conditions are usually excluded from the differential diagnosis based both on their asymmetric distribution and the clinical setting (Weidauer et al., 2004; Chung et al., 2003). Neuroimaging may be a very useful tool in the diagnosis of WE; however, it is important to note that the absence of signs on neuroimaging does not exclude the diagnosis (Antunez et al., 1998; Celik and Kaya, 2004).
Although there is insufficient evidence to suggest the presence of cytotoxic edema in acute WE, there is good evidence for vasogenic edema (Liu et al., 2006). The advantage of DWI over T2-weighted and FLAIR imaging is its ability to better distinguish between cytotoxic and vasogenic edema (Chiossi et al., 2006; Chung et al., 2003). DWI, in conjunction with ADC mapping, is particularly useful, as it is the most sensitive method for diagnosing early injury, i.e. vasogenic edema, before the onset of necrosis, thus facilitating early diagnosis of WE (Doherty et al., 2002; Halavaara et al., 2003). Cytotoxic edema is represented by high intensity signal on DWI with corresponding low signal on ADC mapping. Vasogenic edema, however, will show high signal intensity on both DWI and ADC mapping (Weidauer et al., 2004).
Single-photon emission computed tomography (SPECT) imaging has also been evaluated and at least one study has found it to be useful in cases where conventional MRI may be non-diagnostic (Celik and Kaya, 2004).
Pathology
Wernicke’s encephalopathy, which affects the brainstem, white matter and cortex, has a characteristic appearance on autopsy (Celik and Kaya, 2004). The acute stage of WE is characterized by the inability to maintain proper osmotic gradients of cell membranes, promoting intracellular swelling and red blood cell extravasation into the perivascular space. This stage is distinguished by marked vascular dilatation, endothelial swelling and neuronal demyelinization. The chronic stage is marked by mammillary body atrophy, as well as, loss of neuropil with fibrillary astrocytosis (Weidauer et al., 2004; D’Aprile et al., 2000; Homewood and Bond, 1999). Neurons, however, are generally spared (Liu et al., 2006; Gui et al., 2006; Halavaara et al., 2003). Classic neuropathological findings include petechial hemorrhages of blood vessels and small, symmetric necrotic lesions in the paraventricular areas of the thalamus, hypothalamus, mammillary bodies, periaqueductal area of the midbrain and cerebellum (McEntee, 1997; Chung et al., 2003; Caine et al., 1997). It has been proposed that perhaps the paraventricular regions are more susceptible to thiamine deficiency because they have a higher rate of glucose and oxidative metabolism which require thiamine (Lacasse and Lum, 2004). The most consistent pathological findings, found in 75% of the cases, are mammillary body atrophy and brownish discoloration (Liu et al., 2006; Harper, 1983). Although a small proportion of patients may have normal sized mammillary bodies, almost all have microscopic mammillary body lesions (Charness, 1999). Macrohemorrhage is found in approximately 5% of cases (Mascalchi et al., 1999). Table 14.5 shows Harper’s macroscopic and microscopic autopsy findings in WE (Harper, 1983).
Role of Thiamine
Thiamine (B1) is an essential coenzyme for enzymes involved in Kreb’s cycle (including pyruvate dehydrogenase and alpha ketoglutarate dehydrogenase), lipid metabolism/amino acid production (transketolase) and neurotransmitter synthesis—acetylcholine and GABA (2-oxo-glutarate dehydrogenase) (Chiossi et al., 2006). A commonly found water-soluble vitamin, it is found in lean pork, poultry, fish, eggs, liver, wheat germ, whole grains, beans, peas and nuts. Fruits, vegetable and dairy products are not good sources of thiamine (Table 14.6) Alcoholic beverages have virtually no thiamine (Table 14.7) (Lonsdale, 2006). However, it has been suggested that small amounts of thiamine exist in German and Australian beer (Price and Kerr, 1985). The human daily requirement of thiamine is 1.0–1.5 mg per day but this requirement is increased in states of pregnancy, lactation, thyrotoxicosis and fever. Body stores are approximately 25–30 mg and are found predominantly in skeletal muscles, heart, liver, kidneys and brain (Lacasse and Lum, 2004). These reserves are sufficient for only 2–3 weeks without continued intake (Gui et al., 2006; Kesler et al., 2005). Thiamine, which is excreted in the urine, has a half-life of approximately 10–20 days (Pazirandeh et al., 2006). Loss of thiamine is accelerated by diuretic therapy and may be inactivated by polyphenol containing compounds found in coffee and tea (Lonsdale, 2006). Malabsorption may occur with alcoholism, with gastric surgery and with folate deficiency (Lacasse and Lum, 2004; Price and Kerr, 1985). Alcohol has been found to interfere with the active transport of thiamine in the gastrointestinal system, at least in rodents (Kumar et al., 2000). Thiamine absorption may be significantly decreased in the setting of folate depletion but may return to normal with 4–6 weeks of folate repletion therapy. A deficiency in magnesium, required for the conversion of thiamine to thiamine pyrophosphate, may also cause thiamine deficiency (Bishai and Bozzetti, 1986; Lonsdale, 2006).
Medications postulated to affect body stores of thiamine include: 5-fluorouracil (Heier and Dornish, 1989), loop diuretics (Brady et al., 1995; Seligmann et al., 1991) and dilantin (Patrini et al., 1993; Botez et al., 1993).
Thiamine Absorption
Dietary thiamine exists primarily in the form of thiamine pyrophosphate (TPP), which must be hydrolyzed to free thiamine, before absorption in the small bowel (Dudeja et al., 2001). In the small bowel, thiamine absorption occurs by two processes: passive and active transport. Passive transport occurs only in the presence of high thiamine concentrations, and actually blocks the active transport process. Low doses of thiamine are absorbed by active transport (Rindi and Ventura, 1972). The details of this absorption mechanism are still not completely clear. Dudeja et al. have performed multiple studies evaluating jejunal thiamine absorption at both the brush border membrane (BBM) and the basolateral membrane (BLM). In one study, they found that human intestinal BBM absorption of thiamine is a carrier-mediated process, which is sodium-independent, pH-dependent and amiloride-sensitive. They have also proposed the possibility of a thiamine—/H+ exchange mechanism (Dudeja et al., 2001). In a study of thiamine absorption in jejunal BLM, Dudeja et al. found the transport mechanism to be a pH-dependent and amiloride-sensitive carrier-mediated process (Dudeja et al., 2003). SCL19A2, believed to be a human thiamine transporter, has been shown to be expressed in all gastrointestinal tissues, with the greatest level of expression found in the liver. Reidling et al. have discovered that the minimal promoter region needed for basal activity of SLC19A2 gene is encoded between −356 and −36 (Reidling et al., 2002).
Breen et al. evaluated the influence of acute alcohol perfusion on small bowel absorption of thiamine. They found that alcohol did not significantly decrease thiamine uptake in the jejunum, although there was a trend to lower absorption with alcohol perfusion (Breen et al., 1985). Holzbach evaluated thiamine absorption in patients after 3 days and after 4 weeks of resolution of acute delirium tremens (DT). He found no significant difference in thiamine absorption between normal patients and those with recent delirium tremens. There was, however, a significant increase in thiamine absorption 4 weeks after DT as compared to values obtained shortly (3 days) after DT. They propose the possibility of abnormal thiamine absorption in DT. It is noteworthy that the patients with visual hallucinations had lower thiamine absorption levels than those who did not have this symptom (Holzbach, 1996). Tomasulo studied thiamine deficiency in severely alcoholic patients admitted to the hospital. Forty-three percent of these patients had DT. He measured radioactive thiamine in both urine and stool and found significant differences between controls and alcoholics. The labeled thiamine excreted in 24-hour urine collections of controls and alcoholics were 45.8% and 25.3%, respectively. The reciprocal findings of stool in controls and alcoholics were 4.0% and 21.0%, respectively (Tomasulo et al., 1968). Studies by Thomson et al. (1968) also provide evidence that chronic alcohol abuse may decrease thiamine absorption. These various studies differ, not only in their methodologies, but also in their subject populations. Breen’s study assessed the effects of acute alcohol while the latter three studied chronic alcoholics. This area clearly deserves further research.
Pathogenesis of Wernicke’s Encephalopathy
Serious attempts to determine the mechanism(s) of this disorder have been ongoing for at least 70 years (Peters, 1969). There are many aspects of WE which should make this task relatively easy. First, the clinical picture of this disorder is well characterized, can be readily diagnosed and is relatively specific. Second, the pathology of this entity is elegantly described and imaging by MRI, when present, is characteristic. Third, there are animal models readily available which should permit a biochemical/pathologic dissection of the problem. Fourth, the specific deficiency, a decrease of vitamin B1, responsible for the experimental and clinical findings of WE is well-known. Most importantly, the experimentally induced and clinical syndromes are often readily reversible (if seen early) by the administration of thiamine.
With such an extensive knowledge base, what is the present state of our understanding of the mechanisms of this disorder? Not unexpectedly, initial studies, primarily in experimental animal models, focused on the known metabolic pathways which involve thiamine. Indeed, the classical studies of Peters in 1930 (Peters, 1969) showed lactate accumulation in the brainstem of thiamine deficient birds with normalization of this in vitro when thiamine was added to the tissue. This led to the concept of “the biochemical lesion” of the brain in thiamine deficiency. The enzymes which depend on thiamine are shown in Fig. 14.1. They are transketolase, pyruvate and α-ketoglutarate dehydrogenase. Transketolase is involved in the pentose phosphate pathway needed to form nucleic acids and membrane lipids, including myelin. The ketoacid dehydrogenases are key enzymes of the Krebs cycle needed for energy (ATP) synthesis and also to form acetylcholine via Acetyl CoA synthesis. Decrease in activity of this cycle would result in anaerobic metabolism and lead to lactate formation (i.e., tissue acidosis) (Fig. 14.1).

Enzymes which depend on thiamine: there are transketolase, pyruvate and alpha-ketoglutarate dehydrogenase (see text)
Indeed, studies in animal models of thiamine deficiency and a small number of postmortem human brain specimens have shown that transketolase and α-ketoglutarate dehydrogenase (but not consistently pyruvate dehydrogenase) were depressed. Perhaps the largest and earliest fall was seen in brain transketolase; however, when the neurological signs were reversed with thiamine there was no concomitant improvement in transketolase which rose only slightly (McCandless and Schenker, 1968). Moreover, glucose flux through the pentose phosphate pathway (dependent on transketolase) did not decrease in severe thiamine deficiency and ribose-5-phosphate (a key intermediate in the pentose cycle) did not fall (McCandless et al., 1976; McCandless, 1982). Thus, the current view is that a low transketolase is a marker of thiamine deficiency, but is likely not to be causal in the acute neurological deficits seen in thiamine deficiency (McCandless, 1982; Hazell et al., 1998). The possible effects of prolonged transketolase depression, as with chronic thiamine deficit, are uncertain (Hazell et al., 1998).
Data on the role of acetylcholine deficit in thiamine deficiency are conflicting, but most recent studies do not favor a significant decrease in the synthesis of this neurotransmitter (Hazell et al., 1998; Vorhees et al., 1977). This would be consistent with normal pyruvate dehydrogenase activity in experimental thiamine deficiency, which should not, therefore, result in a lower Acetyl CoA level as a precursor to acetylcholine.
Perhaps the most likely mechanism of low thiamine-induced brain injury has revolved around impairment of the Krebs’ cycle and deficit in available ATP (Desjardins and Butterworth, 2005). This could readily lead to apoptosis and necrosis of neurons, as has been described in such patients (Vorhees et al., 1977). In this context, the data on pyruvate dehydrogenase are somewhat difficult to interpret. Postmortem brain from patients with Wernicke’s encephalopathy did show a major decrease in pyruvate dehydrogenase, albeit in only a few specimens (Butterworth et al., 1993). However, this was not corroborated in experimental models of this syndrome (Desjardins and Butterworth, 2005; Butterworth et al., 1993). By contrast a major decrease in brain α-ketoglutarate dehydrogenase was seen in every type of thiamine deficiency (Desjardins and Butterworth, 2005; Butterworth et al., 1993). Moreover, an impairment in this enzyme could readily explain an increase in brain lactate, due to anaerobic metabolism, and this has been observed uniformly, even as far back as 1930 (Peters, 1969; McCandless and Schenker, 1968; Desjardins and Butterworth, 2005). The major concern about the Krebs cycle deficit concept has been a difficulty in documenting consistently an impairment in brain energy stores, both ATP and phosphocreatine (PCr). Multiple studies in the brain of animals with thiamine deficiency have not shown a decrease in ATP or PCr, even when assayed in brain areas (brainstem and lateral vestibular nucleus) felt to be most affected (McCandless and Schenker, 1968; McCandless, 1982; McEntee, 1997; Holowach et al., 1968). The only exception was a study by Aikawa et al. (1984), who showed a small decrease in ATP and PCr in some parts of rat brain after exposure to pyrithiamine (thiamine antagonist). The functional significance of this small (~10%) drop in ATP is not known, but in our view is unlikely to be important. McCandless has shown increased levels of both ATP and PCr in the lateral vestibular nucleus of such animals, as well as in rats rendered thiamine deficient by dietary means (McCandless, 1982; McCandless and Schwartzenburg, 1981). The higher energy stores reverted to normal on restoration of thiamine. These latter data suggest that energy utilization was impaired during the symptomatic stage. Formal energy turnover studies in critically affected brain areas have not been done, to our knowledge. Clearly, the role of an impaired Krebs cycle in the pathogenesis of Wernicke’s encephalopathy is unresolved.
A number of other mechanisms have been suggested recently for the brain damage caused by thiamine lack. One stipulates that an excess of extracellular glutamate induces increased neurotoxicity (McEntee, 1997). The evidence for this are increased concentrations of glutamate in the extracellular fluid (dialysate) in brains of pyrithiamine-treated rats and decrease in glutamate transporters in astrocytes of these animals (Desjardins and Butterworth, 2005; McEntee, 1997). Another concept is that of oxidative stress via the production of reactive oxygen species and/or increased expression of endothelial nitric oxide synthase (Desjardins and Butterworth, 2005). Finally, neuropathological studies in both animal models and postmortem brain sections in patients with Wernicke’s have shown proliferation of astroglial cells, especially in the early stages of thiamine deficiency (Desjardins and Butterworth, 2005). Based on the known protective effects of astrocytes for neurons, this rather suggests that these cells may be activated in that setting, perhaps to provide GSH as an antioxidant (Rathinam et al., 2006). Overall, it appears that the precise mechanism by which thiamine deficiency causes brain injury is unknown. Conceivably multiple factors may be operative.
Another important question relates to the selective sensitivity of specific brain areas to thiamine deficiency. The basis for this has been discussed in terms of differences in regional metabolism, antioxidant status, or differences in thiamine turnover, but without actual data (Desjardins and Butterworth, 2005). Similar regional sensitivity has been seen with bilirubin and copper deposition/damage without explanation. Much remains to be learned.
It has been suggested that there may be a predisposition to WE in some patients, presumably on a genetic basis. Indeed, a variant of transketolase has been reported in fibroblasts of patients with WE (Blass and Gibson, 1977; Nixon et al., 1984). It was proposed that this may increase the requirements of thiamine in such patients, and thus possibly make them more susceptible to thiamine deficits (Martin et al., 1995). However, this concept has not been further verified (Kaufmann et al., 1987; Blansjaar et al., 1991). The authors are unaware of any reported familial clustering of WE, and transketolase is not felt now to be an enzyme primarily causally involved in the pathogenesis of this cerebral disorder. Studies of possible variants in other enzymes involved in thiamine metabolism have not been reported.
Treatment
Wernicke’s encephalopathy is a potentially fatal but also reversible medical emergency if diagnosed and treated in the acute stage. Treatment includes supportive measures, as well as thiamine replacement; however, the basic questions of thiamine dose, frequency, route and length of treatment remain unclear (Morcos et al., 2004). The Cochrane Collaboration (2007) sought to evaluate the evidence available for the use of thiamine in the treatment of Wernicke-Korsakoff Syndrome due to alcohol abuse. There were actually no studies that addressed this specifically but the Cochrane group identified one published randomized controlled study, by Ambrose et al., that compared the effects of various doses of thiamine therapy in alcoholic patients without overt clinical signs of WKS (Day et al., 2004). In 2001, Ambrose evaluated the effects of differing doses of thiamine hydrochloride (5, 20, 50, 100 and 200 milligrams) given intramuscularly for 2 consecutive days in a group of alcoholic patients, none of whom had any clinical signs of WKS, in an alcohol detoxification center. Post-treatment, patients were compared based on their performance on a delayed alternation task test, established to be sensitive to the cognitive impairments of Wernicke-Korsakoff Syndrome. Patients who received the highest dose of thiamine showed superior performance (Ambrose et al., 2001). However, the initial thiamine status of this patient group was not known. As a result of the paucity of data from randomized clinical trials, the Cochrane Collaboration concluded that currently, there is insufficient data available to provide clinical guidelines regarding the dose, frequency, route or duration of thiamine for the treatment of WKS due to alcoholism (Day et al., 2004).
In various studies/clinical cases, thiamine 100 mg has been given intravenously for several days to two weeks, followed by maintenance doses of 50–100 mg orally per day until the patient is able to eat a well-balanced diet regularly (Lacasse and Lum, 2004; Chiossi et al., 2006). Long-term treatment and prevention should include continued oral thiamine supplementation, alcohol abstinence and a balanced diet (Ogershok et al., 2002), but this program is based on logic and overall good medical care, not data.
Following thiamine therapy in the acute state, one may obtain a dramatic response, which essentially confirms the diagnosis (Squirrell, 2004). Improvement in ophthalmoplegia is often the first sign of treatment benefit and may occur within hours (Koontz et al., 2004; Doherty et al., 2002). Ataxia may take days to weeks but 25% of cases may not improve at all. Residual peripheral neuropathy is also not uncommon (Worden and Allen, 2006; Weidauer et al., 2004; Chiossi et al., 2006). Chiossi, in his review of WE cases due to hyperemesis gravidarum, found that only 29% of patients obtained complete resolution of symptoms, while 53% showed resolution of most signs and symptoms within three months (Chiossi et al., 2006). Improvement in mental status is variable and up to 84% of patients may develop Korsakoff’s psychosis (Harrison et al., 2006; Morcos et al., 2004; Homewood and Bond, 1999). At a two-year follow-up visit, one patient, whose oculomotor and imaging studies had shown improvement, had persistent symptoms of severe cognitive deficits, vertigo and loss of sphincter control (Attard et al., 2006). Improvement of imaging studies may be seen up to four months of treatment with thiamine (Loh et al., 2004). Delay in treatment may result in irreversible neuronal death and possibly death of the patient (Gui et al., 2006).
Prevention
Clearly, one of the most important prevention strategies is physician and patient education in this area. In addition, there has been much debate over thiamine fortification of alcoholic beverages in order to prevent Wernicke’s encephalopathy in alcoholics, the most susceptible population,. In 1987, Australia’s Mental Health Committee recommended fortification of all Australian beer and flagon wine but this was never implemented. In most developed countries, bread (white flour) is enriched with thiamine to restore what is lost from the whole wheat in the process of milling. Australia adopted this plan in 1991, using the same level of enrichment as the United States (6.4 mg thiamine hydrochloride/Kg flour). The incidence of WE in the five years after the above implementation in Australia was 40% lower (perhaps fortuitously) than in the five year period prior to bread fortification. In addition, the post-mortem diagnosis of WE in Sydney, Australia has declined from 2.1% to 1.1% (Truswell, 2000).
Conclusion
There exists a great disparity between the number of patients diagnosed with WE while alive and the number of patients diagnosed post-mortem. This issue can be improved when a high index of suspicion for WE is employed for not only alcoholic patients but any patient with malnutrition or possible thiamine deficiency. Wernicke’s encephalopathy should be considered in the evaluation of any patient found to have one or more of the classic complaints, including confusion, ophthalmoplegia and ataxia, especially in the setting of malnutrition. In patients with acute altered mental status or coma, it is essential to treat empirically with thiamine, which is safe and inexpensive, even prior to the availability of neuroimaging results. We cannot overemphasize that imaging and laboratory data should not delay treatment with thiamine, which should be based initially on clinical assessment (i.e., symptoms/signs).
Since the preparation of this manuscript, a recent paper has reported total tau protein levels in cerebrospinal fluid were elevated in acute WE, but declined at follow up. This may suggest that neuronal cell death occurs transiently in acute WE.
References
Aikawa, H., Watanabe, I.S., Furuse, T., Iwasaki, Y., Satoyoshi, E., Sumi, T., and Moroji, T. (1984). Low energy level in thiamine deficient encephalopathy. J. Neuropathol. Exp. Neurol. 43:276–287.
Ambrose, M.L., Bowden, S.C., and Whelan, G. (2001). Thiamin treatment and working memory function of alcohol-dependent people: preliminary findings. Alcohol. Clin. Exp. Res. 25:112–116.
Antunez, E., Estruch, R., Cardenal, C., Nicholas, J.M., Fernandez-Sola, J., and Urbano-Marquez, A. (1998). Usefulness of CT and MR Imaging in the diagnosis of acute Wernicke’s Encephalopathy. Am. J. Roentgenol. 171:1131–1137.
Attard, O., Dietemann, J.L., Diemunsch, P., Pottecher, T., Meyer, A., and Calon, B.L. (2006). Wernicke encephalopathy: a complication of parenteral nutrition diagnosed by magnetic resonance imaging. Anesthesiology 105:847–848.
Bishai, D.M. and Bozzetti, L.P. (1986). Current progress toward the prevention of the Wernicke-Korsakoff syndrome. Alcohol Alcohol. 21:315–323.
Blansjaar, B.A., Zwang, R., and Blijenberg, B.G. (1991). No transketolase abnormalities in Wernicke-Korsakoff patients. J. Neurol. Sci. 106:88–90.
Blass, J.P. and Gibson, G.E. (1977). Abnormality of a thiamine-requiring enzyme in patients with Wernicke-Korsakoff syndrome. N. Engl. J. Med. 297:1367–1370.
Botez, M.I., Botez, T., Ross-Chouinard, A., and Lalonde, R. (1993). Thiamine and folate treatment of chronic epileptic patients: a controlled study with the Wechsler IQ scale. Epilepsy Res. 16:157–163.
Brady, J.A., Rock, C.L., and Horneffer, M.R. (1995). Thiamin status, diruretic medications, and the management of congestive heart failure. J. Am. Diet. Assoc. 95:541–544.
Breen, K.J., Buttigieg, R., Iossifidis, S., Lourensz, C., and Wood, B. (1985). Jejunal uptake of thiamin hydrocholoride in man: influence of alcoholism and alcohol. Am. J. Clin. Nutr. 42:121–126.
Butterworth, R.F., Kril, J.J., and Harper, C.G. (1993). Thiamine-dependent enzyme changes in the brains of alcoholics: relationship to the Wernicke-Korsakoff syndrome. Alcohol. Clin. Exp. Res. 17:1084–1088.
Caine, D., Halliday, G.M., Kril, J.J., and Harper, C.G. (1997). Operational criteria for the classification of chronic alcoholics identification of Wernicke’s encephalopathy. J. Neurol. Neurosurg. Psychiatr. 62:51–60.
Celik, Y. and Kaya, M. (2004). Brian SPECT findings in Wernicke’s encephalopathy. Neurol. Sci. 25:23–26.
Charness, M.E. (1999). Intracranial voyeurism: revealing the mammillary bodies in alcoholism. Alcohol. Clin. Exp. Res. 23:1941–1944.
Charness, M.E. (2006). Overview of the chronic neurologic complications of alcohol. www.uptodate.com, version 14.3; August 2006.
Chiossi, G., Neri, I., Cavazzuti, M., Basso, G., and Facchinetti, F. (2006). Hyperemesis gravidarum complicated by Wernicke encephalopathy: background, case report, and review of the literature. Obstet. Gynecol. Surv. 61:255–268.
Chung, T.I., Kim, J.S., Park, S.K., Kim, B.S., Ahn, K.J., and Yang, D.W. (2003). Diffusion weighted MR imaging of acute Wernicke’s encephalopathy. Eur. J. Radiol. 45:256–258.
Cirignotta, F., Manconi, M., Mondini, S., Buzzi, G., and Ambrosetto, P. (2000). Wernicke-Korsakoff encephalopathy and polyneuropathy after gastroplasty for morbid obesity. Arch. Neurol. 57:1356–1359.
D’Aprile, P., Tarantino, A., Santoro, N., and Carella, A. (2000). Wernicke’s encephalopathy induced by total parenteral nutrition in patient with acute leukaemia: unusual involvement of caudate nuclei and cerebral cortex on MRI. Neuroradiology. 42:781–783.
Day, E., Bentham, P., Callaghan, R., Kuruvilla, T., and George, S. (2004). Thiamine for Wernicke-Korsakoff Syndrome in people at risk from alcohol abuse. Cochrane Database Systematic Review, Issue 1, Art. No. CD004033.
DeBari, V.A., Frank, O., Baker, H., and Needle, M.A. (1984). Water soluble vitamins in granulocytes, erythrocytes, and plasma obtained from chronic hemodialysis patients. Am. J. Clin. Nutr. 39:410–415.
Desjardins, P. and Butterworth, R.F. (2005). Role of mitochondrial dysfunction and oxidative stress in the pathogenesis of selective neuronal loss in Wernicke’s encephalopathy. Mol. Neurobiol. 31:17–25.
Doherty, M.J., Watson, N.F., Uchino, K., Hallam, D.K., and Cramer, S.C. (2002). Diffusion abnormalities in patients with Wernicke encephalopathy. Neurology 58:655–657.
Dudeja, P.K., Tyagi, S., Gill, R., and Said, H.M. (2003). Evidence of a carrier-mediated mechanism for thiamine transport to human jejunal basolateral membrane vesicles. Dig. Dis. Sci. 48:109–115.
Dudeja, P.K., Tyagi, S., Kavilaveettil, R.J., Gill, R., and Said, H.M. (2001). Mechanism of thiamine uptake by human jejunal brush-border membrane vesicles. Am. J. Physiol. Cell Physiol. 281:C786–C792.
Foster, D., Falah, M., Kadom, N., and Mandler, R. (2005). Wernicke encephalopathy after bariatric surgery: losing more than just weight. Neurology 65:1987.
Giglioli, L., Salani, B., and Mannucci, M. (2004). Case Report: Mental confusion, diplopia, and inability to stand in an 82 year-old-man. (Ltr. to Editor). J. Am. Geriatr. Soc. 52:1218.
Gui, Q.P., Zhao, W.Q., and Wang, L.N. (2006). Wernicek’s encephalopathy in nonalcoholic patients: clinical and pathologic features of three cases and literature reviewed. Neuropathology 26:231–235.
Halavaara, J., Brander, A., Lyytinen, J., Setälä, K., and Kallela, M. (2003). Neuroradiology. 45:519–523.
Harper, C. (1983). The incidence of Wernicke’s encephalopathy in Australia — a neuropathological study of 131 cases. J. Neurol. Neurosurg. Psychiatr. 46:593–598.
Harper, C.G., Giles, M., and Finlay-Jones, R. (1986). Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J. Neurol. Neurosurg. Psychiatr. 49:341–345.
Harrison, R.A., Vu, T., and Hunter, A.J. (2006). Wernicke’s encephalopathy in a patient with schizophrenia. J. Gen. Intern. Med. 21:C8–C11.
Hazell, A.S., Todd, K.G., and Butterworth, R.F. (1998). Mechanisms of neuronal cell death in Wernicke’s encephalopathy. Metab. Brain Dis. 13:97–122.
Heier, M.S. and Dornish, J.M. (1989). Effect of the fluoropyrimidines 5-fluorourcil and Doxifluridine on cellular uptake of thiamin. Anticancer Res. 9:1073–1077.
Heinze, T. and Weber, W. (1990). Determination of thiamine (vitamin B1) in maternal blood during normal pregnancies and pregnancies with intrauterine growth retardation. Zeit. Ernahrungswissensch. 29:39–46.
Hochhalter, A.K. and Joseph, B. (2001). Differential outcomes training facilitates memory in people with Korsakoff and Prader-Willi syndromes. Integr. Physiol. Behav. Sci. 36:196–204.
Holowach, J., Kaufmann, F., Ikossi, M.G., Thomas, C. and McDougal, D.B. (1968). The effects of a thiamine antagonist, pyrithiamine, on levels of selected metabolic intermediates and activities of thiamine dependent enzymes in brain and liver. J. Neurochem. 15:621–631.
Holzbach, E. (1996). Thiamine absorption in alcoholic delirium patients. J. Stud. Alcohol 57:581–584.
Homewood, J., and Bond, N.W. (1999). Thiamin deficiency and Korsakoff’s syndrome: failure to find memory impairments following nonalcoholic Wernicke’s encephalopathy. Alcohol 19:75–84.
Hoyumpa, A.M., Jr. (1980). Mechanisms of thiamin deficiency in chronic alcoholism. Am. J. Clin. Nutr. 33:2750–2761.
Hung, S.C., Hung, S.H., Tarng, D.C., Yang, W.C., Chen, T.W., and Huang, T.P. (2001). Thiamine deficiency and unexplained encephalopathy in hemodialysis and peritoneal dialysis patients. Am. J. Kidney Dis. 38:941–947.
Kaufmann, A., Uhlhaas, S., Friedl, W., and Propping, P. (1987). Human erythrocyte transketolase: no evidence for variants. Clin. Chim. Acta. 162:215–219.
Kesler, A., Stolovitch, C., Hoffmann, C., Avni, I., and Morad, Y. (2005). Acute ophthalmoplegia and nystagmus in infants fed a thiamine-deficient formula: an epidemic of Wernicke encephalopathy. J. Neuroophthalmol. 25:169–172.
Koguchi, K., Nakatsuji, Y., Abe, K., and Sakoda, S. (2004). Wernicke’s encephalopathy after glucose infusion. Neurology 62:512.
Koontz, D.W., Fernandes-Filho, J.A.F., Sagar, S.M., and Rucker, J.C. (2004). Wernicke encephalopathy. Neurology 63:394.
Kumar, D., Nartsupha, C., and West, B.C. (2000). Unilateral internuclear ophthalmoplegia and recovery with thiamine in Wernicke syndrome. Am. J. Med. Sci. 320:278–280.
Kumar, D., Nartsupha, C., and West, B.C. (2008). Unilateral internuclear ophthalmoplegia and recovery with thiamine in Wernicke syndrome. Alcohol Clin Exp Res 32:1091–1095.
Lacasse, L. and Lun, C. (2004). Wernicke encephalopathy in a patient with T-Cell leukemia and severe malnutrition. Can. J. Neurol. Sci. 31:97–98.
Liu, Y.T., Fuh, J.L., Lirng, J.F., Li, A.F.Y., and Ho, D.M.T. (2006). Correlation of magnetic resonance images with neuropathology in acute Wernicke’s encephalopathy. Clin. Neurol. Neurosurg. 108:682–687.
Loh, Y., Watson, W.D., Verma, A., Chang, S.T., Stocker, D.J., and Labutta, R.J. (2004). Acute Wernicke’s encephalopathy following bariatric surgery: clinical course and MRI correlation. Obesity Surg. 14:129–132.
Lonsdale, D. (2006). A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. Evid. Based Complement. Alternat. Med. 3:49–59.
Martin, P.R., McCool, B.A., and Singleton, C.K. (1995). Molecular genetics of transketolase in the pathogenesis of the Wernicke-Korsakoff syndrome. Metab. Brain Dis. 10:45–55.
Mascalchi, M., Simonelli, P., Tessa, C., Giangaspero, F., Petruzzi, P., Bosincu, L., Conti, M., Sechi, G., and Salvi, F. (1999). Do acute lesions of Wernicke’s encephalopathy show contrast enhancement? Report of three cases and review of the literature. Neuroradiology 41:249–254.
Masud, T. (2002). Trace elements and vitamins in renal disease. In (W.E. Mitch, and Klahr, S. eds.), Handbook of Nutrition and the Kidney, 4th edn, Lippincott, Philadelphia, pp. 233–252.
McCandless, D.W. (1982). Energy metabolism in the lateral vestibular nucleus in pryithiamin-induced thiamin deficiency. Ann. N.Y. Acad. Sci. 378:355–364.
McCandless, D.W., Curley, A.D. and Cassidy, C.E. (1976). Thiamine deficiency and the pentose phosphate cycle in rats: intracerebral mechanisms. J. Nutr. 106:1144–1151.
McCandless, D.W. and Schenker, S. (1968). Encephalopathy of thiamine deficiency: studies of intracerebral mechanisms. J. Clin. Invest. 47:2268–2280.
McCandless, D.W. and Schwartzenburg, F.C., Jr. (1981). The effect of thiamine deficiency on energy metabolism in cells of the lateral vestibular nucleus. Res. Comm. Psychol. Psychiat. Behav. 6:183–190.
McEntee, W.J. (1997). Wernicke’s encephalopathy: an excitotoxicity hypothesis. Metab. Brain Dis. 12:183–192.
Merkin-Zaborsky, H., Ifergane, G., Frisher, S., Valdman, S., Herishanu, Y.L., and Wirguin, I. (2001). Thiamine-responsive acute neurological disorders in nonalcoholic patients. Eur. Neurol. 45:34–37.
Morcos, Z., Kerns, S.C., and Shapiro, B.E. (2004). Wernicke encephalopathy. Arch. Neurol. 61:775–776.
Nixon, P.F., Kaczmarek, M.J., Tate, J., Kerr, R.A., and Price, J. (1984). An erythrocyte transketolase isoenzyme pattern associated with the Wernicke-Korsakoff Syndrome. Eur. J. Clin. Invest. 14:278–281.
Ogershok, P.R., Rahman, A., Nestor, S., and Brick, J. (2002). Wernicke encephalopathy in nonalcoholic patients. Am. J. Med. Sci. 323:107–111.
Patrini, C., Perucca, E., Reggiani, C., and Rindi, G. (1993). Effects of phenytoin on the in vivo kinetics of thiamine and its phosphoesters in rat nervous tissues. Brain Res. 628:179–186.
Pazirandeh, S., Lo, C.W., and Burns, D.L. (2006). Overview of water-soluble vitamins. www.uptodate.com, version 14.3.
Peters, R.A. (1969). The biochemical lesion and its historical development. Br. Med. Bull. 25:223–226.
Piccoli, G.B., Burdese, M., Mezza, E., Soragna, G., Tattoli, F., Consiglio, V., Maddalena, E., Bergui, M., Scarzella, G., and Segoloni, G.P. (2006). The suddenly speechless florist on chronic dialysis: the unexpected threats of a flower shop? Nephrol. Dial. Transplant. 21:223–225.
Price, J. and Kerr, R. (1985). Some observations on the Wernicke Korsakoff syndrome in Australia. Br. J. Addict. 80:69–76.
Rathinam, M.L., Watts, L.T., Start, A.A., Mihimainathan, L., Stewart, J., Schenker, S., and Henderson, G.I. (2006). Astrocyte control of fetal cortical neuron glutathione homeostasis: up-regulation by ethanol. J. Neurochem. 96:1289–1300.
Reidling, J.C., Subramanian, V.S., Dudeja, P.K., and Siad, H.M. (2002). Expression and promoter analysis of SLC19A2 in the human intestine. Biochim. Biophys. Acta 1561:180–187.
Reuler, J.B., Girard, D.E., and Cooney, T.G. (1985). Current Concepts: Wernicke’s encephalopathy. N. Eng. J. Med. 312:1035–1039.
Rindi, G. and Ventura, U. (1972). Thiamine intestinal transport. Physiol. Rev. 52:821–827.
Seligmann, H., Halkin, H., Rauchfleisch, S., Kaufmann, N., Motro, M., Vered, Z., and Ezra, D. (1991). Thiamine deficiency in patients with congestive heart failure receiving long-term furosemide therapy: a pilot study. Am. J. Med. 91:151–155.
Shogry, M.E. and Curnes, J.T. (1994). Mammillary body enhancement on MR as the on1ly sign of acute Wernicke encephalopathy. Am. J. Neuroradiol. 15:172–174.
Sivolap, Y.P. (2005). The current state of S.S. Korsakov’s concept of alcoholic polyneuritic psychosis. Neurosci. Behav. Physiol. 35:977–982.
Squirrell, D. (2004). Wernicke encephalopathy. Arch. Ophthalmol. 122:418–419.
The Food Processor SQL. (2006). www.esha.com/foodprocessorsql; version 9.9.0
Thomson, A., Baker, H., and Leevy, C.M. (1968). Thiamine absorption in alcoholism. Am. J. Clin. Nutr. 21:537.
Tomasulo, P.A., Kater, R.M.H., and Iber, F.L. (1968). Impairment of thiamine absorption in alcoholism. Am. J. Clin. Nutr. 21:1340–1344.
Truswell, A.S. (2000). Australian experience with the Wernicke-Korsakoff syndrome. Addiction 95:829–832.
Victor, M., Adams, R.D., and Collins, G.H., eds. (1989). Course in the illness. In: The Wernicke-Korsakoff Syndrome and Related Neurologic Disorders Due to Alcoholism and Malnutrition, Davis, Philadelphia, pp. 31–38.
Vorhees, C.V., Schmidt, D.E., Barrett, R.J., and Schenker, S. (1977). Effect of thiamin deficiency on acetylcholine levels and utilization in vivo in rat brain. J. Nutr. 107:1902–1908.
Weidauer, S., Rosler, A., Zanella, F.E., and Lanfermann, H. (2004). Diffusion-weighted imaging in Wernicke encephalopathy associated with stomach cancer: case report and review of the literature. Eur. Neurol. 51:55–57.
White, M.L., Zhang, Y., Andrew, L.G., and Hadley, W.L. (2005). MR imaging with diffusion-weighted imaging in acute and chronic Wernicke encephalopathy. Am. J. Neuroradiol. 26:2306–2310.
Wood, B., Currie, J., and Breen, K. (1984). Wernicke’s encephalopathy in a metropolitan hospital. A prospective study of incidence, characteristics and outcome. Med. J. Aust. 144:12–16.
Worden, R.W. and Allen, H.M. (2006). Wernicke’s encephalopathy after gastric bypass that masqueraded as acute psychosis: a case report. Curr. Surg. 63:114–116.
Acknowledgement
We thank Amy L. Wyatt, R.D., L.D., affiliated with the University Hospital System in San Antonio, TX, for information provided on the content of thiamine in foods, as listed in Table 14.6. We gratefully acknowledge the excellent reviews by Drs. Roger Butterworth and colleagues (Hazell, 1998; Desjardins and Butterworth, 2005) and W.J. McEntee (1997), which served as valuable source material for this review.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Kashi, M.R., Henderson, G.I., Schenker, S. (2009). Wernicke’s Encephalopathy. In: McCandless, D. (eds) Metabolic Encephalopathy. Springer, New York, NY. https://doi.org/10.1007/978-0-387-79112-8_14
Download citation
DOI: https://doi.org/10.1007/978-0-387-79112-8_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-0-387-79109-8
Online ISBN: 978-0-387-79112-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)