
Abstract:
Neurons are highly integrated both anatomically and metabolically with glial cells, and thus glial cells have a major influence on neuronal survival in ischemia and excitotoxicity. Of the three types of glia in the central nervous system—astrocytes, oligodendrocytes, and microglia—the role of astrocytes in excitotoxicity and ischemia has been best characterized. Under different settings, astrocytes can both limit or contribute to excitotoxic neuronal death. Astrocytes also influence oxidative neuronal injury and contribute to neuronal demise through secretion of nitric oxide and cytokines. Microglia, the resident macrophages of the CNS, can also have both deleterious and salutary effects on neuronal survival. Activated microglia can kill neurons, but on the other hand normal microglial function is probably required for brain remodeling after injury. Interactions between microglia and astrocytes engender an additional layer of complexity to these post‐ischemic processes.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

1 Introduction
Excitotoxic neuronal death is triggered by the sustained activation of glutamate receptors. During brain ischemia, glutamate receptor activation results from massive, sustained elevation in extracellular glutamate concentrations. Neurons are highly integrated both anatomically and metabolically with glial cells, and accordingly the glia have a major impact on neuronal survival in ischemia and excitotoxicity. Of the three types of glia in the central nervous system (CNS)—astrocytes, oligodendrocytes, and microglia—the role of astrocytes in excitotoxicity and ischemia has been best characterized. Under different settings, astrocytes can both limit or contribute to excitotoxic neuronal death. Microglia, the resident macrophages of the CNS, can also contribute to injury resulting from excitotoxicity and stroke, particularly in the postischemic period. The mechanisms by which these glial cell types influence ischemic neuronal survival will be discussed in turn.
2 Excitotoxicity in Stroke
Reductions in blood flow to less than roughly 20% of normal for more than a few minutes leads to a massive increase in extracellular glutamate (Benveniste et al., 1984). The extent and duration of this increase depend on the severity and duration of the ischemic insult. With ischemia of 10‐min duration, there is a transient rise of glutamate followed by a second higher peak of increase during reperfusion (Caragine et al., 1998). Studies with glutamate receptor antagonists have conclusively shown the importance of this cell death pathway in animal models of stroke (Simon et al., 1984; Meldrum et al., 1987; Choi, 1988; Ozyurt et al., 1988; Swan and Meldrum, 1990; Le Peillet et al., 1992). As might be expected, however, the efficacy of these agents is much decreased when administered at time points after glutamate elevations have occurred.
2.1 Mechanisms of Glutamate‐Induced Cell Death
Glutamate binding to neuronal N‐methyl‐d‐aspartate (NMDA) type glutamate receptors triggers an influx of Na+, K+, and Ca2+, which, if sustained, can lead to neuronal death. Key events in the excitotoxic cell death pathway are dysregulation of intracellular calcium homeostasis and the generation of reactive oxygen species. Reactive oxygen species are formed especially during reperfusion after ischemia and include superoxide, nitric oxide, peroxynitrite, and hydrogen peroxide. These agents can damage many critical cell components, but in particular they damage DNA. DNA damage, in turn, can lead to either apoptotic or necrotic cell death.
Glutamate is not the only factor determining NMDA receptor activity. NMDA receptor activity is positively modulated by glycine and d‐serine (White et al., 2000) and neuronal membrane depolarization (Novelli et al., 1988; Greene and Greenamyre, 1996). Conversely, NMDA receptor activity is attenuated by coactivation of neuronal GABA and purinergic receptors (Muir et al., 1996; Ortinau et al., 2003).
3 Astrocyte Modulation of Glutamate Neurotoxicity
3.1 Astrocyte Glutamate Uptake
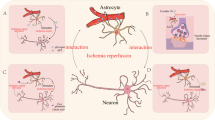
Glutamate uptake is the most well‐characterized mechanism by which astrocytes influence neuronal survival during ischemia, and this topic has been the subject of recent thorough reviews (Schousboe et al., 1997; Robinson, 1998; Vandenberg, 1998; Anderson and Swanson, 2000; Danbolt, 2001). Clearance of glutamate from the extracellular space is accomplished primarily by Na+‐dependent transporters localized on astrocytes (Figure 9-1 ). There are two main subtypes of glutamate transporters expressed by astrocytes. These were originally cloned from the rat brain and termed GLAST; glial glutamate and aspartate transporter and GLT‐1; glial glutamate transporter. Human homologs of these transporters are termed EAAT1 and EAAT2; excitatory amino and transporter 1 and 2 respectively. In cortical cultures, neuronal vulnerability to glutamate is 100‐fold greater in astrocyte‐poor cultures than in cultures with abundant astrocytes (Rosenberg and Aizenman, 1989), suggesting that uptake by astrocytes can be a limiting factor in glutamate neurotoxicity. The dominant role of astrocyte transporters is further supported by studies using genetic downregulation of transporter subtypes. Antisense “knockdown” of GLAST or GLT‐1, but not the neuronal subtype EAAC1, produces excitotoxic neurodegeneration and increased susceptibility to seizures and injury (Rothstein et al., 1996; Tanaka et al., 1997; Watase et al., 1998). Likewise, genetic downregulation of the glial glutamate transporter GLT‐1, but not the neuronal glutamate transporter EAAC1, was shown to exacerbate ischemic neuronal damage in the rat brain (Rao et al., 2001), thereby establishing the importance of astrocyte uptake for neuronal survival during ischemia.

Astrocyte modulation of glutamate excitotoxicity. Neuronal glutamate (Glu) is formed chiefly from astrocyte‐derived glutamine (Gln). Gln is formed in astrocytes from glutamate by glutamine synthase and then released from astrocytes via system N transporters (SN1). Gln is taken up by neurons through Na+‐coupled amino acid transporter (SAT). In neurons, Gln is converted back to Glu by glutaminase. Neuronal Glu is also formed from astrocyte‐derived α‐ketoisocaproate (KIC). KIC is formed in astrocytes by the action of branched amino acid (BCAA) transaminase, whereby leucine (Leu) and α‐ketoglutarate (αKG) are converted to KIC and glutamate. KIC is used by neurons for the reverse reaction as an anaplerotic reaction to replenish tricarboxylic acid (TCA) cycle intermediates that are expended for amino acid neurotransmitter release. Astrocyte uptake of glutamate (Glu) by glutamate transporters (GluT) limits glutamate access to neuronal NMDA receptors (NMDAR). The transporters link glutamate transport to the cotransport of 3 Na+ and 1 H+ and the counter‐transport of 1 K+ ion. Uptake is thus metabolically coupled to the astrocyte plasma membrane Na+/K+‐ATPase. NMDA receptor activity is positively modulated by d‐serine produced by l‐serine racemase in astrocytes, and by glycine, which is actively transported by astrocytes (GlyT). Glutamate binding to NMDA receptor is also positively modulated by neuronal membrane depolarization induced by glutamate activation of AMPA/kainite receptors (AMPA/KAR) and by increased extracellular K+. Astrocyte uptake of glycine, GABA, and glutamate (1) can also proceed in the reversal direction during astrocyte energy failure. Astrocytes can also release glutamate through volume‐sensitive organic anion channels (VSOACs) (2), by Ca2+‐dependent, vesicular release in response to various signaling molecules (3), by ATP‐gated P2x7 receptor channels (4), and by unpaired connexin‐43 hemichannels (Cx‐43) (5). Puryvate (Pyr); alanine (Ala); phosphate (Pi)
Glutamate uptake is energetically costly, requiring roughly 1 ATP per glutamate molecule transported. Given the high energetic cost of glutamate uptake, it is not surprising that complete ischemia leads to abrupt cessation of glutamate uptake and a massive increase in extracellular glutamate concentrations (Benveniste et al., 1984; Choi, 1988; Swanson et al., 1994). During incomplete ischemia, however, continued supply of glucose can fuel continued ATP production by glycolysis, even in the absence of oxygen. Glycolytic ATP production appears sufficient to fuel glutamate uptake in astrocytes (Hakim, 1987; Swanson, 1992; Swanson et al., 1994). However, acidosis is a significant factor determining the ability of astrocytes to maintain ATP levels in the absence of oxygen delivery, and the ability of astrocytes to maintain glutamate uptake during hypoxia is markedly impaired by acidosis over the pH range resulting from ischemia (Swanson et al., 1995). Oxidative stress‐induced activation of poly(ADP‐ribose) polymerase‐1 (PARP‐1) in astrocytes may additionally compromise energy supply, in part, by limiting glycolysis (Ying et al., 2002). PARP‐1 activation has been shown to impair astrocyte glutamate uptake in vitro (Swanson et al., 2002), suggesting that this mechanism may also contribute to impaired glutamate uptake during and after ischemia.
Glutamate uptake capacity is dynamically regulated. Astrocytes in culture can rapidly increase glutamate transport capacity in response to elevated extracellular glutamate concentrations by a process mediated by glutamate‐induced translocation of glutamate transporters from intracellular space to plasma membranes (Duan et al., 1999; Robinson, 2002); however, this process is unlikely to be an important determinant of uptake capacity during ischemia in which energy production is the rate‐limiting factor. On the other hand, several factors can downregulate astrocyte glutamate transport, and these may further compromise glutamate uptake under ischemic conditions. Reactive oxygen species, particularly peroxynitrite, can reduce glutamate transporter function (Pogun et al., 1994; Volterra et al., 1994; Trotti et al., 1996; Trotti et al., 1998; Chen et al., 2000), possibly through interactions with specific protein sulfhydryl groups. Arachidonic acid (Trotti et al., 1995; Zerangue et al., 1995), endothelin‐1 (Leonova et al., 2001), and zinc (Vandenberg et al., 1998) are also released during ischemia and can downregulate or inhibit astrocyte glutamate uptake.
3.2 Astrocyte Glutamate Release
Glutamate transporters, like other transporters, are capable of moving substrates in both inward and outward directions (Szatkowski et al., 1990). In the setting of ATP depletion, as occurs during severe ischemia, the ATP‐supported membrane gradients collapse. This results not only in cessation of uptake but also efflux of glutamate via uptake reversal. ATP depletion has been shown to cause uptake reversal in primary astrocyte cultures (Gemba et al., 1994; Longuemare and Swanson, 1995), retina (Zeevalk et al., 1998), spinal cord and brain slice preparations (Li et al., 1999), and in the intact brain (Seki et al., 1999). These studies suggest that the rise in extracellular glutamate in the brain that occurs during energy failure may be due not only to failure of astrocyte glutamate uptake but also to glutamate efflux from astrocytes via reversal of the astrocyte glutamate transporters. However, there is controversy as to whether it is astrocytes or neurons that are the primary source of glutamate released by uptake reversal during ischemia. Studies in brain slice preparations suggest that glutamate efflux from neurons may be quantitatively more important, particularly at early time points (Rossi et al., 2000; Danbolt, 2001; Hamann et al., 2002). On the other hand, in vivo microdialysis studies have shown that dihydrokainate, which preferentially blocks the astrocyte transporter GLT‐1, significantly attenuates the rise of extracellular glutamate that occurs during brain ischemia (Seki et al., 1999; Dawson et al., 2000).
Glutamate efflux from astrocytes can also occur by several other routes. Astrocyte swelling induced by elevated extracellular potassium or other factors can induce glutamate efflux via volume‐sensitive anion channels (Kimelberg et al., 1990; Rutledge et al., 1998; Longuemare et al., 1999). Astrocytes can also release glutamate in response to bradykinin (Jeftinija et al., 1996) and prostaglandin E2 (Bezzi et al., 1998) in a manner that is sensitive to botulinum B neurotoxin. Glutamate release from astrocytes can be induced by extracellular ATP activation of astrocyte P2X7 receptor channels (Duan et al., 2003). A recent study also reports glutamate release through unpaired connexin‐43 hemichannels (Ye et al., 2003). However, the significance of glutamate release during ischemia by routes other than uptake reversal and volume‐sensitive anion channels remains to be established.
3.3 Astrocyte Support of Neuronal Glutamate Release
Glutamine released by astrocytes is the major precursor for neurotransmitter glutamate synthesis. Glutamate released at neuronal synapses is taken up by surrounding astrocytes (McLennan, 1976; Rothstein et al., 1996) and converted by glutamine synthetase to glutamine. Glutamine is in turn released from astrocytes for uptake by neurons (Bradford et al., 1978; Waniewski and Martin, 1986; Broer and Brookes, 2001; Chaudhry et al., 2002), where it is metabolized back to glutamate and packaged into synaptic vesicles. Pharmacological inhibition of glutamine synthetase reduces brain glutamine levels, reduces K+‐evoked glutamate release (Paulsen and Fonnum, 1989), and has been reported to reduce infarct size in a focal ischemia model of stroke (Swanson et al., 1990). The cycle of glutamate carbon through neurons and astrocytes is supplemented by a similar flux of branched‐chain amino acids in which transamination of leucine and α‐ketoglutarate in astrocytes produces glutamate and α‐ketoisocaproic acid (Yudkoff et al., 1996). α‐Ketoisocaproic acid is released to the extracellular fluid and may be taken up by neurons as a substrate for the reverse reaction.
3.4 Astrocyte Regulation of Extracellular GABA, Glycine, and d‐Serine
GABA is an inhibitory neurotransmitter that can attenuate the effects of glutamate receptor stimulation (Muir et al., 1996). GABA transporters are expressed by astrocytes, and pharmacological inhibitors that target astrocyte GABA uptake exhibit anticonvulsant activity (Schousboe, 2000). However, GABA transporters are also widely expressed by neurons (Palacin et al., 1998; Gadea and Lopez‐Colome, 2001b), and the specific contribution of astrocyte GABA uptake under normal or pathological conditions remains uncertain (Schousboe, 2000).
Glycine and d‐serine act as positive modulators of signal transduction at NMDA‐type glutamate receptors (Wroblewski et al., 1989), and these compounds can significantly influence glutamate excitotoxicity during ischemia (Foster et al., 1990). Astrocytes can both release and take up glycine from the extracellular space through the GlyT1 glycine transporter (Gadea and Lopez‐Colome, 2001a; Supplisson and Roux, 2002). Interestingly, the kinetic properties of GlyT1 differ from those of the dominant neuronal transporter, GlyT2 (Supplisson and Roux, 2002). Glycine uptake by GlyT2 is coupled to the cotransport of 3 Na+ and 1 Cl−, whereas uptake by astrocyte GlyT1 is coupled to 2 Na+ and 1 Cl−. This allows neurons to maintain a higher intracellular glycine concentration, a factor that may facilitate neuronal loading of glycine into synaptic vesicles. Higher neuronal intracellular glycine concentrations are also favored by a kinetic restraint on reverse operation of the GlyT2 transporters. By contrast, astrocyte GlyT1 transporters readily function in “reverse” mode, and because these transporters operate near equilibrium, one function of astrocyte GlyT1 transporters may be to allow efflux of glycine into the extracellular space. d‐Serine is generated from l‐serine by racemase that is exclusively expressed in the protoplasmic astrocytes that typically ensheath synapses (Wolosker et al., 1999; Snyder and Ferris, 2000). Activation of glutamate receptors on astrocytes leads to activation of l‐serine racemase and astrocyte release of d‐serine. Because d‐serine, like glycine, is a positive modulator of neuronal NMDA receptors, this process may contribute to excitotoxic neuronal death (Foster et al., 1990; Snyder and Ferris, 2000).
4 Astrocyte Influences on Downstream Events in Excitotoxic Cell Death
4.1 Astrocyte Influences on Oxidative Neuronal Injury
Reactive oxygen species are generated by several mechanisms during ischemia and ischemia‐reperfusion, and the resulting oxidative stress constitutes a major mechanism of ischemic neuronal injury (Chan, 2001). Glutathione (GSH) is the principal antioxidant in brain (Dringen, 2000), and brains depleted of glutathione are sensitized to ischemic injury (Mizui et al., 1992). Evidence suggests that astrocytes contain greater concentrations of glutathione and enzymes involved in glutathione metabolism than neurons (Slivka et al., 1987; Yudkoff et al., 1990; Makar et al., 1994; Wilson, 1997). Similarly, glucose flux through the pentose phosphate pathway in cultured astrocytes is twice that of cultured neurons and increases three times as much as in neurons during H2O2 exposure (Ben‐Yoseph et al., 1996). These factors suggest that astrocytes are more capable of scavenging reactive oxygen species than neurons and suggest that oxidant‐scavenging mechanisms in astrocytes may function to support neuronal survival. In support of this idea, neurons cultured in the presence of astrocytes are more resistant than neurons cultured alone to injury induced by nitric oxide, hydrogen peroxide, or superoxide (Desagher et al., 1996; Lucius and Sievers, 1996; Tanaka et al., 1999b; Xu et al., 1999). Moreover, astrocytes depleted of glutathione show reduced ability to protect neurons from oxidant injury (Drukarch et al., 1997; McNaught and Jenner, 1999; Chen et al., 2001). The effects of astrocyte glutathione on neuronal resistance to oxidative stress may be mediated, in part, by maintaining neuronal glutathione levels. Astrocytes contribute to neuronal glutathione content by an indirect route: glutathione is released by astrocytes and cleaved to the dipeptide CysGly (Dringen et al., 1999), which in turn is cleaved to free cysteine for uptake into neurons as a substrate for neuronal GSH synthesis (Dringen et al., 2001).
Like glutathione, ascorbate is an important antioxidant that is present in the brain at millimolar concentrations (Rice, 2000). Ascorbate can directly react with oxidants and can also serve as a cofactor for reducing (recycling) oxidized glutathione and α‐tocopherol. Evidence suggests an ascorbate cycle between neurons and astrocytes. Neurons release oxidized ascorbate (dehydroascorbate) for uptake by astrocytes, which then convert it to ascorbate and, in turn, release ascorbate for neuronal uptake (Siushansian and Wilson, 1995; Siushansian et al., 1997; Wilson, 1997; Daskalopoulos et al., 2002). Dehydroascorbate passes across the blood–brain barrier readily and is converted to ascorbate, presumably in astrocytes. Treatment with dehydroascorbate has been shown to attenuate ischemic brain injury, suggesting that this cycle is a significant aspect of astrocyte–neuron interaction during ischemia (Huang et al., 2001).
4.2 Erythropoietin Released by Astrocytes Blocks Ischemic Neuronal Death
The glycoprotein hormone erythropoietin is produced in the CNS as well as in the periphery and has substantial effects on neuronal survival after ischemia (Buemi et al., 2003). Evidence suggests that erythropoietin production in the CNS is localized primarily to astrocytes (Masuda et al., 1994; Nagai et al., 2001; Ruscher et al., 2002), although some studies also report expression by neurons (Bernaudin et al., 2000). Erythropoietin activation of neuronal erythropoietin receptors blocks cell death pathways triggered by excitotoxicity and by combined oxygen/glucose deprivation in neuronal cultures (Morishita et al., 1997; Sakanaka et al., 1998; Ruscher et al., 2002; Wen et al., 2002) and reduces neuronal death after ischemia in vivo (Sakanaka et al., 1998; Siren et al., 2001; Wen et al., 2002). Neurons stimulated by erythropoietin show inhibition of caspase‐9, increased expression of Bcl‐2 family antiapoptotic factors, increased antioxidant capacity, and several other changes that may favor survival during and after ischemia (Calapai et al., 2000; Kawakami et al., 2001; Ruscher et al., 2002; Wen et al., 2002). These effects are mediated by activation of Janus tyrosine kinase‐2 (JAK‐2) pathway, which in turn activates Akt (protein kinase B) and the transcription factors nuclear factor‐kappa B (NF‐κB) and STAT5 (Digicaylioglu and Lipton, 2001; Kawakami et al., 2001; Siren et al., 2001; Ruscher et al., 2002). Astrocyte production of erythropoietin is increased after ischemic stress as a result of hypoxia‐inducible factor‐1 (HIF‐1) activation, and postischemic erythropoietin production has been identified as a mediator of ischemic tolerance induced by sublethal cerebral ischemia (Ruscher et al., 2002; Prass et al., 2003).
5 The Astrocyte Inflammatory Response
The reaction of astrocytes to ischemia and other brain insults is similar to the inflammatory response of peripheral tissues. Within a few hours of virtually any type of brain injury, surviving astrocytes in the affected region begin to exhibit hypertrophy and proliferation (Ridet et al., 1997). This response, termed reactive astrogliosis, is fortified by migration of microglia and macrophages to the damaged area. Reactive astrocytes increase the expression of their structural proteins, GFAP and vimentin (Eng et al., 2000), as well as many other proteins. Cu/Zn superoxide dismutase, glutathione peroxidase, and metallothionein are increased in reactive astrocytes after ischemia (Liu et al., 1993; Takizawa et al., 1994; Neal et al., 1996; Campagne et al., 2000), indicating an enhanced capacity to neutralize reactive oxygen species. Similarly, astrocytes express the inducible form of heme oxygenase‐1 (HO‐1) in response to ischemia and other brain insults (Geddes et al., 1996; Takeda et al., 1996). HO‐1 is the first step of heme metabolism and may be important in preventing heme iron participation in metal‐catalyzed free radical production, particularly after conditions such as trauma or hemorrhagic stroke that liberate hemoglobin into the brain parenchyma. Other aspects of the astrocyte inflammatory response, such as nitric oxide and matrix metalloproteinase (MMP) expression, may be adaptive in settings such as infection but contribute to delayed neuronal death in settings such as stroke. (R)‐(‐)‐2‐propyloctanoic acid, an agent that suppresses the astrocyte inflammatory response, has been shown to reduce infarct size expansion when administered after cerebral ischemia (Matsui et al., 2002; Tateishi et al., 2002), although the detailed mechanism of this effect is not yet established.
5.1 Astrocyte Expression of iNOS and Cytokines
Expression of inducible nitric oxide synthase (iNOS) is a salient aspect of the astrocyte inflammatory response (Endoh et al., 1993). Astrocyte iNOS expression is detectable within a few hours of ischemia and is maximal within 2–3 days (Iadecola et al., 1995; Nakashima et al., 1995). Nitric oxide is a reactive oxygen species that can contribute to neuronal cell death by potentiating glutamate excitotoxicity (Hewett et al., 1994) and by several other mechanisms (Dawson and Dawson, 1998). Mice that are genetically deficient in iNOS exhibited smaller infarct size than control, wild‐type mice (Iadecola et al., 1997), although this effect may also be due to reduced iNOS expression in microglia and invading macrophages.
Astrocytes stimulated by ischemic injury also produce many cytokines, including tumor necrosis factors (TNF‐α, ‐β), interleukins (IL‐1, ‐6, ‐10), and interferons (IFN‐α, ‐β) (Feuerstein et al., 1998; Dong and Benveniste, 2001). The net effect of individual cytokines can be difficult to establish because the effects of many cytokines are strongly influenced by one another and because most cytokines have pleiotropic and cell‐type specific effects. For example, IL‐6 and TNF‐α have been shown to promote demyelination, thrombosis, leukocyte infiltration, and blood–brain barrier disruption (Feuerstein et al., 1998; Dong and Benveniste, 2001). On the other hand, IL‐6 has been shown to protect against ischemic and excitotoxic injury (Maeda et al., 1994; Ali et al., 2000), and hippocampal neurons treated with TNF‐α are less vulnerable to substrate deprivation and excitotoxicity (Cheng et al., 1994). The specific contribution of astrocyte cytokine release to these processes in vivo remains to be established.
A recently identified regulator of inflammatory processes is PARP‐1. PARP‐1 is known to function in DNA repair (Ha and Snyder, 2000) but also functions as a coactivator of NF‐κB (Kameoka et al., 2000; Chiarugi and Moskowitz, 2003), a transcription factor that plays an important role in the expression of inflammatory mediators. The formation of nuclear PARP‐1/NF‐κB complex has been shown to enhance DNA binding of NF‐κB and transcription of NF‐κB‐regulated genes. Whether the enzymatic activity of PARP‐1 is essential in this process remains unsettled (Chang and Alvarez‐Gonzalez, 2001; Ullrich et al., 2001; Chiarugi and Moskowitz, 2003). Downregulation of PARP‐1 causes a large reduction in brain infarct size after ischemia (Eliasson et al., 1997). It is likely that this large effect is due, in part, to an attenuated astrocyte inflammatory response, but there has been little study on this point.
5.2 Astrocyte Release of Matrix Metalloproteases
Cytokine and other proinflammatory stimuli of astrocytes in culture have been shown to induce activation of MMPs. MMPs are endopeptidases that are able to cleave protein components of extracellular matrix and are thus associated with tissue remodeling during developmental and pathological processes (Gottschall and Deb, 1996; Lee et al., 2003). Cytokines also modulate components of the plasminogen activator system, urokinase type plasminogen activator (u‐PA) and tissue‐specific plasminogen activator (t‐PA), which are released by astrocytes and other cell types. These agents promote formation of (active) plasmin from (inactive) plasminogen and thus mediate cleavage of pro‐MMPs into their active form (Faber‐Elman et al., 1995; Tang et al., 2000). MMPs have been shown to not only cleave cytokines like TNF‐α and IL‐1β from their immature pro forms into their active mature forms (Gearing et al., 1995; Schonbeck et al., 1998; English et al., 2000) but also counterbalance the IL‐1β activity by degrading the mature cytokine (Ito et al., 1996). Astrocytes release both MMP‐2 and ‐9 (Rosenberg et al., 2001). Cerebral ischemia has been shown to increase MMP‐2 activity in end‐feet of rat brain astrocytes at time points (3 h and 5–21 days) relevant to changes in blood–brain barrier permeability, suggesting that astrocyte MMP‐2 contributes both to blood–brain barrier opening and to later repair processes (Rosenberg et al., 2001). Astrocytes also express a tissue‐specific inhibitor of metalloproteinase‐1 (TIMP‐1) and inhibitors of plasminogen activators (Tang et al., 2000).
6 Astrocyte Trophic Factor Release
Astrocytes release a variety of trophic factors under normal conditions, and these are likely to influence neuronal survival and plasticity after brain injury. These trophic factors include nerve growth factor (NGF), basic fibroblast growth factor (bFGF), transforming growth factor beta (TGF‐β), platelet‐derived growth factor (PDGF), brain‐derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), and others (Ridet et al., 1997). Reactive astrocytes increase the expression of several of these, notably NGF, bFGF, BDNF, and neuregulins, which can stimulate neurite outgrowth (Schwartz and Nishiyama, 1994; Strauss et al., 1994; Mocchetti and Wrathall, 1995; Tokita et al., 2001). Reactive astrocytes also overexpress neuropilin‐1 and vascular endothelial growth factor (VEGF), which act in concert to promote angiogenesis after cerebral ischemia (Zhang and Chopp, 2002). Although these findings suggest that astrocytes may play an important role in functional recovery after stroke, there has not yet been direct confirmation of this.
The demonstration of accelerated neurogenesis after cerebral ischemia has led to the proposal that newly formed neurons may contribute to functional recovery after stroke (Liu et al., 1998; Jiang et al., 2001; Jin et al., 2001). Factors regulating neurogenesis remain poorly understood, but the essential role of astrocytes in neuronal differentiation during development suggests that astrocytes may similarly be important in regulating neurogenesis. In support of this idea, it has recently been shown that astrocytes induce neurogenesis from adult neural stem cells in culture (Song et al., 2002). Astrocytes are also likely to play a central role in angiogenesis after stroke. Astrocytes promote angiogenesis through the release of VEGF, angiopoietin‐1, and epoxyeicosatrienoic acids (Salhia et al., 2000; Acker et al., 2001; Chow et al., 2001; Zhang and Harder, 2002). Studies of brain development show a close relationship between astrocyte expression of angiogenic factors and microvessel development (Acker et al., 2001), and studies of brain injury confirm a temporal and spatial correlation between reactive astrocytosis, VEGF immunoreactivity, and microvascular density.
7 Microglia Effects on Ischemic Injury
Microglia are the resident macrophages of the CNS. They are derived from bone marrow precursors and have a slow interchange with the circulating macrophage pool (Perry and Gordon, 1988; Kaur et al., 2001). Activated microglia express surface markers similar to macrophages, but microglia have faster reaction time, better mobility, and greater proliferation capacity than macrophages (Giulian et al., 1995; Lyons et al., 2000). Microglia in the normal brain have a highly branched “ramified” morphology that can rapidly transform into activated, amoeboid morphology in response to stressors such as ischemia (Morioka et al., 1991; Kreutzberg, 1996; Raivich et al., 1998). Microglia undergo massive activation after stroke that remains detectable for several weeks (Jorgensen et al., 1993). Ischemia‐induced changes in extracellular ion composition are thought to be a trigger of microglial activation (Boucsein et al., 2000; Kato and Walz, 2000; Schipke et al., 2002). Microglial activation is also induced and modulated by glutamate binding to glutamate receptors (Gottlieb and Matute, 1997; Biber et al., 1999; Noda et al., 2000; Tikka and Koistinaho, 2001; Tikka et al., 2001; Taylor et al., 2003).
7.1 Neurotoxic Effects of Activated Microglia
In vivo and in vitro studies show that microglial activation precedes neuronal death (Morioka et al., 1991; Gehrmann et al., 1992; Tikka and Koistinaho, 2001), and the timing and location of microglial activation correlates with neuronal death (Morioka et al., 1991, 1992; Jorgensen et al., 1993; Zhang and Fedoroff, 1996). Inhibition of microglial activation improves neuronal survival after excitotoxic and ischemic insults (Rogove and Tsirka, 1998; Yrjanheikki et al., 1999). Activated microglia can promote neuronal death by releasing glutamate (Piani et al., 1991) and the NMDA receptor modulator d‐serine (Wu et al., 2004). Microglia also release cytokines, free radicals, and proteases that can affect neuronal viability directly or by modulating function of other brain cells (Table 9-1 ). For example, microglia show early expression of IL‐1β after excitotoxic or ischemic insults (Davies et al., 1999; Pearson et al., 1999). Inhibition of IL‐1β reduces and the administration of IL‐1β increases neuronal death in these settings (Loddick et al., 1996; Lawrence et al., 1998; Davies et al., 1999; Pearson et al., 1999). Activated microglia release nitric oxide through expression of iNOS and may also promote neuron death by release of MMP‐9 (Kauppinen and Swanson, 2004). Phagocytosis of neurons by activated microglia is commonly observed after brain ischemia, but this is likely a result rather than a cause of neuron death because microglia have not been observed to engulf healthy neurons (Kreutzberg, 1996; Streit, 2002). For example, spreading depression in the absence of ischemia does not lead into neuronal damage even though it does induce microglial activation (Gehrmann et al., 1993). Recent studies suggest that neurogenesis after ischemia is also impaired by activated microglia. Inhibition of microglial activation improves neurogenesis and facilitates survival of engrafted progenitor cells in rodent models (Ekdahl et al., 2003; Lacza et al., 2003; Sasaki et al., 2003).
7.2 Trophic and Neuroprotective Functions of Microglia
In addition to their cytotoxic actions, microglia can release a number of trophic factors that promote tissue repair and regeneration (Kreutzberg, 1996) (Table 9-1 ). These factors may be important determinants of outcome after brain ischemia. In cell culture studies, the presence of resting microglia enhances neuron survival and neurite outgrowth (Zhang and Fedoroff, 1996). Resting microglia also improve astrocyte survival in a cell culture model of ischemia through release of GDNF (Lee et al., 2004). In addition, microglia are capable of high‐affinity, high‐velocity glutamate uptake via their glutamate transporters (Kondo et al., 1995; Lopez‐Redondo et al., 2000; Nakajima et al., 2001; van Landeghem et al., 2001), but the biological significance of this during ischemic conditions remains to be established.
In vivo, secretion of the neurotrophic factors GDNF and BDNF by activated microglia promotes dopaminergic sprouting in a model of striatal injury (Batchelor et al., 1999). Similarly, transplantation studies show that engrafted microglial cells in injured spinal cord promote neurite growth, although the mechanism remains to be established (Rabchevsky and Streit, 1997). Microglia increase expression of neurotrophic factors such as BDNF, IL‐6, and TGF‐β after ischemia (Lehrmann et al., 1998; Suzuki et al., 1999; Lee et al., 2002); however, there is as yet no direct evidence that microglia promote neuronal survival in this setting.
7.3 Interactions Between Microglia and Astrocytes
Astrocytes and microglia are both capable of influencing one another (Kim, 1996). Microglia in the vicinity of an ischemic infarct secrete cytokines and chemokines that recruit additional microglia to the infarct site (Kreutzberg, 1996). Microglial activation also induces astrocytic proinflammatory response (Giulian and Baker, 1985; Kreutzberg, 1996; Tanaka et al., 1999a; Hailer et al., 2001), and astrocytic glutamate transport is affected by nitric oxide and TNF‐α released by activated microglia (Trotti et al., 1996; Gegelashvili and Schousboe, 1997; Bezzi et al., 2001). Conversely, astrocytes influence microglial activation and migration. Microglia in coculture with astrocytes or astrocyte‐conditioned medium maintain the resting, ramified morphology, but assume the activated, amoeboid morphology in the absence of astrocytes (Giulian et al., 1995; Kloss et al., 1997; Tanaka et al., 1999a). This affect is mediated by TGF‐β, M‐CSF, and GM‐CSF, as evidenced by the effects of neutralizing antibodies (Schilling et al., 2001). Astrocytes also stimulate microglial proliferation and migration after injury (O'Donnell et al., 2002; Zhang et al., 2003).
8 Summary
Astrocytes and microglia have dynamic interactions with neurons and with each other that influence outcome from stroke and other insults. These interactions involve energy metabolism, redox metabolism, neurotransmitter uptake and release, trophic factor support, and formation of toxic intermediaries. The complexity of these interactions precludes a simple dichotomous classification of glia as positive or negative modulators of brain injury. By the same token, however, these complexities suggest that specific interactions between these cell types may be targeted for therapeutic intervention.
Abbreviations
- AA:
-
arachidonic acid
- Ala:
-
alanine
- AMPA:
-
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid
- AMPA/KAR:
-
AMPA/kainate receptor
- APP:
-
amyloid precursor protein
- ATP:
-
adenosine triphosphate
- BCAA:
-
branched amino acid
- BDNF:
-
brain‐derived neurotrophic factor
- bFGF:
-
basic fibroblast growth factor
- CDNF:
-
ciliary neurotrophic factor
- COX‐2:
-
cyclooxygenase
- cPLA2:
-
cytoplasmic phospholipase A
- DNA:
-
deoxyribonucleic acid
- iNOS:
-
inducible nitric oxide synthase
- GDNF:
-
glial cell‐derived neurotrophic factor
- Glu:
-
glutamate
- Gln:
-
glutamine
- CNS:
-
central nervous system
- EAAT:
-
excitatory amino acid transporter
- GABA:
-
gamma‐aminobutyric acid
- GLAST:
-
glial glutamate and aspartate transporter
- GLT‐1:
-
glial glutamate transporter
- GlyT:
-
glycine transporter
- GM‐CSF:
-
granulocyte‐magrophage colony‐stimulating factor
- GSH:
-
glutathione
- HIF‐1:
-
hypoxia‐inducible factor‐1
- HO‐1:
-
heme oxygenase ‐1
- ICAM‐1:
-
intracellular adhesion molecule‐1
- ICE:
-
interleukin‐1 converting enzyme
- IGF‐I:
-
insulin growth factor I
- IL:
-
interleukin
- JAK‐2:
-
janus tyrosine kinase‐2
- αKG:
-
α‐ketoglutarate
- KIC:
-
α‐ketoisocaproate
- MAPK:
-
mitogen‐activated protein kinase
- M‐CSF:
-
macrophage colony‐stimulating factor
- MHC:
-
major histocompatibility complex
- MIP‐1:
-
macrophage inflammatory protein‐1
- MMP:
-
matrix metalloproteases
- NF‐κB:
-
nuclear factor‐kappaB
- NGF:
-
nerve growth factor
- NMDA:
-
N‐methyl‐D‐aspartate
- NT‐3:
-
neurotrophin‐3
- PA:
-
plasminogen activator
- PARP‐1:
-
poly(ADP‐ribose) polymerase‐1
- PDGF:
-
platelet derived growth factor
- PG:
-
prostaglandin
- PKC:
-
protein kinase C
- PYK2:
-
proline‐rich protein tyrosine kinase
- Pyr:
-
puryvate
- RANTES:
-
regulation on activation normal T‐cell expressed and secreted
- SAT:
-
sodium‐coupled amino acid transporter
- TCA:
-
tricarboxylic acid cycle
- TGF:
-
transforming growth factor
- TIMP:
-
tissue‐specific inhibitor of metalloproteinase
- TNF:
-
tumor necrosis factor
- t‐PA:
-
tissue‐specific plasminogen activator
- u‐PA:
-
urokinase‐plasminogen activator
- VEGF:
-
vascular endothelial growth factor
- VSOAC:
-
volume‐sensitive organic anion channel
References
Acker T, Beck H, Plate KH. 2001. Cell type specific expression of vascular endothelial growth factor and angiopoietin‐1 and ‐2 suggests an important role of astrocytes in cerebellar vascularization. Mech Dev 108: 45–57.
Ali C, Nicole O, Docagne F, Lesne S, Mac Kenzie ET, et al. 2000. Ischemia‐induced interleukin‐6 as a potential endogenous neuroprotective cytokine against NMDA receptor‐mediated excitotoxicity in the brain. J Cereb Blood Flow Metab 20: 956–966.
Aloisi F, Penna G, Cerase J, Menendez Iglesias B, Adorini L. 1997. IL‐12 production by central nervous system microglia is inhibited by astrocytes. J Immunol 159: 1604–1612.
Anderson CM, Swanson RA. 2000. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32: 1–14.
Banati RB, Gehrmann J, Czech C, Monning U, Jones LL, et al. 1993. Early and rapid de novo synthesis of Alzheimer beta A4‐amyloid precursor protein (APP) in activated microglia. Glia 9: 199–210.
Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, et al. 1999. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain‐derived neurotrophic factor and glial cell line‐derived neurotrophic factor. J Neurosci 19: 1708–1716.
Benveniste H, Drejer J, Schousboe A, Diemer NH. 1984. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem 43: 1369–1374.
Ben‐Yoseph O, Boxer PA, Ross BD. 1996. Assessment of the role of the glutathione and pentose phosphate pathways in the protection of primary cerebrocortical cultures from oxidative stress. J Neurochem 66: 2329–2337.
Bernaudin M, Bellail A, Marti HH, Yvon A, Vivien D, et al. 2000. Neurons and astrocytes express EPO mRNA: oxygen‐sensing mechanisms that involve the redox‐state of the brain. Glia 30: 271–278.
Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, et al. 1998. Prostaglandins stimulate calcium‐dependent glutamate release in astrocytes. Nature 391: 281–285.
Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, et al. 2001. CXCR4‐activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 4: 702–710.
Bhat NR, Zhang P, Lee JC, Hogan EL. 1998. Extracellular signal‐regulated kinase and p38 subgroups of mitogen‐activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor‐alpha gene expression in endotoxin‐stimulated primary glial cultures. J Neurosci 18: 1633–1641.
Bhat RV, Di Rocco R, Marcy VR, Flood DG, Zhu Y, et al. 1996. Increased expression of IL‐1beta converting enzyme in hippocampus after ischemia: selective localization in microglia. J Neurosci 16: 4146–4154.
Biber K, Laurie DJ, Berthele A, Sommer B, Tolle TR, et al. 1999. Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem 72: 1671–1680.
Boucsein C, Kettenmann H, Nolte C. 2000. Electrophysiological properties of microglial cells in normal and pathologic rat brain slices. Eur J Neurosci 12: 2049–2058.
Bradford HF, Ward HK, Thomas AJ. 1978. Glutamine: a major substrate for nerve endings. J Neurochem 30: 1453–1459.
Broer S, Brookes N. 2001. Transfer of glutamine between astrocytes and neurons. J Neurochem 77: 705–719.
Buemi M, Cavallaro E, Floccari F, Sturiale A, Aloisi C, et al. 2003. The pleiotropic effects of erythropoietin in the central nervous system. J Neuropathol Exp Neurol 62: 228–236.
Calapai G, Marciano MC, Corica F, Allegra A, Parisi A, et al. 2000. Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur J Pharmacol 401: 349–356.
Campagne MV, Thibodeaux H, van Bruggen N, Cairns B, Lowe DG. 2000. Increased binding activity at an antioxidant‐responsive element in the metallothionein‐1 promoter and rapid induction of metallothionein‐1 and ‐2 in response to cerebral ischemia and reperfusion. J Neurosci 20: 5200–5207.
Caragine LP, Park HK, Diaz FG, Phillis JW. 1998. Real‐time measurement of ischemia‐evoked glutamate release in the cerebral cortex of four and eleven vessel rat occlusion models. Brain Res 793: 255–264.
Chan PH. 2001. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21: 2–14.
Chang WJ, Alvarez‐Gonzalez R. 2001. The sequence‐specific DNA binding of NF‐kappa B is reversibly regulated by the automodification reaction of poly (ADP‐ribose) polymerase 1. J Biol Chem 276: 47664–47670.
Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. 1992. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol 149: 2736–2741.
Chao CC, Hu S, Peterson PK. 1995a. Modulation of human microglial cell superoxide production by cytokines. J Leukoc Biol 58: 65–70.
Chao CC, Hu S, Sheng WS, Peterson PK. 1995b. Tumor necrosis factor‐alpha production by human fetal microglial cells: regulation by other cytokines. Dev Neurosci 7: 97–105.
Chaudhry FA, Schmitz D, Reimer RJ, Larsson P, Gray AT, et al. 2002. Glutamine uptake by neurons: interaction of protons with system a transporters. J Neurosci 22: 62–72.
Chen Y, Vartiainen NE, Ying W, Chan PH, Koistinaho J, et al. 2001. Astrocytes protect neurons from nitric oxide toxicity by a glutathione‐dependent mechanism. J Neurochem 77: 1601–1610.
Chen Y, Ying W, Simma V, Copin JC, Chan PH, et al. 2000. Overexpression of Cu,Zn superoxide dismutase attenuates oxidative inhibition of astrocyte glutamate uptake. J Neurochem 75: 939–945.
Cheng B, Christakos S, Mattson MP. 1994. Tumor necrosis factors protect neurons against metabolic‐excitotoxic insults and promote maintenance of calcium homeostasis. Neuron 12: 139–153.
Chiarugi A, Moskowitz MA. 2003. Poly(ADP‐ribose) polymerase‐1 activity promotes NF‐kappaB‐driven transcription and microglial activation: implication for neurode-generative disorders. J Neurochem 85: 306–317.
Choi DW. 1988. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1: 623–634.
Chow J, Ogunshola O, Fan SY, Li Y, Ment LR, et al. 2001. Astrocyte‐derived VEGF mediates survival and tube stabilization of hypoxic brain microvascular endothelial cells in vitro. Brain Res Dev Brain Res 130: 123–132.
Clemens JA, Stephenson DT, Smalstig EB, Roberts EF, Johnstone EM, et al. 1996. Reactive glia express cytosolic phospholipase A2 after transient global forebrain ischemia in the rat. Stroke 27: 527–535.
Colton C, Wilt S, Gilbert D, Chernyshev O, Snell J, et al. 1996. Species differences in the generation of reactive oxygen species by microglia. Mol Chem Neuropathol 28: 15–20.
Danbolt NC. 2001. Glutamate uptake. Prog Neurobiol 65: 1–105.
Daskalopoulos R, Korcok J, Tao L, Wilson JX. 2002. Accumulation of intracellular ascorbate from dehydroascorbic acid by astrocytes is decreased after oxidative stress and restored by propofol. Glia 39: 124–132.
Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, et al. 1999. The progression and topographic distribution of interleukin‐1beta expression after permanent middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab 19: 87–98.
Dawson LA, Djali S, Gonzales C, Vinegra MA, Zaleska MM. 2000. Characterization of transient focal ischemia‐induced increases in extracellular glutamate and aspartate in spontaneously hypertensive rats. Brain Res Bull 53: 767–776.
Dawson VL, Dawson TM. 1998. Nitric oxide in neurodegeneration. Prog Brain Res 118: 215–229.
Desagher S, Glowinski J, Premont J. 1996. Astrocytes protect neurons from hydrogen peroxide toxicity. J Neurosci 16: 2553–2562.
Digicaylioglu M, Lipton SA. 2001. Erythropoietin‐mediated neuroprotection involves cross‐talk between Jak2 and NF‐kappaB signalling cascades. Nature 412: 641–647.
Dong Y, Benveniste EN. 2001. Immune function of astrocytes. Glia 36: 180–190.
Dringen R. 2000. Metabolism and functions of glutathione in brain. Prog Neurobiol 62: 649–671.
Dringen R, Gutterer JM, Gros C, Hirrlinger J. 2001. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J Neurosci Res 66: 1003–1008.
Dringen R, Pfeiffer B, Hamprecht B. 1999. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci 19: 562–569.
Drukarch B, Schepens E, Jongenelen CA, Stoof JC, Langeveld CH. 1997. Astrocyte‐mediated enhancement of neuronal survival is abolished by glutathione deficiency. Brain Res 770: 123–130.
Duan S, Anderson CM, Keung EC, Chen Y, Swanson RA. 2003. P2X7 receptor‐mediated release of excitatory amino acids from astrocytes. J Neurosci 23: 1320–1328.
Duan S, Farrell K, Guenza JK, Stein BA, Swanson RA. 1999. Glutamate induces a rapid upregulation of astrocyte glutamate transport and redistribution of GLAST. J Neurosci 19: 10193–10200.
Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. 2003. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci USA 100: 13632–13637.
Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, et al. 1997. Poly(ADP‐ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3: 1089–1095.
Elkabes S, DiCicco‐Bloom EM, Black IB. 1996. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci 16: 2508–2521.
Endoh M, Maiese K, Pulsinelli WA, Wagner JA. 1993. Reactive astrocytes express NADPH diaphorase in vivo after transient ischemia. Neurosci Lett 154: 125–128.
Eng LF, Ghirnikar RS, Lee YL. 2000. Glial fibrillary acidic protein: GFAP‐thirty‐one years (1969–2000). Neurochem Res 25: 1439–1451.
English WR, Puente XS, Freije JM, Knauper V, Amour A, et al. 2000. Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor‐alpha convertase activity but does not activate pro‐MMP2. J Biol Chem 275: 14046–14055.
Faber‐Elman A, Miskin R, Schwartz M. 1995. Components of the plasminogen activator system in astrocytes are modulated by tumor necrosis factor‐alpha and interleukin‐1 beta through similar signal transduction pathways. J Neurochem 65: 1524–1535.
Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Collo G, et al. 1997. ATP‐mediated cytotoxicity in microglial cells. Neuropharmacology 36: 1295–1301.
Feuerstein GZ, Wang X, Barone FC. 1998. The role of cytokines in the neuropathology of stroke and neurotrauma. Neuroimmunomodulation 5: 143–159.
Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, et al. 1996. Cyclooxygenase‐2 expression in rat microglia is induced by adenosine A2a‐receptors. Glia 18: 152–160.
Foster AC, Willis CL, Tridgett R. 1990. Protection against N‐methyl‐d‐aspartate receptor‐mediated neuronal degeneration in rat brain by 7‐chlorokynurenate and 3‐amino‐1‐hydroxypyrrolid‐2‐one, antagonists at the allosteric site for glycine. Eur J Neurosci 2: 270–277.
Gadea A, Lopez‐Colome AM. 2001a. Glial transporters for glutamate, glycine, and GABA III Glycine transporters. J Neurosci Res 64: 218–222.
Gadea A, Lopez‐Colome AM. 2001b. Glial transporters for glutamate, glycine, and GABA: II GABA transporters. J Neurosci Res 63: 461–468.
Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements JM, et al. 1995. Matrix metalloproteinases and processing of pro‐TNF‐alpha. J Leukoc Biol 57: 774–777.
Gebicke‐Haerter PJ, Bauer J, Schobert A, Northoff H. 1989. Lipopolysaccharide‐free conditions in primary astrocyte cultures allow growth and isolation of microglial cells. J Neurosci 9: 183–194.
Geddes JW, Pettigrew LC, Holtz ML, Craddock SD, Maines MD. 1996. Permanent focal and transient global cerebral ischemia increase glial and neuronal expression of heme oxygenase‐1, but not heme oxygenase‐2, protein in rat brain. Neurosci Lett 210: 205–208.
Gegelashvili G, Schousboe A. 1997. High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol 52: 6–15.
Gehrmann J, Bonnekoh P, Miyazawa T, Hossmann KA, Kreutzberg GW. 1992. Immunocytochemical study of an early microglial activation in ischemia. J Cereb Blood Flow Metab 12: 257–269.
Gehrmann J, Mies G, Bonnekoh P, Banati R, Iijima T, et al. 1993. Microglial reaction in the rat cerebral cortex induced by cortical spreading depression. Brain Pathol 3: 11–17.
Gemba T, Oshima T, Ninomiya M. 1994. Glutamate efflux via the reversal of the sodium‐dependent glutamate transporter caused by glycolytic inhibition in rat cultured astrocytes. Neuroscience 63: 789–795.
Giulian D, Baker TJ. 1985. Peptides released by ameboid microglia regulate astroglial proliferation. J Cell Biol 101: 2411–2415.
Giulian D, Ingeman JE. 1988. Colony‐stimulating factors as promoters of ameboid microglia. J Neurosci 8: 4707–4717.
Giulian D, Li J, Bartel S, Broker J, Li X, et al. 1995. Cell surface morphology identifies microglia as a distinct class of mononuclear phagocyte. J Neurosci 15: 7712–7726.
Giulian D, Vaca K, Corpuz M. 1993. Brain glia release factors with opposing actions upon neuronal survival. J Neurosci 13: 29–37.
Gottlieb M, Matute C. 1997. Expression of ionotropic glutamate receptor subunits in glial cells of the hippocampal CA1 area following transient forebrain ischemia. J Cereb Blood Flow Metab 17: 290–300.
Gottschall PE, Deb S. 1996. Regulation of matrix metalloproteinase expressions in astrocytes, microglia and neurons. Neuroimmunomodulation 3: 69–75.
Gottschall PE, Yu X, Bing B. 1995. Increased production of gelatinase B (matrix metalloproteinase‐9) and interleukin‐6 by activated rat microglia in culture. J Neurosci Res 42: 335–342.
Greene JG, Greenamyre JT. 1996. Manipulation of membrane potential modulates malonate‐induced striatal excitotoxicity in vivo. J Neurochem 66: 637–643.
Ha HC, Snyder SH. 2000. Poly(ADP‐ribose) polymerase‐1 in the nervous system. Neurobiol Dis 7: 225–239.
Hailer NP, Wirjatijasa F, Roser N, Hischebeth GT, Korf HW, et al. 2001. Astrocytic factors protect neuronal integrity and reduce microglial activation in an in vitro model of N‐methyl‐d‐aspartate‐induced excitotoxic injury in organotypic hippocampal slice cultures. Eur J Neurosci 14: 315–326.
Hakim AM. 1987. The cerebral ischemic penumbra. Can J Neurol Sci 14: 557–559.
Hamann M, Rossi DJ, Marie H, Attwell D. 2002. Knocking out the glial glutamate transporter GLT‐1 reduces glutamate uptake but does not affect hippocampal glutamate dynamics in early simulated ischaemia. Eur J Neurosci 15: 308–314.
Hetier E, Ayala J, Bousseau A, Prochiantz A. 1991. Modulation of interleukin‐1 and tumor necrosis factor expression by beta‐adrenergic agonists in mouse ameboid microglial cells. Exp Brain Res 86: 407–413.
Hewett SJ, Csernansky CA, Choi DW. 1994. Selective potentiation of NMDA‐induced neuronal injury following induction of astrocytic Inos. Neuron 13: 487–494.
Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, et al. 1996. Human microglia convert l‐tryptophan into the neurotoxin quinolinic acid. Biochem J 320 (Pt. 2): 595–597.
Honda S, Nakajima K, Nakamura Y, Imai Y, Kohsaka S. 1999. Rat primary cultured microglia express glial cell line‐derived neurotrophic factor receptors. Neurosci Lett 275: 203–206.
Hu S, Chao CC, Ehrlich LC, Sheng WS, Sutton RL, et al. 1999. Inhibition of microglial cell RANTES production by IL‐10 and TGF‐beta. J Leukoc Biol 65: 815–821.
Huang J, Agus DB, Winfree CJ, Kiss S, Mack WJ, et al. 2001. Dehydroascorbic acid, a blood‐brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc Natl Acad Sci USA 98: 11720–11724.
Iadecola C, Xu X, Zhang F, el‐Fakahany EE, Ross ME. 1995. Marked induction of calcium‐independent nitric oxide synthase activity after focal cerebral ischemia. J Cereb Blood Flow Metab 15: 52–59.
Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. 1997. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci 17: 9157–9164.
Ito A, Mukaiyama A, Itoh Y, Nagase H, Thogersen IB, et al. 1996. Degradation of interleukin 1beta by matrix metalloproteinases. J Biol Chem 271: 14657–14660.
Janabi N, Hau I, Tardieu M. 1999. Negative feedback between prostaglandin and alpha‐ and beta‐chemokine synthesis in human microglial cells and astrocytes. J Immunol 162: 1701–1706.
Jeftinija SD, Jeftinija KV, Stefanovic G, Liu F. 1996. Neuroligand‐evoked calcium‐dependent release of excitatory amino acids from cultured astrocytes. J Neurochem 66: 676–684.
Jiang W, Gu W, Brannstrom T, Rosqvist R, Wester P. 2001. Cortical neurogenesis in adult rats after transient middle cerebral artery occlusion. Stroke 32: 1201–1207.
Jin K, Minami M, Lan JQ, Mao XO, Batteur S, et al. 2001. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci USA 98: 4710–4715.
Jorgensen MB, Finsen BR, Jensen MB, Castellano B, et al. 1993. Microglial and astroglial reactions to ischemic and kainic acid‐induced lesions of the adult rat hippocampus. Exp Neurol 120: 70–88.
Kameoka M, Ota K, Tetsuka T, Tanaka Y, Itaya A, et al. 2000. Evidence for regulation of NF‐kappaB by poly(ADP‐ribose) polymerase. Biochem J 346 (Pt. 3): 641–649.
Kato H, Walz W. 2000. The initiation of the microglial response. Brain Pathol 10: 137–143.
Kauppinen TM, Swanson RA. 2005. Poly(ADP‐ribose)polymerase‐1 promotes microglial activation, proliferation, and matrix metalloproteinase‐9‐mediated neuron death. J Immunol, 174: 2288–2296.
Kaur C, Hao AJ, Wu CH, Ling EA. 2001. Origin of microglia. Microsc Res Tech 54: 2–9.
Kawakami M, Sekiguchi M, Sato K, Kozaki S, Takahashi M. 2001. Erythropoietin receptor‐mediated inhibition of exocytotic glutamate release confers neuroprotection during chemical ischemia. J Biol Chem 276: 39469–39475.
Kim JS. 1996. Cytokines and adhesion molecules in stroke and related diseases. J Neurol Sci 137: 69–78.
Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. 1990. Swelling‐induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci 10: 1583–1591.
Kloss CU, Kreutzberg GW, Raivich G. 1997. Proliferation of ramified microglia on an astrocyte monolayer: characterization of stimulatory and inhibitory cytokines. J Neurosci Res 49: 248–254.
Kondo K, Hashimoto H, Kitanaka J, Sawada M, Suzumura A, et al. 1995. Expression of glutamate transporters in cultured glial cells. Neurosci Lett 188: 140–142.
Koponen S, Goldsteins G, Keinanen R, Koistinaho J. 2000. Induction of protein kinase Cdelta subspecies in neurons and microglia after transient global brain ischemia. J Cereb Blood Flow Metab 20: 93–102.
Kreutzberg GW. 1996. Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19: 312–318.
Lacza Z, Horvath EM, Komjati K, Hortobagyi T, Szabo C, et al. 2003. PARP inhibition improves the effectiveness of neural stem cell transplantation in experimental brain trauma. Int J Mol Med 12: 153–159.
Lawrence CB, Allan SM, Rothwell NJ. 1998. Interleukin‐1beta and the interleukin‐1 receptor antagonist act in the striatum to modify excitotoxic brain damage in the rat. Eur J Neurosci 10: 1188–1195.
Lee GA, Lin CH, Jiang HH, Chao HJ, Wu CL, et al. 2004. Microglia‐derived glial cell line‐derived neurotrophic factor could protect Sprague‐Dawley rat astrocyte from in vitro ischemia‐induced damage. Neurosci Lett 356: 111–114.
Lee TH, Kato H, Chen ST, Kogure K, Itoyama Y. 2002. Expression disparity of brain‐derived neurotrophic factor immunoreactivity and mRNA in ischemic hippocampal neurons. Neuroreport 13: 2271–2275.
Lee WJ, Shin CY, Yoo BK, Ryu JR, Choi EY, et al. 2003. Induction of matrix metalloproteinase‐9 (MMP‐9) in lipopolysaccharide‐stimulated primary astrocytes is mediated by extracellular signal‐regulated protein kinase 1/2 (Erk1/2). Glia 41: 15–24.
Lehrmann E, Kiefer R, Christensen T, Toyka KV, Zimmer J, et al. 1998. Microglia and macrophages are major sources of locally produced transforming growth factor‐beta1 after transient middle cerebral artery occlusion in rats. Glia 24: 437–448.
Leonova J, Thorlin T, Aberg ND, Eriksson PS, Ronnback L, et al. 2001. Endothelin‐1 decreases glutamate uptake in primary cultured rat astrocytes. Am J Physiol Cell Physiol 281: C1495–C1503.
Le Peillet E, Arvin B, Moncada C, Meldrum BS. 1992. The non‐NMDA antagonists, NBQX and GYKI 52466, protect against cortical and striatal cell loss following transient global ischaemia in the rat. Brain Res 571: 115–120.
Li S, Mealing GA, Morley P, Stys PK. 1999. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+‐dependent glutamate transport. J Neurosci (Online) 19: RC16.
Liu J, Solway K, Messing RO, Sharp FR. 1998. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci 18: 7768–7778.
Liu XH, Kato H, Nakata N, Kogure K, Kato K. 1993. An immunohistochemical study of copper/zinc superoxide dismutase and manganese superoxide dismutase in rat hippocampus after transient cerebral ischemia. Brain Res 625: 29–37.
Loddick SA, Mac Kenzie A, Rothwell NJ. 1996. An ICE inhibitor, z‐VAD‐DCB attenuates ischaemic brain damage in the rat. Neuroreport 7: 1465–1468.
Longuemare MC, Swanson RA. 1995. Excitatory amino acid release from astrocytes during energy failure by reversal of sodium‐dependent uptake. J Neurosci Res 40: 379–386.
Longuemare MC, Rose CR, Farrell K, Ransom BR, Waxman SG, et al. 1999. K(+)‐induced reversal of astrocyte glutamate uptake is limited by compensatory changes in intracellular Na+. Neuroscience 93: 285–292.
Lopez‐Redondo F, Nakajima K, Honda S, Kohsaka S. 2000. Glutamate transporter GLT‐1 is highly expressed in activated microglia following facial nerve axotomy. Brain Res Mol Brain Res 76: 429–435.
Lucius R, Sievers J. 1996. Postnatal retinal ganglion cells in vitro: protection against reactive oxygen species (ROS)‐induced axonal degeneration by cocultured astrocytes. Brain Res 743: 56–62.
Lyons SA, Pastor A, Ohlemeyer C, Kann O, Wiegand F, et al. 2000. Distinct physiologic properties of microglia and blood‐borne cells in rat brain slices after permanent middle cerebral artery occlusion. J Cereb Blood Flow Metab 20: 1537–1549.
Maeda A, Sobel RA. 1996. Matrix metalloproteinases in the normal human central nervous system, microglial nodules, and multiple sclerosis lesions. J Neuropathol Exp Neurol 55: 300–309.
Maeda Y, Matsumoto M, Hori O, Kuwabara K, Ogawa S, et al. 1994. Hypoxia/reoxygenation‐mediated induction of astrocyte interleukin 6: a paracrine mechanism potentially enhancing neuron survival. J Exp Med 180: 2297–2308.
Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, et al. 1994. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem 62: 45–53.
Mallat M, Houlgatte R, Brachet P, Prochiantz A. 1989. Lipopolysaccharide‐stimulated rat brain macrophages release NGF in vitro. Dev Biol 133: 309–311.
Masuda S, Okano M, Yamagishi K, Nagao M, Ueda M, et al. 1994. A novel site of erythropoietin production. Oxygen‐dependent production in cultured rat astrocytes. J Biol Chem 269: 19488–19493.
Matsui T, Mori T, Tateishi N, Kagamiishi Y, Satoh S, et al. 2002. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part I: Enhanced astrocytic synthesis of s‐100beta in the periinfarct area precedes delayed infarct expansion. J Cereb Blood Flow Metab 22: 711–722.
McLennan H. 1976. The autoradiographic localization of l‐[3h]glutamate in rat brain tissue. Brain Res 115: 139–144.
McNaught KS, Jenner P. 1999. Altered glial function causes neuronal death and increases neuronal susceptibility to 1‐methyl‐4‐phenylpyridinium‐ and 6‐hydroxydopamine‐induced toxicity in astrocytic/ventral mesencephalic co‐cultures. J Neurochem 73: 2469–2476.
Meldrum BS, Evans MC, Swan JH, Simon RP. 1987. Protection against hypoxic/ischaemic brain damage with excitatory amino acid antagonists. Med Biol 65: 153–157.
Minghetti L, Levi G. 1995. Induction of prostanoid biosynthesis by bacterial lipopolysaccharide and isoproterenol in rat microglial cultures. J Neurochem 65: 2690–2698.
Mizui T, Kinouchi H, Chan PH. 1992. Depletion of brain glutathione by buthionine sulfoximine enhances cerebral ischemic injury in rats. Am J Physiol 262: H313–H317.
Mocchetti I, Wrathall JR. 1995. Neurotrophic factors in central nervous system trauma. J Neurotrauma 12: 853–870.
Moller T, Hanisch UK, Ransom BR. 2000. Thrombin‐induced activation of cultured rodent microglia. J Neurochem 75: 1539–1547.
Morioka T, Kalehua AN, Streit WJ. 1991. The microglial reaction in the rat dorsal hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab 11: 966.
Morioka T, Kalehua AN, Streit WJ. 1992. Progressive expression of immunomolecules on microglial cells in rat dorsal hippocampus following transient forebrain ischemia. Acta Neuropathol (Berl) 83: 149–157.
Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R. 1997. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate‐induced neuronal death. Neuroscience 76: 105–116.
Muir JK, Lobner D, Monyer H, Choi DW. 1996. GABAA receptor activation attenuates excitotoxicity but exacerbates oxygen‐glucose deprivation‐induced neuronal injury in vitro. J Cereb Blood Flow Metab 16: 1211–1218.
Murphy GM Jr, Jia XC, Song Y, Ong E, Shrivastava R, et al. 1995. Macrophage inflammatory protein 1‐alpha mRNA expression in an immortalized microglial cell line and cortical astrocyte cultures. J Neurosci Res 40: 755–763.
Nagai A, Nakagawa E, Choi HB, Hatori K, Kobayashi S, et al. 2001. Erythropoietin and erythropoietin receptors in human CNS neurons, astrocytes, microglia, and oligodendrocytes grown in culture. J Neuropathol Exp Neurol 60: 386–392.
Nakajima K, Tohyama Y, Kohsaka S, Kurihara T. 2001. Ability of rat microglia to uptake extracellular glutamate. Neurosci Lett 307: 171–174.
Nakajima K, Tsuzaki N, Nagata K, Takemoto N, Kohsaka S. 1992a. Production and secretion of plasminogen in cultured rat brain microglia. FEBS Lett 308: 179–182.
Nakajima K, Tsuzaki N, Shimojo M, Hamanoue M, Kohsaka S. 1992b. Microglia isolated from rat brain secrete a urokinase‐type plasminogen activator. Brain Res 577: 285–292.
Nakashima MN, Yamashita K, Kataoka Y, Yamashita YS, Niwa M. 1995. Time course of nitric oxide synthase activity in neuronal, glial, and endothelial cells of rat striatum following focal cerebral ischemia. Cell Mol Neurobiol 15: 341–349.
Neal JW, Singhrao SK, Jasani B, Newman GR. 1996. Immunocytochemically detectable metallothionein is expressed by astrocytes in the ischaemic human brain. Neuropathol Appl Neurobiol 22: 243–247.
Noda M, Nakanishi H, Nabekura J, Akaike N. 2000. AMPA‐kainate subtypes of glutamate receptor in rat cerebral microglia. J Neurosci 20: 251–258.
Novelli A, Reilly JA, Lysko PG, Henneberry RC. 1988. Glutamate becomes neurotoxic via the N‐methyl‐d‐aspartate receptor when intracellular energy levels are reduced. Brain Res 451: 205–212.
O'Donnell SL, Frederick TJ, Krady JK, Vannucci SJ, Wood TL. 2002. IGF‐I and microglia/macrophage proliferation in the ischemic mouse brain. Glia 39: 85–97.
Ortinau S, Laube B, Zimmermann H. 2003. ATP inhibits NMDA receptors after heterologous expression and in cultured hippocampal neurons and attenuates NMDA‐mediated neurotoxicity. J Neurosci 23: 4996–5003.
Ozyurt E, Graham DI, Woodruff GN, McCulloch J. 1988. Protective effect of the glutamate antagonist, MK‐801 in focal cerebral ischemia in the cat. J Cereb Blood Flow Metab 8: 138–143.
Paakkari I, Lindsberg P. 1995. Nitric oxide in the central nervous system. Ann Med 27: 369–377.
Palacin M, Estevez R, Bertran J, Zorzano A. 1998. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev 78: 969–1054.
Paulsen RE, Fonnum F. 1989. Role of glial cells for the basal and Ca2+‐dependent K+‐evoked release of transmitter amino acids investigated by microdialysis. J Neurochem 52: 1823–1829.
Pearson VL, Rothwell NJ, Toulmond S. 1999. Excitotoxic brain damage in the rat induces interleukin‐1beta protein in microglia and astrocytes: correlation with the progression of cell death. Glia 25: 311–323.
Perry VH, Gordon S. 1987. Modulation of CD4 antigen on macrophages and microglia in rat brain. J Exp Med 166: 1138–1143.
Perry VH, Gordon S. 1988. Macrophages and microglia in the nervous system. Trends Neurosci 11: 273–277.
Piani D, Frei K, Do KQ, Cuenod M, Fontana A. 1991. Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett 133: 159–162.
Pogun S, Dawson V, Kuhar MJ. 1994. Nitric oxide inhibits 3H‐glutamate transport in synaptosomes. Synapse 18: 21–26.
Prass K, Scharff A, Ruscher K, Lowl D, Muselmann C, et al. 2003. Hypoxia‐induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 34: 1981–1986.
Rabchevsky AG, Streit WJ. 1997. Grafting of cultured microglial cells into the lesioned spinal cord of adult rats enhances neurite outgrowth. J Neurosci Res 47: 34–48.
Raivich G, Haas S, Werner A, Klein MA, Kloss C, et al. 1998. Regulation of MCSF receptors on microglia in the normal and injured mouse central nervous system: a quantitative immunofluorescence study using confocal laser microscopy. J Comp Neurol 395: 342–358.
Rao VL, Dogan A, Todd KG, Bowen KK, Kim BT, et al. 2001. Antisense knockdown of the glial glutamate transporter GLT‐1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia‐induced neuronal damage in rat brain. J Neurosci 21: 1876–1883.
Rice ME. 2000. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci 23: 209–216.
Ridet JL, Malhotra SK, Privat A, Gage FH. 1997. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 20: 570–577.
Robinson MB. 1998. The family of sodium‐dependent glutamate transporters: a focus on the GLT‐1/EAAT2 subtype. Neurochem Int 33: 479–491.
Robinson MB. 2002. Regulated trafficking of neurotransmitter transporters: common notes but different melodies. J Neurochem 80: 1–11.
Rogove AD, Tsirka SE. 1998. Neurotoxic responses by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol 8: 19–25.
Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, et al. 2001. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP‐9 linked to stromelysin‐1 and microglia in cell cultures. Brain Res 893: 104–112.
Rosenberg PA, Aizenman E. 1989. Hundred‐fold increase in neuronal vulnerability to glutamate toxicity in astrocyte‐poor cultures of rat cerebral cortex. Neurosci Lett 103: 162–168.
Rossi DJ, Oshima T, Attwell D. 2000. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403: 316–321.
Rothstein JD, Dykes‐Hoberg M, Pardo CA, Bristol LA, Jin L, et al. 1996. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686.
Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, et al. 2002. Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci 22: 10291–10301.
Rutledge EM, Aschner M, Kimelberg HK. 1998. Pharmacological characterization of swelling‐induced d‐[3H]aspartate release from primary astrocyte cultures. Am J Physiol 274: C1511–C1520.
Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, et al. 1998. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA 95: 4635–4640.
Salhia B, Angelov L, Roncari L, Wu X, Shannon P, et al. 2000. Expression of vascular endothelial growth factor by reactive astrocytes and associated neoangiogenesis. Brain Res 883: 87–97.
Sasaki T, Kitagawa K, Sugiura S, Omura‐Matsuoka E, Tanaka S, et al. 2003. Implication of cyclooxygenase‐2 on enhanced proliferation of neural progenitor cells in the adult mouse hippocampus after ischemia. J Neurosci Res 72: 461–471.
Sawada M, Suzumura A, Itoh Y, Marunouchi T. 1993. Production of interleukin‐5 by mouse astrocytes and microglia in culture. Neurosci Lett 155: 175–178.
Schilling T, Nitsch R, Heinemann U, Haas D, Eder C. 2001. Astrocyte‐released cytokines induce ramification and outward K+ channel expression in microglia via distinct signalling pathways. Eur J Neurosci 14: 463–473.
Schipke CG, Boucsein C, Ohlemeyer C, Kirchhoff F, Kettenmann H. 2002. Astrocyte Ca2+ waves trigger responses in microglial cells in brain slices. FASEB J 16: 255–257.
Schonbeck U, Mach F, Libby P. 1998. Generation of biologically active IL‐1 beta by matrix metalloproteinases: a novel caspase‐1‐independent pathway of IL‐1 beta processing. J Immunol 161: 3340–3346.
Schousboe A. 2000. Pharmacological and functional characterization of astrocytic GABA transport: a short review. Neurochem Res 25: 1241–1244.
Schousboe A, Sonnewald U, Civenni G, Gegelashvili G. 1997. Role of astrocytes in glutamate homeostasis. Implications for excitotoxicity. Adv Exp Med Biol 429: 195–206.
Schroeter M, Jander S, Huitinga I, Stoll G. 2001. CD8+ phagocytes in focal ischemia of the rat brain: predominant origin from hematogenous macrophages and targeting to areas of pannecrosis. Acta Neuropathol (Berl) 101: 440–448.
Schwab JM, Nguyen TD, Meyermann R, Schluesener HJ. 2001. Human focal cerebral infarctions induce differential lesional interleukin‐16 (IL‐16) expression confined to infiltrating granulocytes, CD8+ T‐lymphocytes and activated microglia/macrophages. J Neuroimmunol 114: 232–241.
Schwartz JP, Nishiyama N. 1994. Neurotrophic factor gene expression in astrocytes during development and following injury. Brain Res Bull 35: 403–407.
Seki Y, Feustel PJ, Keller RW Jr, Tranmer BI, Kimelberg HK. 1999. Inhibition of ischemia‐induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke 30: 433–440.
Sheng WS, Hu S, Kravitz FH, Peterson PK, Chao CC. 1995. Tumor necrosis factor alpha upregulates human microglial cell production of interleukin‐10 in vitro. Clin Diagn Lab Immunol 2: 604–608.
Shimojo M, Nakajima K, Takei N, Hamanoue M, Kohsaka S. 1991. Production of basic fibroblast growth factor in cultured rat brain microglia. Neurosci Lett 123: 229–231.
Simon RP, Swan JH, Griffiths T, Meldrum BS. 1984. Blockade of N‐methyl‐d‐aspartate receptors may protect against ischemic damage in the brain. Science 226: 850–852.
Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, et al. 2001. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA 98: 4044–4049.
Siushansian R, Wilson JX. 1995. Ascorbate transport and intracellular concentration in cerebral astrocytes. J Neurochem 65: 41–49.
Siushansian R, Tao L, Dixon SJ, Wilson JX. 1997. Cerebral astrocytes transport ascorbic acid and dehydroascorbic acid through distinct mechanisms regulated by cyclic. AMP J Neurochem 68: 2378–2385.
Slivka A, Mytilineou C, Cohen G. 1987. Histochemical evaluation of glutathione in brain. Brain Res 409: 275–284.
Snyder SH, Ferris CD. 2000. Novel neurotransmitters and their neuropsychiatric relevance. Am J Psychiatry 157: 1738–1751.
Song H, Stevens CF, Gage FH. 2002. Astroglia induce neurogenesis from adult neural stem cells. Nature 417: 39–44.
Strauss S, Otten U, Joggerst B, Pluss K, Volk B. 1994. Increased levels of nerve growth factor (NGF) protein and mRNA and reactive gliosis following kainic acid injection into the rat striatum. Neurosci Lett 168: 193–196.
Streit WJ. 2002. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40: 133–139.
Supplisson S, Roux MJ. 2002. Why glycine transporters have different stoichiometries. FEBS Lett 529: 93–101.
Suzuki S, Tanaka K, Nogawa S, Nagata E, Ito D, et al. 1999. Temporal profile and cellular localization of interleukin‐6 protein after focal cerebral ischemia in rats. J Cereb Blood Flow Metab 19: 1256–1262.
Swan JH, Meldrum BS. 1990. Protection by NMDA antagonists against selective cell loss following transient ischaemia. J Cereb Blood Flow Metab 10: 343–351.
Swanson RA. 1992. Astrocyte glutamate uptake during chemical hypoxia in vitro. Neurosci Lett 147: 143–146.
Swanson RA, Chen J, Graham SH. 1994. Glucose can fuel glutamate uptake in ischemic brain. J Cereb Blood Flow Metab 14: 1–6.
Swanson RA, Farrell K, Simon RP. 1995. Acidosis causes failure of astrocyte glutamate uptake during hypoxia. J Cereb Blood Flow Metab 15: 417–424.
Swanson RA, Shiraishi K, Morton MT, Sharp FR. 1990. Methionine sulfoximine reduces cortical infarct size in rats after middle cerebral artery occlusion. Stroke 21: 322–327.
Swanson RA, Suh SW, Anderson CM, Ying W, Sevigny MB, et al. 2002. Effects of PARP activation on astrocyte‐neuron interactions. Pharmacology of Cerebral Ischemia. Krieglstein J, Klumpp S, editors. Suttgart: Medpharm Scientific Publishers; pp. 89–95.
Szatkowski M, Barbour B, Attwell D. 1990. Non‐vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 348: 443–446.
Takeda A, Kimpara T, Onodera H, Itoyama Y, Shibahara S, et al. 1996. Regional difference in induction of heme oxygenase‐1 protein following rat transient forebrain ischemia. Neurosci Lett 205: 169–172.
Takizawa S, Matsushima K, Shinohara Y, Ogawa S, Komatsu N, Utsunomiya H, et al. 1994. Immunohistochemical localization of glutathione peroxidase in infarcted human brain. J Neurol Sci 122: 66–73.
Tanaka J, Toku K, Sakanaka M, Maeda N. 1999a. Morphological differentiation of microglial cells in culture: involvement of insoluble factors derived from astrocytes. Neurosci Res 34: 207–215.
Tanaka J, Toku K, Zhang B, Ishihara K, Sakanaka M, et al. 1999b. Astrocytes prevent neuronal death induced by reactive oxygen and nitrogen species. Glia 28: 85–96.
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, et al. 1997. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT‐1. Science 276: 1699–1702.
Tang H, Fu WY, Ip NY. 2000. Altered expression of tissue‐type plasminogen activator and type 1 inhibitor in astrocytes of mouse cortex following scratch injury in culture. Neurosci Lett 285: 143–146.
Tateishi N, Mori T, Kagamiishi Y, Satoh S, Katsube N, et al. 2002. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part II: Suppression of astrocytic activation by a novel agent (R)‐(‐)‐2‐propyloctanoic acid (ONO‐2506) leads to mitigation of delayed infarct expansion and early improvement of neurologic deficits. J Cereb Blood Flow Metab 22: 723–734.
Taylor DL, Diemel LT, Pocock JM. 2003. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J Neurosci 23: 2150–2160.
Thery C, Hetier E, Evrard C, Mallat M. 1990. Expression of macrophage colony‐stimulating factor gene in the mouse brain during development. J Neurosci Res 26: 129–133.
Tian D, Litvak V, Lev S. 2000. Cerebral ischemia and seizure induce tyrosine phosphorylation of PYK2 in neurons and microglial cells. J Neurosci 20: 6478–6487.
Tikka TM, Koistinaho JE. 2001. Minocycline provides neuroprotection against N‐methyl‐d‐aspartate neurotoxicity by inhibiting microglia. J Immunol 166: 7527–7533.
Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. 2001. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21: 2580–2588.
Tokita Y, Keino H, Matsui F, Aono S, Ishiguro H, et al. 2001. Regulation of neuregulin expression in the injured rat brain and cultured astrocytes. J Neurosci 21: 1257–1264.
Trotti D, Danbolt NC, Volterra A. 1998. Glutamate transporters are oxidant‐vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol Sci 19: 328–334.
Trotti D, Rossi D, Gjesdal O, Levy LM, Racagni G, et al. 1996. Peroxynitrite inhibits glutamate transporter subtypes. J Biol Chem 271: 5976–5979.
Trotti D, Volterra A, Lehre KP, Rossi D, Gjesdal O, et al. 1995. Arachidonic acid inhibits a purified and reconstituted glutamate transporter directly from the water phase and not via the phospholipid membrane. J Biol Chem 270: 9890–9895.
Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. 1997. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci 17: 543–552.
Ullrich O, Diestel A, Eyupoglu IY, Nitsch R. 2001. Regulation of microglial expression of integrins by poly(ADP‐ribose) polymerase‐1. Nat Cell Biol 3: 1035–1042.
Vandenberg RJ. 1998. Molecular pharmacology and physiology of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol 25: 393–400.
Vandenberg RJ, Mitrovic AD, Johnston GA. 1998. Molecular basis for differential inhibition of glutamate transporter subtypes by zinc ions. Mol Pharmacol 54: 189–196.
van Landeghem FK, Stover JF, Bechmann I, Bruck W, Unterberg A, et al. 2001. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia 35: 167–179.
Volterra A, Trotti D, Tromba C, Floridi S, Racagni G. 1994. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J Neurosci 14: 2924–2932.
Walker DG, Kim SU, McGeer PL. 1995. Complement and cytokine gene expression in cultured microglial derived from postmortem human brains. J Neurosci Res 40: 478–493.
Waniewski RA, Martin DL. 1986. Exogenous glutamate is metabolized to glutamine and exported by rat primary astrocyte cultures. J Neurochem 47: 304–313.
Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, et al. 1998. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci 10: 976–988.
Wen TC, Sadamoto Y, Tanaka J, Zhu PX, Nakata K, et al. 2002. Erythropoietin protects neurons against chemical hypoxia and cerebral ischemic injury by up‐regulating Bcl‐xL expression. J Neurosci Res 67: 795–803.
White BC, Sullivan JM, De Gracia DJ, O'Neil BJ, Neumar RW, et al. 2000. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 179: 1–33.
Wilson JX. 1997. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol 75: 1149–1163.
Wolosker H, Blackshaw S, Snyder SH. 1999. Serine racemase: a glial enzyme synthesizing d‐serine to regulate glutamate‐N‐methyl‐d‐aspartate neurotransmission. Proc Natl Acad Sci USA 96: 13409–13414.
Wroblewski JT, Fadda E, Mazzetta J, Lazarewicz JW, Costa E. 1989. Glycine and d‐serine act as positive modulators of signal transduction at N‐methyl‐d‐aspartate sensitive glutamate receptors in cultured cerebellar granule cells. Neuropharmacology 28: 447–452.
Wu SZ, Bodles AM, Porter MM, Griffin WS, Basile AS, et al. 2004. Induction of serine racemase expression and d‐serine release from microglia by amyloid beta‐peptide. J Neuroinflamm 1: 2.
Xu L, Lee JE, Giffard RG. 1999. Overexpression of bcl‐2, bcl‐XL or hsp70 in murine cortical astrocytes reduces injury of co‐cultured neurons. Neurosci Lett 277: 193–197.
Ye ZC, Wyeth MS, Baltan‐Tekkok S, Ransom BR. 2003. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci 23: 3588–3596.
Ying W, Chen Y, Alano CC, Swanson RA. 2002. Tricarboxylic acid cycle substrates prevent PARP‐mediated death of neurons and astrocytes. J Cereb Blood Flow Metab 22: 774–779.
Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, et al. 1999. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA 96: 13496–13500.
Yudkoff M, Daikhin Y, Grunstein L, Nissim I, Stern J, et al. 1996. Astrocyte leucine metabolism: significance of branched‐chain amino acid transamination. J Neurochem 66: 378–385.
Yudkoff M, Pleasure D, Cregar L, Lin ZP, Nissim I, et al. 1990. Glutathione turnover in cultured astrocytes: studies with [15N]glutamate. J Neurochem 55: 137–145.
Zeevalk GD, Davis N, Hyndman AG, Nicklas WJ. 1998. Origins of the extracellular glutamate released during total metabolic blockade in the immature retina. J Neurochem 71: 2373–2381.
Zerangue N, Arriza JL, Amara SG, Kavanaugh MP. 1995. Differential modulation of human glutamate transporter subtypes by arachidonic acid. J Biol Chem 270: 6433–6435.
Zhang C, Harder DR. 2002. Cerebral capillary endothelial cell mitogenesis and morphogenesis induced by astrocytic epoxyeicosatrienoic acid. Stroke 33: 2957–2964.
Zhang J, Geula C, Lu C, Koziel H, Hatcher LM, et al. 2003. Neurotrophins regulate proliferation and survival of two microglial cell lines in vitro. Exp Neurol 183: 469–481.
Zhang P, Hogan EL, Bhat NR. 1998. Activation of JNK/SAPK in primary glial cultures: II Differential activation of kinase isoforms corresponds to their differential expression. Neurochem Res 23: 219–225.
Zhang SC, Fedoroff S. 1996. Neuron‐microglia interactions in vitro. Acta Neuropathol (Berl) 91: 385–395.
Zhang Z, Chopp M. 2002. Vascular endothelial growth factor and angiopoietins in focal cerebral ischemia. Trends Cardiovasc Med 12: 62–66.
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2007 Springer Science+Business Media, LLC
About this entry
Cite this entry
Kauppinen, T.M., Swanson, R.A. (2007). The Role of Glia in Excitotoxicity and Stroke. In: Lajtha, A., Chan, P.H. (eds) Handbook of Neurochemistry and Molecular Neurobiology. Springer, New York, NY. https://doi.org/10.1007/978-0-387-30383-3_9
Download citation
DOI: https://doi.org/10.1007/978-0-387-30383-3_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-0-387-30352-9
Online ISBN: 978-0-387-30383-3
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences